

Abstract
Background: Senescence marker protein-30 (SMP30) decreases with aging, and SMP30 knockout (KO) mice show a short life with increased oxidant stress. Aims: We assessed the effect of oxidant stress with SMP30 deficiency in coronary artery spasm and clarify its underlying mechanisms. Results: We measured vascular responses to acetylcholine (ACh) and sodium nitroprusside (SNP) of isolated coronary arteries from SMP30 KO and wild-type (WT) mice. In SMP30 KO mice, ACh-induced vasoconstriction occurred, which was changed to vasodilation by dithiothreitol (DTT), a thiol-reducing agent. However, Nω-nitro-l-arginine-methyl ester, nitric oxide (NO) synthase inhibitor, or tetrahydrobiopterin did not change the ACh response. In isolated coronary arteries of WT mice, ACh-induced vasodilation occurred. Inhibition of glutathione reductase by 1, 3-bis(2-chloroethyl)-1-nitrosourea decreased ACh-induced vasodilation (n=10, p<0.01), which was restored by DTT. To evaluate the thiol oxidation, we measured the fluorescence of monochlorobimane (MCB) in coronary arteries, which covalently labels the total. The fluorescence level to MCB decreased in SMP30 KO mice, but with DTT treatment restored to a level comparable to that of WT mice. The reduced glutathione and total thiol levels were also low in the aorta of SMP30 KO mice compared with those of WT mice. Administration of ACh into the aortic sinus in vivo of SMP30 KO mice induced coronary artery spasm. Innovation: The thiol redox state is a key regulator of endothelial NO synthase activity, and thiol oxidation was associated with endothelial dysfunction in the SMP30 deficiency model. Conclusion: These results suggest that chronic thiol oxidation by oxidant stress is a trigger of coronary artery spasm, resulting in impaired endothelium-dependent vasodilation. Antioxid. Redox Signal. 19, 1063–1073.
Introduction
Coronary artery spasm plays an important role in ischemic heart conditions, especially in Japan, in forms such as variant angina (35). It has become a clinical problem in the Western world, leading to a rise in complex and aggressive of coronary interventions (25, 30). Recent advances in research on the pathogenesis of coronary artery spasms indicate the presence of endothelial dysfunction, which may be mediated by an impaired endothelial nitric oxide (NO) synthase (eNOS) activity or primarily smooth muscle cell contraction with Rho-kinase activation in the spasm site (21, 28, 38). Moreover, aging associated with oxidant stress elevation is an important risk factor for the development of ischemic heart disease (10). In the clinical setting, a shift in the redox equilibrium to a more oxidative state reportedly exists in patients with coronary artery spasm (27). However, the underlying mechanism of coronary artery spasm and age-related oxidant stress remains elusive.
The oxidation of the thiol group, including cysteine, is involved in many biological processes. Thiol oxidation induces protein conformation changes by converting free thiols (−SH) into sulfenic acids (SO−), sulfinic acids (SOO−), sulfonic acids (SOOO−), and disulfide bridges (S–S) (34). Thiol oxidation is involved in many cellular processes, such as eNOS glutathionylation (6), increased ryanodine receptor activity, inhibition of SERCA activity (46), and pulmonary artery vasoconstriction (26). Recently, we observed that reactive oxygen species (ROS) regulate the coronary vascular tone mediated by thiol oxidation (33). Thus, we hypothesized that thiol oxidation with chronic oxidant stress plays a key role in age-related coronary artery spasm. Senescence marker protein-30 (SMP30) is an aging marker molecule. SMP30 knockout (KO) mice cannot synthesize vitamin C in vivo and have a short life. Humans are similarly incapable of synthesizing vitamin C. With aging, SMP30 content decreases in the liver, kidney, and lung in human, and such a SMP30 deficiency increases superoxide production (19, 20). Therefore, SMP30 KO mouse is a suitable model to test this hypothesis. Then, we determined the role of oxidative stress related to thiol oxidation in the redox-dependent regulation of coronary vascular tone associated with NO generation, including coronary vasospasm using SMP30 KO mouse coronary arteries.
Innovation.
Age-related oxidant stress with a decrease of senescence marker protein-30 (SMP30) plays a pivotal role in coronary artery spasm, which is a major health problem. Nitric oxide (NO) generation in endothelial NO synthase (eNOS) is of critical importance in maintaining proper coronary circulation. Chronic oxidant stress, through NADPH oxidase activation with the SMP30 deficiency, induced thiol oxidation in eNOS, and attenuated bioactive NO generation resulted in coronary artery spasm. The specifying of thiol redox state as a key regulator of the eNOS activity provides a new platform for strategies to prevent coronary artery spasm and alleviate the underlying process of aging-induced coronary artery disease.
Results
NO concentration induced by acetylcholine and vitamin C level
We hypothesized that SMP30 deficiency decreases NO biosynthesis activity in the artery, because endothelium-dependent dilation, namely, derived from NO, is an important mechanism of coronary flow regulation. Therefore, we assessed the NO generation induced by acetylcholine (ACh, 1 μM) in the aorta of SMP30 KO and wild-type (WT) mice. The NO levels in the aortas of the SMP30 KO mice were lower than those of the WT mice (2.6±4.5 nM vs. 82.8±9.6 nM, n=10 each, p<0. 01) (Fig. 1). On the other hand, vitamin C levels in the aortas did not change between the SMP30 KO and WT mice (0.86±0.12 μmol/g tissue vs. 0.98±0.16 μmol/g tissue; n=10 each). These results suggest that SMP30 deficiency impairs NO biosynthesis from eNOS in the coronary artery, which is not dependent of the radical-scavenging effect of vitamin C.
FIG. 1.

Nitric oxide (NO) generation in aorta. Representative electrochemical detection tracing of NO concentration in isolated aortas from the senescence marker protein-30 (SMP30) knockout (KO) and wild type (WT) mice in an organ chamber. NO concentration is directly proportional to the current (pA) by a free-radical analyzer (Apollo 4000). ACh (1 μM)-stimulated NO release was attenuated in SMP30 KO mice vs. WT mice (p<0.01). ACh indicates acetylcholine administration.
Concentration-dependent vasomotion of coronary arteries by ACh
We next assessed the coronary vascular response to ACh (1×10−10–1×10−5 M), which has endothelium-dependent vasodilative and vascular smooth muscle-dependent vasoconstrictive properties, in isolated pressurized coronary arteries of mice. In the SMP30 KO mice, ACh constricted the coronary arteries dose dependently (n=10, Fig. 2A). ACh-induced vasoconstriction did not change with Nω-nitro-l-arginine-methyl ester (L-NAME, 0.3 mM, n=10), an NOS inhibitor (Fig. 2A), or tetrahydrobiopterin (BH4, 1 μM, n=10, Fig. 2B). With pretreatment of dithiothreitol (DTT, 0.1 μM), a thiol-reducing agent, the ACh response to coronary arteries changed from vasoconstriction to vasodilation in the SMP30 KO mice (n=10, p<0.01) (Fig. 2C). In the WT mice, ACh dilated coronary arteries dose dependently, and ACh-induced vasodilation was blunted by L-NAME (n=10, each, Fig. 2A). ACh-induced vasodilation in the coronary arteries of WT mice decreased with pretreatment of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU, 80 μM), an inhibitor of glutathione reductase (n=10, p<0.01), which was restored by DTT (n=10, Fig. 2D). Vasodilation of sodium nitroprusside (SNP, 1×10−10–1×10−5 M), an endothelium-independent vasodilator, was comparable between SMP30 KO and WT mice (n=10, each, Fig. 3). These results indicated that the NOS activity was deteriorated in the coronary arteries with SMP30 deficiency. Redox signaling is a candidate for regulation of the coronary vascular tone accompanied with thiol residue oxidation induced by ACh.
FIG. 2.

Isolated coronary artery responses to Ach (1×10−10–1×10−5 M ) in SMP30 KO and WT mice in an organ chamber. ACh-induced vasoconstriction appeared in the SMP30 KO mice. The NOS inhibitor, Nω-nitro-l-arginine-methyl ester (L-NAME) (0.3 mM), did not further enhance this response. In the WT mice, ACh-induced vasodilation appeared, blunted with L-NAME (A). BH4 (1 μM) did not change ACh-induced vasoconstriction in the SMP30 KO mice (B). ACh-induced vasoconstriction in the SMP30 KO mice changed to vasodilation with the thiol-reducing agent, dithiothreitol (DTT) (0.1 μM) (C). Inhibition of glutathione reductase with 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) (80 μM) decreased ACh-induced vasodilation, which was restored by DTT in the WT mice (D). Values were expressed as means±S.E.M., n=10, each, *p<0.01.
FIG. 3.

Sodium nitroprusside (SNP) response in isolated coronary arteries from SMP30 KO and WT mice in an organ chamber. SNP (1×10−10–1×10−5 M) induced vasodilation dose dependently, which was comparable between the SMP30 and WT mice. Values were expressed as means±S.E.M., n=10, each.
Generation of hydrogen peroxide
We assessed the hydrogen peroxide (H2O2) generation in the aorta of SMP30 KO and WT mice. SMP30 KO mice had a more than threefold increase in H2O2 levels of that measured in WT mice (18.1±1.8 vs. 4.6±0.7 pmol/mg protein/min for SMP30 KO mice vs. WT mice, n=10 each, p<0.01, Fig. 4).
FIG. 4.

Hydrogen peroxide (H2O2) generation in the aorta. H2O2 production determined by the Amplex-Red assay in the aortic tissue of WT and SMP30 KO mice. SMP30 KO mice had a more than threefold increase in H2O2 levels. Values were expressed as means±S.E.M., n=10, each, *p<0.01.
Concentration of asymmetric dimethylarginine
Asymmetric dimethylarginine (ADMA), an endogenous inhibitor of NO synthase, plays a pivotal role in endothelial dysfunction (23). Plasma and aorta ADMA levels were higher in SMP30 KO mice than WT mice (plasma ADMA 1.3±0.1 vs. 0.7±0.1 nmol/ml, Fig. 5A; aorta ADMA 6.2±0.8 vs. 2.6±0.3 nmol/ml, n=10 each, p<0.01, respectively, Fig. 5B). These results suggest the possibility that SMP30 deficiency may impact l-arginine metabolism of eNOS, and ADMA may contribute to the increase in superoxide generation.
FIG. 5.

Plasma and aorta asymmetric dimethylarginine (ADMA) levels. Plasma ADMA was higher in SMP30 KO mice than WT mice (A). Aorta ADMA was similarly higher in SMP30 KO mice (B). Values were expressed as means±S.E.M., n=10, each, *p<0.01.
Total thiol and glutathione levels in aorta
To determine how SMP30 deficiency affects thiols, total thiols and reduced (GSH) and oxidized glutathione (GSSG) levels were measured in aortic tissue of SMP30 KO and WT mice. In the aortic tissue of SMP30 KO mice, decreases of total thiols, GSH level, and GSH/GSSG with increase of the GSSG level appeared compared to those in WT mice (total thiols 6.3±1.6 vs. 33.1±4.8 nmol/mg protein, Fig. 6A; GSH 5.1±0.8 vs. 18.6±2.1 nmol/mg protein, Fig. 6B; GSSG 1.9±0.3 vs. 1.1±0.2 nmol/mg protein, Fig. 6C; GSH/GSSG 2.8±0.5 vs. 14.8±1.8 in SMP30 KO mice vs. WT mice, n=10 each, p<0.01 respectively, Fig. 6D). From these results, depletion of SMP30 may affect the total thiol level in the aortic tissue.
FIG. 6.

Aortic tissue total thiols and glutathione levels. In aortic tissue, the levels of total thiols (A) and of reduced glutathione (GSH) (B) decreased in SMP30 KO mice compared to those in WT mice. In contrast, the level of oxidized glutathione (GSSG) increased in SMP30 KO mice (C). Deficiency of SMP30 led to a decrease in the ratio of reduced to oxidized glutathione (GSH/GSSG) (D). The values were expressed as means±S.E.M., n=10, each, *p<0.01.
Generation of O2•−in coronary artery
As for the isolated coronary arteries, the production of superoxide anion radical (O2•−) was measured by staining with dihydroethidium (DHE 10 μM). We then examined ROS generation in the coronary arteries of the SMP30 KO mice with the hypothesis that SMP30 deficiency exacerbates ROS generation. The signal of DHE staining was enhanced in the coronary arteries with SMP30 deficiency [54±6.2 fluorescence intensity/100 μm2 (arbitrary units), n=10] compared to that of WT mice (p<0.01). Apocynin (0.3 mM, n=10), an NADPH oxidase inhibitor, decreased this signal in the coronary arteries of the SMP30 KO mice (DHE 9.2±1.1 fluorescence intensity/100 μm2, n=10 p<0.01 vs. without agent) (Fig. 7). This result suggests that superoxide generation in SMP30 deficiency depends on NADPH oxidase in the coronary arteries.
FIG. 7.
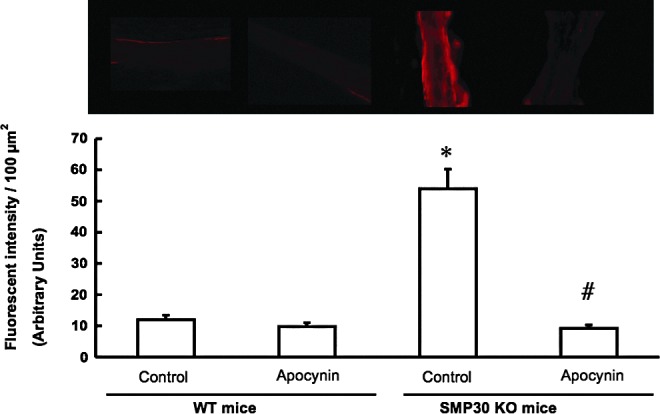
DHE staining in coronary arteries. Production of superoxide anion radical was measured by staining with 10 μM dihydroethidium (DHE) in the isolated coronary arteries. Upper panel shows representative DHE, and the lower panel shows summary data of DHE fluorescent intensity (in arbitrary units) in the coronary arteries. The signal of DHE was potent with SMP30 deficiency, which was attenuated with apocynin (0.3 mM). The values were expressed as means±S.E.M. *p<0.01 vs. without any treatment in WT mice, #p<0.01, vs. without treatment in SMP30 KO mice, n=10, each.
Oxidation of thiols in coronary arteries
To assess thiol oxidation in the coronary arteries, the location of thiols was determined by administration of fluorochrome monochlorobimane (MCB, 20 μM) or monobromotrimethyl ammoniobimane (MBB, 20 μM). Although these compounds covalently react with thiols, if the thiols are oxidized, the compounds do not bind. Because MCB is permeable to the cell membrane and MBB is not, the total thiols and extracellular thiols can be, respectively, labeled. Thus, a reduction in the fluorescence intensity means that the thiols were oxidized, and that there was less binding of either fluorochrome (33). We investigated the modification of thiol oxidation by ACh (1 μM) and DTT (0.1 μM). ACh treatment decreased the fluorescence levels due to MCB in the coronary arteries of the WT mice (MCB: 2.4±1.2 fluorescence intensity/100 μm2, p<0.01 vs. without treatment, n=10, each). The fluorescence levels due to MCB or MBB in the coronary arteries decreased in the SMP30 KO mice (MCB 4.2±1.2, MBB 24±4.4 fluorescence intensity/100 μm2, n=10 each, p<0.01, 0.05 vs. WT mice, respectively), and the level was restored with DTT treatment to a level comparable to that of the WT mice (SMP30 KO mice with DTT vs. WT mice: MCB 69±7.8 vs. 75±8.6, MBB 52±7.5 vs. 48±5.6, fluorescence intensity/100 μm2, n=10, each). The degree of fluorescence intensity reduction with ACh in the coronary arteries was potent in MCB staining compared to that of MBB staining. These results suggest that total thiols were oxidized in the SMP30 KO mice. Elevation of fluorescence of MCB or MBB by DTT indicates reduction of oxidized thiols. Ach oxidized intracellular thiols with NO generation in the WT mouse coronary artery. Deficiency of SMP30 extinguishes additional thiol oxidation with ACh (Fig. 8A, B). Under high levels of total thiols and GSH as seen in the WT mouse artery, ACh can release bioactive NO. However, deficiency of SMP30 shifts total thiols, including glutathione, reduced to oxidized, so ACh cannot release bioactive NO probably due to S-glutathionylation by oxidant stress in eNOS. From these results, we speculate that total thiol oxidation makes generation of bioactive NO in eNOS difficult.
FIG. 8.

Oxidation of thiols in coronary arteries. We investigated the modification of thiol oxidation with ACh (1 μM) or DTT (0.1 μM) in the isolated coronary arteries. Total or extracellular thiols were labeled with either monochlorobimane (MCB) (20 μM, A) or monobromotrimethyl ammoniobimane (MBB) (20 μM, B), respectively. Upper panels show fluorescent images of total reduced or extracellular reduced free thiol groups, and the lower panels show the summary data of fluorescent intensity (in arbitrary units) in isolated coronary arteries. A decrease in fluorescence indicates thiol oxidation with less binding of either fluorochrome. Fluorescence from MCB or MBB was decreased (increased thiol oxidation) in SMP30 KO mice compared to those of the WT mice. Ach decreased the MCB signal in the coronary arteries of the WT mice. Fluorescence to either indicator increased after administration of DTT (0.1 μM). Decrease of the fluorescent intensity was greater in MCB than in MBB of the SMP30 KO mice, indicating the effect on intracellular thiol oxidation. The values were expressed as means±S.E.M., n=10, each. *p<0.01, **p<0.05 versus without any treatment of WT mice. #p<0.01, ##p<0.05 versus without any treatment of SMP30 KO mice.
The ST-T segment change in electrocardiogram
Finally, we examined whether intra-aortic sinus administration of ACh induces coronary artery spasm in SMP30 KO mice. We determined that administration of 2 μg ACh is a suitable dose for examination of the coronary artery response in the in vivo condition. The duration and magnitude of the ST-T segment change by administration of 0.2 μg ACh were too small. In addition, the administration of 20 μg ACh decreased the blood pressure to 72%±8% as well as bradycardia. We thus administered 2 μg ACh to determine the effect of ACh on the ST-T segment change in the electrocardiogram. In the SMP30 KO mice, ischemic ST-T segment elevation appeared from 30 s to 1 min after ACh administration [0.35±0.06 mV (0.2–0.5 mV)] with reciprocal ST-T segment depression, and spontaneously returned to the baseline after around 2 min. In the WT mice, the ST-T segment did not change with ACh administration (Fig. 9A, B). These results implied that SMP30 KO mouse is a suitable model for the investigation of coronary artery spasm.
FIG. 9.

Ach-induced coronary artery spasm. Intra-aortic sinus administration of ACh (2 μg) to the SMP30 KO mice induced transient ST-T segment elevation and reciprocal ST-T segment depression in electrocardiogram (A). In the WT mice, ACh-induced ST-T segment change did not appear (B). Before, after 1, 2, and 5 min; before, after 1-, 2-, and 5-min intra-aortic sinus administration of ACh, respectively.
Discussion
To our knowledge, this is the first study indicating that thiol oxidation inhibits NO production and causes coronary artery spasm under SMP 30 deficiency. The new findings bear on the understanding of the mechanisms by which thiol oxidation modifies the coronary artery tone, and reveal how age-associated oxidant stress is a major risk factor of coronary vasospasm. First of all, coronary artery vasoconstriction or vasodilation is redox sensitive. A large fluorescent signal from either MCB or MBB in coronary arteries and high levels of total thiol and GSH in aorta appeared in WT mice. On the other hand, the signal for MCB was reduced in the coronary arteries of SMP30 KO mice (nearly eliminated), even more than for MBB with decreases of GSH and total thiols in the aorta. This suggests that the largest component of thiols oxidized (and thus less binding) is intracellular. The principal decrease of fluorescence was prevented or attenuated by DTT. Moreover, vasoconstriction in SMP30 KO mouse coronary arteries induced by ACh, which is widely used to investigate the bioavailability of NO and coronary artery spasm in clinical setting (2), was inhibited by DTT. BCNU, an inhibitor of glutathione reductase, attenuated vasodilation induced by ACh in WT mouse coronary arteries, which was reversed by DTT.
These results suggest that the redox condition of intracellular thiols mediates coronary vasospasm. Second, SMP30 has a protective action against oxidant damage that does not influence the antioxidant enzyme status (36). Increased levels of ROS, emphasized NADPH oxidase, and myeloperoxidase activities appeared in SMP30 KO mice (37). Therefore, SMP30 KO mice are suitable chronic oxidant stress models. Deficiency of SMP30 has a deletion of vitamin C biosynthesis (37). In this study, we fed vitamin C, including chow, and the level of vitamin C was not different among SMP30 KO and WT mice. Thus, the ACh-induced vascular response is unaffected by the level of vitamin C. However, the long-term effect of vitamin C treatment remains unknown because of previous data suggesting that there was improvement of NOS activity with an overdose chronic administration of vitamin C (8). Thus, further study is needed to clarify the effect of vitamin C on thiol oxidation.
Taken together, our results support the conclusion that redox-dependent vasoconstriction has an important role in the pathogenesis of coronary artery spasm that comes with aging. The present data are supported by a number of previous observations. NO synthesized in the presence of l-arginine and BH4 in eNOS has a critical role in the regulation of vascular function (31, 40). In the absence of BH4, NO synthesis is abrogated, and superoxide is generated instead. While NO dysfunction occurs increasingly with redox stresses such as aging, BH4 repletion only partly restores NOS activity and NOS-dependent vasodilation (9). It appeared in this study that pretreatment of BH4 did not attenuate ACh-induced vasoconstriction in vitro. Accordingly, coronary vasospasm in SMP30 KO mice induced by ACh is not due to the deletion of BH4. The protein thiol can undergo thiol oxidation, a reversible protein modification involved in cellular signaling and adaptation (12). Under oxidant stress, thiol oxidation such as S-glutathionylation in cysteine residues, which are critical for maintenance of eNOS function, occurs through a thiol–disulfide exchange with GSSG, which can only occur when the cellular GSH/GSSG ratio is low as shown in SMP30 KO mice (3, 11, 41), or a reaction of oxidant-induced protein thiyl radicals with reduced glutathione (GSH) (45).
Recently, it has been reported that S-glutathionylation of eNOS reversibly decreases the NOS activity with an increase in superoxide generation primarily from the reductase. In this state, two highly conserved cysteine residues are identified as the sites of S-glutathionylation, and found to be critical for redox regulation of eNOS function (6). It has been reported that GSH synthase inhibition without oxidation by buthionine-(S, R)-sulfoximine had no effect on NO bioactivity (16). However, thiol depletion in vivo greatly reduces NO generation from eNOS (7, 14, 15). Decrease of NO bioactivity with GSH depletion under oxidant stress has been reported (44), and thiol-oxidizing agents such as diamide decreased both the GSH level and NO bioactivity (16). Therefore, we speculate that decreases of GSH and total thiols, including cysteine residues in the eNOS domain, by oxidative stress may contribute to impairment of NO generation in SMP30 KO mice. However, the possibility that ADMA, which increased in plasma and aortic tissue of SMP30 KO mice, has a role in the deterioration of endothelium-dependent vasodilation under SMP30 deficiency cannot be ruled out.
Although the main source of superoxide production in SMP30 KO mouse coronary arteries is undefined, we speculate that it is an NAD(P)H oxidase-dependent O2•− production. There is increasing evidence that NAD(P)H oxidase is a major source of O2•− in the vasculature, and that O2•− from this enzyme serves as an important physiological redox-signaling molecule, participating in the regulation of vascular function associated with a novel Ca2+-signaling pathway (13, 29, 47). In addition, deficiency of SMP30, also reported as a regucalcin, induces the lack of vitamin C biosynthesization, impairs the scavenging effect of ROS, and increases the NADPH oxidase activity in vessels (22, 42). From this evidence, we propose that an augmentation of NADPH oxidase activity might trigger an increase in intracellular Ca2+ stress. This will lead to a coronary vasospasm in the SMP30 KO model, though further study is needed.
A limitation of our present study is that we did not observe coronary angiograms during vasospasm attacks, although ischemic ST-T elevation and reciprocal ST-T segment depression in electrocardiograms were transient, and SNP was effective in releasing the ST-T segment change. Moreover, a similar transient ST-T segment change appeared with intravenous administration of 5-hydroxytryptamine (data not shown). Thus, it is possible that the coronary artery spasm in this model is constant, not provocatively agent dependent. Therefore, SMP30 KO mouse may be a useful model to investigate oxidant stress-mediated coronary artery spasm.
In conclusion, thiol oxidation is a pivotal switch for inducing endothelial dysfunction and coronary artery spasm, providing a redox modification of the coronary vascular tone in aging.
Materials and Methods
Animal condition
SMP30 KO mice were bred from C57BL/6 mice by a gene-targeting technique, as described previously (17). WT C57BL/6 and SMP30 KO mice (age 8–10 W, B.W., 22.5±2.6 g) were housed and bred in a room at 22°C±3°C, with a relative humidity of 50%±10% and a 12-h light–dark cycle. They were given food, including vitamin C 21 mg/100 g (CLEA Japan, Tokyo, Japan), and water ad libitum. This investigation conformed to the Guidelines on Animal Experiments of Fukushima Medical University, the Japanese Government Animal Protection and Management Law (No. 105), and the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996).
All reagents were obtained from Sigma-Aldrich.
Measurements of NO and vitamin C level
After the mice were anesthetized by an intraperitoneal injection of sodium pentobarbital (50 mg/kg) and mid-sternotomies were performed, the aortas were isolated. In the aortas of the SMP30 KO mice (n=10, 45.5±7.8 mg) and WT mice (n=10, 49.3±5.8 mg), NO concentrations induced by ACh were measured by a free-radical analyzer (Apollo 4000; WPI Co., Ltd.) (32). Vitamin C in the aortas was measured by a high-performance liquid chromatography with an electrochemical detection method (18).
Experimental procedures
The mice were heparinized (400 units/kg i.p.) and anesthetized with sodium pentobarbital (50 mg/kg, i.p.). The hearts were removed and placed in a buffered physiological salt solution (PSS) to pH 7.4 at 4°C, and prepared for vessel dissection. The bathing solution used for vessel dissection had the following composition (in mM): NaCl 145.0, KCl 4.7, CaCl2 2.0, MgSO4 1.17, NaH2PO4 1.2, glucose 5.0, pyruvate 2.0, EDTA 0.02, 3-N-morpholino-propane sulfonic acid buffer (MOPS) 3.0, and 1% bovine serum albumin (1 g/100 ml).
Left anterior descending arteries or circumflex coronary arteries from the left ventricle [84±8 (70–112) μm in diameter] were isolated under a microscope (24, 33, 34). Each artery with its surrounding ventricular muscle was excised, transferred to a temperature-controlled dissection dish (4°C) containing PSS, and dissected free of muscle tissue. The side branches were tied off using an 11-0 suture. The vessels were transferred to a Lucite chamber and cannulated at both ends using micropipettes and pressurized at 60 mmHg. The arteries were tied to each pipette using an 11-0 suture. The PSS, bubbled with 20% O2, 5% CO2, and 75% N2, used to perfuse the vessels during the experiments was buffered to pH 7.4 at 37°C. The preparation was then transferred to the stage of an inverted microscope. To assess leaks, the pressure at zero flow was measured, which should be equal to that of the inflow reservoir pressure when there are no leaks. Any preparations showing leaks were discarded. The vessels were slowly warmed to 37°C and allowed to develop a spontaneous tone.
Measurement of vessel diameter
First, the control inner diameter of the isolated coronary artery from the SMP30 KO and WT mice was measured at 37°C in a vessel chamber (2 ml) with an Olympus IX71 inverted microscope. In the SMP30 KO mice, vasodilative responses to increasing concentrations of ACh with or without DTT, L-NAME, and BH4, were measured. In the WT mice, vasodilative responses to increasing concentrations of ACh with or without BCNU, DTT, or L-NAME were measured. In the SMP30 KO and WT mice, vasodilative responses to increasing concentrations of SNP were measured.
Amplex-red assay for H2O2 production
Freshly isolated aortic rings (4×2 mm) were used for assessment of H2O2 production using a fluorometric horseradish peroxidase assay (Amplex-Red assay; Molecular Probes). Fluorescence was measured (excitation 530 nm and emission 590 nm) after 1 h of incubation at 37°C in the dark against background fluorescence of the buffer. The polyethylene glycol-conjugated catalase (300 units/ml; Sigma) inhibitable fraction reflects a specific H2O2 signal. The rate of H2O2 production was presented as picomoles per milligram protein per minute after calculation, according to a standard curve generated using fresh H2O2 in the reaction buffer (5).
ADMA concentration
Plasma and tissue ADMA concentration was determined by using a commercially available enzyme-linked immunosorbent assay kit (DLD Diagnostika GmbH) according to the manufacturer's instructions (23).
Tissue glutathione and total thiol concentrations in aorta
Total thiols were determined in the aorta homogenates by measuring the absorbance of 5-thio-2-nitrobenzoic acid, the reaction product of the sulfhydryl groups with 5, 5′-dithiobis-2-nitrobenzoic acid (Ellman's reagent). An equal volume of 10% metaphosphoric acid was added to the samples; the resulting precipitated proteins were pelleted by centrifugation; and the supernatant was neutralized with 50 μl/ml of 4 M triethanolamine. Thiols were then measured by adding 200 μl Ellman's reagent from a commercially available assay to 50 μl of neutralized supernatant. The absorbance of the Ellman's reagent adduct was measured at 405 nm (39). Tissue concentrations of glutathione (total, reduced, and oxidized) were measured in tissue homogenates (10% wt/vol) after deproteinization with metaphosphoric acid in an enzymatic-recycling method with glutathione reductase as provided by a commercially available assay (Cayman Chemical Co.) (1). Values were normalized to protein concentration in the homogenate.
Measurement of fluorescence
The isolated arteries were placed on a slide glass and incubated for 5 min at 37°C with DHE or dichlorodihydrofluorescein fluorescence (DCF). To assess the distribution of DHE or DCF, the arteries were scanned by an Olympus IX71 inverted microscope (Olympus CCD). DHE or DCF staining was also performed with apocynin (20 min). MCB or MBB was administered to the isolated coronary arteries without any treatment, or after the administration of ACh or DTT in the conditioned buffer (5 min). The vessels were exposed to the fluorochromes for 30 min. Fluorescence was measured using the fluorescein isothiocyanate excitation/emission spectra from digitized images, and normalized to the vessel area (expressed as intensity/100 μm2). All camera settings were maintained constant throughout the image analyses (33). We measured the fluorescence intensities of five isolated coronary arteries in each heart, and averaged the data (43).
Measurement of ST-T segment changes in electrocardiogram
After the mice were anesthetized with an intraperitoneal injection of 2,2,2-tribromoethanol (250 mg/kg), ACh was administered by inserting a 27-G catheter from the cervical artery to the aortic sinus in the SMP30 KO and WT mice. The standard limb leads, aVR, aVL, and aVF, of the electrocardiogram were recorded by an electrocardiograph during 1-min intervals, and were constantly monitored (ECG-9122; Nihon Kohden). Because the T-wave is very close to the QRS complex in rodents, the end of the QRS complex of each signal was considered to be the point at which the T-wave started. Changes in the ST-T segment were measured by manual verification. Therefore, ST height represented the distance (in mV) from the baseline to the line where the T-wave started (4).
Data analysis and statistics
All statistical analyses were performed using StatView software (Abacus Concept). The percentage change in the diameter was calculated from the diameter change induced by an intervention as a percentage of the total amount of active tone (maximal diameter – baseline diameter). A plus value indicates vasodilation, and a minus value indicates vasoconstriction. A two-way ANOVA for repeated measures, followed by Tukey's post hoc test, was used to determine the differences in vasodilation resulting from the various interventions. A one-way ANOVA was used to determine the changes in biochemical data and fluorescence intensities, followed by Tukey's test for multiple comparisons. A probability value of<0.05 was used to determine the statistical significance.
Abbreviations Used
- ACh
acetylcholine
- ADMA
asymmetric dimethylarginine
- BCNU
1,3-bis(2-chloroethyl)-1-nitrosourea
- BH4
tetrahydrobiopterin
- DCF
dichlorodihydro-fluorescein fluorescence
- DHE
dihydroethidium
- DTT
dithiothreitol
- eNOS
endothelial nitric oxide synthase
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- H2O2
hydrogen peroxide
- KO
knockout
- L-NAME
Nω-nitro-l-arginine-methyl ester
- MBB
monobromotrimethyl ammoniobimane
- MCB
monochlorobimane
- NO
nitric oxide
- PSS
physiological salt solution
- ROS
reactive oxygen species
- SMP30
senescence marker protein-30
- SNP
sodium nitroprusside
- WT
wild type
Acknowledgments
This study was supported in part by grants-in-aid for Scientific Research (Nos. 21590935 and 24591100) from the Japan Society for the Promotion of Science.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Baker MA. Cerniglia GJ. Zaman A. Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal Biochem. 1990;190:360–365. doi: 10.1016/0003-2697(90)90208-q. [DOI] [PubMed] [Google Scholar]
- 2.Beltrame JF. Sasayama S. Maseri A. Racial heterogeneity in coronary artery vasomotor reactivity: differences between Japanese and Caucasian patients. J Am Coll Cardiol. 1999;33:1442–1452. doi: 10.1016/s0735-1097(99)00073-x. [DOI] [PubMed] [Google Scholar]
- 3.Biswas S. Chida AS. Rahman I. Redox modifications of protein-thiols: emerging roles in cell signaling. Biochem Pharmacol. 2006;71:551–564. doi: 10.1016/j.bcp.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 4.Bostick B. Yue Y. Duan D. Phenotyping cardiac gene therapy in mice. Methods Mol Biol. 2011;709:91–104. doi: 10.1007/978-1-61737-982-6_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai H. McNally JS. Weber M. Harrison DG. Oscillatory shear stress upregulation of endothelial nitric oxide synthase requires intracellular hydrogen peroxide and CaMKII. J Mol Cell Cardiol. 2004;37:121–125. doi: 10.1016/j.yjmcc.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 6.Chen CA. Wang TY. Varadharaj S. Reyes LA. Hemann C. Talukder MAH. Chen YR. Druhan LJ. Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–1121. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuzzocrea S. Zingarelli B. O'Connor M. Salzman AL. Szabó C. Effect of L-buthionine-(S,R)-sulphoximine, an inhibitor of gamma-glutamylcysteine synthetase on peroxynitrite- and endotoxic shock-induced vascular failure. Br J Pharmacol. 1998;123:525–537. doi: 10.1038/sj.bjp.0701612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.d'Uscio LV. Milstien S. Richardson D. Smith R. Katusic Z. Long-term vitamin C treatment increases vascular tetrahydrobiopterin levels and nitric oxide synthase activity. Circ Res. 2003;92:88–95. doi: 10.1161/01.res.0000049166.33035.62. [DOI] [PubMed] [Google Scholar]
- 9.Dumitrescu C. Biondi R. Xia Y. Cardounel AJ. Druhan LJ. Ambrosio G. Zweier JL. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci U S A. 2007;104:15081–15086. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finkel T. Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 11.Gallogly MM. Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 12.Glustarini D. Rossi R. Milzani A. Colombo R. Dalle-Donne I. S-glutathionylation: from redox regulation of protein functions to human disease. J Cell Mol Med. 2004;8:201–212. doi: 10.1111/j.1582-4934.2004.tb00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griendling KK. Sorescu D. Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 14.Harbrecht BG. Di Silvio M. Chough V. Kim YM. Simmons RL. Billiar TR. Glutathione regulates nitric oxide synthase in cultured hepatocytes. Ann Surg. 1997;225:76–87. doi: 10.1097/00000658-199701000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hothersall JS. Cunha FQ. Neild GH. Norohna-Dutra AA. Induction of nitric oxide synthesis in J774 cells lowers intracellular glutathione: effect of modulated glutathione redox status on nitric oxide synthase induction. Biochem J. 1997;322:477–481. doi: 10.1042/bj3220477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang A. Xiao H. Samii JM. Vita JA. Contrasting effects of thiol-modulating agents on endothelial NO bioactivity. Am J Physiol Cell Physiol. 2001;281:C719–C725. doi: 10.1152/ajpcell.2001.281.2.C719. [DOI] [PubMed] [Google Scholar]
- 17.Ishigami A. Fujita T. Handa S. Shirasawa T. Koseki H. Kitamura T. Senescence marker protein-30 knock-out mouse liver is highly susceptible to tumor necrosis factor-alpha and Fas-mediated apotosis. Am J Pathol. 2002;16:1273–1281. doi: 10.1016/s0002-9440(10)64404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwama M. Shimokado K. Maruyama N. Ishigami A. Time course of vitamin C distribution and absorption after oral administration in SMP30/GNL knockout mice. Nutrition. 2010;30:40916. doi: 10.1016/j.nut.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 19.Kashio Amano A. Kondo Y. Sakamoto T. Iwamura H. Suzuki M. Ishigami A. Yamasoba T. Effect of vitamin C depletion on age-related hearing loss in SMP30/GNL knockout mice. Biochem Biophys Res Commun. 2009;390:394–398. doi: 10.1016/j.bbrc.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Kondo Y. Sasaki T. Sato Y. Amano A. Aizawa S. Iwama M. Handa S. Shimada N. Fukuda M. Akita M. Lee J. Jeong KS. Maruyama N. Ishigami A. Vitamin C depletion increases superoxide generation in brains of SMP30/GNL knockout mice. Biochem Biophy Res Commun. 2008;377:291–296. doi: 10.1016/j.bbrc.2008.09.132. [DOI] [PubMed] [Google Scholar]
- 21.Kugiyama K. Ohsugi M. Motoyama T. Sugiyama S. Ogawa H. Yoshimura M. Inobe Y. Hirashima O. Kawano H. Soejima H. Yasue H. Nitric oxide-mediated flow-dependent dilation is impaired in coronary arteries in patients with coronary spastic angina. J Am Coll Cardiol. 1997;30:920–926. doi: 10.1016/s0735-1097(97)00236-2. [DOI] [PubMed] [Google Scholar]
- 22.Lai P. Yip NC. Michelangeli F. Regucalcin (RGN/SMP30) alters agonist—and thapsigagin—induced cytosolic [Ca2+] transients in cells by increasing SERCA Ca2+ ATPase levels. FEBS Lett. 2011;585:142902. doi: 10.1016/j.febslet.2011.05.058. [DOI] [PubMed] [Google Scholar]
- 23.Li Volti G. Salomone S. Sorrenti V. Mangiameli A. Urso V. Siarkos I. Galvano F. Salamone F. Effect of silibinin on endothelial dysfunction and ADMA levels in obese diabetic mice. Cardiovasc Diabetol. 2011;10:62. doi: 10.1186/1475-2840-10-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Machii H. Saitoh S. Kaneshiro T. Takeishi Y. Aging impairs myocardium-induced dilation in coronary arterioles: role of hydrogen peroxide and angiotensin. Mech Age Dev. 2010;131:710–717. doi: 10.1016/j.mad.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Maekawa K. Kawamoto K. Fuke S. Yoshioka R. Saito H. Saito T. Hioka T. Images cardiovascular medicine. Severe endothelial dysfunction after sirolimus-eluting stent implantation. Circulation. 2006;113:e850–e851. doi: 10.1161/CIRCULATIONAHA.105.597948. [DOI] [PubMed] [Google Scholar]
- 26.Mingone CJ. Gupte SA. Ali N. Oeckler RA. Wollin MS. Thiol oxidation inhibits nitric oxide-mediated pulmonary artery relaxation and guanylate cyclase stimulation. Am J Physiol Lung Cell Mol Physiol. 2006;290:l549–1557. doi: 10.1152/ajplung.00331.2005. [DOI] [PubMed] [Google Scholar]
- 27.Miwa K. Kishimoto C. Nakamura H. Makita T. Ishii K. Okuda N. Taniguchi A. Shioji K. Yodoi J. Sasayama S. Increased oxidative stress with elevated serum thioredoxin level in patients with coronary spastic angina. Clin Cardiol. 2003;26:177–181. doi: 10.1002/clc.4960260406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyata K. Shimokawa H. Yamawaki T. Kunihiro I. Zhou X. Higo T. Tanaka E. Katsumata N. Egashira K. Takeshita A. Endothelial vasodilator function is preserved at the spastic/inflammatory coronary lesions in pigs. Circulation. 1999;100:1432–1437. doi: 10.1161/01.cir.100.13.1432. [DOI] [PubMed] [Google Scholar]
- 29.Mohazzab KM. Wolin MS. Sites of superoxide anion production detected by lucigenin in calf pulmonary artery smooth muscle. Am J Physiol Lung Cell Mol Physiol. 1994;267:L815–L822. doi: 10.1152/ajplung.1994.267.6.L815. [DOI] [PubMed] [Google Scholar]
- 30.Moukarbel GV. Dakik HA. Diffuse coronary artery spasm induced by guidewire insertion. J Invasive Cardiol. 2003;15:353–354. [PubMed] [Google Scholar]
- 31.Palmer RM. Ashton DS. Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 32.Picchi A. Gao X. Belmadani S. Potter BJ. Focardi M. Chilian WM. Zhang C. Tumor necrosis factor-α induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res. 2006;99:69–77. doi: 10.1161/01.RES.0000229685.37402.80. [DOI] [PubMed] [Google Scholar]
- 33.Saitoh S. Kiyooka T. Rocic P. Rogers PA. Zhang C. Swafford A. Dick GM. Viswanathan C. Park Y. Chilian WM. Redox-dependent coronary metabolic dilation. Am J Physiol Heart Circ Physiol. 2007;293:H3720–H3725. doi: 10.1152/ajpheart.00436.2007. [DOI] [PubMed] [Google Scholar]
- 34.Saitoh S. Zhang C. Tune JD. Potter B. Kiyooka T. Rogers PA. Knudson JD. Dick GM. Swafford A. Chilian WM. Hydrogen peroxide: a feed-forward dilator that couples myocardial metablism to coronary flow. Arterioscler Thromb Vasc Biol. 2006;26:2614–2621. doi: 10.1161/01.ATV.0000249408.55796.da. [DOI] [PubMed] [Google Scholar]
- 35.Shimokawa H. Cellular and molecular mechanisms of coronary artery spasm. Jpn Circ J. 2000;64:1–12. doi: 10.1253/jcj.64.1. [DOI] [PubMed] [Google Scholar]
- 36.Son TG. Kim SJ. Kim K. Kim MS. Chung HY. Lee J. Cytoprotective roles of senescence marker protein 30 against intracellular calcium elevation and oxidative stress. Arch Pharm Res. 2008;31:872–877. doi: 10.1007/s12272-001-1240-3. [DOI] [PubMed] [Google Scholar]
- 37.Son TG. Zou Y. Jung KJ. Yu BP. Ishigami A. Maruyama N. Lee J. SMP30 deficiency causes increased oxidant stress in brain. Mech Age Dev. 2006;127:451–457. doi: 10.1016/j.mad.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Teragawa H. Kato M. Kurosawa J. Yamagata T. Matsuura H. Chayama K. Endothelial dysfunction is an independent factor responsible for vasospastic angina. Clin Sci (Lond) 2001;101:707–713. [PubMed] [Google Scholar]
- 39.Weiss N. Heydrick S. Zhang YY. Bierl C. Cap A. Loscalzo J. Cellular redox state and endothelial dysfunction in mildly hyperhomocysteinemic cystathionine β-synthase-deficient mice. Arterioscler Thromb Vasc Biol. 2002;22:34–41. doi: 10.1161/hq1201.100456. [DOI] [PubMed] [Google Scholar]
- 40.Werner ER. Gorren AC. Heller R. Werner-Felmayer G. Mayer B. Tetrahydrobiopterin and nitric oxide: mechanistic and pharmacological aspects. Exp Biol Med. 2003;228:1291–1300. doi: 10.1177/153537020322801108. [DOI] [PubMed] [Google Scholar]
- 41.Winterbourn CC. Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 42.Wison JX. Wu F. Vitamin C in sepsis. Subcell Biochem. 2012;56:67–83. doi: 10.1007/978-94-007-2199-9_5. [DOI] [PubMed] [Google Scholar]
- 43.Yamaguchi O. Kaneshiro T. Saitoh S. ishibashi T. Maruyama Y. Takeishi Y. regulation of coronary vascular tone via redox modulation in the α1-adrenergic-angiotensin-endothelin-axis of the myocardium. Am J Physiol Heart Circ Physiol. 2009;296:H226–H232. doi: 10.1152/ajpheart.00480.2008. [DOI] [PubMed] [Google Scholar]
- 44.Yan J. Tie G. Messina LM. Tetrahydrobiopterin, L-arginine and vitamin C act synergistically to decrease oxidative stress, increase nitricoxide and improve blood flow after induction of hindlimb ischemia in the rat. Mol Med. 2012;18:676–684. doi: 10.2119/molmed.2011.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ying J. Clavreul N. Sethuraman M. Adachi T. Cohen RA. Thiol oxidation in signaling and response to stress detection and quantification of physiological and pathophysiological thiol modifications. Free Radic Biol Med. 2006;43:1099–1108. doi: 10.1016/j.freeradbiomed.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zima AV. Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 47.Zulueta JJ. Yu FS. Hertig IA. Thannickal VJ. Hassoun PM. Release of hydrogen peroxide in response to hypoxia-reoxygenation: role of an NAD(P)H oxidase-like enzyme in endothelial cell plasma membrane. Am J Respir Cell Mol Biol. 1995;12:41–49. doi: 10.1165/ajrcmb.12.1.7529030. [DOI] [PubMed] [Google Scholar]