

Summary
Aurora kinases play an important role in the control of the cell cycle and have been implicated in tumourigenesis in a number of cancers. Among the haematological malignancies, overexpression of Aurora kinases has been reported in acute myeloid leukaemia, chronic myeloid leukaemia, acute lymphoblastic leukaemia, multiple myeloma, aggressive non-Hodgkin lymphoma and Hodgkin lymphoma. A large number of Aurora kinase inhibitors are currently in different stages of clinical development. In addition to varying in their selectivity for the different Aurora kinases, some also have activity directed at other cellular kinases involved in important molecular pathways in cancer cells. This review summarizes the biology of Aurora kinases and discusses why they may be good therapeutic targets in different haematological cancers. We describe preclinical data that has served as the rationale for investigating Aurora kinase inhibitors in different haematological malignancies, and summarize published results from early phase clinical trials. While the anti-tumour effects of Aurora kinase inhibitors appear promising, we highlight important issues for future clinical research and suggest that the optimal use of these inhibitors is likely to be in combination with cytotoxic agents already in use for the treatment of various haematological cancers.
Keywords: cell cycle, myeloid leukaemia, lymphoma, myeloma
Introduction
Collectively, haematological malignancies account for approximately 10% of newly diagnosed cancers (Jemal, et al 2010). The ability to induce efficient killing of cancer cells using combination chemotherapy has significantly improved the survival rates for patients with leukaemia, lymphoma, and multiple myeloma (MM) (Lichtman, 2008). Also, targeted therapies using small molecules, including tyrosine kinase inhibitors (TKI), proteasome inhibitors, and immunomodulatory drugs, have changed the natural history of some diseases, such as chronic myeloid leukaemia (CML) and MM. While representing significant progress, primary or acquired resistance agents, as well as toxicity, remain problematic in many patients, indicating the need for continued investigation of novel agents.
Of the different cellular processes targeted by small molecule inhibitors, a significant number of novel anti-cancer drugs being developed target protein kinases, particularly those involved in signal transduction and cell cycle control (Noble et al, 2004; Zhang et al, 2009). Dysregulated protein kinase activity is important in many cancers, including haematological malignancies (Zhang, et al 2009). Four groups of protein kinases are generally recognized. First, the receptor tyrosine kinases, which include the epidermal growth factor receptor (EGFR), insulin-like growth factor-1 receptor (IGF1R), vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) 1, FGFR3 and FGFR4, FMS-like tyrosine kinase (FLT3) and c-KIT (Noble et al, 2004; Zhang et al, 2009). A second group is the non-receptor tyrosine kinases, including c-SRC, ABL, Janus kinase 2 (JAK2), c-YES, and focal adhesion kinase (FAK) (Noble et al, 2004; Paul & Mukhopadhyay, 2004; Zhang et al, 2009). Third are the lipid kinases, such as phosphatidylinositol 3-kinase (PI3K). A key downstream effector of PI3K is the serine-threonine kinase AKT, which phosphorylates and regulates the activity of a number of targets including other kinases, transcription factors and other regulatory molecules. The PI3K/AKT pathway plays an important role in cell growth and survival, and is activated in various cancers (Hennessy et al, 2005). Finally, the serine/threonine kinases, which include proteins such as AKT, ataxia telangiectasia mutated (ATM), mammalian target of rapamycin (mTOR), S6K, b-RAF, and cell cycle control kinases, such as cyclin-dependent kinases (CDK), Aurora kinases, and Polo-like kinases (Zhang et al, 2009).
The efficacy of drugs that interfere with cell division has long been recognized in the treatment of the haematological malignancies. The Aurora kinases are intimately involved in the process of mitosis, and, as discussed below, are over-expressed in a number of tumours compared to normal tissues. Therefore, inhibitors of these kinases are now being investigated in a number of cancers. Herein, we review on the function of Aurora kinases and focus on the potential role of Aurora kinase inhibitors in the treatment of the haematological malignancies.
Mitosis and cell division: functional roles of Aurora kinases
Cell proliferation is characterized visually by division of the nucleus (mitosis) and physical splitting of the cell into two daughter cells (cytokinesis); both occurring during the M phase of the cell cycle. After completion of M phase, and in preparation for the next mitosis, a cell replicates its nuclear material (S phase). Before DNA synthesis begins and after it ends, however, two intervals or gap (G) phases intervene where important checkpoints occur (Weinberg 2007). In G1, a checkpoint for DNA integrity blocks a cell’s advance into S phase until DNA damage is repaired. Similarly, in G2 a checkpoint occurs to ensure that all DNA replication is complete before the cell advances to M phase. M phase includes four subphases, including prophase, metaphase, anaphase, and telophase, and culminates in cytokinesis.
Aurora kinases are serine/threonine kinases that are essential for cell proliferation, regulating cell cycle transit from G2, formation of the mitotic spindle, centrosome maturation and separation, and cytokinesis (Andrews et al, 2003; Hirota et al, 2003; Vader et al, 2006). There are three human homologues of Aurora kinases, A, B, and C, which have a high degree of sequence homology, particularly in their carboxy-terminal catalytic domains (Bischoff et al, 1998; Giet & Prigent, 1999). Aurora kinases A and B are expressed in most normal cells, although their localization and activation during the cell cycle differ (Fig 1). Aurora C, initially thought to be restricted to the testicular tissue where it plays a role in meiosis, is now also known to have overlapping functions with Aurora B kinase in mitosis (Sasai et al, 2004; Yan et al, 2005), although its role is less well understood and will not be considered further in this review.
Figure 1. Cellular localization of Aurora kinases A and B in mitosis.

In G1, the levels of both Aurora A (green squares) and Aurora B (red circles) kinases are markedly reduced, but increase with different localization during M phase. In prophase, Aurora A is located around the centrosomes, whereas Aurora B is nuclear. In metaphase, Aurora A is on the microtubules near the spindle poles, whereas Aurora B is located in the inner centromere. In anaphase, most Aurora A is on the polar microtubules, but some might also be located in the spindle midzone. Aurora B is concentrated in the spindle midzone and at the cell cortex at the site of cleavage-furrow ingression. In cytokinesis, both kinases are concentrated in the midbody. Reprinted by permission from Nature Publishing Group: Nature Reviews Molecular Cell Biology 4, 842–854, Carmena & Earnshaw (2003).
Aurora kinase A
The localization and activation of Aurora A is tightly regulated during the cell cycle. Aurora A activity depends on the phosphorylation of a threonine residue (T288) in the activation loop (Littlepage et al, 2002; Satinover et al, 2004). The level and activity of Aurora A is low in G1. By late S/early G2, Aurora A levels increase and the kinase associates with the duplicated, separating centrosomes, where its function is to recruit multiple proteins, including γ-tubulin, required for centrosome maturation and spindle assembly (Carmena and Earnshaw 2003). The mechanism of recruitment of Aurora A to the centrosome and its activation is incompletely understood, but involves a complex interaction with several proteins and a combination of feedback loops (Fig 2) (Lens et al, 2010; Lok et al, 2010). Briefly, in late G2, Aurora A autophosphorylates, and also activates polo-like kinase 1 (PLK1), which in turn promotes the binding of Aurora A to centrosomin. The activity of Aurora A is enhanced by several co-factors, including BORA, Ajuba, and TPX2. BORA is specifically required for activation of Aurora A-induced phosphorylation of PLK1 (Macurek et al, 2008). Ajuba is located on the centrosomes and promotes Aurora A autophosphorylation (Hirota et al, 2003).
Figure 2. Function of Aurora A during late G2 and mitosis.
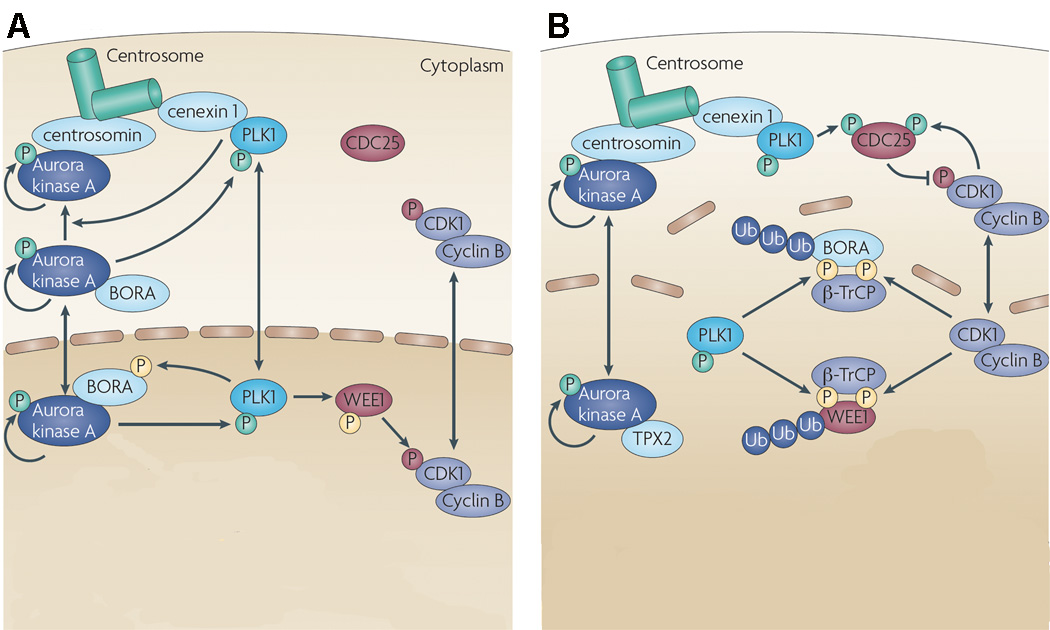
(A) In late G2, Aurora A autophosphorylates and also activates PLK1, which in turn promotes the binding of Aurora A to centrosomin. PLK1 is recruited to the centrosome by cenexin 1. While Aurora A can self-activate, its activity is enhanced by several cofactors, including TPX2 and BORA. Active PLK1 phosphorylates WEE1, a kinase that negatively regulates cyclin B-CDK1 complexes. The cell division cycle 25 (CDC25) phosphatases that antagonize WEE1 are largely inactive in G2. (B) Mitotic entry is triggered by a steep increase in cyclin B-CDK1 activity. On the centrosome, Aurora A and PLK1 promote recruitment and activation of CDC25B, which in turn, activates cyclin B-CDK1. Cyclin B–CDK1 complexes further phosphorylate WEE1 and BORA, which are then recognized by the F-box protein β-transducin repeat containing (β-TrCP) that promotes polyubiquitylation (Ub) and degradation of WEE1 and BORA. Freed of BORA, Aurora kinase A is then able to interact with TPX2, thus facilitating the role of the kinase in spindle assembly (see text). Green phosphates (P) are activating, red phosphates are inhibitory and yellow phosphates prime a protein for degradation. Reprinted by permission from Nature Publishing Group: Nature Reviews Cancer 10, 825–841, Lens et al (2010).
After late G2, mitotic entry is triggered by a steep increase in cyclin B-cyclin-dependent kinase (CDK) 1 activity (Lindqvist et al, 2009). Aurora A contributes to this event directly, as well as indirectly by activating PLK1 (Watanabe et al, 2004). On the centrosome, Aurora A promotes recruitment of cyclin B1 and phosphorylates CDK-activating phosphatase cell division cyclin (CDC) 25B (Dutertre et al, 2004), which activates cyclin B-CDK1 activity. Also, Aurora A activates PLK1, which in turn phosphorylates the CDK-inhibitory kinase WEE1, a negative regulator of CDK1, targeting it for degradation. Activated PLK1 also activates the CDK-activating phosphatase CDC25C, which antagonizes WEE1. Therefore, activation of CDC25B and Inhibition and degradation of WEE1 lead to the increase in cyclin B-CDK1 activity. The increase in cyclin B-CDK1 activity phosphorylates BORA, resulting in BORA being recognized for polyubiquitination and degradation (Chan et al, 2008). Freed of BORA, Aurora A then interacts with cofactor TPX2, stimulating Aurora A autophosphorylation and protecting against its dephosphorylation by inhibiting protein phosphatase PP1, and thus facilitating the role of the kinase in spindle assembly (Eyers et al, 2003; Kufer et al, 2002). Recent data suggests that while Aurora-A activity accelerates mitotic entry in normal cells, it is essential for mitotic entry only in DNA-damaged cells (Macurek et al, 2008; van Vugt et al, 2004).
Throughout mitosis Aurora A localizes mainly near the spindle poles and on the polar microtubules, consistent with its pivotal role in spindle formation (Barr & Gergely 2007). While the mechanisms involved in spindle formation are unclear, it is known that activated Aurora A promotes the formation of a large multi-protein complex that is important for spindle formation by recruiting proteins required for microtubule nucleation, stabilization, bundling, and motor activity (Barr & Gergely 2007, Koffa et al, 2006). Aurora A inhibition, therefore, results in mitotic arrest with formation of abnormal spindles. Finally, in late M/early G1, Aurora A is degraded by the anaphase-promoting complex-ubiquitin-proteasome pathway (Honda et al, 2000).
In addition to the functions of Aurora A in mitosis, Aurora A also regulates the functions of TP53 (p53) and NF-κB, which may have important therapeutic implications. Aurora A phosphorylates TP53 at Ser315 leading to its degradation by ubiquitination by MDM2 and proteolysis (Katayama et al, 2004). As TP53 is important in activating the pro-apoptotic machinery of the cell, loss of TP53 function can render cells resistant to DNA damaging agents (Ozaki & Nakagawara 2011). Indeed, cells depleted of Aurora A kinase activity appear to be more sensitive to cisplatin-induced apoptosis, and elevated expression of Aurora A kinase abolishes this response (Katayama et al, 2004). Finally, Aurora A has been shown to phosphorylate IκB (an antagonist of NF-κB), thereby increasing NF-κB activity (Sun et al, 2007), which is a potent anti-apoptotic effector that contributes to chemotherapy resistance. Inhibition of Aurora A has been shown to downregulate NF-κB and enhance the efficacy of cytotoxic drugs (Sun et al, 2007).
Aurora kinase B
Aurora B is the catalytic component of the chromosomal passenger complex, which consists of three additional noncatalytic subunits that direct its activity: inner centromere protein (INCENP), survivin and borealin (Ruchaud et al, 2007; Vader et al, 2008), and is involved in accurate segregation of chromatids during mitosis, histone modification, and cytokinesis (Lens et al, 2006). The expression and activity of Aurora B is low in G1 and increases at G2/M transition, with maximal activity reached during mitosis (Bischoff et al, 1998; Terada et al, 1998). Aurora B is first detected on chromatin in late G2. In prophase, Aurora B localizes along the chromosome arms and inner centromere region, and by metaphase it is found only at the inner centromere region. In anaphase and telophase, Aurora B localizes on the central spindle, cortex and midbody to regulate cytokinesis (Carmena and Earnshaw 2003). The localization of Aurora B in different phases in mitosis correlates with its different functions (Fig 1).
In later G2 and early prophase, Aurora B promotes chromatin modification and facilitates chromosomal condensation by phosphorylating histone H3 at Ser10 and Ser28 (Monier et al, 2007) and promoting the dissociation of heterochromatin proteins 1 from chromatin (Fischle et al, 2005; Hirota et al, 2005). The activation of Aurora B requires additional cofactors, including the components of the chromosome passenger complex (Klein et al, 2006). INCENP undergoes autophosphorylation and induces a conformational change in the catalytic pocket of Aurora B, promoting its activity (Sessa et al, 2005). Survivin probably plays a related regulatory role, although the mechanism is poorly defined (Andrews 2005). Borealin does not activate Aurora B, but together with INCENP and survivin, targets the complex to the centromere early in mitosis, and to the spindle midzone and cleavage furrow late in mitosis (Gassmann et al, 2004).
During prometaphase and metaphase, Aurora B promotes the localization of centromeric proteins, ensuring the appropriate attachment of centromeres with bipolar spindles (Murata-Hori & Wang 2002). In this role, Aurora B is said to play a mitotic spindle checkpoint function (Fig 3). After nuclear envelope breakdown in prometaphase, chromosomes that have duplicated in S phase (sister chromatids) make attachments to a nearby spindle pole via one sister kinetochore, the protein structure on chromosomes where the spindle fibres attach during cell division to pull the chromosomes apart. The free kinetochore then captures a microtubule from the opposite pole resulting in a bipolar attachment that ultimately results in the positioning of the chromosome midway between the spindle poles (Carmena & Earnshaw 2003). Microtubules that attach in a non-bipolar manner to the kinetochore do not generate tension across the centromere, and therefore, the kinetochore remains in close proximity Aurora B located at the inner centromere region (Andrews et al, 2004; Liu et al, 2009). Close proximity to Aurora B allows it to phosphorylate several kinetochore-associated proteins, known as the KMN network (comprised of KNL1, MIS12, and NDC80), which serves as the main microtubule-binding unit at the kinetochore (Welburn et al, 2010). In addition, phosphorylation of KNL1 by Aurora B prevents the binding of a counteracting protein phosphatase PP1γ to the kinetochore (Liu et al, 2009). Thus, phosphorylation of KMN components and blocking of PP1γ binding to the kinetochore significantly reduces kinetochore affinity for microtubules. Attachment of the kinetochore to the microtubules, therefore, is broken, preventing chromatid separation until the appropriate attachments are reassembled (Ditchfield et al, 2003). On the other hand, when there is correct bipolar attachment and tension is generated, the kinetochore is physically separated from Aurora B and PP1γ is not inhibited, resulting in dephosphorylation of the KMN network components and stabilization of the microtubule-kinetochore attachment. Once proper chromosome alignment is established and chromatids have separated, Aurora B is translocated to the spindle midzone, where it resides in anaphase until cytokinesis (Carmena & Earnshaw 2003). Aurora B does not appear to be essential for initiation of cytokinesis, but plays a more important role in completion of the process (Burkard et al, 2007; Guse et al, 2005; Petronczki et al, 2007). In this respect, Aurora B is required for localization and stabilization of a tetrameric protein complex, called central spindilin, that is important for contractile ring formation and cleavage furrow ingression (Burkard et al, 2009; Petronczki et al, 2007; Wolfe et al, 2009). Inhibition of Aurora B activity, therefore, leads to incomplete cytokinesis, resulting in unviable polyploid cells (Yokoyama et al, 2005).
Figure 3. Aurora B regulation of kinetochore-microtubule interactions.

The affinity of the microtubules to the kinetochore is affected by a phosphorylation gradient of Aurora B. (A) In prophase, Aurora B, present at the inner centromere, is in close proximity to the kinetochore due to a lack of tension allowing Aurora B to phosphorylate all components of the KMN network (comprising KNL1, MIS12 and NDC80 (also known as highly expressed in cancer [HEC1]), which serves as the main microtubule-binding unit at the kinetochore; consequently, the kinetochore has low affinity for microtubules. Similarly, non-bipolar attachment of microtubules to the kinetochore does not generate tension across the centromere, and therefore the kinetochore remains in close proximity to Aurora B and the KMN complex is phosphorylated by Aurora B. Also, phosphorylation of KNL1 by Aurora B prevents the binding of PP1γ (which dephosphorylates KMN substrates) to the kinetochore. Phosphorylation of KMN components and blocking of PP1γ binding to the kinetochore significantly reduce kinetochore affinity for microtubules. Attachment of the kinetochore to the microtubules, therefore, is broken, preventing chromatid separation until the appropriate attachments are reassembled. As the cell progresses through prometaphase (B) and metaphase (C), and when there is correct bipolar attachment of microtubules, tension is generated and the kinetochore is physically separated from Aurora B and PP1γ binding is not inhibited, resulting in dephosphorylation of the KMN components. Further, Aurora B also recruits the kinetochore-associated checkpoint kinase BURB1 where it is phosphorylated by PLK1, which in turn results in reduced Aurora B activity, also promoting attachment of microtubules to the kinetochore. Reprinted by permission from Nature Publishing Group: Nature Reviews Cancer 10, 825–841, Lens et al (2010).
Role of Aurora kinases in cancer and their potential as therapeutic targets
Aurora kinases A and B have been recently recognized as potential targets for cancer therapy (Gautschi et al, 2008; Katayama et al, 2003). Over-expression of wild type Aurora A is oncogenic in murine models (Bischoff et al, 1998; Zhou et al, 1998). Also, the gene encoding for Aurora A, AURKA, is located at chromosome position 20q13, a site that is amplified in a number of solid tumour types and is associated with a poor prognosis (Bischoff et al, 1998; Bodvarsdottir et al, 2007; Klein et al, 2004; Nishida et al, 2007; Reichardt et al, 2003; Sen et al, 1997; Sen et al, 2002; Tatsuka et al, 2005). Unlike AURKA, however, amplification of the AURKB gene, located on chromosome 17p13.1, has not been reported. Over-expression of both Aurora kinase A and B independently of gene amplification, however, has been reported in a wide range of tumour types, although this may be related to rapid cell division rather than being a cause of the malignant phenotype (Gautschi et al, 2008; Keen & Taylor 2009; Mountzios et al, 2008). Furthermore, Aurora A and B overexpression has been associated with higher grades of malignancy, higher proliferation rates, and poor prognosis in several tumours (Araki et al, 2004; Gritsko et al, 2003; Jeng et al, 2004; Kurai et al, 2005; Miyoshi et al, 2001; Smith et al, 2005). While AURKA gene duplication has not been reported in the haematological malignancies, overexpression occurs in a number of these cancers.
Beyond the direct effect of Aurora kinases A and B on mitosis and cell division, they also participate in other cellular pathways important in cancer. For example, Aurora A is a downstream target of MAPK1/ERK, and constitutive activation of MAPK1 in pancreatic cancer has been reported to result in overexpression of Aurora A (Furukawa et al, 2006). Aurora A binds to c-Myc sites on the promoter of telomerase reverse transcriptase and increases its expression and activity, which in turn may confer a survival advantage to cancer cells (Yang et al, 2004). Finally, Aurora A upregulates the PI3K/AKT pathway by enhancing phosphorylation of AKT at Ser473 (Dar et al, 2008; Yang et al, 2006), and possibly GSK-3β (Dar et al, 2009), thereby enhancing cell survival and proliferation. The extent to which these effects might be important in specific cancer types, including the haematological malignancies, is uncertain.
Aurora kinases inhibitors in haematological malignancies
A large number of small molecule inhibitors of Aurora kinases have been developed for the treatment of cancer. These small molecules inhibit Aurora kinases by binding to the ATP binding pocket of the kinase, and thus compete with ATP substrate. As the ATP binding pocket is highly homologous across the members protein kinase family (Scapin 2006), many of these inhibitors are not specific for one or other of the Aurora kinases, and also some have additional “off target” effects that may contribute to their clinical efficacy. Differences in molecular structure of Aurora kinase inhibitors that allow them to interact differently with regions adjacent to the ATP binding site determine to a large extent the kinase specificities of the inhibitors (Pollard & Mortimore 2009). Early inhibitors of the Aurora kinases included Hesperadin (specific for Aurora B) and ZM447439 (inhibitor of Aurora A and B), which did not enter clinical development but are available as research tools (Hauf et al, 2003). VX680 (MK0457) is relatively specific inhibitor of Aurora A, as well as of FLT3 and BCR-ABL1, which entered clinical trials but was discontinued after reports of QTc prolongation (Gizatullin et al, 2006; Harrington et al, 2004; Young et al, 2006). The Aurora kinase inhibitors currently in clinical development in the haematological malignancies together with their targets are listed in Table 1 and are discussed below.
Table 1.
Target profiles of Aurora kinase inhibitors currently in clinical investigation in the haematological malignancies
| Drug | Manufacturer | Target kinase inhibited by drug* |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aurora A |
Aurora B |
FLT3 | ABL1 | ABL1 (T315I) |
KIT | VEGFR | JAK2 | FGFR3 | SYC | LYN | IGF1R | ||
| AZD1152 | Astra Zeneca | 1369 | 0.37 | - | - | - | - | - | - | - | - | - | - |
| MLN8237 | Millennium | 1 | 1100 | - | - | - | - | - | - | - | - | - | - |
| ENMD-2076 | EntreMed | 14 | 290 | 3 | 295 | 81 | 126 | 36 | 120 | 92 | - | - | - |
| AS703569 | Merck Sereno | 4 | 4.8 | 99% inhibition at 100 nM |
92% inhibition at 100 nM |
- | - | - | - | - | - | - | - |
| KW-2449 | Kyowa Hakko Kirin Pharma |
48 | 48 | 6.6 | 14 | 4 | 300 | - | 150 | - | - | - | - |
| AT9283 | Astex Therapeutics |
3 | 3 | 10 | 110 | 4 | - | 10–30 | 1.2 | 10–30 | - | - | - |
| Danusertib | Nerviano | 13 | 70 | - | 21 | 4 | - | - | - | - | - | - | - |
| XL228 | Exelixis | 3.1 | 0.6 | - | 5 | 1.4 | - | - | - | 3 | 6.1 | 5 | 1.2 |
Concentration inhibiting target kinase activity by 50% (IC50) shown (nM) unless otherwise indicated.
Acute Myeloid Leukaemia (AML)
Recent studies have reported aberrantly increased expression of Aurora A and B in human myeloid leukaemia cell lines and fresh leukaemic cells obtained from AML patients. Aurora A and B transcript levels were reported to be significantly higher in freshly isolated leukaemia cells obtained from 44 AML cases compared with peripheral blood mononuclear cells or bone marrow cells obtained from healthy volunteers (Ikezoe et al, 2007). Also, in the same study, the dual Aurora A and B inhibitor ZM447439, which has no activity against ABL1 or FLT3 kinase, inhibited the proliferation, induced polyploidy, apoptosis, and inhibited the clonogenic growth of freshly isolated leukaemia cells from AML patients, including those who were refractory to conventional chemotherapy (Ikezoe et al, 2007). In another study that examined for Aurora A protein expression by Western blotting, leukaemic blasts from 98 de novo AML patients showed markedly increased Aurora A expression compared with negligible expression in bone marrow mononuclear cells obtained from normal donors, with expression considered high in about two thirds of cases as defined by >30% of blasts showing strong cytoplasmic granular immunocytostaining (Huang et al, 2008). Importantly, VX680, a relatively specific Aurora A inhibitor, induced apoptosis preferentially in leukaemic blasts with high expression of Aurora A compared to blasts with low expression and bone marrow cells from normal donors. VX680-induced apoptosis was associated with destruction of normal bipolar spindles, G2/M cell cycle arrest, reduction of p-AKT, activation of caspases, and a dose-dependent increase in the Bax/Bcl-2 ratio (Huang et al, 2008).
A number of Aurora kinase inhibitors have entered early phase clinical trials in patients with AML (Table 2). AZD1152 is a highly selective Aurora B inhibitor, which in vitro blocked phosphorylation of histone H3, increased the population of tetraploid cells, and induced apoptosis of human leukaemic cell lines (Yang et al, 2007). In a Phase I/II trial, AZD1152 was administered at doses of 50–1600 mg/day for 7 days every 21 days by continuous intravenous (CIV) infusion to 32 patients with advanced AML (Lowenberg et al, 2009). The maximum tolerated dose (MTD) was 1200 mg/day, with the most commonly reported grade 3–4 toxicities being febrile neutropenia and stomatitis. Of the 32 patients enrolled in the dose-escalation phase, 1 patient at the 1200 mg dose level achieved a complete response (CR), and 2 patients treated at 400 mg and 800 mg, respectively, achieved CR with incomplete count recovery (CRi). In a Phase II extension of the trial at the MTD, 5 of 32 additional AML patients achieved CR (n=2) or CRi (n=3) with similar toxicity to that observed in the Phase I portion (Lowenberg et al, 2009). Based on its observed clinical activity, AZD1152 is being evaluated in combination with low dose cytarabine in newly diagnosed AML patients 60 years and older who are unfit for intensive induction chemotherapy (Kantarjian et al, 2010). In a preliminary report, the MTD of AZD1152 was 1000 mg/day (CIV infusion for 7 days) when combined with cytarabine 20 mg subcutaneously twice daily for 10 days, every 28 days, with stomatitis being dose-limiting. Of 21 patients reported, CR was observed in 7 and CRi in 2 patients, for a total response rate of 43%. A Phase II trial of this combination is currently ongoing to better define the response rate.
Table 2.
Aurora kinase inhibitors currently in clinical development in AML
| Drug | Clinical trial |
Patients included |
N | Route | Dose | Main toxicity reported |
Responses | Reference |
|---|---|---|---|---|---|---|---|---|
| AZD1152 | Phase I/II | Advanced AML | 64 | CIV | 50–1600 mg/d×7d q 21 days (MTD 1200 mg) | Febrile neutropenia, stomatitis. | Phase I: 1 CR, 2 CRi Phase II: 2 CR, 3 CRi |
Lowenberg et al, (2009) |
| Phase I (with low-dose AraC) | AML (≥60 years) | 21 | CIV | MTD 1000 mg/d ×7d (with AraC 20 mg SC bid×10 d) q 28d | Stomatitis. | 7 CR, 2 CRi | Kantarjian et al, (2010) | |
| MLN8237 | Phase II | Advanced AML/MDS |
67 | PO | 50 mg bid×7d q 21 days | Febrile neutropenia, thrombocytopenia, anaemia, fatigue, somnolence | 1 CR, 5 PR | Goldberg et al, (2010) |
| ENMD-2076 | Phase I | Relapsed or refractory AML | 20 | PO | 225–325 mg/d (MTD 225 mg) |
Fatigue, typhlitis, syncope | 1 CRi, 3 morphological leukaemia-free state | Yee et al, (2010) |
| AS703569 | Phase I | AML, MDS, CML, MPD |
45 | PO | 3–47 mg/m2/d×6d q 21 days | Neutropenia, thrombocytopenia, mucositis, diarrhoea, infection | 3/33 (AML) PR 1/5 (MDS) PR |
Sonet et al, (2008) |
| KW-2449 | Phase II | AML, MDS, ALL, CML | 29 | PO | 12.5–500 mg bid (Trial stopped early; MTD not reached) | Pneumonia, dyspnea, cardiac arrhythmia, cardiac ischaemia. | 5/11 FLT3 ITD AML achieved transient reduction in blasts. | Pratz et al, (2009) |
| Phase I/I | Relapsed or refractory AML (AML FLT3 ITD in Phase II | NR | PO | NR | NR | NR | NCT00779480* | |
| AT9283 | Phase I/II | Refractory AML, MDS, ALL, CML, MF | NR | IV | NR | NR | NR | NCT00522990* |
AML, acute myeloid leukaemia; MDS, myelodysplasia; CML, chronic myeloid leukaemia; MPD, myeloproliferative disease; MF, myelofibrosis; ALL, acute lymphoblastic leukaemia; CIV, continuous intravenous infusion; IV intravenous injection; PO, oral; SC, subcutaneous injection; bid, twice daily; MTD, maximum tolerated dose; CR, complete response; CRi, complete response with incomplete count recovery; PR, partial response; NR, not reported; Ara-C, cytarabine; FLT3-ITD, FLT3 internal tandem duplication.
ClinicalTrials.gov Identifier number (cited where no report is available).
MLN8237 is a highly selective inhibitor of Aurora A, which has undergone clinical evaluation in adults with advanced AML or intermediate-2/high-risk myelodysplasia (MDS). In a multicentre Phase II trial, 67 patients of median age 72 (range, 46–85) years, with AML (n=46) or MDS (n=21), received MLN8237 at 50 mg orally twice daily for seven days followed by 14 days rest until progression (Goldberg et al, 2010). Patients received a median of 2 (range, 1–16) cycles. One AML patient who did not receive any prior therapy achieved a CR that lasted 1 year, and an additional 5 AML patients achieved partial responses (PR) for an overall response rate of 16%. Treatment-related grade 3–4 adverse events included febrile neutropenia, thrombocytopenia, anaemia, fatigue, neutropenia, and somnolence, with treatment discontinued in 14 patients (25%) because of excessive toxicity.
Some of the Aurora kinase inhibitors also inhibit other kinases, including FLT3. FLT3 activating internal tandem duplication (ITD) or tyrosine kinase domain (TDK) mutations are found in approximately 20–30% of AML cases and predict a poor prognosis (Kottaridis et al, 2001; Yamamoto et al, 2001). The additional inhibitory activity against FLT3 may be an important secondary mechanism of anti-leukaemic activity. In preclinical studies, while FLT3 mutational status did not predict drug sensitivity to VX680 in AML blasts with high Aurora A expression, blasts with FLT3-ITD mutation that had a lower expression of Aurora A were sensitive to VX680-induced apoptosis (Huang et al, 2008). In another study, the inhibition of clonogenic potential of primary AML blasts by AZD1152 was significantly higher in AML samples with FLT3-ITD compared with that in FLT3 wild-type samples (Grundy et al, 2010), a finding which has also been confirmed in vivo in AML xenografts using AS703569 (McLaughlin et al, 2010). These studies suggest that analysis of both Aurora kinase expression and FLT3-ITD mutational status may be valuable in identifying patients who may benefit from Aurora kinase inhibitors with concomitant FLT3 kinase inhibitory activity.
As shown in Table 2, four Aurora kinase inhibitors with FLT3 inhibitory activity have entered clinical trials in patients with leukaemia. ENMD-2076 is an oral inhibitor of Aurora A and B, as well as FLT3 and multiple other tyrosine kinases (Fletcher et al, 2011). and preliminary results from a Phase I trial in relapsed or refractory AML patients have been reported (Yee et al, 2010). In 27 patients, of median age 69 (range, 43–84) years, who received daily doses of ENMD-2076 at 225 mg, 375 mg, 325 mg and 275 mg, dose-limiting toxicity included fatigue, typhlitis, and syncope. The 225 mg dose was best tolerated and was recommended as the Phase II dose for further evaluation. Of 20 evaluable patients, one achieved a CRi and 3 achieved a morphological leukaemia-free state. In the patient achieving a CRi, striking decreases in the stimulation of ERK, AKT, STAT5 and S6 in leukaemic cells were observed (Yee et al, 2010). It was not reported, however, if the patient who achieved CRi had FLT3 ITD+ leukaemia.
AS703569 is another inhibitor of Aurora A and B, with additional activity against FLT3 and ABL1 kinases (McLaughlin et al, 2010). In preclinical studies in vitro and in vivo, AML cells with FLT3 ITD mutations were up to 10-fold more sensitive to AS703569 than those with wild type FLT3, and in xenograft models, sustained inhibition of both FLT3 and histone H3 phosphorylation was observed in tumour lysates up to 48 h after the last dose was administered, suggesting that inhibition of both kinases might have been important for anti-leukaemic activity (McLaughlin et al, 2010). In a Phase I trial of AS703569 in 45 patients with AML, MDS, CML and myeloproliferative disease, significant reductions in bone marrow and/or peripheral blood blasts were observed in 3 of 33 AML patients who received 5–7 cycles, and a PR was observed in 1 of 5 patients with MDS (Sonet et al, 2008). Frequently observed toxicities included neutropenia, thrombocytopenia, infection, mucositis and diarrhoea, but the MTD is yet to be defined.
Of the Aurora kinase inhibitors with additional kinase inhibitory activity, KW-224 has the most potent activity against FLT3 (Shiotsu et al, 2009). In preclinical studies, KW-224 inhibited proliferation and induced apoptosis of AML cell lines that harbored FLT3 ITD as well as AML cells with wild type FLT3 (Shiotsu et al, 2009). In FLT3 ITD+ AML cell lines, KW-224 also reduced the phosphorylation of FLT3 and STAT5, and in cells with wild type FLT3, inhibition of Aurora kinase activity was also demonstrated by reduced phosphorylation of histone H3 and G2/M arrest (Shiotsu et al, 2009). In a Phase I trial in patients with leukaemia, that included AML, KW-224 was administered orally at 7 dose levels, escalating from 12.5 mg to 250 mg twice daily (Pratz et al, 2009). Only modest clinical activity was observed, with transient reductions in peripheral blood blasts by 50% or more noted in 5 of 11 patients with FLT3 ITD+ AML. However, using this dosing schedule, FLT3 phosphorylation was only transiently inhibited by greater than 80% at the highest dose level, suggesting that the failure to fully inhibit FLT3 in sustained fashion may have been the reason for the minimal success observed (Pratz et al, 2009). Therefore, the study was terminated before the MTD was reached, and a new study with a 3–4 times per day dosing strategy, aimed at continually abrogating FLT3 autophosphorylation, is currently ongoing in FLT3 ITD+ AML patients (ClinicalTrials.gov Identifier: NCT00779480).
While Aurora kinase inhibitors have significant therapeutic potential in AML, single agent activity appears to be uniformly modest. It is likely, however, that Aurora kinase inhibitors will find their place in combination therapy, as suggested by the improved response rates to the combination of AZD1152 with low dose cytarabine (Kantarjian et al, 2010). In this respect, preclinical studies have also shown synergy of Aurora kinase inhibitors with both daunorubicin (Yang et al, 2007) and etoposide (Huang et al, 2008), which are commonly used drugs in the frontline treatment of AML. Also, as larger trials of Aurora kinase inhibitors are conducted in the future, it will be important to identify those AML patients who may most benefit from treatment by correlating responses to Aurora kinase inhibitors with Aurora A or B expression in leukaemic blasts, and possibly FLT3 mutational status, as suggested by results of preclinical studies (Grundy et al, 2010; Huang et al, 2008; McLaughlin et al, 2010).
Acute lymphoblastic leukaemia (ALL) and chronic myeloid leukaemia
Unlike in AML, there is minimal data on the level of expression of Aurora kinases in fresh blasts obtained from patients with ALL or CML, although aberrantly high expression levels have been noted in some cell lines (Ikezoe et al, 2007; Yang et al, 2007). Also, while direct inhibition of Aurora kinases may be important in ALL (Maris et al, 2010), most research has centred on evaluating the potential of inhibitors with additional activity against BCR-ABL1 kinase in Ph+ ALL and CML, and in particular, their ability to overcome TKI resistance due to mutations of the ABL1 kinase domain. Point mutations of the ABL1 kinase domain, particularly the T315I mutation, occur in approximately 43% of Ph+ leukaemia patients during TKI therapy, with the highest rates of mutations observed in blast crisis CML and Ph+ ALL (Quintas-Cardama et al, 2007; Soverini et al, 2006). Importantly, for many of the Aurora kinases with additional activity against ABL1, activity against mutated ABL1 kinase isoforms (including the T315I mutant) is higher than for the wild-type kinase (Table 1). For example, the Aurora kinase inhibitors, MK0457, AT9283, Danusertib (formerly PHA-739358), XL228, and KW2449, inhibit the T315I ABL1-mutant at lower concentrations than wild type ABL1 (Moore et al, 2010), making them of potential benefit for Ph+ ALL and CML patients who have failed first and second line TKI therapy.
A number of the Aurora kinase inhibitors are currently undergoing clinical evaluation in patients with Ph+ ALL or CML (Table 3). AT9283 is an Aurora kinase A and B inhibitor with inhibitory activity against ABL1, FLT3, JAK2, FGFR and VEGFR, and has shown preclinical in vivo efficacy in murine xenograft models using T315I or E255K mutant BCR-ABL1 transfected BaF3 cells and in vitro efficacy against primary CML cells with E255K mutated BCR-ABL1 (Tanaka et al, 2008). Consistent with these studies, AT9283 showed clinical activity against CML in a Phase I trial in patients with refractory leukaemia (Foran et al, 2008). Using a 72-h CIV infusion at eight different dose levels, two patients with accelerated phase CML refractory to the TKI inhibitors imatinib and dasatinib achieved haematological responses, including a partial cytogenetic response in one patient. Pharmacodynamic studies demonstrated a reduction in phosphorylated histone H3 and of p-STAT5, which is downstream of BCR-ABL1 (as well as FLT3 and JAK2) signalling, suggesting multiple kinase inhibition. A trial utilizing a different schedule is currently ongoing (ClinicalTrials.gov Identifier: NCT00522990)
Table 3.
Aurora kinase inhibitors currently in clinical development in CML and Ph+ ALL
| Drug | Clinical trial |
Patients included |
N | Route | Dose | Main toxicity reported |
Responses | Reference |
|---|---|---|---|---|---|---|---|---|
| AT9283 | Phase I/II | Refractory AML, MDS, ALL, CML, MF | NR | IV | NR | NR | NR | NCT00522990* |
| Danusertib | Phase I | Advanced-stage CML**, Ph+ ALL** | 23 | IV | 90–200 mg/m2 /day × 7d q 2 weeks (MTD not reached) | Diarrhoea, nausea, pyrexia, fainting, headache | 5 HR, 1 CCR, 1 PCR, 1 MCR, 1 CMolR | Cortes-Franco et al, (2009) |
| Phase II | CML, relapsed after prior RTK inhibitors | NR | IV | NR | NR | NR | NCT00335868* | |
| XL228 | Phase I | Refractory CML**, Ph+ ALL | 27 | IV | 0.45–10.8 mg/kg weekly or twice weekly (MTD not reached) | Syncope, hyperglycaemia, nausea, vomiting, fatigue. | 3 (2 with T315I) decrease BCR-ABL1 by > 1 logarithm | Cortes et al, (2008a) |
| AS703569 | Phase I | AML, MDS, CML**, MPD | 45 | PO | 3–47 mg/m2/d×6d q 21 days | Neutropenia, thrombocytopenia, mucositis, diarrhoea, infection | 1/6 CML with T315I cytogenetic response | Sonet et al, (2008) |
| KW-2449 | Phase I | AML, MDS, ALL, CML** | 29 | PO | 12.5–500 mg bid (Trial stopped early; MTD not reached) | Pneumonia, dyspnea, cardiac arrhythmia, cardiac ischaemia. | 1/5 CML disappearance of T315I clone. | Pratz et al, (2009) |
AML, acute myeloid leukaemia; MDS, myelodysplasia; CML, chronic myeloid leukaemia; MPD, myeloproliferative disease; MF, myelofibrosis; ALL, acute lymphoblastic leukaemia; IV intravenous injection; PO, oral; bid, twice daily; MTD, maximum tolerated dose; HR, haematological remission; CCR, complete cytogenetic response; PCR, partial cytogenetic response; MCR, minor cytogenetic response; CMolR, complete molecular response; NR, not reported.
ClinicalTrials.gov Identifier number (cited where no report is available).
Trials that have reported clinical activity in CML patients with T315I mutations (see text).
Danusertib is an Aurora kinase A, B and C inhibitor with additional activity against ABL1, FLT3, and VEGFR3 (Steeghs et al, 2010), which has demonstrated in vitro activity against CML cells with wild-type BCR-ABL1 and cells possessing the ABL1 T315I mutation (Gontarewicz et al, 2008). Preliminary results of a Phase I clinical trial in 23 patients with advanced stage CML and Ph+ ALL, refractory or intolerant to TKI therapy (15 of which had the T315I mutation), have recently been reported (Cortes-Franco et al, 2009). Danusertib was administered as a 3-h intravenous infusion for 7 days every 2 weeks at five dose levels. To date, the MTD has not been reached. However, 5 patients have achieved a haematological response and three have demonstrated cytogenetic (one complete, one partial, one minimum) responses. Of note, one additional patient with extensively pre-treated ALL with T315I mutation achieved a complete molecular response.
XL228 is a multi-kinase inhibitor with activity against Aurora A, ABL1, IGF1R, SYC and LYN, which is currently being evaluated in patients with refractory leukaemia. In a phase I trial involving 27 CML and Ph+ ALL patients, 10 of whom had the T315I mutation, XL228 was administered as a 1-h intravenous infusion once or twice weekly (Cortes et al, 2008a). In patients receiving at least 3.6 mg/kg, objective responses were observed with decreases in white blood cell count and/or > 1 log reduction in the level of BCR-ABL1 transcripts (Cortes et al, 2008a). KW-2449, an Aurora kinase A and B inhibitor with additional activity against ABL1 (including T315I mutant), FLT3 and FGFR1, also shows activity in vitro (Shiotsu et al, 2009), and clinically against CML (Cortes et al, 2008b). Of five imatinib-resistant CML patients included in a Phase I trial, treatment with KW-2449 resulted in disappearance of the T315I clone and an improvement in blast count in one patient with blast crisis CML (Cortes et al, 2008b). Finally, in a phase I trial of AS703569 in myeloid malignancies, 1 of 6 CML patients with a T315I mutation achieved a haematological and cytogenetic response (Sonet et al, 2008).
While results of early phase clinical trials suggest a potential therapeutic role for Aurora kinase inhibitors in Ph+ ALL and CML, it remains uncertain whether efficacy is simply related to inhibition of BCR-ABL1 signalling or to the combined inhibition of Aurora and BCR-ABL1. Recent data suggests that the contribution of Aurora inhibition is important. Low concentrations of AS703569, a dual inhibitor of Aurora kinase B and ABL1 kinase, induced a phenotype consistent with inhibition of Aurora B and induced apoptosis of BCR-ABL1 transformed cells while not inhibiting BCR-ABL1 (Seitz et al, 2009). Similarly, MK0457 suppressed Aurora kinase activity and induced apoptosis in imatinib-resistant CML cells expressing the T315I and other BCR-ABL1 mutations but without affecting BCR-ABL1 kinase activity (Donato et al, 2010). Finally, Danusertib and MLN8237 have shown synergistic activity with imatinib and nilotinib, respectively, in preclinical studies, suggesting that the inhibition of both kinases may be important (Gontarewicz et al, 2008; Kelly et al, 2010; Nawrocki et al, 2008), and that combination therapy should be a new avenue of clinical research in Ph+ leukaemias. On the other hand, the potential importance of Aurora kinase inhibition as a mechanism of anti-leukaemia activity, independent of the effect on BCR-ABL1, suggests that Aurora kinase inhibitors may have activity in Ph negative ALL. While patients with Ph negative ALL are included with patients with other haematological malignancies in ongoing trials of Aurora kinase inhibitors (see Tables 2 and 3), the activity of Aurora kinase inhibitors in Ph negative ALL remains to be better defined.
Multiple Myeloma
The Aurora kinases have been validated as potential targets for therapy in multiple myeloma (MM) (Manfredi et al, 2007). As MM is characterized by genetic instability with numerous chromosomal abnormalities (Fonseca et al, 2004), it was suggested that there must exist disruption of checkpoints that would normally arrest the MM cells at the G2/M transition or at mitosis to allow potential repair of DNA damage or spindle abnormalities (Manfredi et al, 2007). Such deficient checkpoints would render the MM cells susceptible to apoptotic death in mitosis if further disruption of the mitotic pathway were induced, as with inhibition of Aurora kinases (Manfredi et al, 2007). Further, the Aurora kinase inhibitor, VX680, induced tetraploidy in 48 h, followed by apoptosis at 72 h in MM cells, which correlated with inhibition of Aurora activity as demonstrated by a reduction of histone H3 phosphorylation (Manfredi et al, 2007). Also, ectopic expression of Aurora A by stable transfection of OPM-2 myeloma cells protected against the proapoptotic action of Aurora inhibitors, but not against bortezomib, indicating target specificity (Manfredi et al, 2007). Finally, it was recently reported that 24% of previously untreated MM patients expressed Aurora A, and that myeloma cells with detectable Aurora A expression showed significantly higher proliferation rates compared with those without detectable Aurora-A expression (Hose et al, 2009).
A number of Aurora kinase inhibitors have been investigated in MM cells (Evans et al, 2008a, Evans et al, 2008b; Gorgun et al, 2010; Hose et al, 2009; Negri et al, 2009; Santo et al, 2011). Some of these inhibitors also have additional off target effects that may contribute to anti-myeloma activity. Of particular interest, ENMD-2076, an inhibitor of Aurora kinase and multiple tyrosine kinases, has been shown to have anti-myeloma activity in vitro and in vivo and appears to interfere with multiple pathways that are important in MM (Wang et al, 2010). ENMD-2076 induces apoptosis of MM cell lines and primary MM cells within 6 h in a dose-dependent manner, with downregulation of MCL1, survivin and XIAP, inhibition of the PI3K/AKT pathway, and induction of G2/M arrest with decreases of cyclins A and B (Wang et al, 2010). A potentially important off target effect of ENMD-2076 is inhibition of FGFR3, which is ectopically expressed in approximately 15% of MM cases associated with translocation t(4;14) (Chesi et al, 2001; Chesi et al, 1997; Otsuki et al, 1999). MM cases with t(4;14) have a particularly poor outcome (Chng et al, 2008; Moreau et al, 2007). The t(4;14) abnormality causes dysregulation of the FGFR3 by its translocation from chromosome 4p16.3 to chromosome 14q32, adjacent to the immunoglobulin heavy chain locus (IGH@) (Chesi et al, 1997), resulting in ectopic expression of functional FGFR3 on MM cells that otherwise do not express it (Chesi et al, 2001; Chesi et al, 1997). The FGF receptors, including FGFR3, are tyrosine kinase receptors and signal primarily through the ERK pathway (Hart et al, 2000; Kanai et al, 1997). Ectopic expression of FGFR3 on MM cells stimulates myeloma cell proliferation and prevents apoptosis (Plowright et al, 2000), and MM cells ectopically expressing FGFR3 are sensitive to the inhibitory and pro-apoptotic effects of specific FGFR inhibitors (Paterson et al, 2004), suggesting that FGFR3 may be a good therapeutic target in MM with t(4;14). ENMD-2076 has been shown to inhibit the phosphorylation of FGFR3 in H929 plasmacytoma xenografts in a dose-dependent manner (Wang et al, 2010), and may therefore be a useful drug in this subset of MM.
Aurora kinase inhibitors under clinical investigation in MM are listed in Table 4. Interim results from an ongoing Phase I trial of ENMD-2076 in patients with relapsed or refractory MM indicates that ENMD-2076 has clinical activity in heavily pretreated patients (Farag et al, 2010). ENMD-2076 was given orally for 28 days at once daily doses of 150, 225or 325 mg. In the first 10 patients, activity was observed in 3 of 4 patients treated at the highest dose level of 325 mg/day; one patient achieved a 60% reduction urinary monoclonal (M) protein excretion and a 75% reduction in volume of extramedullary plasmacytomas, and 2 additional patients achieved 21% and 19% reductions in serum M-protein, respectively, after one cycle. Of note, however, none of the patients had t(4;14) MM. Dose-limiting toxicity consisting of grade 3 thrombocytopenia in one patient and posterior reversible encephalopathy syndrome in another, however, has been observed at the 325 mg/day dose. Other toxicities included hypertension, anorexia, nausea, diarrhoea, fatigue, asymptomatic elevation of amylase and lipase, leucopenia, and proteinuria, although none were dose limiting. The trial is currently ongoing, enrolling patients on a reduced dose of 275 mg/day. While the interim results from the Phase I clinical trial indicate that ENMD-2076 has some clinical activity in MM, this appears to occur at a dose where toxicity may be problematic. Further study at the lower dose of 275 mg/day will determine if clinically useful activity will still be observed with this agent when administered singly.
Table 4.
Aurora kinase inhibitors currently in clinical development in MM
| Drug | Clinical trial |
Patients included |
N | Route | Dose | Main toxicity reported |
Responses | Reference |
|---|---|---|---|---|---|---|---|---|
| ENMD-2076 | Phase I | Relapsed or refractory MM | 18 | PO | 150–325 mg/d (MTD 275 mg) | Thrombocytopenia, PRES, anorexia, hypertension, fatigue, proteinuria, elevated lipase and amylase, diarrhoea. | To date: 2 /10 had reduction in M protein; 1/10 had 75% reduction in plasmacytoma | Farag et al, (2010) |
| MLN8237 | Phase I | Haematological malignancies, (including relapsed MM) | 29 | PO | 35–90 mg bid day 1, then once daily×14 d every 28 days (MTD 90 mg) | Neutropenia, thrombocytopenia, asthenia. | 1/8 MM had stabilization of M protein. | Padmanabhan et al, (2010) |
| Phase I (with bortezomib) | Relapsed or refractory MM | NR | PO | NR | NR | NR | NCT01034553* | |
| AT9283 | Phase II | Relapsed or refractory MM | NR | IV | NR | NR | NR | NCT01145989* |
| Danusertib | Phase II | Relapsed or refractory MM with t(4;14). | NR | IV | NR | NR | NR | NCT00872300* |
MM, multiple myeloma; IV intravenous injection; PO, oral; bid, twice daily; MTD, maximum tolerated dose; PRES, posterior reversible encephalopathy syndrome; NR, not reported.
ClinicalTrials.gov Identifier number (cited where no report is available).
Preliminary results from a Phase I trial of MLN8237 in 29 patients with a variety of haematological malignancies have also been reported (Padmanabhan et al, 2010). Of 8 relapsed or refractory MM patients included, one patient was observed to have stabilization at the MTD (90 mg/day for 14 days each 28-day cycle), although no objective responses were observed. Phase II trials of AT9273 and Danusertib are currently ongoing in patients with relapsed or refractory MM, and clinical activity of these agents in MM remains to be determined.
Preclinical studies suggest that the optimal use of Aurora kinase inhibitors in MM may be in combination with other drugs. In this respect, MLN8237 has shown in vitro synergistic or additive activity in combination with melphalan, dexamethasone, and bortezomib (Gorgun et al, 2010). A Phase I trial of MLN8237 in combination with bortezomib is currently ongoing (ClinicalTrial.gov Identifier: NTC01034553). We have also shown in vitro and in vivo synergy of ENMD-2076 with lenalidomide against MM cell lines and plasmacytoma xenografts (Zhang & Farag 2011). Similarly, AT9283 showed potent in vitro anti-myeloma activity in combination with lenalidomide in MM.1S cells cultured with bone marrow stromal cells (Santo et al, 2010). Collectively, these results provide rationale for future evaluation of combination therapy in MM. An important issue in future trials in MM, however, is identifying patients who might benefit most from Aurora kinase inhibitors. A recent report indicated that centrosome amplification is common in MM, and that a gene-expression based centrosome index (CI), which correlates with centrosome amplification, correlated with Aurora expression in MM cells (Chng et al, 2008). Furthermore, MM cell lines with higher CI were more responsive to Aurora inhibitors. The CI, therefore, may be a promising biomarker and should be prospectively investigated in trials of Aurora kinases inhibitors in MM.
Non-Hodgkin lymphoma (NHL) and Hodgkin lymphoma (HL)
Overexpression of Aurora kinases occurs in NHL and, to a lesser extent, in HL. In NHL, Aurora A and B overexpression has been observed largely in aggressive subtypes. Early studies indicated that Aurora A transcripts are overexpressed in NHL cell lines and Epstein-Barr virus-transformed B cell lines compared with normal B lymphocytes (Hamada et al, 2003; Yakushijin et al, 2004). Furthermore, in single tumour cell suspensions from patients with different B-cell NHL, Aurora A transcripts were only expressed at high levels in histologically aggressive types, including diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma (BL), while cells from patients with follicular lymphoma (FL) and normal B cells did not show overexpression (Hamada et al, 2003; Yakushijin et al, 2004). Also, DLBCL cases with a high International Prognostic Index (IPI) expressed higher levels than those with lower IPI scores, indicating that Aurora A expression also correlated with disease activity in NHL. Immunohistochemical studies have also indicated that Aurora B overexpression occurs in 48% and 86% of DLBCL and BL cases, respectively, with only 1 of 20 FL cases showing overexpression (Ikezoe et al, 2009). The mechanism by which Aurora A and B are upregulated in aggressive NHL is not well understood as gene amplification has not been observed (Hamada et al, 2003). However, at least in some cases, upregulation of Aurora kinases may be related to overexpression of MYC (den Hollander et al, 2010; Yang et al, 2010).
Overexpression of Aurora kinases has also been reported in mantle cell lymphoma (MCL). In a study of 26 cases of MCL, the highest levels of AURKA transcripts were observed in blastoid variant cases compared with other variants, and were correlated strongly with Ki-67 expression (Camacho et al, 2006). In another study, high levels of AURKA expression was observed in 88% and AURKB in 75% of MCL cases (Qi et al, 2011). Furthermore, using the Leukaemia/Lymphoma Molecular Profiling Project database, AURKA and AURKB expression was studied in 92 MCL patients, and correlated inversely with survival. Patients with AURKA and AURKB expression in the highest quartiles had significantly reduced overall survival (Qi et al, 2011). While systematic evaluation of Aurora kinase expression in other histological subtypes of lymphoma has not been reported, small studies have suggested that overexpression of AURKA and AURKB may occur in a proportion of patients with HL (Mori et al, 2011), and of AURKA in some cases of adult T-cell leukaemia/lymphoma (Tomita et al, 2010).
The overexpression of Aurora kinases in different lymphoma subtypes has provided rationale for investigating Aurora kinase inhibitors in these diseases. In preclinical studies, the selective Aurora B inhibitor AZD1152 induced growth arrest and apoptosis in BL cell lines in a caspase-dependent and independent manner, and showed synergistic anti-lymphoma activity in association with vincristine (Ikezoe et al, 2009). In another study, AZD1152 also showed antiproliferative effects, inhibited the phosphorylation of histone H3, induced polyploidy with cells with >4N DNA content, caspase-dependent apoptosis, and inhibited survivin expression in BL and HL cell lines (Mori et al, 2011). AT9283, a pan-Aurora/JAK2 kinase inhibitor also induced apoptosis of B-cell lymphoma cell lines, and while modest in vivo anti-lymphoma activity in a mouse xenograft MCL model was observed, significant synergy was observed when AT9283 was combined with docetaxel (Mahadevan et al, 2010). Finally, MLN8237, an Aurora kinase inhibitor with relative specificity for Aurora A, also inhibited proliferation and induced apoptosis in aggressive B cell lymphoma cell lines (Huck et al, 2008), and showed in vivo synergy with docetaxel in a mouse MCL xenograft (Qi et al, 2011).
Only a limited number of Aurora kinase inhibitors have been clinically investigated in NHL (Table 5). In a Phase I trial of MLN8237 in patients with advanced haematological malignancies that also included patients with aggressive NHL, anti-tumour activity was observed in 2 of 19 lymphoma patients; one patient with relapsed and refractory DLBCL showed a reduction in tumour volume, and another achieved a PR that has lasted more than 1 year (Padmanabhan et al, 2010). Phase II trials in patients with relapsed or refractory aggressive NHL are currently ongoing with MLN8237 (ClinicalTrial.gov Identifier: NTC00807495) and AZD1152 (ClinicalTrials.gov Identifier: NTC01354392). It is likely, however, that the greatest potential for Aurora kinase inhibitors in aggressive NHL will be in combination with other cytotoxic agents, including DNA-damaging and microtubule-damaging agents, already used in the treatment of lymphoma, and potentially with histone deacetylase inhibitors that may modulate MYC expression (Kretzner et al, 2011).
Table 5.
Effects of Aurora kinase inhibitors investigated in NHL
| Drug | Clinical trial |
Patients included |
N | Route | Dose | Main toxicity reported |
Responses | Reference |
|---|---|---|---|---|---|---|---|---|
| MLN8237 | Phase I | Haematological malignancies, (including relapsed NHL) | 29 | PO | 35–90 mg bid day 1, then once daily×14 d every 28 days (MTD 90 mg) | Neutropenia, thrombocytopenia, asthenia. | 1/19 DLBCL achieved PR. | Padmanabhan et al, (2010) |
| Phase II | Relapsed or refractory aggressive NHL | NR | PO | NR | NR | NR | NCT00807495* | |
| AZD1152 | Phase II | Relapsed or refractory DLBCL or transformed NHL | NR | IV | NR | NR | NR | NCT01354392* |
NHL, non-Hodgkin’s lymphoma; DLBCL, diffuse large B-cell lymphoma; IV intravenous injection; PO, oral; bid, twice daily; MTD, maximum tolerated dose; NR, not reported.
ClinicalTrials.gov Identifier number (cited where no report is available).
Conclusion
Aurora kinases are aberrantly expressed in many cancers and have emerged as important therapeutic targets. Overexpression of Aurora kinases A and B is observed in a significant proportion of patients with AML, Ph+ leukaemia, aggressive NHL and HL, and in some cases higher expression is associated with an adverse outcome. Currently a large number of Aurora kinase inhibitors are in various stages of development in the haematological malignancies. However, Aurora target specificity among these inhibitors varies significantly, and many inhibitors exhibit effects against other kinases. Indeed, effects against kinases such as FLT3, BCR-ABL1 mutants, JAK2, and FGFR3 may in some cases contribute to added activity of these agents against the specific malignancies where these targets are important drivers. On the other hand, the additional off target effects might also contribute to increased toxicity. Resolving issues, such as which of the Aurora kinases is the more important target, as well as the relative importance of inhibiting the Aurora kinases versus other kinases in different haematological malignancies, will be important in setting some priority in developing the large number of inhibitors currently available. However, other differences between the inhibitors will also be important, including route of administration and pharmacokinetic profile. While many of the Aurora kinase inhibitors tested in early phase clinical trials have shown anti-tumour activity, future clinical investigation should focus on identifying patient subsets in each of the haematological cancers who might benefit most from treatment. Preclinical data suggests that baseline Aurora kinase expression may predict response to these inhibitors, and this should be further investigated in clinical trials. Similarly, expression of other kinases targeted should be also correlated with response wherever relevant. Finally, it is likely that the optimal use of Aurora kinase inhibitors will be in combination with cytotoxic agents already in use. In this respect, the combination with existing DNA damaging agents should be a priority for future trials. A potentially important limitation of the aurora kinase inhibitors, however, is the possibility of inducing of second cancers. Indeed, sobering data has emerged from studies of Aurora A knockout mice (Lu et al, 2008). While the Aurora A null (A−/−) genotype was lethal, Aurora A heterozygous (A+/−) mice were born healthy with no obvious effects except for a slight decrease in Aurora A levels compared with Aurora A wild-type mice. Importantly, with up to 70 weeks follow-up, Aurora A+/− mice had a three-fold higher incidence of tumours compared with Aurora A wild-type mice (25% vs 8.8%; P<0.001) (Lu et al, 2008). Approximately half of the tumours were of haematopoeitic origin, including lymphoma, leukaemia and myeloma, although a variety of epithelial malignancies were also observed. It is likely that chromosomal instability associated with deficiency of Aurora A led to tumorigenesis. While second cancers have yet not been reported in patients treated with Aurora kinase inhibitors in clinical trials, possibly because most have succumbed to their primary cancer, close vigilance for development of secondary cancers in long-term survivors will be important. The coming decade will determine if the promise of the Aurora kinase inhibitors will be realized in the haematological malignancies.
Acknowledgements
This work was supported in part by the National Cancer Institute of the United States grant CA141404.
Footnotes
Disclosures
The author has received research support from EntreMed, Inc.
References
- Andrews PD. Aurora kinases: shining lights on the therapeutic horizon? Oncogene. 2005;24:5005–5015. doi: 10.1038/sj.onc.1208752. [DOI] [PubMed] [Google Scholar]
- Andrews PD, Knatko E, Moore WJ, Swedlow JR. Mitotic mechanics: the auroras come into view. Curr Opin Cell Biol. 2003;15:672–683. doi: 10.1016/j.ceb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Andrews PD, Ovechkina Y, Morrice N, Wagenbach M, Duncan K, Wordeman L, Swedlow JR. Aurora B regulates MCAK at the mitotic centromere. Dev Cell. 2004;6:253–268. doi: 10.1016/s1534-5807(04)00025-5. [DOI] [PubMed] [Google Scholar]
- Araki K, Nozaki K, Ueba T, Tatsuka M, Hashimoto N. High expression of Aurora-B/Aurora and Ipll-like midbody-associated protein (AIM-1) in astrocytomas. J Neurooncol. 2004;67:53–64. doi: 10.1023/b:neon.0000021784.33421.05. [DOI] [PubMed] [Google Scholar]
- Barr AR, Gergely F. Aurora-A: the maker and breaker of spindle poles. J Cell Sci. 2007;120:2987–2996. doi: 10.1242/jcs.013136. [DOI] [PubMed] [Google Scholar]
- Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, Chan CS, Novotny M, Slamon DJ, Plowman GD. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. Embo J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodvarsdottir SK, Hilmarsdottir H, Birgisdottir V, Steinarsdottir M, Jonasson JG, Eyfjord JE. Aurora-A amplification associated with BRCA2 mutation in breast tumours. Cancer Lett. 2007;248:96–102. doi: 10.1016/j.canlet.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Burkard ME, Randall CL, Larochelle S, Zhang C, Shokat KM, Fisher RP, Jallepalli PV. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc Natl Acad Sci U S A. 2007;104:4383–4388. doi: 10.1073/pnas.0701140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard ME, Maciejowski J, Rodriguez-Bravo V, Repka M, Lowery DM, Clauser KR, Zhang C, Shokat KM, Carr SA, Yaffe MB, Jallepalli PV. Plk1 self-organization and priming phosphorylation of HsCYK-4 at the spindle midzone regulate the onset of division in human cells. PLoS Biol. 2009;7:e1000111. doi: 10.1371/journal.pbio.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho E, Bea S, Salaverria I, Lopez-Guillermo A, Puig X, Benavente Y, de Sanjose S, Campo E, Hernandez L. Analysis of Aurora-A and hMPS1 mitotic kinases in mantle cell lymphoma. Int J Cancer. 2006;118:357–363. doi: 10.1002/ijc.21370. [DOI] [PubMed] [Google Scholar]
- Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–854. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- Chan EH, Santamaria A, Sillje HH, Nigg EA. Plk1 regulates mitotic Aurora A function through betaTrCP-dependent degradation of hBora. Chromosoma. 2008;117:457–469. doi: 10.1007/s00412-008-0165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, Bergsagel PL. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Brents LA, Ely SA, Bais C, Robbiani DF, Mesri EA, Kuehl WM, Bergsagel PL. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;97:729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- Chng WJ, Braggio E, Mulligan G, Bryant B, Remstein E, Valdez R, Dogan A, Fonseca R. The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood. 2008;111:1603–1609. doi: 10.1182/blood-2007-06-097774. [DOI] [PubMed] [Google Scholar]
- Cortes J, Paquette R, Talpaz M, Pinilla J, Asatiani E, Wetzler M, Lipton JH, Kasap C, Bui LA, Clary DO, Shah N. Preliminary Clinical Activity in a Phase I Trial of the BCR-ABL/IGF-1R/Aurora Kinase Inhibitor XL228 in Patients with Ph++ Leukemias with Either Failure to Multiple TKI Therapies or with T315I Mutation. Blood (ASH Annual Meeting Abstracts) 2008a;112:3232. [Google Scholar]
- Cortes J, Roboz GJ, Kantarjian HM, Feldman EJ, Karp JE, Pratz KW, Rao NS, Akinaga S, Levis MJ. A Phase I Dose Escalation Study of KW-2449, An Oral Multi-Kinase Inhibitor against FLT3, Abl, FGFR1 and Aurora in Patients with Relapsed/Refractory, AML, ALL and MDS or Resistant/Intolerant CML. Blood (ASH Annual Meeting Abstracts) 2008b;112:2967. [Google Scholar]
- Cortes-Franco J, Dombret H, Schafhausen P, Brummendorf TH, Boissel N, Latini F, Capolongo L, Laffranchi B, Comis S. Danusertib Hydrochloride (PHA-739358), a Multi-Kinase Aurora Inhibitor, Elicits Clinical Benefit in Advanced Chronic Myeloid Leukemia and Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia. Blood (ASH Annual Meeting Abstracts) 2009;114:864. [Google Scholar]
- Dar AA, Zaika A, Piazuelo MB, Correa P, Koyama T, Belkhiri A, Washington K, Castells A, Pera M, El-Rifai W. Frequent overexpression of Aurora Kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer. 2008;112:1688–1698. doi: 10.1002/cncr.23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AA, Belkhiri A, El-Rifai W. The aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene. 2009;28:866–875. doi: 10.1038/onc.2008.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, Kremer M, Graf N, Scheerer M, Hall MA, Goga A, von Bubnoff N, Duyster J, Peschel C, Cleveland JL, Nilsson JA, Keller U. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116:1498–1505. doi: 10.1182/blood-2009-11-251074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato NJ, Fang D, Sun H, Giannola D, Peterson LF, Talpaz M. Targets and effectors of the cellular response to aurora kinase inhibitor MK-0457 (VX-680) in imatinib sensitive and resistant chronic myelogenous leukemia. Biochem Pharmacol. 2010;79:688–697. doi: 10.1016/j.bcp.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, Mirey G, Bouche JP, Theis-Febvre N, Schmitt E, Monsarrat B, Prigent C, Ducommun B. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J Cell Sci. 2004;117:2523–2531. doi: 10.1242/jcs.01108. [DOI] [PubMed] [Google Scholar]
- Evans R, Naber C, Steffler T, Checkland T, Keats J, Maxwell C, Perry T, Chau H, Belch A, Pilarski L, Reiman T. Aurora A kinase RNAi and small molecule inhibition of Aurora kinases with VE-465 induce apoptotic death in multiple myeloma cells. Leuk Lymphoma. 2008a;49:559–569. doi: 10.1080/10428190701824544. [DOI] [PubMed] [Google Scholar]
- Evans RP, Naber C, Steffler T, Checkland T, Maxwell CA, Keats JJ, Belch AR, Pilarski LM, Lai R, Reiman T. The selective Aurora B kinase inhibitor AZD1152 is a potential new treatment for multiple myeloma. Br J Haematol. 2008b;140:295–302. doi: 10.1111/j.1365-2141.2007.06913.x. [DOI] [PubMed] [Google Scholar]
- Eyers PA, Erikson E, Chen LG, Maller JL. A novel mechanism for activation of the protein kinase Aurora A. Curr Biol. 2003;13:691–697. doi: 10.1016/s0960-9822(03)00166-0. [DOI] [PubMed] [Google Scholar]
- Farag S, Zhang S, Suvannasankha A, Liang J, O'Bryant R, Lisa W, Gupta S, Bray M, Sidor CF, Abonour R. Clinical Activity of a Novel Multiple Tyrosine Kinase and Aurora Kinase Inhibitor, ENMD-2076, Against Multiple Myeloma: Interim Phase I Trial Results. ASH Annual Meeting Abstracts. 2010;116:1957. [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- Fletcher GC, Brokx RD, Denny TA, Hembrough TA, Plum SM, Fogler WE, Sidor CF, Bray MR. ENMD-2076 Is an Orally-Active Kinase Inhibitor with Antiangiogenic and Antiproliferative Mechanisms of Action. Mol Cancer Ther. 2011;10:126–137. doi: 10.1158/1535-7163.MCT-10-0574. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel PL, Chesi M, Davies FE, Drach J, Greipp PR, Kirsch IR, Kuehl WM, Hernandez JM, Minvielle S, Pilarski LM, Shaughnessy JD, Jr., Stewart AK, Avet-Loiseau H. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 2004;64:1546–1558. doi: 10.1158/0008-5472.can-03-2876. [DOI] [PubMed] [Google Scholar]
- Foran JM, Ravandi F, O'Brien SM, Borthakur G, Rios M, Boone P, Worrell J, Mallett KH, Squires M, Fazal LH, Kantarjian HM. Phase I and pharmacodynamic trial of AT9283, an aurora kinase inhibitor, in patients with refractory leukemia. ASCO Meeting Abstracts. 2008;26:2518. [Google Scholar]
- Furukawa T, Kanai N, Shiwaku HO, Soga N, Uehara A, Horii A. AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene. 2006;25:4831–4839. doi: 10.1038/sj.onc.1209494. [DOI] [PubMed] [Google Scholar]
- Gassmann R, Carvalho A, Henzing AJ, Ruchaud S, Hudson DF, Honda R, Nigg EA, Gerloff DL, Earnshaw WC. Borealin: a novel chromosomal passenger required for stability of the bipolar mitotic spindle. J Cell Biol. 2004;166:179–191. doi: 10.1083/jcb.200404001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN, Jr., Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14:1639–1648. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- Giet R, Prigent C. Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine-threonine kinases. J Cell Sci. 1999;112(Pt 21):3591–3601. doi: 10.1242/jcs.112.21.3591. [DOI] [PubMed] [Google Scholar]
- Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006;66:7668–7677. doi: 10.1158/0008-5472.CAN-05-3353. [DOI] [PubMed] [Google Scholar]
- Goldberg SL, Fenaux P, Craig MD, Gyan E, Lister J, Kassis J, Pigneux A, Schiller GJ, Jung J, Leonard EJ, Fingert H, Westervelt P. Phase 2 Study of MLN8237, An Investigational Aurora A Kinase (AAK) Inhibitor In Patients with Acute Myelogenous Leukemia (AML) or Myelodysplastic Syndromes (MDS) Blood (ASH Annual Meeting Abstracts) 2010;116:3273. [Google Scholar]
- Gontarewicz A, Balabanov S, Keller G, Colombo R, Graziano A, Pesenti E, Benten D, Bokemeyer C, Fiedler W, Moll J, Brummendorf TH. Simultaneous targeting of Aurora kinases and Bcr-Abl kinase by the small molecule inhibitor PHA-739358 is effective against imatinib-resistant BCR-ABL mutations including T315I. Blood. 2008;111:4355–4364. doi: 10.1182/blood-2007-09-113175. [DOI] [PubMed] [Google Scholar]
- Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, Ikeda H, Bianchi G, Hu Y, Cirstea D, Santo L, Tai YT, Nahar S, Zheng M, Bandi M, Carrasco RD, Raje N, Munshi N, Richardson P, Anderson KC. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115:5202–5213. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, Fiorica JV, Nicosia SV, Cheng JQ. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003;9:1420–1426. [PubMed] [Google Scholar]
- Grundy M, Seedhouse C, Shang S, Richardson J, Russell N, Pallis M. The FLT3 internal tandem duplication mutation is a secondary target of the aurora B kinase inhibitor AZD1152-HQPA in acute myelogenous leukemia cells. Mol Cancer Ther. 2010;9:661–672. doi: 10.1158/1535-7163.MCT-09-1144. [DOI] [PubMed] [Google Scholar]
- Guse A, Mishima M, Glotzer M. Phosphorylation of ZEN-4/MKLP1 by aurora B regulates completion of cytokinesis. Curr Biol. 2005;15:778–786. doi: 10.1016/j.cub.2005.03.041. [DOI] [PubMed] [Google Scholar]
- Hamada M, Yakushijin Y, Ohtsuka M, Kakimoto M, Yasukawa M, Fujita S. Aurora2/BTAK/STK15 is involved in cell cycle checkpoint and cell survival of aggressive non-Hodgkin's lymphoma. Br J Haematol. 2003;121:439–447. doi: 10.1046/j.1365-2141.2003.04311.x. [DOI] [PubMed] [Google Scholar]
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- Hart KC, Robertson SC, Kanemitsu MY, Meyer AN, Tynan JA, Donoghue DJ. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene. 2000;19:3309–3320. doi: 10.1038/sj.onc.1203650. [DOI] [PubMed] [Google Scholar]
- Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, Hatakeyama K, Saya H. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–598. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- Hirota T, Lipp JJ, Toh BH, Peters JM. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- Honda K, Mihara H, Kato Y, Yamaguchi A, Tanaka H, Yasuda H, Furukawa K, Urano T. Degradation of human Aurora2 protein kinase by the anaphase-promoting complex-ubiquitin-proteasome pathway. Oncogene. 2000;19:2812–2819. doi: 10.1038/sj.onc.1203609. [DOI] [PubMed] [Google Scholar]
- Hose D, Reme T, Meissner T, Moreaux J, Seckinger A, Lewis J, Benes V, Benner A, Hundemer M, Hielscher T, Shaughnessy JD, Jr., Barlogie B, Neben K, Kramer A, Hillengass J, Bertsch U, Jauch A, De Vos J, Rossi JF, Mohler T, Blake J, Zimmermann J, Klein B, Goldschmidt H. Inhibition of aurora kinases for tailored risk-adapted treatment of multiple myeloma. Blood. 2009;113:4331–4340. doi: 10.1182/blood-2008-09-178350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XF, Luo SK, Xu J, Li J, Xu DR, Wang LH, Yan M, Wang XR, Wan XB, Zheng FM, Zeng YX, Liu Q. Aurora kinase inhibitory VX-680 increases Bax/Bcl-2 ratio and induces apoptosis in Aurora-A-high acute myeloid leukemia. Blood. 2008;111:2854–2865. doi: 10.1182/blood-2007-07-099325. [DOI] [PubMed] [Google Scholar]
- Huck JJ, Zhang M, Hyer ML, Manfredi MG. Anti-Tumor Activity of the Aurora a Inhibitor MLN8237 in Diffuse Large B-Cell Lymphoma Preclinical Models. Blood (ASH Annual Meeting Abstracts) 2008;112 Abstract 1592. [Google Scholar]
- Ikezoe T, Takeuchi T, Yang J, Adachi Y, Nishioka C, Furihata M, Koeffler HP, Yokoyama A. Analysis of Aurora B kinase in non-Hodgkin lymphoma. Lab Invest. 2009;89:1364–1373. doi: 10.1038/labinvest.2009.106. [DOI] [PubMed] [Google Scholar]
- Ikezoe T, Yang J, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, Komatsu N, Bandobashi K, Togitani K, Koeffler HP, Taguchi H. A novel treatment strategy targeting Aurora kinases in acute myelogenous leukemia. Mol Cancer Ther. 2007;6:1851–1857. doi: 10.1158/1535-7163.MCT-07-0067. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Jeng YM, Peng SY, Lin CY, Hsu HC. Overexpression and amplification of Aurora-A in hepatocellular carcinoma. Clin Cancer Res. 2004;10:2065–2071. doi: 10.1158/1078-0432.ccr-1057-03. [DOI] [PubMed] [Google Scholar]
- Kanai M, Goke M, Tsunekawa S, Podolsky DK. Signal transduction pathway of human fibroblast growth factor receptor 3. Identification of a novel 66-kDa phosphoprotein. J Biol Chem. 1997;272:6621–6628. doi: 10.1074/jbc.272.10.6621. [DOI] [PubMed] [Google Scholar]
- Kantarjian HM, Sekeres MA, Ribrag V, Rousselot P, Garcia-Manero G, Jabbour E, Owen K, Stockman P, Oliver S. Phase I Study to Assess the Safety and Tolerability of AZD1152 In Combination with Low Dose Cytosine Arabinoside In Patients with Acute Myeloid Leukemia (AML) Blood (ASH Annual Meeting Abstracts) 2010;116:656. [Google Scholar]
- Katayama H, Brinkley WR, Sen S. The Aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev. 2003;22:451–464. doi: 10.1023/a:1023789416385. [DOI] [PubMed] [Google Scholar]
- Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA, Sen S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55–62. doi: 10.1038/ng1279. [DOI] [PubMed] [Google Scholar]
- Keen N, Taylor S. Mitotic drivers--inhibitors of the Aurora B Kinase. Cancer Metastasis Rev. 2009;28:185–195. doi: 10.1007/s10555-009-9184-9. [DOI] [PubMed] [Google Scholar]
- Kelly KR, Ecsedy J, Medina E, Mahalingam D, Padmanabhan S, Nawrocki ST, Giles FJ, Carew JS. The novel Aurora A kinase inhibitor MLN8237 is active in resistant chronic myeloid leukemia and significantly increases the efficacy of nilotinib. J Cell Mol Med. 2010 doi: 10.1111/j.1582-4934.2010.01218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A, Reichardt W, Jung V, Zang KD, Meese E, Urbschat S. Overexpression and amplification of STK15 in human gliomas. Int J Oncol. 2004;25:1789–1794. [PubMed] [Google Scholar]
- Klein UR, Nigg EA, Gruneberg U. Centromere targeting of the chromosomal passenger complex requires a ternary subcomplex of Borealin, Survivin, and the N-terminal domain of INCENP. Mol Biol Cell. 2006;17:2547–2558. doi: 10.1091/mbc.E05-12-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffa MD, Casanova CM, Santarella R, Kocher T, Wilm M, Mattaj IW. HURP is part of a Ran-dependent complex involved in spindle formation. Curr Biol. 2006;16:743–754. doi: 10.1016/j.cub.2006.03.056. [DOI] [PubMed] [Google Scholar]
- Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett AK, Goldstone AH, Linch DC. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–1759. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- Kretzner L, Scuto A, Dino PM, Kowolik CM, Wu J, Ventura P, Jove R, Forman SJ, Yen Y, Kirschbaum MH. Combining Histone Deacetylase Inhibitor Vorinostat with Aurora Kinase Inhibitors Enhances Lymphoma Cell Killing with Repression of c-Myc, hTERT, and microRNA Levels. Cancer Res. 2011;71:3912–3920. doi: 10.1158/0008-5472.CAN-10-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufer TA, Sillje HH, Korner R, Gruss OJ, Meraldi P, Nigg EA. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol. 2002;158:617–623. doi: 10.1083/jcb.200204155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurai M, Shiozawa T, Shih HC, Miyamoto T, Feng YZ, Kashima H, Suzuki A, Konishi I. Expression of Aurora kinases A and B in normal, hyperplastic, and malignant human endometrium: Aurora B as a predictor for poor prognosis in endometrial carcinoma. Hum Pathol. 2005;36:1281–1288. doi: 10.1016/j.humpath.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Lens SM, Rodriguez JA, Vader G, Span SW, Giaccone G, Medema RH. Uncoupling the central spindle-associated function of the chromosomal passenger complex from its role at centromeres. Mol Biol Cell. 2006;17:1897–1909. doi: 10.1091/mbc.E05-08-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10:825–841. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- Lichtman MA. Battling the hematological malignancies: the 200 years' war. Oncologist. 2008;13:126–138. doi: 10.1634/theoncologist.2007-0228. [DOI] [PubMed] [Google Scholar]
- Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlepage LE, Wu H, Andresson T, Deanehan JK, Amundadottir LT, Ruderman JV. Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proc Natl Acad Sci U S A. 2002;99:15440–15445. doi: 10.1073/pnas.202606599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Vader G, Vromans MJ, Lampson MA, Lens SM. Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science. 2009;323:1350–1353. doi: 10.1126/science.1167000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok W, Klein RQ, Saif MW. Aurora kinase inhibitors as anti-cancer therapy. Anticancer Drugs. 2010;21:339–350. doi: 10.1097/CAD.0b013e3283350dd1. [DOI] [PubMed] [Google Scholar]
- Lowenberg B, Rousselot P, Martinelli G, Goudie A, Stockman P, Kantarjian H. Phase I/II Study to Assess the Safety and Efficacy of the Aurora B Kinase Inhibitor, AZD1152, in Patients with Advanced Acute Myeloid Leukemia. Blood (ASH Annual Meeting Abstracts) 2009;114:2080. [Google Scholar]
- Lu L, Wood J, Minter-Dykhouse K, Saunders T, Yu X, Chen J. Aurora A is essential for early embryonic development and tumor suppression. J Biol Chem. 2008;283:31785–31790. doi: 10.1074/jbc.M805880200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455:119–123. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- Mahadevan D, Liu X, Persky DO, Miller TP, Squires MS, Qi W. AT9283, a Novel Pan-Aurora/JAK-2 Kinase Inhibitor Suppresses Tumor Growth In Aggressive B-Cell Non-Hodgkin's Lymphoma. Blood (ASH Annual Meeting Abstracts) 2010;116 Abstract 3930. [Google Scholar]
- Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, Galvin KM, Hoar KM, Huck JJ, LeRoy PJ, Ray ET, Sells TB, Stringer B, Stroud SG, Vos TJ, Weatherhead GS, Wysong DR, Zhang M, Bolen JB, Claiborne CF. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A. 2007;104:4106–4111. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, Keir ST, Reynolds CP, Kang MH, Wu J, Smith MA, Houghton PJ. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP) Pediatr Blood Cancer. 2010;55:26–34. doi: 10.1002/pbc.22430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, Markovtsov V, Li H, Wong S, Gelman M, Zhu Y, Franci C, Lang DW, Pali E, Lasaga J, Low C, Zhao F, Chang B, Gururaja TL, Xu W, Baluom M, Sweeny D, Carroll D, Sran A, Thota S, Parmer M, Romane A, Clemens G, Grossbard E, Qu K, Jenkins Y, Kinoshita T, Taylor V, Holland SJ, Argade A, Singh R, Pine P, Payan DG, Hitoshi Y. Preclinical characterization of Aurora kinase inhibitor R763/AS703569 identified through an image-based phenotypic screen. J Cancer Res Clin Oncol. 2010;136:99–113. doi: 10.1007/s00432-009-0641-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi Y, Iwao K, Egawa C, Noguchi S. Association of centrosomal kinase STK15/BTAK mRNA expression with chromosomal instability in human breast cancers. Int J Cancer. 2001;92:370–373. doi: 10.1002/ijc.1200. [DOI] [PubMed] [Google Scholar]
- Monier K, Mouradian S, Sullivan KF. DNA methylation promotes Aurora-B-driven phosphorylation of histone H3 in chromosomal subdomains. J Cell Sci. 2007;120:101–114. doi: 10.1242/jcs.03326. [DOI] [PubMed] [Google Scholar]
- Moore AS, Blagg J, Linardopoulos S, Pearson AD. Aurora kinase inhibitors: novel small molecules with promising activity in acute myeloid and Philadelphia-positive leukemias. Leukemia. 2010;24:671–678. doi: 10.1038/leu.2010.15. [DOI] [PubMed] [Google Scholar]
- Moreau P, Attal M, Garban F, Hulin C, Facon T, Marit G, Michallet M, Doyen C, Leyvraz S, Mohty M, Wetterwald M, Mathiot C, Caillot D, Berthou C, Benboubker L, Garderet L, Chaleteix C, Traulle C, Fuzibet JG, Jaubert J, Lamy T, Casassus P, Dib M, Kolb B, Dorvaux V, Grosbois B, Yakoub-Agha I, Harousseau JL, Avet-Loiseau H. Heterogeneity of t(4;14) in multiple myeloma. Long-term follow-up of 100 cases treated with tandem transplantation in IFM99 trials. Leukemia. 2007;21:2020–2024. doi: 10.1038/sj.leu.2404832. [DOI] [PubMed] [Google Scholar]
- Mori N, Ishikawa C, Senba M, Kimura M, Okano Y. Effects of AZD1152, a selective Aurora B kinase inhibitor, on Burkitt's and Hodgkin's lymphomas. Biochem Pharmacol. 2011;81:1106–1115. doi: 10.1016/j.bcp.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Mountzios G, Terpos E, Dimopoulos MA. Aurora kinases as targets for cancer therapy. Cancer Treat Rev. 2008;34:175–182. doi: 10.1016/j.ctrv.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Murata-Hori M, Wang YL. The kinase activity of aurora B is required for kinetochore-microtubule interactions during mitosis. Curr Biol. 2002;12:894–899. doi: 10.1016/s0960-9822(02)00848-5. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Medina E, Smith S, Swords R, Kelly K, Padmanabhan S, Ecsedy J, Giles FJ, Carew J. The Aurora Kinase Inhibitor MLN8237 Has Potent Anticancer Activity in CML and Ph+ ALL Models and Significantly Increases the Efficacy of Nilotinib. Blood (ASH Annual Meeting Abstracts) 2008;112 Abstract 3198. [Google Scholar]
- Negri JM, McMillin DW, Delmore J, Mitsiades N, Hayden P, Klippel S, Hideshima T, Chauhan D, Munshi NC, Buser CA, Pollard J, Richardson PG, Anderson KC, Mitsiades CS. In vitro anti-myeloma activity of the Aurora kinase inhibitor VE-465. Br J Haematol. 2009;147:672–676. doi: 10.1111/j.1365-2141.2009.07891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N, Nagasaka T, Kashiwagi K, Boland CR, Goel A. High copy amplification of the Aurora-A gene is associated with chromosomal instability phenotype in human colorectal cancers. Cancer Biol Ther. 2007;6:525–533. doi: 10.4161/cbt.6.4.3817. [DOI] [PubMed] [Google Scholar]
- Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- Otsuki T, Yamada O, Yata K, Sakaguchi H, Kurebayashi J, Nakazawa N, Taniwaki M, Yawata Y, Ueki A. Expression of fibroblast growth factor and FGF-receptor family genes in human myeloma cells, including lines possessing t(4;14)(q16.3;q32. 3) and FGFR3 translocation. Int J Oncol. 1999;15:1205–1212. doi: 10.3892/ijo.15.6.1205. [DOI] [PubMed] [Google Scholar]
- Ozaki T, Nakagawara A. p53: the attractive tumor suppressor in the cancer research field. J Biomed Biotechnol. 2011;2011:603925. doi: 10.1155/2011/603925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan S, Shea TC, Vose JM, Reeder CB, Berdeja JG, McDonagh KT, Goy A, Kelly KR, Zhou X, Liu H, Fingert H, Fowler N. Phase I Study of An Investigational Aurora A Kinase Inhibitor MLN8237 In Patients with Advanced Hematologic Malignancies. Blood (ASH Annual Meeting Abstracts) 2010;116:2799. [Google Scholar]
- Paterson JL, Li Z, Wen XY, Masih-Khan E, Chang H, Pollett JB, Trudel S, Stewart AK. Preclinical studies of fibroblast growth factor receptor 3 as a therapeutic target in multiple myeloma. Br J Haematol. 2004;124:595–603. doi: 10.1111/j.1365-2141.2004.04814.x. [DOI] [PubMed] [Google Scholar]
- Paul MK, Mukhopadhyay AK. Tyrosine kinase - Role and significance in Cancer. Int J Med Sci. 2004;1:101–115. doi: 10.7150/ijms.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronczki M, Glotzer M, Kraut N, Peters JM. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev Cell. 2007;12:713–725. doi: 10.1016/j.devcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Plowright EE, Li Z, Bergsagel PL, Chesi M, Barber DL, Branch DR, Hawley RG, Stewart AK. Ectopic expression of fibroblast growth factor receptor 3 promotes myeloma cell proliferation and prevents apoptosis. Blood. 2000;95:992–998. [PubMed] [Google Scholar]
- Pollard D, Mortimore M. Discovery and development of aurora kinase inhibitors as anticancer agents. J Med Chem. 2009;52:2629–2651. doi: 10.1021/jm8012129. [DOI] [PubMed] [Google Scholar]
- Pratz KW, Cortes J, Roboz GJ, Rao N, Arowojolu O, Stine A, Shiotsu Y, Shudo A, Akinaga S, Small D, Karp JE, Levis M. A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response. Blood. 2009;113:3938–3946. doi: 10.1182/blood-2008-09-177030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Cooke LS, Liu X, Rimsza L, Roe DJ, Manziolli A, Persky DO, Miller TP, Mahadevan D. Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity in mantle cell lymphoma. Biochem Pharmacol. 2011;81:881–890. doi: 10.1016/j.bcp.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834–848. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- Reichardt W, Jung V, Brunner C, Klein A, Wemmert S, Romeike BF, Zang KD, Urbschat S. The putative serine/threonine kinase gene STK15 on chromosome 20q13.2 is amplified in human gliomas. Oncol Rep. 2003;10:1275–1279. [PubMed] [Google Scholar]
- Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- Santo L, Hideshima T, Cirstea D, Bandi ML, Nelson EA, Ikeda H, Vallet S, Pozzi S, Patel K, Gorgun G, Mimura N, Fabre C, Hu Y, Chauhan D, Squires MS, Munshi NC, Anderson KC, Raje N. AT9283, a Small Molecule Multi-Targeted Kinase Inhibitor with Potent Activity Against Aurora Kinase and STAT3 In Combination with Lenalidomide Results In Synergistic Anti-Myeloma Activity. Blood (ASH Annual Meeting Abstracts) 2010;116:2994. [Google Scholar]
- Santo L, Hideshima T, Cirstea D, Bandi M, Nelson EA, Gorgun G, Rodig SJ, Vallet S, Pozzi S, Patel K, Unitt CL, Squires MS, Hu Y, Chauhan D, Mahindra A, Munshi NC, Anderson KC, Raje NS. Anti-myeloma activity of a multi targeted kinase inhibitor, AT9283, via potent Aurora Kinase and STAT3 inhibition either alone or in combination with lenalidomide. Clin Cancer Res. 2011;17:3259–3271. doi: 10.1158/1078-0432.CCR-10-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai K, Katayama H, Stenoien DL, Fujii S, Honda R, Kimura M, Okano Y, Tatsuka M, Suzuki F, Nigg EA, Earnshaw WC, Brinkley WR, Sen S. Aurora-C kinase is a novel chromosomal passenger protein that can complement Aurora-B kinase function in mitotic cells. Cell Motil Cytoskeleton. 2004;59:249–263. doi: 10.1002/cm.20039. [DOI] [PubMed] [Google Scholar]
- Satinover DL, Leach CA, Stukenberg PT, Brautigan DL. Activation of Aurora-A kinase by protein phosphatase inhibitor 2, a bifunctional signaling protein. Proc Natl Acad Sci U S A. 2004;101:8625–8630. doi: 10.1073/pnas.0402966101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapin S, Carneiro F, Alves A, Medrano F, Guimaraes B, Zanchin N. The crystal structure of the small GTPase Rab11b reveals critical differences relative to the Rab11a isoform. J Struct Biol. 2006;154:260–268. doi: 10.1016/j.jsb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Seitz AK, von Bubnoff N, Sarno SM, Peschel C, Duyster J. Activity of Serono-AS703569, a Dual Inhibitor of Bcr-Abl and Aurora Kinases in Bcr-Abl Transformed Cells, Is Dependent On Aurora B Inhibition, and Is Not Affected by the Presence of the Highly Imatinib Resistant Bcr-Abl Mutation T315I. Blood (ASH Annual Meeting Abstracts) 2009;114:3247. [Google Scholar]
- Sen S, Zhou H, White RA. A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene. 1997;14:2195–2200. doi: 10.1038/sj.onc.1201065. [DOI] [PubMed] [Google Scholar]
- Sen S, Zhou H, Zhang RD, Yoon DS, Vakar-Lopez F, Ito S, Jiang F, Johnston D, Grossman HB, Ruifrok AC, Katz RL, Brinkley W, Czerniak B. Amplification/overexpression of a mitotic kinase gene in human bladder cancer. J Natl Cancer Inst. 2002;94:1320–1329. doi: 10.1093/jnci/94.17.1320. [DOI] [PubMed] [Google Scholar]
- Sessa F, Mapelli M, Ciferri C, Tarricone C, Areces LB, Schneider TR, Stukenberg PT, Musacchio A. Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell. 2005;18:379–391. doi: 10.1016/j.molcel.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Shiotsu Y, Kiyoi H, Ishikawa Y, Tanizaki R, Shimizu M, Umehara H, Ishii K, Mori Y, Ozeki K, Minami Y, Abe A, Maeda H, Akiyama T, Kanda Y, Sato Y, Akinaga S, Naoe T. KW-2449, a novel multikinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation. Blood. 2009;114:1607–1617. doi: 10.1182/blood-2009-01-199307. [DOI] [PubMed] [Google Scholar]
- Smith SL, Bowers NL, Betticher DC, Gautschi O, Ratschiller D, Hoban PR, Booton R, Santibanez-Koref MF, Heighway J. Overexpression of aurora B kinase (AURKB) in primary non-small cell lung carcinoma is frequent, generally driven from one allele, and correlates with the level of genetic instability. Br J Cancer. 2005;93:719–729. doi: 10.1038/sj.bjc.6602779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonet A, Graux C, Maertens J, Hartog C-M, Duyster J, Gotze K, Greiner J, Hutter M-L, Gratwohl A, Heim D, Hess D, Chalandon Y, Gianella-Borradori A, Rejeb N, Ottmann O. Phase I Dose-Escalation Study of 2 Dosing Regimens of AS703569, An Inhibitor of Aurora and Other Kinases, Administered Orally in Patients with Advanced Hematological Malignancies. Blood (ASH Annual Meeting Abstracts) 2008;112:2963. [Google Scholar]
- Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, Iacobucci I, Amabile M, Abruzzese E, Orlandi E, Radaelli F, Ciccone F, Tiribelli M, di Lorenzo R, Caracciolo C, Izzo B, Pane F, Saglio G, Baccarani M, Martinelli G, Leukemia GWPoCM. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12:7374–7379. doi: 10.1158/1078-0432.CCR-06-1516. [DOI] [PubMed] [Google Scholar]
- Steeghs N, Mathijssen RH, Wessels JA, de Graan AJ, van der Straaten T, Mariani M, Laffranchi B, Comis S, de Jonge MJ, Gelderblom H, Guchelaar HJ. Influence of pharmacogenetic variability on the pharmacokinetics and toxicity of the aurora kinase inhibitor danusertib. Invest New Drugs. 2010;29:953–962. doi: 10.1007/s10637-010-9405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Chan F, Briassouli P, Linardopoulos S. Aurora kinase inhibition downregulates NF-kappaB and sensitises tumour cells to chemotherapeutic agents. Biochem Biophys Res Commun. 2007;352:220–225. doi: 10.1016/j.bbrc.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Tanaka R, Squires MS, Kimura S, Yokota A, Mallett K, Smyth T, Thompson NT, Nagao R, Yamauchi T, Ushiki T, Lyons JF, Maekawa T. Activity of the Multi-Targeted Kinase Inhibitor, AT9283 on Imatinib-Resistant CML Models. Blood (ASH Annual Meeting Abstracts) 2008;112:1104. [Google Scholar]
- Tatsuka M, Sato S, Kitajima S, Suto S, Kawai H, Miyauchi M, Ogawa I, Maeda M, Ota T, Takata T. Overexpression of Aurora-A potentiates HRAS-mediated oncogenic transformation and is implicated in oral carcinogenesis. Oncogene. 2005;24:1122–1127. doi: 10.1038/sj.onc.1208293. [DOI] [PubMed] [Google Scholar]
- Terada Y, Tatsuka M, Suzuki F, Yasuda Y, Fujita S, Otsu M. AIM-1: a mammalian midbody-associated protein required for cytokinesis. Embo J. 1998;17:667–676. doi: 10.1093/emboj/17.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita M, Tanaka Y, Mori N. Aurora kinase inhibitor AZD1152 negatively affects the growth and survival of HTLV-1-infected T lymphocytes in vitro. Int J Cancer. 2010;127:1584–1594. doi: 10.1002/ijc.25178. [DOI] [PubMed] [Google Scholar]
- Vader G, Medema RH, Lens SM. The chromosomal passenger complex: guiding Aurora-B through mitosis. J Cell Biol. 2006;173:833–837. doi: 10.1083/jcb.200604032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vader G, Maia AF, Lens SM. The chromosomal passenger complex and the spindle assembly checkpoint: kinetochore-microtubule error correction and beyond. Cell Div. 2008;3:10. doi: 10.1186/1747-1028-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vugt MA, Bras A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell. 2004;15:799–811. doi: 10.1016/j.molcel.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Wang X, Sinn AL, Pollok K, Sandusky G, Zhang S, Chen L, Liang J, Crean CD, Suvannasankha A, Abonour R, Sidor C, Bray MR, Farag SS. Preclinical activity of a novel multiple tyrosine kinase and aurora kinase inhibitor, ENMD-2076, against multiple myeloma. Br J Haematol. 2010;150:313–325. doi: 10.1111/j.1365-2141.2010.08248.x. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A. 2004;101:4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The biology of cancer. New York, NY: Garland Science; 2007. [Google Scholar]
- Welburn JP, Vleugel M, Liu D, Yates JR, 3rd, Lampson MA, Fukagawa T, Cheeseman IM. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell. 2010;38:383–392. doi: 10.1016/j.molcel.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe BA, Takaki T, Petronczki M, Glotzer M. Polo-like kinase 1 directs assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex to initiate cleavage furrow formation. PLoS Biol. 2009;7:e1000110. doi: 10.1371/journal.pbio.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakushijin Y, Hamada M, Yasukawa M. The expression of the aurora-A gene and its significance with tumorgenesis in non-Hodgkin's lymphoma. Leuk Lymphoma. 2004;45:1741–1746. doi: 10.1080/10428190410001683615. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, Akiyama H, Saito K, Nishimura M, Motoji T, Shinagawa K, Takeshita A, Saito H, Ueda R, Ohno R, Naoe T. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–2439. doi: 10.1182/blood.v97.8.2434. [DOI] [PubMed] [Google Scholar]
- Yan X, Wu Y, Li Q, Cao L, Liu X, Saiyin H, Yu L. Cloning and characterization of a novel human Aurora C splicing variant. Biochem Biophys Res Commun. 2005;328:353–361. doi: 10.1016/j.bbrc.2004.12.168. [DOI] [PubMed] [Google Scholar]
- Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci U S A. 2010;107:13836–13841. doi: 10.1073/pnas.1008366107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ou CC, Feldman RI, Nicosia SV, Kruk PA, Cheng JQ. Aurora-A kinase regulates telomerase activity through c-Myc in human ovarian and breast epithelial cells. Cancer Res. 2004;64:463–467. doi: 10.1158/0008-5472.can-03-2907. [DOI] [PubMed] [Google Scholar]
- Yang H, He L, Kruk P, Nicosia SV, Cheng JQ. Aurora-A induces cell survival and chemoresistance by activation of Akt through a p53-dependent manner in ovarian cancer cells. Int J Cancer. 2006;119:2304–2312. doi: 10.1002/ijc.22154. [DOI] [PubMed] [Google Scholar]
- Yang J, Ikezoe T, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, Komatsu N, Bandobashi K, Togitani K, Koeffler HP, Taguchi H, Yokoyama A. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood. 2007;110:2034–2040. doi: 10.1182/blood-2007-02-073700. [DOI] [PubMed] [Google Scholar]
- Yee KWL, Chen H-WT, Brandwein J, Chow S, Sanfelice D, Hedley D, Gupta V, Schimmer A, Sidor CF, Fletcher G, Arnott J, Bray M, Minden MD, Schuh A. A Phase I Study of ENMD-2076 In Patients with Relapsed or Refractory Leukemia. Blood (ASH Annual Meeting Abstracts) 2010;116:3307. [Google Scholar]
- Yokoyama T, Goto H, Izawa I, Mizutani H, Inagaki M. Aurora-B and Rho-kinase/ROCK, the two cleavage furrow kinases, independently regulate the progression of cytokinesis: possible existence of a novel cleavage furrow kinase phosphorylates ezrin/radixin/moesin (ERM) Genes Cells. 2005;10:127–137. doi: 10.1111/j.1365-2443.2005.00824.x. [DOI] [PubMed] [Google Scholar]
- Young MA, Shah NP, Chao LH, Seeliger M, Milanov ZV, Biggs WH, 3rd, Treiber DK, Patel HK, Zarrinkar PP, Lockhart DJ, Sawyers CL, Kuriyan J. Structure of the kinase domain of an imatinib-resistant Abl mutant in complex with the Aurora kinase inhibitor VX-680. Cancer Res. 2006;66:1007–1014. doi: 10.1158/0008-5472.CAN-05-2788. [DOI] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Farag SS. From cell biology to therapy: ENMD-2076 in the treatment of multiple myeloma. Expert Opin Investig Drugs. 2011;20:1015–1028. doi: 10.1517/13543784.2011.584869. [DOI] [PubMed] [Google Scholar]
- Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, Brinkley BR, Sen S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]