

Abstract
Significance: The role of reactive oxygen species (ROS) in angiotensin II (AngII) induced endothelial dysfunction, cardiovascular and renal remodeling, inflammation, and fibrosis has been well documented. The molecular mechanisms of AngII pathophysiological activity involve the stimulation of NADPH oxidases, which produce superoxide and hydrogen peroxide. AngII also increases the production of mitochondrial ROS, while the inhibition of AngII improves mitochondrial function; however, the specific molecular mechanisms of the stimulation of mitochondrial ROS is not clear. Recent Advances: Interestingly, the overexpression of mitochondrial thioredoxin 2 or mitochondrial superoxide dismutase attenuates AngII-induced hypertension, which demonstrates the importance of mitochondrial ROS in AngII-mediated cardiovascular diseases. Critical Issues: Although mitochondrial ROS plays an important role in normal physiological cell signaling, AngII, high glucose, high fat, or hypoxia may cause the overproduction of mitochondrial ROS, leading to the feed-forward redox stimulation of NADPH oxidases. This vicious cycle may contribute to the development of pathological conditions and facilitate organ damage in hypertension, atherosclerosis, and diabetes. Future Directions: The development of antioxidant strategies specifically targeting mitochondria could be therapeutically beneficial in these disease conditions. Antioxid. Redox Signal. 19, 1085–1094.
Introduction
Angiotensin II (AngII) has been shown to participate in both physiological processes, such as sodium and water homeostasis and vascular contraction, and pathophysiological processes, including atherosclerosis and hypertension (25). AngII effects are mediated by complex signaling events that are initiated by G-protein-coupled receptor type 1 (AT1R) and type 2 (AT2R). AT2R is thought to counter-regulate the AT1R function. AT1R activates at least two discrete cell signaling axes (6), where one, represented by Erk1/2 and its downstream targets, is redox independent, and the another involves the activation of redox-dependent pathways, including the activation of Rac, c-Src, Akt, or AMPK. The activation of these redox-dependent pathways involves the stimulation of NADPH oxidases, which produce reactive oxygen species (ROS) such as superoxide (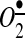 ) and hydrogen peroxide (H2O2). Interestingly, the expression of AT1R is redox dependent, and, therefore, the overproduction of ROS may result in the overstimulation of AT1R-mediated pathways and result in oxidative stress. These pathophysiological effects of AngII result in mitogenic, proinflammatory, and profibrotic actions, causing hypertrophic cell growth, cell senescence, endothelial dysfunction, and cardiovascular and renal remodeling, which, in turn, lead to organ damage, hypertension, atherosclerosis, pathological heart hypertrophy, kidney, and brain dysfunctions. Unfortunately, the AngII-induced ROS production is not completely understood and still requires extensive studies in order to provide pharmacological treatment for AngII-related diseases.
) and hydrogen peroxide (H2O2). Interestingly, the expression of AT1R is redox dependent, and, therefore, the overproduction of ROS may result in the overstimulation of AT1R-mediated pathways and result in oxidative stress. These pathophysiological effects of AngII result in mitogenic, proinflammatory, and profibrotic actions, causing hypertrophic cell growth, cell senescence, endothelial dysfunction, and cardiovascular and renal remodeling, which, in turn, lead to organ damage, hypertension, atherosclerosis, pathological heart hypertrophy, kidney, and brain dysfunctions. Unfortunately, the AngII-induced ROS production is not completely understood and still requires extensive studies in order to provide pharmacological treatment for AngII-related diseases.
Nonphagocytic NADPH oxidases provide major sources of ROS in the cardiovascular and renal systems (34). It is a complex that is composed of membrane-associated proteins, gp91phox (Nox2) and p22phox, which require cytosolic components, p47phox and p67phox, and the regulatory protein Rac (34). The discovery of Nox2 homolog, Nox1, was followed by other family members Nox3, Nox4, and Nox5 (Fig. 1). All cells in the cardiovascular system, including cardiomyocytes, endothelial cells, vascular smooth muscle cells, fibroblasts, and kidney tissue, express various catalytic subunits of NADPH oxidase (NOX) components. AngII strongly stimulates NOX activity and expression.
FIG. 1.

Upstream role of NADPH oxidase in the stimulation of mitochondrial reactive oxygen species (ROS) by angiotensin II (AngII). AngII binds to membrane G-protein coupled receptor type 1 (AT1R) and initiates signaling events, including PKC, c-Src activation that is required for superoxide (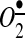 ) production by the catalytic subunit of NADPH oxidases NOX1 and NOX2. In parallel, the AngII-mediated increase of cytoplasm Ca2+ activates NOX5 to generate H2O2 and possibly
) production by the catalytic subunit of NADPH oxidases NOX1 and NOX2. In parallel, the AngII-mediated increase of cytoplasm Ca2+ activates NOX5 to generate H2O2 and possibly 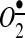 .
. 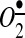 rapidly scavenges NO• to produce peroxynitrite (ONOO−). Hydrogen peroxide (H2O2) and ONOO− impact the mitochondrial matrix and stimulate the production of mitochondrial
rapidly scavenges NO• to produce peroxynitrite (ONOO−). Hydrogen peroxide (H2O2) and ONOO− impact the mitochondrial matrix and stimulate the production of mitochondrial 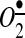 , which dismutates to H2O2 by mitochondrial manganese superoxide dismutase (SOD2). Mitochondrial H2O2 can pass through the mitochondrial membrane and provide the feed-forward stimulation of cytoplasmic NADPH oxidase. PKC, protein kinase C.
, which dismutates to H2O2 by mitochondrial manganese superoxide dismutase (SOD2). Mitochondrial H2O2 can pass through the mitochondrial membrane and provide the feed-forward stimulation of cytoplasmic NADPH oxidase. PKC, protein kinase C.
AngII activates NADPH oxidases by protein kinase C (PKC) and c-Src-dependent pathways (Fig. 1) (36). The initial activation of the AT1R leads to PKC-mediated phosphorylation of p47phox. This leads to c-Src activation and stimulation of the epidermal growth factor receptor, which evokes phosphatidylinositol 3-kinase-dependent production of phosphatidylinositol (3,4,5)-trisphosphate and, in turn, activates the Rac1 subunit of NADPH oxidase (55). Nox4 and Nox5 do not require p47phox or Rac1 subunits (43). Thus, in vascular cells, AngII primarily increases the activity of Nox1 or Nox2 (Fig. 1) (35). The activation of c-Src is redox sensitive and stimulated by H2O2 (60), which appears to represent a feed-forward mechanism whereby the H2O2-mediated activation of c-Src amplifies the NADPH oxidase activity of Nox1 and Nox2. It is important that Nox isoforms not only have different regulations and specific subcellular localization but also generate distinct ROS. For example, Nox4 is responsible for the basal production of H2O2, (19, 59), while Nox1 and Nox2 generates 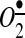 (19), and Nox5 produces H2O2 in a Ca2+-dependent fashion (24).
(19), and Nox5 produces H2O2 in a Ca2+-dependent fashion (24).
Stimulation of Mitochondrial ROS by NADPH Oxidases
We have previously reported that AngII increases the production of mitochondrial ROS and decreases mitochondrial membrane potential, respiratory control ratio, and low-molecular-weight thiol content. The depletion of p22phox, an essential component for NADPH oxidase function, led to a significant decrease in ROS production in mitochondria isolated from AngII-treated cells. The inhibition of NADPH oxidases by apocynin or selective PKC inhibitor chelerythrine completely prevented AngII-induced mitochondrial dysfunction and attenuated the production of mitochondrial ROS (Fig. 1) (21). Interestingly, treatment with the mitochondrial ATP-sensitive potassium channels (mitoKATP) blocker 5-hydroxydecanoic acid or glibenclamide prevented the increase in mitochondrial H2O2, attenuated the decrease in mitochondrial membrane potential, and preserved respiratory control ratio and low-molecular-weight thiol content induced by AngII (21). This can be explained by the recently reported redox sensitivity of mitoKATP (51). Taken together, these results suggest that the stimulation of mitochondrial ROS by AngII requires the full enzymatic activity of NADPH oxidases and may depend on the activation of mitoKATP.
It has been recently reported that Nox4 is expressed in the mitochondria of rat kidney cortex (5) and in the mitochondria of cardiac myocytes (33). Ago et al. reported a higher expression of Nox4 in the mitochondrial fraction of cardiac myocytes compared with the microsomal fraction (1). Confocal microscopy showed significant co-localization of Nox4 with mitochondrial F1F0-ATP synthase, as well as the p22phox subunit of NADPH oxidases. These studies, however, remain highly controversial, as they were not able to directly demonstrate Nox4 activity in mitochondrial preparations. Our studies did not show the presence of Nox1, Nox2, Nox4, and p22phox subunits in the mitochondria of endothelial cells and vascular tissue, arguing against the mitochondrial localization of NADPH oxidases in these tissues (21). It has been previously shown that Nox4 is specifically localized in focal adhesions, along stress fibers, and in the nucleus (26, 41). It is possible that the mitochondrial localization of Nox4 reported by Block et al. (5) and Ago et al. (1) differs from previous publications (26, 41) due to the distinct Nox4 antibodies used for immunostaining, as many authors have raised concerns regarding the specificity of some Nox4 antibodies. The difference in Nox4 localization could be also due to the fact that these groups have investigated different cell types, and Nox4 localization in mitochondria may be cell-type specific. Although it may be intriguing to suggest the role of Nox4 in mitochondrial oxidative stress, the lack of data on mitochondrial p22phox and the absence of specific measurements of mitochondrial Nox4 activity have challenged this hypothesis. It is also important that mitochondria do not require any Nox isoform to produce ROS as just described, and ROS production by mitochondria can significantly surpass the amount of ROS produced by Nox4, particularly in the heart. It is conceivable that cytoplasmic Nox4 may contribute to the redox-sensitive upregulation of mitochondrial ROS production. Considering the controversy and inconsistent observations, the mitochondrial expression of Nox4 and its functional significance should be taken with caution and requires additional studies.
Mitochondria are a major source of ROS and play an important role in cellular redox signaling under normal physiological conditions. The stimulation of mitochondrial ROS by AngII is accompanied by mild uncoupling and decreases in ATP synthesis (21). The overproduction of mitochondrial ROS, however, leads to oxidative stress and a further decline in mitochondrial ATP. This may cause mitochondrial dysfunction and apoptosis or necrosis (Fig. 2). AngII-induced production of mitochondrial ROS in various pathophysiological conditions and different tissues is discussed next.
FIG. 2.

Inverse relationship of mitochondrial ATP synthesis and ROS production. An increased production of mitochondrial ROS under redox signaling is coupled with reduced ATP production. The overproduction of mitochondrial ROS may result in the impairment of mitochondrial respiration, leading to a further decrease in ATP and mitochondrial oxidative stress, which may trigger programmed cell death apoptosis. Apoptosis requires ATP; thus, a further decline in ATP may result in ATP-independent cell death necrosis.
Hypertension
Hypertension promotes mitochondrial dysfunction in the brain, heart, vasculature, and kidney (16). These organs are involved in the development of hypertension, and mitochondrial dysfunction may contribute toward retaining hypertension as well as tissue damage observed in hypertension. It has been previously reported that AngII blockade improves mitochondrial function in the kidney of spontaneously hypertensive rats (SHRs). Elevated systolic blood pressure in SHR was accompanied by a reduced kidney mitochondrial membrane potential and an increased production of mitochondrial H2O2 compared with control animals. The treatment of SHR animals with AT1R antagonist candesartan normalized the mitochondrial membrane potential and inhibited the production of mitochondrial H2O2 (16). Interestingly, the treatment of SHR with Ca2+-channel blocker amlodipine reduced the blood pressure but did not affect the mitochondrial dysfunction, while the AT1R antagonist losartan improved both mitochondrial function and reduced mitochondrial H2O2 (17). This difference between amlodipine and losartan was likely due to the inability of Ca2+-channel blocker to inhibit ROS production, because the amount of oxidized glutathione in SHR+amlodipine animals was higher than in control or SHR+losartan rats (17). These data indicate an important role of AT1R signaling in the regulation of mitochondrial ROS.
We have previously reported that the stimulation of endothelial cells with AngII significantly oxidized mitochondrial reduced glutathione (GSH), and the inhibition of NADPH oxidase with apocynin attenuated the loss of GSH, and prevented the stimulation of mitochondrial ROS (21). Furthermore, depletion of the NADPH subunit p22phox abolished the AngII-mediated increase in mitochondrial ROS (Fig. 3). It should be noted that the NADPH subunit p22phox and NADPH oxidase complex were not localized in mitochondria but in the endoplasmic membrane fraction. These data indicate a key role of NADPH oxidases in the AngII-mediated modulation of the mitochondrial redox status, which may be important in the stimulation of mitochondrial ROS. Indeed, Widder et al. have recently shown that the overexpression of a key regulator of mitochondrial redox status thioredoxin 2 blocks the AngII-induced production of mitochondrial ROS. Interestingly, the overexpression of thioredoxin 2 in mice significantly attenuated vascular 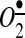 and the expression of NADPH oxidase subunits in response to AngII infusion, improved aortic endothelium-dependent relaxation, and attenuated AngII-induced hypertension. These data suggest an important role of mitochondrial ROS in endothelial dysfunction and hypertension.
and the expression of NADPH oxidase subunits in response to AngII infusion, improved aortic endothelium-dependent relaxation, and attenuated AngII-induced hypertension. These data suggest an important role of mitochondrial ROS in endothelial dysfunction and hypertension.
FIG. 3.

Cross-talk between NADPH oxidase and mitochondria in endothelial dysfunction and hypertension. AngII stimulates NOX2, which triggers the production of mitochondrial ROS. Mitochondria release H2O2, providing the feed-forward stimulation of NOX2. The overproduction of cytoplasmic ROS uncouples endothelial isoform of nitric oxide synthase (eNOS) and results in endothelial dysfunction.
The role of mitochondrial 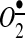 in the regulation of vascular NADPH oxidases and the development of hypertension have been investigated in AngII and DOCA salt-induced hypertension using mitochondria-targeted SOD2 (manganese containing mitochondrial superoxide dismutase) mimetic mitoTEMPO and SOD2 overexpression (Fig. 3). Co-infusion of mitochondria-targeted antioxidant mitoTEMPO and AngII attenuated hypertension, decreased mitochondrial
in the regulation of vascular NADPH oxidases and the development of hypertension have been investigated in AngII and DOCA salt-induced hypertension using mitochondria-targeted SOD2 (manganese containing mitochondrial superoxide dismutase) mimetic mitoTEMPO and SOD2 overexpression (Fig. 3). Co-infusion of mitochondria-targeted antioxidant mitoTEMPO and AngII attenuated hypertension, decreased mitochondrial 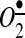 , reduced cellular NADPH oxidase activity, inhibited vascular
, reduced cellular NADPH oxidase activity, inhibited vascular 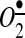 production, and prevented the loss of endothelial nitric oxide (NO•) (20). The treatment of mice with mitoTEMPO significantly decreased blood pressure by 30 mm Hg after the establishment of both AngII-induced and DOCA-salt hypertension, while a similar dose of non-targeted TEMPOL was not effective. In vivo, mitoTEMPO decreased the vascular
production, and prevented the loss of endothelial nitric oxide (NO•) (20). The treatment of mice with mitoTEMPO significantly decreased blood pressure by 30 mm Hg after the establishment of both AngII-induced and DOCA-salt hypertension, while a similar dose of non-targeted TEMPOL was not effective. In vivo, mitoTEMPO decreased the vascular 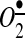 produced by NADPH oxidases, increased vascular NO• production, and improved endothelial-dependent vasorelaxation. Interestingly, transgenic mice overexpressing mitochondrial SOD2 demonstrated attenuated AngII-induced hypertension and reduced vascular oxidative stress similar to mice treated with mitoTEMPO (20), while SOD2+/− mice were predisposed to both age-related and salt-induced hypertension (52). Taken together, these studies show that mitochondrial
produced by NADPH oxidases, increased vascular NO• production, and improved endothelial-dependent vasorelaxation. Interestingly, transgenic mice overexpressing mitochondrial SOD2 demonstrated attenuated AngII-induced hypertension and reduced vascular oxidative stress similar to mice treated with mitoTEMPO (20), while SOD2+/− mice were predisposed to both age-related and salt-induced hypertension (52). Taken together, these studies show that mitochondrial 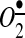 is important for the development of hypertension and that the antioxidant strategies specifically targeting this organelle could have a therapeutic benefit (20).
is important for the development of hypertension and that the antioxidant strategies specifically targeting this organelle could have a therapeutic benefit (20).
Diabetes and Metabolic Syndrome
AngII is involved in the development and pathological changes in diabetes and metabolic syndrome. Under these conditions, the involvement of mitochondria seems to be critical. It has been recently shown that the AT1R blockade protected kidney mitochondria in streptozotocin-induced type 1 diabetes (13). In streptozotocin-treated rats, mitochondrial H2O2 production rate was higher and uncoupling protein-2 content, cytochrome c oxidase activity, and renal glutathione level were lower than in streptozotocin+Losartan and control groups, indicating an important role of AT1R signaling in the diabetes-induced deterioration of mitochondria (13). The AT1R blockade protects kidney mitochondria and kidney structure in diabetes, independently of blood pressure and glycemia (Fig. 4).
FIG. 4.

Potential role of mitochondrial ROS and angiotensin in metabolic syndrome. Hyperglycemia and hyperlipidemia may increase mitochondrial ROS due to the over-reduction of mitochondria and mitochondrial impairment. Mitochondrial H2O2 stimulates the redox-dependent expression of angiotensinogen and AT1R, leading to the AngII-mediated activation of NOX1 and NOX2. These, in turn, provide the feed-forward stimulation of mitochondrial ROS.
The role of the renin-angiotensin system (RAS) in the diabetes and metabolic syndrome was emphasized in a work by Chan's group (28). They showed that high glucose stimulates angiotensinogen gene expression via ROS generation in rat kidney proximal tubular cells (28). These data, however, do not provide clear mechanisms and primary sources of ROS. We suggest that diabetes and metabolic syndrome may initially cause an increase in mitochondrial ROS production, which stimulates AngII production and the expression of AT1R (Fig. 4). This, in turn, will stimulate ROS production by NADPH oxidases and enhance mitochondrial ROS. Therefore, mitochondrial ROS can be both an initiating factor and a target of ROS under these conditions.
The oxidative stress mediated by the hyperglycemia-induced generation of ROS significantly contributes to the development and progression of diabetes and related vascular complications (22). Many studies emphasized the role of mitochondrial dysfunction and mitochondrial ROS in diabetes (8). Brownlee suggested that the mitochondrial electron transport chain plays a key role in the hyperglycemia-induced overproduction of 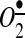 and the development of secondary complications such as endothelial dysfunction (7). In endothelial cells, removal of the mitochondrial electron transport chain completely inhibited hyperglycemia-induced ROS production. These results suggest that diabetes-induced defects in the electron transport chain promote ROS overproduction (8). On the other hand, the overproduction of ROS by NADPH oxidases leads to mitochondrial dysfunction (22). Interestingly, diabetes may be associated with the increased opening of mitoKATP (50), which may be important in reduced insulin secretion and ischemic preconditioning (48). These data support the presence of feed-forward interactions between NADPH oxidases and mitochondria in the settings of hyperglycemia and diabetes, which could be mediated by the activation of mitoKATP, as described in endothelial cells (20). The pathophysiological role of this cross-talk in diabetes has not been fully investigated.
and the development of secondary complications such as endothelial dysfunction (7). In endothelial cells, removal of the mitochondrial electron transport chain completely inhibited hyperglycemia-induced ROS production. These results suggest that diabetes-induced defects in the electron transport chain promote ROS overproduction (8). On the other hand, the overproduction of ROS by NADPH oxidases leads to mitochondrial dysfunction (22). Interestingly, diabetes may be associated with the increased opening of mitoKATP (50), which may be important in reduced insulin secretion and ischemic preconditioning (48). These data support the presence of feed-forward interactions between NADPH oxidases and mitochondria in the settings of hyperglycemia and diabetes, which could be mediated by the activation of mitoKATP, as described in endothelial cells (20). The pathophysiological role of this cross-talk in diabetes has not been fully investigated.
Heart
AngII impacts heart functions and remodeling in many ways. As a result of the systematic negative action on vasoconstriction, thrombosis, and inflammation, AngII induces left ventricular hypertrophy, fibrosis, diastolic dysfunction, and heart failure. At the molecular level, AngII directly acts on cardiomyocytes and through signaling events, it modulates hypertrophy, cell survival/apoptosis, mitochondrial dysfunctions, and autophagy. In contrast to other tissues, in the heart, AngII may induce protective responses preventing injury. Both positive and negative actions of AngII in the heart are mediated by AT1R-initiated signaling with ROS as a crucial contributor to these signaling events. AngII induces ROS generation in the cytoplasm and mitochondria of cardiomyocytes. It has been shown that under ischemic conditions, the pharmacological inhibition of AT1R preserves the energy state of mitochondria, indicating the significance of AT1R signaling in the pathology of ischemia-induced heart damage, and links AT1R signaling and mitochondria functions under stress conditions.
While excessive mitochondrial and/or cytoplasmic H2O2 triggers degenerative changes, moderate and controlled mitochondrial ROS induces an adaptive response which is known as a preconditioning that results in raising the heart's resistance to stress conditions, among others, through an increased expression of antioxidant proteins. In a similar fashion, moderate RAS activation by acute AngII stimulation leads to heart preconditioning through NADPH oxidase and mitochondria-dependent mechanisms (30). Prolonged exposure overwhelms the system and initiates destructive pathological changes. We and others have reported that AngII may utilize the same pathways as those activated on preconditioning (21). The role of the mitoKATP channel has been implicated in this adaptive response to moderate stress and other stress factors. Numerous studies have shown that acute stress leads to the opening of mitoKATP channels, which is prevented by NADPH oxidase inhibition, mitoKATP channel inhibitor 5-HD, or antioxidant treatment. The opening of mitoKATP releases mitochondrial ROS, which stimulates various signaling pathways, including those that are critical for survival and antioxidant protein expression, namely AMPK, p38AMPK, PI3 kinase, and Akt (46). Moreover, Akt activation facilitates NO• production by NOS and NO•-dependent signaling events. AngII is a powerful inducer of ROS and NO•; therefore, protective preconditioning actions of AngII might be, at least in part, mediated by ROS/NO•-dependent signaling events (Fig. 5). We have shown that moderate H2O2 activates an endothelial isoform of nitric oxide synthase (eNOS) that triggers an adaptive response (9). However, excessive H2O2 decreases tetrahydrobiopterin availability and uncouples eNOS and the neuronal isoform of nitric oxide synthase (nNOS) (56). When respiring, mitochondria continuously produce H2O2. The release of H2O2 can be potentially controlled by enzymatic systems such as thioredoxin reductase-2/thioredoxin 2 (57). The H2O2 released from mitochondria may increase the activity of cytoplasmic NADPH oxidases by c-Scr activation that is required for cytoplasmic NOX activity. NOX1, NOX2, and NOX4 have been shown to be expressed in the heart (Fig. 5); however, NOX2 is the most abundant isoforms, and NOX1 is barely detectable (27). It is conceivable that through similar pathways as those just described for heart preconditioning (3), NOX2 initiates a number of pathological conditions, such as AngII-induced hypertrophy (27), and is critical for key processes underlying the development of myocardial infarction, contractile dysfunction, and remodeling (39, 65). The fact that increased NOX2 expression and activity is observed in tissues of the dysfunctional heart indicates the NOX2 contribution to heart disease (32). In line with this evidence, the silencing of Rac, an NOX2 complex component, or NOX2 depletion abolishes both AngII-stimulated ROS generation and cellular hypertrophy in primary neonatal cardiomyocytes (27).
FIG. 5.

Stimulation of mitochondrial ROS by AngII in the heart. Three major isoforms of NADPH oxidase are expressed in the heart: NOX1, NOX2, and NOX4. AngII not only directly activates NOX1 and NOX2 but also increases the expression of cardiac NOX4. ROS production by NADPH oxidases results in eNOS and the neuronal isoform of nitric oxide synthase (nNOS) uncoupling and contributes to mitochondrial oxidative stress.
While the role of NOX2 seems to better described, NOX4 contribution to the heart functions, and its pathology is more complex and, in some cases, controversial. It has been previously reported that NOX4 generates H2O2, while NOX2 produces 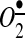 (19). This leads to distinct cellular redox signaling responses (2). Shah group reported that Nox4-generated ROS is beneficial during cardiac remodeling after load-induced stress (64). In response to pathological stress such as pressure overload, myocardial infarction, or hypoxia, as a part of adaptive response enabling angiogenesis, endogenous Nox4 expression increases (64). These NOX4-mediated adaptive stress responses activate the Nrf2-regulated pathway, and suggest a potential role for the Nox4 in the regulation of GSH redox in cardiomyocyes (6) that is important for mitochondrial function.
(19). This leads to distinct cellular redox signaling responses (2). Shah group reported that Nox4-generated ROS is beneficial during cardiac remodeling after load-induced stress (64). In response to pathological stress such as pressure overload, myocardial infarction, or hypoxia, as a part of adaptive response enabling angiogenesis, endogenous Nox4 expression increases (64). These NOX4-mediated adaptive stress responses activate the Nrf2-regulated pathway, and suggest a potential role for the Nox4 in the regulation of GSH redox in cardiomyocyes (6) that is important for mitochondrial function.
In contrast to results showing the protective role of NOX4, Sadoshima group reported that NOX4 depletion improved the mitochondrial functions measured by reduced mitochondrial swelling, cytochrome c release, and decreases in both mitochondrial DNA and aconitase activity in response to pressure overload (1). It has been shown that cardiac-specific Nox4 knockout can attenuate cardiac hypertrophy, interstitial fibrosis, and apoptosis, and show better cardiac function when compared with wild-type mice (33). Due to contradicting results shown in literature and unanswered questions, the pathophysiological role of NOX4 in the heart remains unclear.
We suggest that an initial adaptive response mediated by NOX4 can be transformed to maladaptive response due to ROS overproduction. It is conceivable that the physiological role of NOX4 may include the redox regulation of eNOS activity, GSH synthesis, and mitochondria biogenesis. However, NOX4 overexpression and mislocalization may contribute to pathological processes.
Various approaches providing antioxidant defenses to mitochondria resulted in improved mitochondrial functions. Interestingly, mitochondria-targeted antioxidant prevents not only AngII-induced mitochondrial oxidative stress, cardiac hypertrophy, diastolic dysfunction, and fibrosis but also attenuated AngII-induced NOX4 up-regulation (11). AngII induces the pathological hypertrophic growth of cardiac tissue, leading to a significant increase in heart weight that is prevented by the administration of a mitochondria-targeted antioxidant (11). Similar protective effects were observed when mitochondria-targeted catalase was overexpressed in mice chronically infused with AngII (11). Constitutive autophagy in the heart is a homeostatic mechanism that contributes to the mechanisms maintaining cardiomyocytes size and cardiac tissue structure and functions. Significantly, autophagy alterations have been observed in a variety of heart diseases, including cardiac hypertrophy and heart failure. The upregulation of autophagy in pathological heart conditions is an adaptive response that protects cells from stress (45). Autophagy is regulated by ROS-sensitive mechanisms; therefore, mitochondrial ROS may regulate autophagy. Mice overexpressing catalase targeted to mitochondria challenged by AngII infusion are resistant to cardiac hypertrophy, fibrosis and mitochondrial damage, biogenesis, and autophagy induced by AngII (12).
Despite controversy around NADPH oxidase localization in the heart tissue, an analysis of literature indicates a close interrelationship between cytoplasmic and mitochondrial ROS and its role in heart pathological conditions and remodeling. Further studies are required to reveal the precise mechanisms and mediators of NADPH oxidases-mitochondria signaling.
Kidney
The pathophysiological actions of AngII on the kidney have a broad spectrum on different types of cells, including fibroblasts, endothelial cells, vascular smooth muscle cells, mesangial cells, tubular cells, and podocytes. AngII induced pathological changes of kidney morphology, and its functions are primarily associated with the loss of redox homeostasis. The NOX1, NOX2, NOX4, and NOX regulatory subunits are widely expressed in kidney tissue and have been considered key contributors to kidney fibrosis, loss of podocytes, or inflammation. The infusion of AngII to animals increases ROS production in kidneys and the overexpression of NOX isoforms (62). Increased NOX4 expression has been associated with proteinuria and hypertension in rats (47). Consistently with findings showing the critical role of ROS in the development of kidney disease, knockout NOX components prevents AngII-induced kidney damage (4). Significantly, a link between AT1R and kidney mitochondrial functions and mitochondrial ROS has been established. Several reports show that AngII induces the mitochondrial dysfunctions of kidney cells and/or mitochondria-dependent cell dysfunctions/cell death (58). The mitochondria of SHRs generate significantly higher levels of H2O2. The inhibition of AT1R prevents mitochondria dysfunction and morphological changes (16). Combination of a high salt diet and AngII infusion increases NOX2 expression and lowers SOD1 and SOD2 expression (31). It remains unclear whether mitochondria-targeted antioxidants are able to prevent AngII-induced kidney dysfunctions. However, studies with other cells and tissue showing AngII-induced mitochondrial dysfunction suggest a similar mechanism where NOX and mitochondria signal to each other to generate ROS (Figs. 1 and 5). Additional studies focusing on kidney dysfunction are required to test this hypothesis and show the potential physiological effects of mitochondria-targeted treatments.
Brain
In the central nervous system, AngII triggers intra-neuronal signaling events that lead to neuronal activation (66). Signals of AngII detected by AT1R in neurons are communicated to brain regions, such as the subfornical organ (SFO) or paraventricular nucleus (PVN), in order to initiate sympathetic nervous system responses and restore body fluid and cardiovascular system homeostasis. In pathological conditions, this neuronal response becomes critical for the development of cardiovascular pathology, including hypertension or heart failure. ROS is a critical mediator of neuronal activation that not only facilitates physiological signaling events but also importantly orchestrates the development of disease. 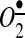 has been recognized as a key mediator of AngII action in the central nervous system. Early studies conducted on AngII action on neurons show that the overexpression of SOD2 and CuZnSOD in the brain, specifically in the SFO, prevents AngII-induced changes in blood pressure and heart rate (67). Increased
has been recognized as a key mediator of AngII action in the central nervous system. Early studies conducted on AngII action on neurons show that the overexpression of SOD2 and CuZnSOD in the brain, specifically in the SFO, prevents AngII-induced changes in blood pressure and heart rate (67). Increased 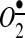 in the central and peripheral nervous system and the SFO has been recognized as a specific site for elevating the
in the central and peripheral nervous system and the SFO has been recognized as a specific site for elevating the 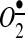 associated with developing hypertension (37, 38). These meaningful data show that AngII signals can be potentially suppressed at various levels, in cytoplasm or in mitochondria, and both sources of
associated with developing hypertension (37, 38). These meaningful data show that AngII signals can be potentially suppressed at various levels, in cytoplasm or in mitochondria, and both sources of 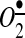 are equally important for the propagation of the AngII signal; thus, both sources of
are equally important for the propagation of the AngII signal; thus, both sources of 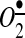 might serve as potential targets for pharmacological treatment. A later work from the same group shows a potential role of NADPH oxidases in AngII signaling in neurons and cellular
might serve as potential targets for pharmacological treatment. A later work from the same group shows a potential role of NADPH oxidases in AngII signaling in neurons and cellular 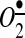 as a mediator and regulator of intracellular Ca2+ (68). They show that the expression of a dominant-negative isoform of Rac1, a critical component for NADPH oxidase activation and
as a mediator and regulator of intracellular Ca2+ (68). They show that the expression of a dominant-negative isoform of Rac1, a critical component for NADPH oxidase activation and 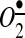 production, significantly inhibited the increase in intracellular Ca2+ after AngII stimulation.
production, significantly inhibited the increase in intracellular Ca2+ after AngII stimulation.
AngII-related neuro-cardiovascular diseases are associated with excessive redox sensitive sympathoexcitation, which can be potentially counteracted by angiotensin-1-7 via an NO• pathway (66). Although AngII directly stimulates Nox1 and Nox2, it has been recently reported that Nox4 also contributes to redox signaling in the PVN, leading to sympathetic overactivation and a decline in cardiac function (29). In a number of pathological conditions such as SOD1 mutations, malignant gliomas and cerebral ischemia brain tissue can be overwhelmed with excessive 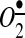 production, leading to increasing reactions with NO• to produce cytotoxic peroxynitrite (ONOO−). Interestingly, a sustained blockade of brain AT1 receptors before and after focal cerebral ischemia reduces neuronal injury, apoptosis, and inflammatory responses (40). One of the major cytoplasmic ROS sources in neurons is NOX2, which generates
production, leading to increasing reactions with NO• to produce cytotoxic peroxynitrite (ONOO−). Interestingly, a sustained blockade of brain AT1 receptors before and after focal cerebral ischemia reduces neuronal injury, apoptosis, and inflammatory responses (40). One of the major cytoplasmic ROS sources in neurons is NOX2, which generates 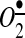 (Fig. 6). Additionally, nNOS once uncoupled by ONOO− mediated reactions also significantly contributes to the pull of cytoplasmic
(Fig. 6). Additionally, nNOS once uncoupled by ONOO− mediated reactions also significantly contributes to the pull of cytoplasmic 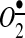 . When ONOO− is generated in close vicinity to mitochondria or within mitochondria, it has a major impact on the number of mitochondrial proteins, including SOD2 and complex I (Fig. 6). ONOO− may cause the oxidation of cysteine residues and tyrosine nitration. These changes within neuronal tissue may promote the development of degenerative disease (53).
. When ONOO− is generated in close vicinity to mitochondria or within mitochondria, it has a major impact on the number of mitochondrial proteins, including SOD2 and complex I (Fig. 6). ONOO− may cause the oxidation of cysteine residues and tyrosine nitration. These changes within neuronal tissue may promote the development of degenerative disease (53).
FIG. 6.

Stimulation of mitochondrial ROS by AngII in the brain. NOX2 and NOX4 are the primary NADPH oxidase isoforms in the brain. The activation of these NOXs results in nNOS uncoupling and the stimulation of mitochondrial ROS via reverse electron transfer (RET), which is attenuated by SOD2. The release of mitochondrial H2O2 provides a feed-forward stimulation of cytoplasmic NOXs and may contribute to neurodegeneration.
Aging
Aging is a complex and not fully understood process that leads to the progressive loss of tissues and organs without the capability to regenerate. In normal healthy humans, aging is associated with progressive endothelial dysfunction, remodeling of the small and large arteries, leading to arteriolosclerosis, kidney, and heart dysfunctions. Clinical factors, including hypertension, diabetes mellitus, and local and systemic inflammatory processes or tissue factors such as AngII, oxidative stress and mitochondrial dysfunction have been linked with aging-related deterioration. A clear connection between RAS and aging provides an opportunity for possible interventions that prevent some of the degenerative processes. The pharmacological inhibition of the AngII converting enzyme or AT1R prevents numerous age-related changes in animal studies (14). Furthermore, AT1R deficiency in mice promotes a longevity that is associated with decreased cardiac, vascular, renal, and pancreatic injury; reduced oxidative stress; and upregulation of the prosurvival gene sirtuin 3 (Sirt3) (4). These authors, in line with previous reports, have also shown in vitro that AngII decreases prosurvival and antioxidant enzyme expression. These changes are observed along with a decline of mitochondrial biogenesis, mitochondrial energy production, and its antioxidant defense mechanisms that result in apoptosis and senescence. The deleterious effects of AngII on mitochondrial function and biogenesis leading to degenerative changes in tissue can be prevented by the pretreatment with the AT1R blockers or mitochondria-targeted antioxidants. Mitochondrial ROS and mitochondria dysfunction initiated by AT1R-dependent cytoplasmic NADPH oxidases may represent a vicious cycle that may contribute to progressive cell and tissue degeneration. Since AT1R blockers have much more broad effects and prolonged treatment may not be the most efficient and targeted approach, targeting dysfunctional mitochondria may prove to be more therapeutically beneficial.
Conclusions
Hypertension is a common disease that affects one-third of adults in Western societies (10), and approximately 60% of the population is affected by cardiovascular conditions (54, 61). Oxidative stress is strongly implicated in the pathogenesis of these cardiovascular diseases (23). Mitochondria are one of the most important sources of ROS, and mitochondrial dysfunction is a prominent feature of most cardiovascular diseases (42, 49); however, the role of mitochondrial ROS is not completely understood.
Recently, it has been proposed that mitochondrial dysfunction along with endothelial dysfunction represents an important early step in the chain of events leading to atherosclerotic disease (49), and mitochondrial dysfunction in response to AngII could have direct ramifications for the development of endothelial dysfunction (21). Interestingly, angiotensin-converting enzyme inhibitors and AT1R blockers reduce age-related mitochondrial dysfunction, attenuate hypertension-induced renal mitochondrial dysfunction, and protect against cardiac mitochondrial dysfunction in the setting of acute ischemia (15, 17, 44). These findings suggest that AngII can alter mitochondrial function presumably by NADPH oxidases. Indeed, the inhibition of NADPH oxidases by apocynin and chelerythrine or the depletion of p22phox, an essential NADPH oxidase complex component, completely prevented mitochondrial dysfunction and attenuated mitochondrial ROS in response to AngII (21). On the other hand, consistent with the concept of NADPH oxidase-mitochondrial crosstalk (18), SOD2 overexpression and mitochondria-targeted SOD-mimetic, mitoTEMPO attenuated the AngII stimulation of NADPH oxidase and reduced hypertension (20). Taken together, these studies indicate that the interplay between mitochondrial and NADPH oxidase-derived 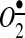 constitutes a feed-forward cycle in which the NADPH oxidases increase mitochondrial ROS, which further activates the cytoplasmic NADPH oxidases and increases cellular ROS, diminishing NO• bioavailability and leading to eNOS uncoupling.
constitutes a feed-forward cycle in which the NADPH oxidases increase mitochondrial ROS, which further activates the cytoplasmic NADPH oxidases and increases cellular ROS, diminishing NO• bioavailability and leading to eNOS uncoupling.
During the past decade, it has become apparent that crosstalk between mitochondria and NADPH oxidases plays a critical role in the genesis of many cardiovascular diseases (18). Since mitochondria are both a target and source of ROS, they play an important role in ROS-induced ROS production. Interestingly, the scavenging of mitochondrial ROS-attenuated responses to AngII in the brain, kidney, and endothelial cells. These data indicate that the stimulation of mitochondrial ROS by AngII is an important amplification of redox signaling, which can be important for normal physiological functions. However, diminished SOD2 expression (20), decreased redox mitochondrial status (63), or metabolic syndrome may result in the uncontrolled amplification of AngII signaling, leading to a feed-forward viscous cycle of ROS production by mitochondria and NADPH oxidases that can be pharmacologically targeted by AT1R blockers or mitochondria-targeted antioxidants. The use of antioxidant strategies specifically targeting mitochondria (11, 20) is a new promising approach for the treatment of many pathological conditions, including aging, atherosclerosis, diabetes, hypertension, and degenerative neurological disorders in which mitochondrial oxidative stress seems to play a critical role. Additional studies conducted on the AngII-mediated production of mitochondrial ROS are required to reveal the detailed mechanisms and specific mediators that could serve as novel targets for pharmacological treatments.
Abbreviations Used
- AngII
angiotensin II
- AT1R
angiotensin II receptor type 1
- AT2R
angiotensin II receptor type 2
- eNOS
endothelial isoform of nitric oxide synthase
- GSH
reduced glutathione
- H2O2
hydrogen peroxide
- nNOS
neuronal isoform of nitric oxide synthase
- NO•
nitric oxide
- NOX
catalytic subunit of NADPH oxidases
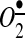
superoxide
- ONOO−
peroxynitrite
- PKC
protein kinase C
- PVN
paraventricular nucleus
- RAS
renin-angiotensin system
- RET
reverse electron transfer
- ROS
reactive oxygen species
- SFO
subfornical organ
- SHR
spontaneously hypertensive rats
- SOD2
manganese containing mitochondrial superoxide dismutase
Acknowledgment
This work was supported by funding from the National Institute of Health grant HL094469.
References
- 1.Ago T. Kuroda J. Pain J. Fu C. Li H. Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anilkumar N. Weber R. Zhang M. Brewer A. Shah AM. Nox4 and nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler Thromb Vasc Biol. 2008;28:1347–1354. doi: 10.1161/ATVBAHA.108.164277. [DOI] [PubMed] [Google Scholar]
- 3.Bell RM. Cave AC. Johar S. Hearse DJ. Shah AM. Shattock MJ. Pivotal role of NOX-2-containing NADPH oxidase in early ischemic preconditioning. FASEB J Off Publ Fed Am Soc Exp Biol. 2005;19:2037–2039. doi: 10.1096/fj.04-2774fje. [DOI] [PubMed] [Google Scholar]
- 4.Benigni A. Corna D. Zoja C. Sonzogni A. Latini R. Salio M. Conti S. Rottoli D. Longaretti L. Cassis P. Morigi M. Coffman TM. Remuzzi G. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Investig. 2009;119:524–530. doi: 10.1172/JCI36703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Block K. Gorin Y. Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brewer AC. Murray TV. Arno M. Zhang M. Anilkumar NP. Mann GE. Shah AM. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic Bio Med. 2011;51:205–215. doi: 10.1016/j.freeradbiomed.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 8.Bugger H. Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res. 2010;88:229–240. doi: 10.1093/cvr/cvq239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai H. Li Z. Dikalov S. Holland SM. Hwang J. Jo H. Dudley SC., Jr. Harrison DG. NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin II. J Biol Chem. 2002;277:48311–48317. doi: 10.1074/jbc.M208884200. [DOI] [PubMed] [Google Scholar]
- 10.Chobanian AV. Bakris GL. Black HR. Cushman WC. Green LA. Izzo JL., Jr. Jones DW. Materson BJ. Oparil S. Wright JT., Jr. Roccella EJ. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. Jama. 2003;289:2560–2572. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 11.Dai DF. Chen T. Szeto H. Nieves-Cintron M. Kutyavin V. Santana LF. Rabinovitch PS. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai DF. Rabinovitch P. Mitochondrial oxidative stress mediates induction of autophagy and hypertrophy in angiotensin-II treated mouse hearts. Autophagy. 2011;7:917–918. doi: 10.4161/auto.7.8.15813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Cavanagh EM. Ferder L. Toblli JE. Piotrkowski B. Stella I. Fraga CG. Inserra F. Renal mitochondrial impairment is attenuated by AT1 blockade in experimental Type I diabetes. Am J Physiol Heart Circ Physiol. 2008;294:H456–H465. doi: 10.1152/ajpheart.00926.2007. [DOI] [PubMed] [Google Scholar]
- 14.de Cavanagh EM. Inserra F. Ferder L. Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc Res. 2011;89:31–40. doi: 10.1093/cvr/cvq285. [DOI] [PubMed] [Google Scholar]
- 15.de Cavanagh EM. Piotrkowski B. Basso N. Stella I. Inserra F. Ferder L. Fraga CG. Enalapril and losartan attenuate mitochondrial dysfunction in aged rats. FASEB J. 2003;17:1096–1098. doi: 10.1096/fj.02-0063fje. [DOI] [PubMed] [Google Scholar]
- 16.De Cavanagh EM. Toblli JE. Ferder L. Piotrkowski B. Stella I. Fraga CG. Inserra F. Angiotensin II blockade improves mitochondrial function in spontaneously hypertensive rats. Cell Mol Biol. 2005;51:573–578. [PubMed] [Google Scholar]
- 17.de Cavanagh EM. Toblli JE. Ferder L. Piotrkowski B. Stella I. Inserra F. Renal mitochondrial dysfunction in spontaneously hypertensive rats is attenuated by losartan but not by amlodipine. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1616–R1625. doi: 10.1152/ajpregu.00615.2005. [DOI] [PubMed] [Google Scholar]
- 18.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dikalov SI. Dikalova AE. Bikineyeva AT. Schmidt HH. Harrison DG. Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. 2008;45:1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dikalova AE. Bikineyeva AT. Budzyn K. Nazarewicz RR. McCann L. Lewis W. Harrison DG. Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doughan AK. Harrison DG. Dikalov SI. Molecular mechanisms of angiotensin II mediated mitochondrial dysfunction. Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 22.Gao L. Mann GE. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res. 2009;82:9–20. doi: 10.1093/cvr/cvp031. [DOI] [PubMed] [Google Scholar]
- 23.Griendling KK. Sorescu D. Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 24.Guzik TJ. Chen W. Gongora MC. Guzik B. Lob HE. Mangalat D. Hoch N. Dikalov S. Rudzinski P. Kapelak B. Sadowski J. Harrison DG. Calcium-dependent NOX5 NADPH oxidase contributes to vascular oxidative stress in human coronar artery disease. J Am Coll Cardiol. 2008;52:1803–1809. doi: 10.1016/j.jacc.2008.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanna IR. Taniyama Y. Szocs K. Rocic P. Griendling KK. NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid Redox Signal. 2002;4:899–914. doi: 10.1089/152308602762197443. [DOI] [PubMed] [Google Scholar]
- 26.Hilenski LL. Clempus RE. Quinn MT. Lambeth JD. Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 27.Hingtgen SD. Tian X. Yang J. Dunlay SM. Peek AS. Wu Y. Sharma RV. Engelhardt JF. Davisson RL. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics. 2006;26:180–191. doi: 10.1152/physiolgenomics.00029.2005. [DOI] [PubMed] [Google Scholar]
- 28.Hsieh TJ. Zhang SL. Filep JG. Tang SS. Ingelfinger JR. Chan JS. High glucose stimulates angiotensinogen gene expression via reactive oxygen species generation in rat kidney proximal tubular cells. Endocrinology. 2002;143:2975–2985. doi: 10.1210/endo.143.8.8931. [DOI] [PubMed] [Google Scholar]
- 29.Infanger DW. Cao X. Butler SD. Burmeister MA. Zhou Y. Stupinski JA. Sharma RV. Davisson RL. Silencing nox4 in the paraventricular nucleus improves myocardial infarction-induced cardiac dysfunction by attenuating sympathoexcitation and periinfarct apoptosis. Circ Res. 2010;106:1763–1774. doi: 10.1161/CIRCRESAHA.109.213025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimura S. Zhang GX. Nishiyama A. Shokoji T. Yao L. Fan YY. Rahman M. Suzuki T. Maeta H. Abe Y. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005;45:860–866. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- 31.Kitiyakara C. Chabrashvili T. Chen Y. Blau J. Karber A. Aslam S. Welch WJ. Wilcox CS. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol. 2003;14:2775–2782. doi: 10.1097/01.asn.0000092145.90389.65. [DOI] [PubMed] [Google Scholar]
- 32.Krijnen PA. Meischl C. Hack CE. Meijer CJ. Visser CA. Roos D. Niessen HW. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J Clin Pathol. 2003;56:194–199. doi: 10.1136/jcp.56.3.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuroda J. Ago T. Matsushima S. Zhai P. Schneider MD. Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 35.Lassegue B. Sorescu D. Szocs K. Yin Q. Akers M. Zhang Y. Grant SL. Lambeth JD. Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 36.Lavoie JL. Sigmund CD. Minireview: overview of the renin-angiotensin system—an endocrine and paracrine system. Endocrinology. 2003;144:2179–2183. doi: 10.1210/en.2003-0150. [DOI] [PubMed] [Google Scholar]
- 37.Lob HE. Marvar PJ. Guzik TJ. Sharma S. McCann LA. Weyand C. Gordon FJ. Harrison DG. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 2010;55:277–283. doi: 10.1161/HYPERTENSIONAHA.109.142646. 6p following 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lob HE. Vinh A. Li L. Blinder Y. Offermanns S. Harrison DG. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension. 2011;58:232–239. doi: 10.1161/HYPERTENSIONAHA.111.172718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Looi YH. Grieve DJ. Siva A. Walker SJ. Anilkumar N. Cave AC. Marber M. Monaghan MJ. Shah AM. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension. 2008;51:319–325. doi: 10.1161/HYPERTENSIONAHA.107.101980. [DOI] [PubMed] [Google Scholar]
- 40.Lou M. Blume A. Zhao Y. Gohlke P. Deuschl G. Herdegen T. Culman J. Sustained blockade of brain AT1 receptors before and after focal cerebral ischemia alleviates neurologic deficits and reduces neuronal injury, apoptosis, and inflammatory responses in the rat. J Cereb Blood Flow Metab. 2004;24:536–547. doi: 10.1097/00004647-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 41.Lyle AN. Deshpande NN. Taniyama Y. Seidel-Rogol B. Pounkova L. Du P. Papaharalambus C. Lassegue B. Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madamanchi NR. Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–473. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- 43.Martyn KD. Frederick LM. von Loehneysen K. Dinauer MC. Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 44.Monteiro P. Duarte AI. Goncalves LM. Providencia LA. Valsartan improves mitochondrial function in hearts submitted to acute ischemia. Eur J Pharmacol. 2005;518:158–164. doi: 10.1016/j.ejphar.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 45.Nakai A. Yamaguchi O. Takeda T. Higuchi Y. Hikoso S. Taniike M. Omiya S. Mizote I. Matsumura Y. Asahi M. Nishida K. Hori M. Mizushima N. Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 46.Nishino Y. Miura T. Miki T. Sakamoto J. Nakamura Y. Ikeda Y. Kobayashi H. Shimamoto K. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc Res. 2004;61:610–619. doi: 10.1016/j.cardiores.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 47.Nistala R. Wei Y. Sowers JR. Whaley-Connell A. Renin-angiotensin-aldosterone system-mediated redox effects in chronic kidney disease. Transl Res. 2009;153:102–113. doi: 10.1016/j.trsl.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pomerantz BJ. Robinson TN. Heimbach JK. Calkins CM. Miller SA. Banerjee A. Harken AH. Selective mitochondrial KATP channel opening controls human myocardial preconditioning: too much of a good thing? Surgery. 2000;128:368–373. doi: 10.1067/msy.2000.107423. [DOI] [PubMed] [Google Scholar]
- 49.Puddu P. Puddu GM. Galletti L. Cravero E. Muscari A. Mitochondrial dysfunction as an initiating event in atherogenesis: a plausible hypothesis. Cardiology. 2005;103:137–141. doi: 10.1159/000083440. [DOI] [PubMed] [Google Scholar]
- 50.Quast U. Stephan D. Bieger S. Russ U. The impact of ATP-sensitive K+ channel subtype selectivity of insulin secretagogues for the coronary vasculature and the myocardium. Diabetes. 2004;53(Suppl 3):S156–S164. doi: 10.2337/diabetes.53.suppl_3.s156. [DOI] [PubMed] [Google Scholar]
- 51.Queliconi BB. Wojtovich AP. Nadtochiy SM. Kowaltowski AJ. Brookes PS. Redox regulation of the mitochondrial K(ATP) channel in cardioprotection. Biochim Biophys Acta. 2011;1813:1309–1315. doi: 10.1016/j.bbamcr.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez-Iturbe B. Sepassi L. Quiroz Y. Ni Z. Wallace DC. Vaziri ND. Association of mitochondrial SOD deficiency with salt-sensitive hypertension and accelerated renal senescence. J Appl Physiol. 2007;102:255–260. doi: 10.1152/japplphysiol.00513.2006. [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez-Pallares J. Rey P. Parga JA. Munoz A. Guerra MJ. Labandeira-Garcia JL. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol Dis. 2008;31:58–73. doi: 10.1016/j.nbd.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 54.Rossi R. Cioni E. Nuzzo A. Origliani G. Modena MG. Endothelial-dependent vasodilation and incidence of type 2 diabetes in a population of healthy postmenopausal women. Diabetes Care. 2005;28:702–707. doi: 10.2337/diacare.28.3.702. [DOI] [PubMed] [Google Scholar]
- 55.Seshiah PN. Weber DS. Rocic P. Valppu L. Taniyama Y. Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–413. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 56.Silberman GA. Fan TH. Liu H. Jiao Z. Xiao HD. Lovelock JD. Boulden BM. Widder J. Fredd S. Bernstein KE. Wolska BM. Dikalov S. Harrison DG. Dudley SC., Jr Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010;121:519–528. doi: 10.1161/CIRCULATIONAHA.109.883777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stanley BA. Sivakumaran V. Shi S. MacDonald I. Lloyd D. Watson WH. Aon MA. Paolocci N. Thioredoxin reductase 2 is essential for keeping low levels of H2O2 emission from isolated heart mitochondria. J Biol Chem. 2011;286:33669–33677. doi: 10.1074/jbc.M111.284612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun L. Xiao L. Nie J. Liu FY. Ling GH. Zhu XJ. Tang WB. Chen WC. Xia YC. Zhan M. Ma MM. Peng YM. Liu H. Liu YH. Kanwar YS. p66Shc mediates high-glucose and angiotensin II-induced oxidative stress renal tubular injury via mitochondrial-dependent apoptotic pathway. Am J Physiol Renal Physiol. 2010;299:F1014–F1025. doi: 10.1152/ajprenal.00414.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takac I. Schroder K. Zhang L. Lardy B. Anilkumar N. Lambeth JD. Shah AM. Morel F. Brandes RP. The E-loop Is Involved in Hydrogen Peroxide Formation by the NADPH Oxidase Nox4. J Biol Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ushio-Fukai M. Griendling KK. Becker PL. Hilenski L. Halleran S. Alexander RW. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:489–495. doi: 10.1161/01.atv.21.4.489. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y. Wang QJ. The prevalence of prehypertension and hypertension among US adults according to the new joint national committee guidelines: new challenges of the old problem. Arch Intern Med. 2004;164:2126–2134. doi: 10.1001/archinte.164.19.2126. [DOI] [PubMed] [Google Scholar]
- 62.Whaley-Connell A. Habibi J. Nistala R. Cooper SA. Karuparthi PR. Hayden MR. Rehmer N. DeMarco VG. Andresen BT. Wei Y. Ferrario C. Sowers JR. Attenuation of NADPH oxidase activation and glomerular filtration barrier remodeling with statin treatment. Hypertension. 2008;51:474–480. doi: 10.1161/HYPERTENSIONAHA.107.102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Widder JD. Fraccarollo D. Galuppo P. Hansen JM. Jones DP. Ertl G. Bauersachs J. Attenuation of Angiotensin II-Induced Vascular Dysfunction and Hypertension by Overexpression of Thioredoxin 2. Hypertension. 2009;54:338–344. doi: 10.1161/HYPERTENSIONAHA.108.127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang M. Brewer AC. Schroder K. Santos CX. Grieve DJ. Wang M. Anilkumar N. Yu B. Dong X. Walker SJ. Brandes RP. Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao Y. McLaughlin D. Robinson E. Harvey AP. Hookham MB. Shah AM. McDermott BJ. Grieve DJ. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 2010;70:9287–9297. doi: 10.1158/0008-5472.CAN-10-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zimmerman MC. Angiotensin II and angiotensin-1–7 redox signaling in the central nervous system. Curr Opin Pharmacol. 2011;11:138–143. doi: 10.1016/j.coph.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zimmerman MC. Lazartigues E. Lang JA. Sinnayah P. Ahmad IM. Spitz DR. Davisson RL. Superoxide Mediates the Actions of Angiotensin II in the Central Nervous System. Circ Res. 2002;91:1038–1045. doi: 10.1161/01.res.0000043501.47934.fa. [DOI] [PubMed] [Google Scholar]
- 68.Zimmerman MC. Sharma RV. Davisson RL. Superoxide mediates angiotensin II-induced influx of extracellular calcium in neural cells. Hypertension. 2005;45:717–723. doi: 10.1161/01.HYP.0000153463.22621.5e. [DOI] [PubMed] [Google Scholar]