

Abstract
Context:
We describe the clinical investigation of the first generation aldosterone synthase inhibitor, LCI699, in patients with essential, uncontrolled, resistant, or secondary hypertension. LCI699 competitively reduced blood pressure at lower doses yet counterintuitive effects were observed at higher doses.
Objective and method:
An extensive endocrine biomarker analysis was performed to better understand the pharmacological mechanism of the drug.
Results:
The interference of LCI699 in the renin–angiotensin–aldosterone system occurred with limited target selectivity, as a dose-dependent compensatory stimulation of the hypothalamic-pituitary-adrenal feedback axis was discovered. Thus, LCI699 affected two endocrine feedback loops that converged at a single point, inhibiting the 11β-hydroxylase reaction in the adrenal gland, leading to supraphysiological levels of 11-deoxycortiscosterone. The accumulation of this potent mineralocorticoid may explain the blunted blood pressure response to LCI699.
Conclusion:
Future aldosterone synthase inhibitors may improve their target selectivity by sparing the 11β-hydroxylase reaction and preferentially inhibiting one of the two other enzymatic reactions mediated by aldosterone synthase.
Keywords: aldosterone synthase, endocrine, feedback, hypertension, hypothalamo-hypophyseal system
INTRODUCTION
Inhibition of the effects of aldosterone represents an important objective for cardiovascular disease management. The mineralocorticoid receptor blockers, spironolactone and eplerenone, demonstrated impressive outcome benefits in heart failure [1–4]. However, blockade of the mineralocorticoid receptors leads to a reactive increase of aldosterone secretion that may enhance nongenomic effects reported to regulate the contractility of the heart and coronary arteries [5,6]; affect tissues not protected by the receptor blocker such as the brain; and require compensatory higher doses of the blocker. Therefore, at least conceptually, the inhibition of aldosterone synthesis, similar to the successful development of aromatase inhibitors for estrogen suppression and hormone-dependent breast cancer prevention, would appear to represent a preferable alternative to receptor blockade [7].
Indeed, the concept of aldosterone synthase inhibition originated from the development of the nonsteroidal aromatase inhibitor fadrozole (CGS16949), which in a dose-escalation study revealed aldosterone-suppressing activity [8,9]. LCI699 is a proprietary, first-generation aldosterone synthase inhibitor that was structurally derived from FAD286, the enantiomer of fadrozole that harbors minimal aromatase activity while retaining potent aldosterone synthase inhibitory activity [10].
Aldosterone secretion from the adrenal gland, like blood pressure (BP) control, is circadian and governed by several factors that exert complex regulatory interactions. In humans, the circadian pacemaker activity mediates the secretion of adrenocorticotropic hormone (ACTH) and renin in the early morning, which controls the release of aldosterone with a peak level around waking (Fig. 1b–d). Subsequently, the renin–angiotensin–aldosterone system (RAAS) and potassium are the major regulators of aldosterone synthesis in the adrenal gland, whereas ACTH and sodium remain as minor modulators. In contrast, ACTH tightly controls the release of cortisol [11].
FIGURE 1.
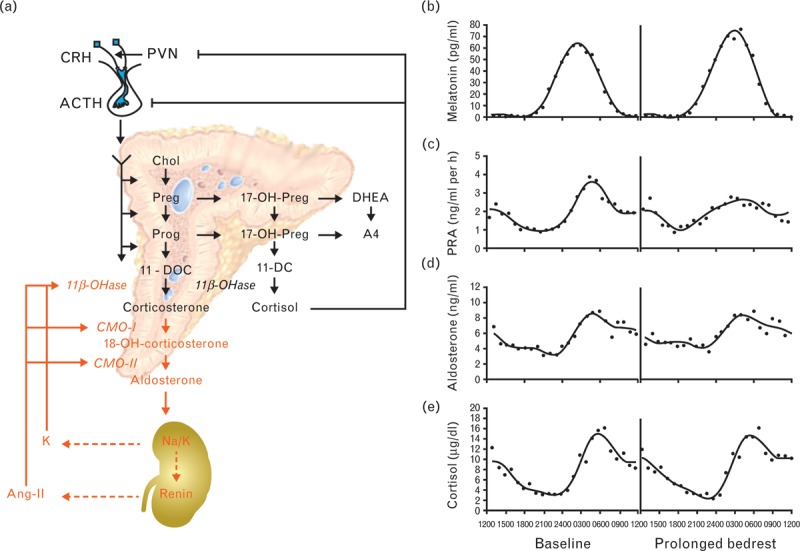
(a) Regulatory endocrine feedback loops that control early and late adrenal steroidogenesis. (b–e) Diurnal variation of aldosterone secretion. Baseline hormone peak times of melatonin (b), plasma renin activity (PRA) (c), aldosterone (d), and cortisol (e) [11,12]. 11β-OHase, 11β-hydroxylase; 11-DC, 11-deoxycortisol; 11-DOC, 11-deoxycorticosterone; 17-OH-Preg, 17-hydroxypregnenolone; 17-OH-Prog, 17-hydroxyprogesterone; 18-OH-corticosterone, 18-hydroxycorticosterone; A4, androstenedione; ACTH, adrenocorticotropin; Ang, angiotensin; Chol, cholesterol; CMO, corticosterone methyl oxidase; CRH, corticotropin-releasing hormone; DHEA, dehydroepiandrosterone; K, potassium; Na, sodium; Preg, pregnenolone; Prog, progesterone; PVN, paraventricular nucleus; RAAS, renin–angiotensin–aldosterone system. Figure 1b–e reproduced with permission [11].
The synthesis of aldosterone is controlled by the biosynthetic activity of the adrenal gland in general and the activity of the rate-limiting enzyme aldosterone synthase in particular. Specifically, aldosterone production can be regulated by modulating either one or both of two biosynthetic steps [12]. The early step is the conversion of cholesterol to pregnenolone and the late step is the conversion of deoxycorticosterone to aldosterone (Fig. 1a). The late step converts the three substrates of aldosterone synthase [cytochrome p450 (CYP) 11B2], 11-deoxycorticosterone, corticosterone, and 18-hydroxycorticosterone to aldosterone via three distinct reactions: an 11β-hydroxylase followed by an 18-hydroxylase and a final 18-isomerase reaction. In contrast, the late synthetic step from 11-deoxycortisol to cortisol is only mediated by an 11β-hydroxylase reaction (CYP11B1). As the pharmacological target CYP11B2 shares an enzymatic 11β-hydroxylase reaction and, therefore, a high sequence homology with CYP11B1, a dose-dependent and time-dependent monitoring of the mediators of the aldosterone and cortisol endocrine feedback loops was necessary to determine the selectivity, efficacy, and safety of LCI699.
Careful investigation of a new hormonal therapy provides an opportunity to analyze the components of the affected endocrine systems in order to characterize the mode of action of the drug, its target and potential related off-target pharmacodynamic properties as well as the short- and long-term adaptations of these systems that may affect the efficacy and the safety of the therapy. Given that eplerenone has a plasma half-life similar to LCI699, it was believed that evaluating both once-daily and twice-daily applications of LCI699 was warranted. Indeed, a twice-daily application of LCI699 may better prevent the circadian morning surge of aldosterone. The therapeutic responsiveness of a disease condition characterized by an absolute or relative aldosterone excess (i.e., relative to the renin activity) or even in the absence of an apparent excess was matter of conceptual debate. Therefore, the pharmacological profile of LCI699 was explored in four clinical phase II studies in patients with primary aldosteronism (absolute excess), resistant and uncontrolled hypertension (relative excess), and in essential hypertension (no apparent excess).
PATIENTS AND METHODS
Four clinical phase II studies with LCI699 in patients with hypertension have been sponsored by Novartis Pharma AG in Basel and are registered at http://www.clinicaltrials.gov. For all studies, the study was described by a nurse, co-ordinator, or investigator and informed consent was obtained from each participant in writing before randomization.
Study CLCI699A2201
Study CLCI699A2201 is a placebo and active controlled dose-finding study to evaluate the efficacy and safety of LCI699 in patients with essential hypertension (NCT00758524) [13]. This was a multicenter, randomized, double-blind, placebo-controlled and active-controlled, parallel group study to evaluate the efficacy and safety of LCI699 compared with placebo after 8 weeks’ treatment in patients with essential hypertension, and the methods have been presented elsewhere in more detail [13]. The primary objective was to evaluate the reduction in mean sitting DBP (MSDBP) 23–26 h postdose (11–14 h after twice-daily dosing).
Study CLCI699A2206
Study CLCI699A2206 is a proof-of-concept study for the aldosterone synthase inhibitor LCI699 in patients with primary hyperaldosteronism (NCT00732771) [14]. This was a 4-week treatment single-blind, placebo-controlled, sequential, and forced-titration study in 14 patients (18–70 years) diagnosed with primary aldosteronism within the past 3 years, and the methods have been presented elsewhere in more detail [14]. The main objectives of the study were to determine whether LCI699 would decrease aldosterone production, lower the mean 24-h ambulatory BP monitoring (ABPM), and increase the plasma potassium concentration compared with baseline. Plasma electrolytes and blood hormone levels were measured after patients had rested in the supine position for 1 h.
Study CLCI699A2215
Study CLCI699A2215 is an evaluation of the effects of LCI699 on cortisol in patients with hypertension (NCT00817414) [15]. This was a 6-week prospective, randomized, double-blind, placebo-controlled study of LCI699 in patients (18–75 years) with an established diagnosis of hypertension (>140/90 mmHg and <180/110 mmHg) and taking at least one antihypertensive treatment [15]. The primary objective of this study was to determine the maximally tolerated dose of LCI699 with respect to cortisol suppression following ACTH stimulation in patients with hypertension. The secondary objectives were to characterize the LCI699 exposure–response relationship on cortisol levels following ACTH stimulation.
Study CLCI699A2216
Study CLCI699A2216 is an evaluation of the effects of LCI699 on safety and efficacy in individuals with resistant hypertension (NCT00817635) [16]. This was a randomized, double-blind, placebo and active controlled, parallel-group, multicenter, dose-ranging study to evaluate the efficacy and safety of LCI699 compared with placebo and eplerenone after 8 weeks’ treatment in patients (18–75 years) with resistant hypertension [elevated mean sitting SBP (MSSBP) ≥140 and ≤180 mmHg taking three or more antihypertensive drugs, including a nonpotassium-sparing diuretic, at optimal doses] [16]. The primary objective of this study was to explore the efficacy of three LCI699 dose regimens with respect to the change from baseline in MSSBP compared with placebo.
Blood sampling and biomarker analysis
In order to further characterize the target and off-target pharmacodynamic effects of LCI699 in these studies, and to assess the response of endocrine feedback to the exposure of LCI699, morning plasma samples were subjected to an extensive biomarker analysis. Blood samples in all four clinical studies were drawn in the morning between 0700 and 1000 h from patients who had fasted overnight and after resting in seated position for approximately 15 min. The blood samples were sent to a central laboratory for analysis of biochemical markers, such as sodium and potassium, as well as hormonal markers, such as cortisol, 11-deoxycortisol, 11-deoxycorticosterone, ACTH, aldosterone, plasma renin concentration, or plasma renin activity. Cortisol, 11-deoxycortisol, and 11-deoxycorticosterone were measured using a standardized high performance liquid chromatography/tandem mass spectrometry (LC-MS/MS) method. Aldosterone, ACTH, renin concentration, and renin activity were measured using radioimmunoassay systems. Descriptive statistics for biochemical and hormonal markers were provided for all studies. The means and descriptive measures displayed here were obtained from the corresponding tables prepared for study reports.
RESULTS
Study CLCI699A2201: LCI699 in essential hypertension
LCI699 treatment resulted in both diastolic and systolic office BP reductions that were statistically significant compared with placebo [13]. Furthermore, LCI699 once daily showed a dose-dependent lowering in MSSBP with placebo-subtracted reductions of 6.07 ± 2.07 mmHg for the 0.25 mg dose (two-sided P value for change from baseline of 0.0035), 7.76 ± 1.87 for the 0.5 mg dose (P <0.0001), and 9.01 ± 2.11 for the 1 mg dose (P <0.0001). The 1 mg once-daily LCI699 dose was numerically noninferior to eplerenone at 50 mg twice daily and resulted in a placebo-subtracted reduction in MSSBP of 10.42 ± 2.28 mmHg (P <0.0001). Surprisingly, the placebo-subtracted MSSBP reduction of 6.19 ± 2.26 mmHg (P = 0.0063) achieved with LCI699 0.5 mg twice daily appeared to be inferior to dosing of 1.0 and 0.5 mg once daily, even though plasma aldosterone levels were further suppressed with this dose. As expected, the LCI699 twice-daily regimen effectively lowered plasma aldosterone levels in contrast to eplerenone, which raised plasma aldosterone levels due to its pharmacological mechanism of action (Fig. 2a). In addition, the other plasma biomarkers characterizing the aldosterone endocrine feedback loop showed the anticipated pharmacologically induced changes within reported reference values: that is, raised plasma renin activity, raised serum potassium, and decreased serum sodium, similar to treatment with eplerenone (Fig. 2b–d).
FIGURE 2.
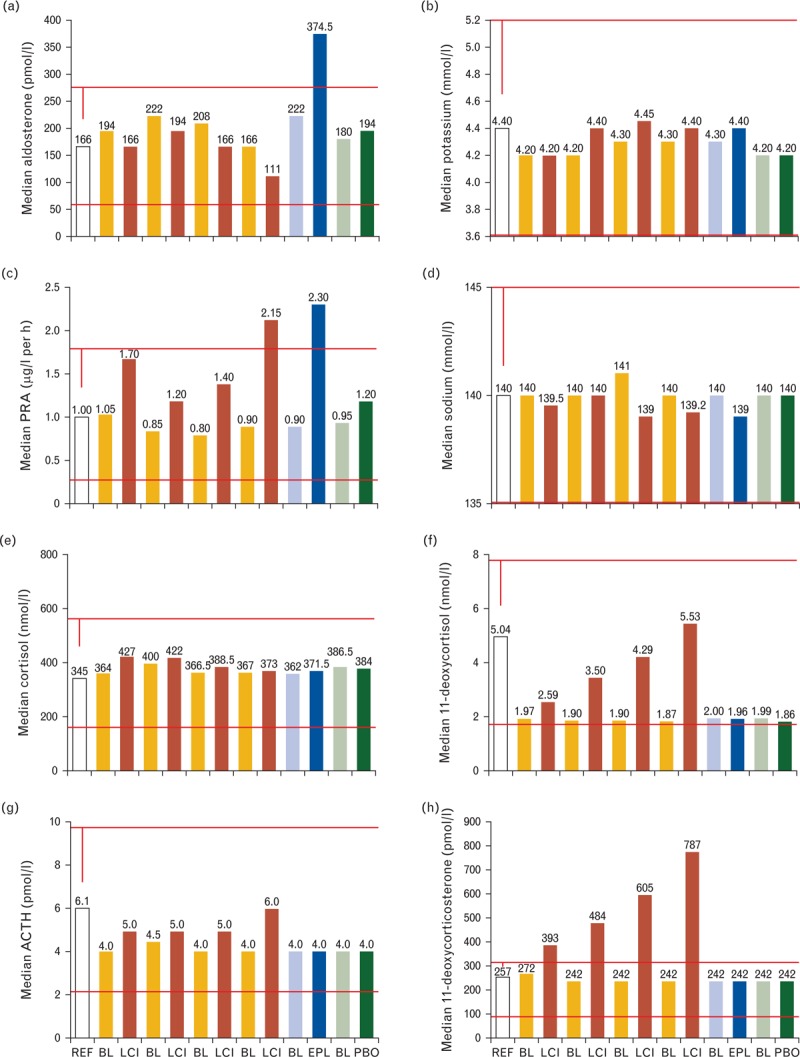
Plasma biomarkers derived from study CLCI6992201 in essential hypertension. (a) Aldosterone; (b) potassium; (c) plasma renin activity (PRA); (d) sodium; (e) cortisol; (f) 11-deoxycortisol; (g) adrenocorticotrophic hormone (ACTH); (h) 11-deoxycorticosterone. BL, baseline value for the respective adjacent study arm to the right; EPL, eplerenone 50 mg twice daily; LCI, LCI699 applied at 0.25 mg once daily, 0.5 mg once daily, 1.0 mg once daily, 0.5 mg twice daily from left to right; PBO, placebo; REF, clinical reference value.
The assessment of the hypothalamic-pituitary-adrenal (HPA) feedback axis revealed stable plasma cortisol levels (Fig. 2e). These values most likely reflect a compensatory endocrine feedback mechanism as LCI699 dose-dependently inhibits 11β-hydroxylase, as revealed by increasing enzymatic substrate levels of 11-deoxycortisol. The dose-dependent rise in ACTH levels confirms the induction of an endocrine feedback loop that results in the stimulation of adrenal steroidogenesis to compensate for the impaired cortisol synthesis. However, the adrenal compensation in the presence of LCI699 leads to a significant elevation of circulating 11-deoxycorticosterone beyond the normal reference values for this substrate of aldosterone synthase. Not surprisingly, in view of the ACTH response, the LCI699-induced increase of 11-deoxycorticosterone showed a clear dose–response relationship with 0.5 mg twice-daily dosing superior to 1.0 mg once-daily dosing (Fig. 2h).
Study CLCI699A2206: LCI699 in primary hyperaldosteronism
The primary endpoint of BP reduction was met, although BP reductions were modest [14]. The mean 24-h and day-time ambulatory SBP (ASBP) was reduced by only 4.2 mmHg (P = 0.0441 and P = 0.0355) compared with baseline. The anticipated marked BP reduction was not observed in spite of the complete normalization of the excessive plasma aldosterone levels and the normalization of hypokalemia (Supplemental digital content: Fig. 1). The increase in renin activity and a slight decrease in serum sodium further reflected the inhibitory effect on aldosterone synthesis.
The assessment of the HPA feedback axis revealed stable plasma cortisol values yet increased 11-deoxycortisol and ACTH values again indicating inhibition of 11β-hydroxylase and compensatory induction of the endocrine feedback loop (Supplemental digital content: Fig. 1). In the presence of LCI699, the stimulated adrenal steroidogenesis led to a dramatic increase of the aldosterone synthase substrate, 11-deoxycorticosterone. The excessive values for deoxycorticosterone returned to normal within a week of LCI699 withdrawal (Supplemental digital content: Fig. 1).
Study CLCI699A2215: LCI699 on cortisol in hypertension
The study showed a clear dose-dependence of the ACTH stress test for LCI699 and indicated a time-dependence for the maximal onset and reversal [15]. Furthermore, a dose-dependent treatment effect on BP was apparent for once-daily dosing (mean change in MSSBP from baseline to day 43 were –11.2, –12.4, –14.9, and –13.3 mmHg for LCI699 0.5 mg once daily, 1.0 mg once daily, 1.0 mg twice daily, and 2.0 mg once daily, respectively, vs.–2.4 mmHg with placebo). The plasma biomarkers for aldosterone, serum potassium, plasma renin activity, and serum sodium revealed pharmacologically induced changes consistent with an inhibition of aldosterone synthase (Supplemental digital content: Fig. 2).
Once again, the changes in the HPA feedback axis reflected the observed dose-dependent impairment of the ACTH stress test (Supplemental digital content: Fig. 1). The increased 11-deoxycortisol levels in the presence of stable cortisol values are indicative of an inhibitory effect on 11β-hydroxylase and explain the inferior cortisol excursions upon exogenous ACTH stimulation. The compensatory induction of endogenous ACTH stimulates adrenal steroidogenesis and mediates the accumulation of 11-deoxycorticosterone in the presence of aldosterone synthase inhibition. The administration of LCI699 1 mg twice daily induced increases in 11-deoxycorticosterone, 11-deoxycortisol, and ACTH beyond the normal reference ranges (Supplemental digital content: Fig. 2).
Study CLCI699A2216: LCI699 in resistant hypertension
In this study, the reported BP-lowering effects of LCI699 were inferior to eplerenone (change from baseline MSSBP: LCI699 0.25 mg twice daily, –11.4 mmHg; 0.5/1.0 mg twice daily, –12.5 mmHg; 1.0 mg once daily, –13.1 mmHg; eplerenone 50 mg twice daily, –18.7 mmHg; placebo –8.8 mmHg) [16]. The plasma biomarkers for potassium, renin activity, and sodium revealed pharmacologically induced changes similar to eplerenone (Fig. 3b–d). However, plasma aldosterone levels decreased upon LCI699 treatment, which was in contrast to eplerenone exposure (Fig. 3a). The biomarker assessment of the HPA axis confirmed an inhibitory effect on 11β-hydroxylase and a compensatory stimulation of adrenal steroidogenesis leading to the excessive accumulation of 11-deoxycorticosterone (Fig. 3e–h).
FIGURE 3.
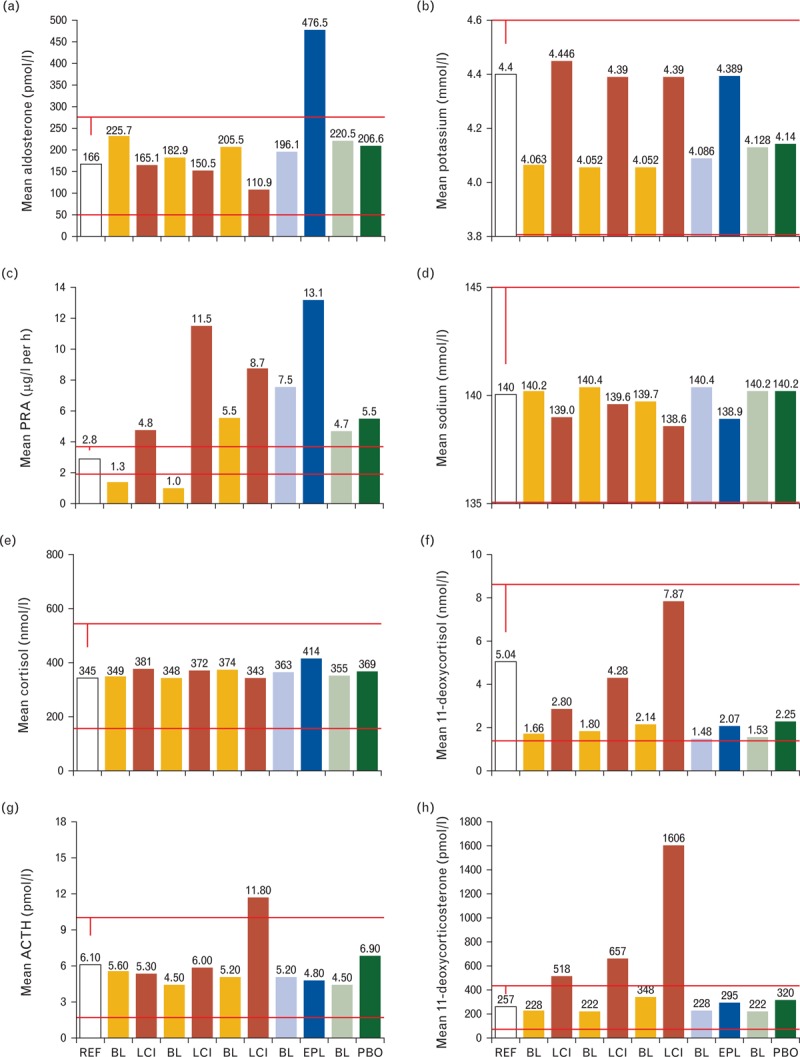
Plasma biomarkers derived from study CLCI699A2216 in resistant hypertension. (a) Aldosterone; (b) potassium; (c) plasma renin activity (PRA); (d) sodium; (e) cortisol; (f) 11-deoxycortisol; (g) adrenocorticotrophic hormone (ACTH); (h) 11-deoxycorticosterone. BL, baseline value for the respective adjacent study arm to the right; EPL, eplerenone 50 mg twice daily; LCI, LCI699 applied at 0.25 mg twice daily, 1.0 mg once daily, 0.5 mg/1.0 mg twice daily from left to right; PBO, placebo; REF, clinical reference value.
Summary
The four phase II clinical studies in patients with various forms of hypertension revealed a consistent interference of LCI699 in the aldosterone and HPA endocrine feedback loops. The accumulation of 11-deoxycorticosterone was excessive beyond the normal reference ranges and shows a clear dose-dependence, with enhanced sensitivity to twice-daily dosing regimens (Fig. 4). An indirect comparison of LCI699 on the induction of 11-deoxycorticosterone and reduction of plasma aldosterone levels with fadrozole shows that treatment of healthy volunteers with fadrozole 2 mg twice daily for 2 weeks impaired aldosterone synthase, clearly reduced plasma aldosterone, and only slightly increased substrate values [17]. However, the increase of 11-deoxycorticosterone upon fadrozole exposure was markedly less than the levels observed with the administration of LCI699 at relatively similar levels of aldosterone suppression.
FIGURE 4.
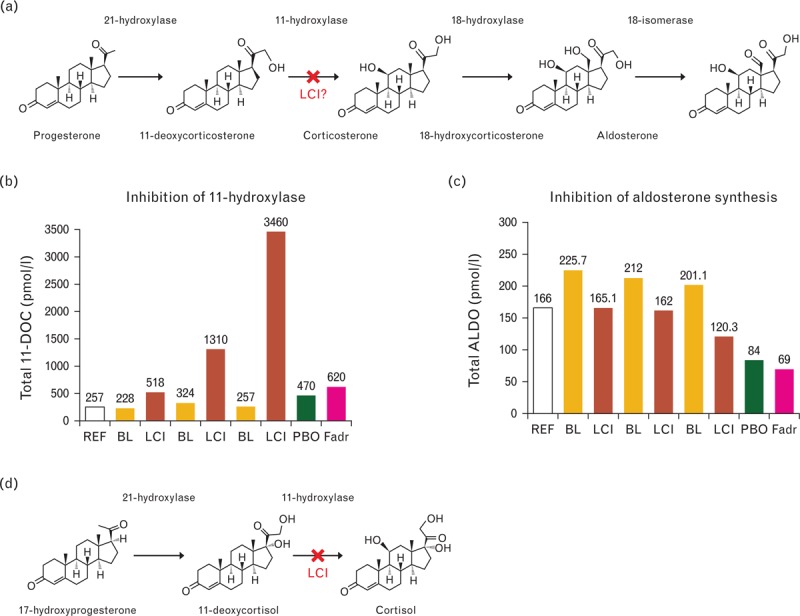
LCI699, in contrast to fadrozole, is primarily an 11β-hydroxylase inhibitor. X indicates the suspected enzyme inhibitory step for LCI699. The comparison of LCI699 with fadrozole is a dose compilation of clinical studies with LCI699 described herein and fadrozole reference data. BL, baseline value for the respective adjacent LCI699 study arm to the right; Fadr, fadrozole applied at 2.0 mg twice daily (derived from a healthy volunteer study published by Trunet et al. 1992 [17]); LCI, LCI699 applied at 0.25 mg twice daily (derived from study CLCI6992216), 0.5 mg twice daily (derived from study CLCI699A2201), and 1.0 mg twice daily (derived from study CLCI699A2215); PBO, placebo control for the fadrozole study arm; REF, clinical reference value.
DISCUSSION
LCI699 established a consistent pharmacological profile across all four hypertension studies demonstrating a dose-dependent and administration-dependent (once or twice daily) reduction in BP. However, further investigation revealed interference with two endocrine feedback loops that converge at the adrenal gland: the RAAS and the HPA axis. Inhibition of aldosterone synthase and the resulting suppression of aldosterone levels was demonstrated by the expected stimulation of the RAAS feedback axis with a slight decrease in plasma sodium and an increase in plasma potassium levels as well as stimulation of the plasma renin concentration and activity. Unfortunately, the observed increase in 11-deoxycortisol levels indicated off-target inhibition of the 11β-hydroxylase activity of CYP11B1 that resulted in the stimulation of the HPA feedback axis. The resulting increase in ACTH levels stimulated adrenal steroidogenesis to compensate for the inhibited cortisol secretion. The consequences of an ACTH-stimulated adrenal gland in the presence of a pharmacologically inhibited aldosterone synthase, thus, produced a supraphysiological increase of the aldosterone synthase substrate, 11-deoxycorticosterone.
The biological activity of steroid hormones can be explained by their unbound availability. At physiological concentration, 96% of circulating cortisol or 11-dexocorticosterone is protein-bound. Conversely, aldosterone is weakly protein bound (63%) and most of the rest is free [18,19]. The steroid-binding plasma proteins determine the biological hormone activity as the intracellular steroid concentration – and therefore the induced transcriptional activity – is proportional to the free steroid in the plasma and not the total [12]. As such, the total (free and protein-bound) plasma concentration of aldosterone is in the range of 11-deoxycorticosterone, yet significantly lower than the concentration of corticosterone. If one compares the free plasma steroid fractions, aldosterone dominates the level of 11-deoxycorticosterone yet remains lower than corticosterone. However, the biological activity of corticosterone is limited by its weak mineralocorticoid activity, which is likely two orders of magnitude lower than aldosterone and 11-deoxycorticosterone. Therefore, the biological activity of aldosterone is assured by the tight protein binding of the potent mineralocorticoid 11-deoxycorticosterone and the weak mineralocorticoid activity of corticosterone [20,21].
The 11-deoxycorticosterone plasma levels induced by LCI699 increased up to 10-fold from baseline. Should the free steroid fraction remain constant, LCI699 would induce significant levels of biologically active 11-deoxycorticosterone. The relative mineralocorticoid receptor stimulation of 11-deoxycorticosterone would further increase in the presence of reduced aldosterone levels. The unselective steroidogenesis inhibitor metyrapone, used for Cushing's disease, has been reported to increase 11-deoxycorticosterone levels, and this has been associated with BP increases and the occurrence of hypokalemia [22,23]. Thus, the LCI699-induced increase in 11-deoxycorticosterone levels may very well explain the observed disappointing BP reductions achieved with the compound at higher doses and particularly upon twice-daily administration. The potassium-sparing activity of LCI699, however, seems less affected by increasing 11-deoxycorticosterone levels. Consistently, the lower and once-daily applied doses of LCI699, despite modest plasma aldosterone reductions, resulted in relatively better BP reductions as observed in study CLCI699A2201 in untreated patients with essential hypertension.
The fact that urinary sodium and potassium excretion was not measured to quantitate the overall mineralocorticoid effect might be considered a certain limitation of these efficacy trials. However, under steady state conditions, when the present measurements were carried out, electrolytes in the urine may predominantly reflect the amount of sodium and potassium ingested rather than the prevailing mineralocorticoid effect. Careful cumulative balance studies would have been required to quantitate the overall mineralocorticoid effect and these were not possible in the framework of these investigations.
What mechanistic profile should an aldosterone inhibitor ideally exhibit to be therapeutically successful? We propose the following features based on the clinical studies with LCI699 in hypertension, the mechanistic observations with fadrozole in a dose-escalation study, and two genetic inborn errors in the terminal step of aldosterone synthesis.
First, the clinical data with LCI699 clearly indicate that a compound must exhibit a significant separation between aldosterone synthase inhibition on CYP11B2 and 11β-hydroxylase inhibition on CYP11B1. Even without apparent changes in plasma cortisol levels, increasing concentrations of the enzyme substrate 11-deoxycortisol indicate 11β-hydroxylase inhibitory activity and subsequent induction of the HPA feedback axis to compensate for an impairment of cortisol synthesis via stimulation of adrenal steroidogenesis. Because ACTH stimulates the first enzymatic steps in adrenal steroidogenesis, it stimulates the synthesis of glucocorticoids, mineralocorticoids, as well as adrenal androgens and long-term hypertrophy of the adrenal gland (Fig. 1).
Second, an aldosterone synthase inhibitor should have a longer plasma elimination half-life than LCI699, as the aldosterone wake surge should be suppressed with either an evening or a morning drug administration. In addition, a compound with a longer half-life allows the establishment of narrower therapeutic drug plasma levels; that is, it avoids high peak–trough drug level ranges that affect the compound selectivity at the peak level and the compound efficacy at the trough level.
Third, an ideal aldosterone synthesis inhibitor would inhibit the conversion of a weak mineralocorticoid precursor into aldosterone. As aldosterone synthase catalyzes three enzymatic steps, the enzyme offers potentially three target sites for an enzyme inhibitor with different substrate:product ratios. Fadrozole treatment increased the ratios of plasma 18-hydroxycorticosterone/aldosterone as well as urinary 18-hydroxy-tetrahydroaldosterone/tetrahydroaldosterone [24]. As these ratio alterations were nearly identical to those previously observed in members of a family with corticosterone methyloxidase type II defect, the authors concluded that fadrozole inhibits aldosterone synthesis primarily via inhibition of the 18-isomerase reaction (corticosterone methyloxidase type II). In this case, the conversion of an enzyme substrate of weak mineralocorticoid activity is inhibited with fadrozole. Therefore, we conclude that a pharmacological agent for aldosterone synthase inhibition should preferentially inhibit the 18-isomerase step in order to build up the weak mineralocorticoids18-hydroxycorticosterone and corticosterone.
Has the objective of selective aldosterone synthesis inhibition been achieved? Unfortunately not yet, as LCI699 has demonstrated its limitations. However, based on the observations made with LCI699, fadrozole, and FAD286, the therapeutic strategy remains viable and technically feasible. A selective aldosterone synthesis inhibitor may be complementary or an alternative to spironolactone for the treatment of conditions that require high spironolactone doses, such as in primary aldosteronism or hepatic cirrhosis as doses beyond 50 mg per day have been clearly associated with endocrine side-effects. A selective aldosterone synthase inhibitor in conditions treated with lower doses of spironolactone or eplerenone, such as chronic heart failure and hypertension, may allow a better dose-dependent normalization of aldosterone and potassium levels.
ACKNOWLEDGEMENTS
The authors are grateful to Georgina Bermann for the statistical evaluation of the biomarker concentrations and thank Teresa Gerlock for critically reviewing the article.
They would like to thank Graham Allcock of CircleScience for providing editorial assistance, which was funded by Novartis.
C.S. is a full time employee of Novartis and contributed to the data evaluation, interpretation, and drafting of the article. R.S. contributed to the data evaluation and interpretation, and critical reviewing of the article. H.B. contributed to the data interpretation and drafting of the article.
Clinical trial registrations: CLCI699A2201 = NCT00758524; CLCI699A2206 = NCT00732771; CLCI699A2215 = NCT00817414; CLCI699A2216 = NCT00817635 (http://www.clinicaltrials.gov)
Conflicts of interest
The clinical trials were sponsored by Novartis.
R.S. is a former employee of Novartis and has received consulting fees from it.
H.B. has also received consulting fees from Novartis.
Supplementary Material
Reviewers’ Summary Evaluations
Referee 1
Schumacher et al. describe the clinical investigation of a first generation aldosterone synthase inhibitor (LCI699) in patients with essential hypertension, primary aldosteronism and resistant arterial hypertension. The inhibition of aldosterone synthase is an interesting approach in order to interfere with aldosterone effects on the cardiovascular system, reduce nongenomic effects on target organ, and protect tissues potentially without the known limitations of mineralocorticoid receptor blockers. Unfortunately, LCI699 was found to indirectly affect the hypothalamic-pituitary-adrenal axis thus leading to compensatory accumulation of ACTH and 11-deoxycortisterone. As a consequence of this, the lower, once daily administered dose of LCI699 resulted in grater BP reductions.
Referee 2
This is an interesting hypothesis-generating analysis of clinical studies of an aldosterone synthase inhibitor. The authors provide evidence that 11-deoxycorticosterone accumulates when LCI699 is given twice a day and hypothesize that activation of the mineralocorticoid receptor by this precursor could reduce the beneficial effects of decreased aldosterone formation. The study would have been strengthened by inclusion of urinary sodium and potassium data.
Footnotes
Correspondence to Hans R. Brunner, Bahnhofstrasse 50, 4125 Riehen, Switzerland. Tel: +41 61 641 2510; e-mail: hrbrunner13@gmail.com
Abbreviations: ABPM, ambulatory blood pressure monitoring; ACTH, adrenocorticotropic hormone; ASBP, ambulatory SBP; BP, blood pressure; HPA, hypothalamic-pituitary-adrenal; LC-MS/MS, liquid chromatography/tandem mass spectrometry; MSDBP, mean sitting DBP; MSSBP, mean sitting SBP; RAAS, renin–angiotensin–aldosterone system
REFERENCES
- 1.Brunner HR, Laragh JH, Baer L, Newton MA, Goodwin FT, Krakoff LR, et al. Essential hypertension: renin and aldosterone, heart attack and stroke. N Engl J Med 1972; 286:441–449 [DOI] [PubMed] [Google Scholar]
- 2.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717 [DOI] [PubMed] [Google Scholar]
- 3.Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321erratum 348: 2271 [DOI] [PubMed] [Google Scholar]
- 4.Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21 [DOI] [PubMed] [Google Scholar]
- 5.Chai W, Danser AH. Why are mineralocorticoid receptor antagonists cardioprotective? Naunyn Schmiedebergs Arch Pharmacol 2006; 374:153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schiffrin EL. Effects of aldosterone on the vasculature. Hypertension 2006; 47:312–318 [DOI] [PubMed] [Google Scholar]
- 7.Santen RJ, Brodie H, Simpson ER, Siiteri PJ, Brodie A. History of aromatase: saga of an important biological mediator and therapeutic target. Endocr Rev 2009; 30:343–375 [DOI] [PubMed] [Google Scholar]
- 8.Browne LJ, Gude C, Rodriguez H, Steele RE, Bhatnager A. Fadrozole hydrochloride: a potent, selective, nonsteroidal inhibitor of aromatase for the treatment of estrogen-dependent disease. J Med Chem 1991; 34:725–736 [DOI] [PubMed] [Google Scholar]
- 9.Lipton A, Harvey HA, Demers LM, Hanagan JR, Mulagha MT, Kochak GM, et al. A phase I trial of CGS 16949A. A new aromatase inhibitor. Cancer 1990; 65:1279–1285 [DOI] [PubMed] [Google Scholar]
- 10.Ménard J, Pascoe L. Can the dextroenantiomer of the aromatase inhibitor fadrozole be useful for clinical investigation of aldosterone-synthase inhibition? J Hypertens 2006; 24:993–997 [DOI] [PubMed] [Google Scholar]
- 11.Hurwitz S, Cohen RJ, Williams GH. Diurnal variation of aldosterone and plasma renin activity: timing relation to melatonin and cortisol and consistency after prolonged bed rest. J Appl Physiol 2004; 96:1406–1414 [DOI] [PubMed] [Google Scholar]
- 12.Orth DN, Kovacs WJ. Wilson JD, Foster DW, Kronenberg HM, Larsen PR. The adrenal cortex. Williams textbook of endocrinology 9th edPhiladelphia, PA:WB Saunders; 1998. 517–518 [Google Scholar]
- 13.Calhoun DA, White WB, Krum H, Guo W, Bermann G, Trapani A, et al. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: results of a randomized, double-blind, placebo- and active-controlled phase 2 trial. Circulation 2011; 124:1945–1955 [DOI] [PubMed] [Google Scholar]
- 14.Amar L, Azizi M, Ménard J, Peyrard S, Watson C, Plouin PF. Aldosterone synthase inhibition with LCI699: a proof-of-concept study in patients with primary aldosteronism. Hypertension 2010; 56:831–838 [DOI] [PubMed] [Google Scholar]
- 15.Andersen K, Hartman D, Peppard T, Hermann D, Van Ess P, Lefkowitz M, Trapani A. The effects of aldosterone synthase inhibition on aldosterone and cortisol in patients with hypertension: a phase II, randomized, double-blind, placebo-controlled, multicenter study. J Clin Hypertens (Greenwich) 2012; 14:580–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karns AD, Bral JM, Hartmann D, Peppard T, Schumacher C. Study of aldosterone synthase inhibition as an add-on therapy in resistant hypertension. J Clin Hypertens (Greenwich) 2013; 15:186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trunet PF, Mueller PH, Girard F, Aupetit B, Bhatnagar AS, Zognbi F, et al. The effects of fadrozole hydrochloride on aldosterone secretion in healthy male subjects. J Clin Endocrinol Metab 1992; 74:571–576 [DOI] [PubMed] [Google Scholar]
- 18.Dunn JF, Nisula BC, Rodbard D. Transport of steroid hormones: binding of 21 endogenous steroids to both testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab 1981; 53:58–68 [DOI] [PubMed] [Google Scholar]
- 19.Mendel CM. The free hormone hypothesis: a physiologically based mathematical model. Endocr Rev 1989; 10:232–274 [DOI] [PubMed] [Google Scholar]
- 20.Rupprecht R, Reul JM, van Steensel B, Spengler D, Söder M, Berning B, et al. Pharmacological and functional characterization of human mineralocorticoid and glucocorticoid receptor ligands. Eur J Pharmacol 1993; 247:145–154 [DOI] [PubMed] [Google Scholar]
- 21.Hellal-Levy C, Couette B, Fagart J, Souque A, Gomez-Sanchez C, Rafestin-Oblin M. Specific hydroxylations determine selective corticosteroid recognition by human glucocorticoid and mineralocorticoid receptors. FEBS Lett 1999; 464:9–13 [DOI] [PubMed] [Google Scholar]
- 22.Biller BM, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertheret J, et al. Treatment of adrenocorticotropin-dependent Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab 2008; 93:2454–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonino N, Boscaro M. Medical therapy for Cushing's disease. Endocrinol Metab Clin North Am 1999; 28:211–222 [DOI] [PubMed] [Google Scholar]
- 24.Demers LM, Melby JC, Wilson TE, Lipton A, Harvey HA, Santen RJ. The effects of CGS 16949A, an aromatase inhibitor on adrenal mineralocorticoid biosynthesis. J Clin Endocrinol Metab 1990; 70:1162–1166 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.