

Abstract
In a phase II trial, 16 patients with relapsed refractory multiple myeloma received temsirolimus 25 mg IV weekly until progression. One partial response and 5 minor responses were observed for a total response rate of 38%. The median time to progression was 138 days. Grade 3–4 toxicity included fatigue (n=3), neutropenia (n=2), thrombocytopenia (n=2), interstitial pneumonitis (n=1), stomatitis (n=1) and diarrhea (n=1). Clinical activity was associated with a higher area under the curve (AUC) and maximal reduction in phosphorylated p70S6K and 4EBP1 in peripheral blood mononuclear cells. At the dose and schedule used, temsirolimus had low single agent activity. Investigation of alternate dosing schedules and use in combinations is indicated.
Keywords: Multiple myeloma, mTOR, temsirolimus, pharmacokinetics
INTRODUCTION
In spite of recent improvements in outcome, treatment of multiple myeloma (MM) remains essentially palliative. Although high-dose chemotherapy has improved long-term outcome, almost all patients still relapse [1,2]. Furthermore, while recently approved drugs such as thalidomide, bortezomib, and lenalidomide improve response rates and delay progression compared to more conventional cytotoxic agents [3–5], relapses invariably occur indicating the need for continued investigation of novel agents in this disease.
The PI3K/Akt/mTOR pathway is important for mediating survival, growth and resistance to apoptosis signals in myeloma cells [6,7]. Although survival responses may be the result of phosphorylation of one or more of many different substrates, stimulation of proliferation is a result of downstream signaling through mTOR, which results in phosphorylation of the p70S6 kinase (p70S6k) and of the 4EBP1 translational repressor. p70S6k activation results in increased phosphorylation of the 40S ribosomal S6 protein, and 4EBP1 phosphorylation disrupts its interaction with the eIF-4E initiation factor, allowing eIF-4E to participate in assembly of a translation initiation complex. These lead to up-regulation of proteins needed for cell cycle transit [8]. Inhibition of the PI3K/Akt/mTOR pathway may therefore be a therapeutically useful target in MM.
Temsirolimus is an ester of sirolimus and is a signal transduction inhibitor with antitumor properties. Temsirolimus reacts with the ubiquitous intracellular FK506-binding protein 12, and the complex specifically inhibits mTOR, a Ser/Thr kinase located predominantly in the nuclear fraction of both neoplastic and normal cells. Pre-clinical data indicated that temsirolimus is active against MM cell lines [9,10]. In an animal model of myeloma, injection of temsirolimus inhibited the growth of 8662 cells transplanted into NOD/SCID mice, and this was associated with in vivo inhibition of p70S6k [10]. Based on the above data, we conducted a phase II trial to assess the response rate of temsirolimus in patients with relapsed or refractory MM.
PATIENTS AND METHODS
Eligibility Criteria
Patients were eligible if aged 18 years or older, had MM as defined by World Health Organization criteria [11], a measurable serum or urine monoclonal (M)-protein, stage I-IIIA defined by the Salmon-Durie system [12], and progressive disease after at least one prior systemic treatment. In addition, patients were required to have an Eastern Cooperative Oncology Group performance score of 0 to 2, acceptable hematologic and biochemical parameters defined as absolute neutrophil count (ANC) ≥ 1.2 × 109/l, platelets > 75 × 109/l, serum creatinine ≤ 1.5 mg/dl, total bilirubin ≤ 1.5 mg/dl, AST and ALT ≤ 2.5 × upper limit of normal, fasting serum cholesterol ≤ 350 mg/dl and triglycerides ≤ 400 mg/dl. The protocol was approved by the Institutional Review Board of the Ohio State University, and all patients gave written informed consent prior to initiating protocol procedures.
Study Treatment and Monitoring
Temsirolimus was administered at a dose of 25 mg intravenously (IV) over 30 minutes every week. Patients were pre-medicated with diphenhydramine 25–50 mg IV 30 minutes before infusion to prevent idiosyncratic hypersensitivity reactions; no prophylactic corticosteroids were used. A treatment cycle was defined as 4 weeks. Patients were monitored for toxicity weekly, which was graded according the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0. Treatment was delayed for an ANC < 1.0 ×109/l or platelet count < 75 ×109/l, and for grade 3 or 4 non-hematological toxicity. Upon recovery, the dose of temsirolimus was reduced by 20% for subsequent doses. Toxicities manageable with standard supportive therapy, including electrolyte replacement for electrolyte disturbances and statin drugs for hyperlipidemia, did not require dose-reduction. Patients were taken off study for grade 3 or 4 toxicities that did not recover to at least grade 2 within 3 weeks, and if more than 3 dose reductions were required. A minimum of 2 cycles were required. Patients with progressive disease after 2 cycles were taken off study. Otherwise, patients continued until evidence of disease progression.
Assessment of Response
Disease response or progression was assessed at the beginning of each treatment cycle (every 4 weeks) according to the criteria of the European Group for Blood and Marrow Transplantation (EBMT) [13]. Serum and urine M-protein quantification was performed every 4 weeks by protein electrophoresis. Responses or progression required confirmation by an assessment at least 4 weeks later. Patients continued to receive temsirolimus until confirmation of progression was established.
Statistical Analysis
The primary endpoint was the best overall response (complete response [CR] or partial response [PR]). Although minor responses (MR) are noted as recommended recently for phase I/II trials of novel agents in patients with relapsed refractory MM [14,15], they were not considered as an endpoint in the original statistical design of the trial. The trial was designed to test the null hypothesis that the true overall response rate was at most 10%. An overall response rate of 30% or greater was considered sufficient to indicate that the regimen was worthy of further study. The study design was based on the parameters and assumptions of a two-stage Simon min max design [16] with types I and II errors set at 0.1. This required a maximum of 25 assessable patients, where an interim analysis was performed after the accrual of 16 evaluable patients who had been followed for at least 2 cycles. At least 2 responses, defined as CR or PR, in the first 16 patients were required in the interim analysis to continue accrual. Time to progression (TTP) was defined as the time from initial administration of temsirolimus to first documented progression or censored at the time of last seen. The TTP was estimated using the Kaplan-Meier method [17]. Statistical analysis was performed using S-Plus 6.1 (Insightful Corporation, Seattle, WA).
Pharmacokinetic Analysis
Temsirolimus pharmacokinetic (PK) analysis was performed on plasma samples obtained immediately before treatment and at 2, 3, 4, 6, 8, and 24 hours after the first dose of temsirolimus of cycles 1 and 2. Plasma levels were quantified by a liquid chromatography and tandem mass spectroscopy (LC-MS/MS) method established at the Ohio State University by Dr. Kenneth Chan. Assays were performed on a TSQ Quantum Triple Quadruple mass spectrometer (Thermo, San Jose, CA) with a lower limit of quantification at 0.01 ng/ml. Briefly, 200 μl of plasma were spiked with a constant amount of the internal standard AP3 (10 ng/ml) and the samples were extracted with 1 ml ethyl acetate. The supernatant was separated from the aqueous layer and the ethyl acetate was evaporated under a stream of nitrogen and the residue was reconstituted in 90% methanol buffered with 5 mM ammonium acetate and a 10 μL aliquot of the reconstituted solution was injected into a Shimadzu HPLC system (Shimadzu, Columbia, MD), which was equipped with a CBM-20A system controller, an LC-20 AD pump, a SIL-20AC autosampler, CTO-20A Column Oven, DGU-20A5 degasser and FCV-11AL Valve Unit. CCI-779 and the internal standard AP3 were separated on a Betabasic C8 column (2.1 mm I.D. × 50 mm, 5 μm; Thermo Hypersil-Keystone, Bellefonte, PA) coupled to a 2 μm pre-column filter (Thermo Hypersil-Keystone, Bellefonte, PA) using a 2.5-minute isocratic elution (90% methanol/5 mM ammonium acetate) at the flow rate of 0.2 ml/min. Temsirolimus and AP3 were monitored using the ionic transitional channels m/z 1047.51 >979.37 and 635.14>547.15, respectively. The temsirolimus plasma concentration versus time data were analyzed using noncompartmental and compartmental techniques to estimate the peak concentration (Cmax), half-life (t1/2), area under the curve (AUC), volume of distribution (Vdss), and clearance (CL).
Pharmacodynamic Analysis
The phosphorylation status of p70S6k and 4EBP1 in peripheral blood mononuclear cells (PBMC) was used to evaluate the pharmacodynamic activity of temsirolimus. Blood samples were collected at baseline, 4, 24, and 48 hours after the first dose of temsirolimus. Mononuclear cells were isolated by gradient centrifugation, washed with ice-cold phosphate buffered saline, and then resuspended in lysis buffer containing the phosphatase inhibitors sodium orthovanadate (1 mM) and microcystin LR (1 μM) (both from Sigma, St. Louis, MO). Lysates were collected after centrifugation at 13,000 rpm for 10 minutes. Equivalent amounts of protein (50 μg) from each lysate were resolved in 4–20% sodium dodecyl sulphate-polyacrilamide gel electrophoresis (SDS-PAGE), transferred to 0.2 μm nitrocellulose membranes (Schleicher & Schuell, Keene, NH), and the blots probed with primary antibody specific for the following proteins at the indicated dilutions: phospho-p70 S6K-(Thr389) (1:1000), phospho-4E-BP1(Thr37/46) (1:500), 4E-BP1 (1:500) (Cell Signaling Technology, Beverly, MA); p70 S6K (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA); β-actin (1:2000) (Novus Biologicals, Littleton, CO). Following incubation with antibody, the proteins were detected with chemiluminescent substrate (SuperSignal, Pierce, Rockford, IL). Protein bands were quantified by integration of the chemiluminescence signals on AlphaEase FC (Flurochem SP) software (Alpha Innotech, San Leandro, CA) with autobackground correction.
RESULTS
Patients
A total of 17 patients were enrolled from March 2004 to May 2006. One patient did not receive any treatment due acute pulmonary edema before initiation of therapy and was considered ineligible. Sixteen patients received at least two cycles of treatment and were considered evaluable for response and toxicity; their characteristics are shown in Table 1.
Table 1.
Patient Characteristics
| Patient | Age (years) | Gender | Subtype | ISS Stage | PS | No. prior regimens | Previous transplant | β2-mg (g/dl) | Albumin (g/dl) | Se M-protein (mg/dl)* | U M-protein (g/24 h)* | Karyotype (FISH) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 41 | F | IgG.κ | I | 1 | 3 | Yes | 2.5 | 3.6 | 3880 | - | Normal |
| 2 | 55 | M | IgG.κ | II | 2 | 2 | No | 4.4 | 2.0 | 6650 | 525 | Normal |
| 3 | 65 | M | IgG. κ | II | 1 | 1 | No | 4.6 | 3.7 | 2059 | - | +15 |
| 4 | 65 | M | IgA. κ | II | 1 | 1 | No | 3.4 | 2.9 | 3750 | 526 | Hyperdiploid |
| 5 | 55 | F | IgA. κ | II | 0 | 2 | Yes | 2.7 | 3.6 | 2910 | - | 13q- |
| 6 | 63 | F | IgG. κ | III | 1 | 2 | Yes | 7.9 | 4.1 | 2636 | 1195 | Complex, 13q- |
| 7 | 61 | F | IgG. κ | II | 0 | 3 | Yes | 4.7 | 3.4 | 1920 | - | Hyperdiploid |
| 8 | 58 | F | IgG. κ | I | 0 | 2 | No | 3.2 | 3.7 | 1469 | - | Hyperdiploid |
| 9 | 71 | M | IgG. κ | I | 0 | 2 | No | 3.2 | 4.0 | 1138 | - | Hyperdiploid |
| 10 | 62 | M | IgA. κ | I | 0 | 5 | Yes | 3.2 | 4.5 | 128 | 3681 | Hyperdiploid |
| 11 | 75 | F | IgG. κ | II | 1 | 1 | No | 3.5 | 3.8 | 3119 | - | Normal |
| 12 | 74 | M | free λ | I | 0 | 3 | Yes | 3.0 | 3.8 | - | 536 | Normal |
| 13 | 73 | M | free κ | II | 0 | 1 | No | 5.1 | 3.4 | - | 2732 | 13q- |
| 14 | 57 | M | IgG. λ | II | 1 | 1 | No | 2.9 | 2.4 | 4210 | 242 | t(11;14) |
| 15 | 58 | F | IgG. λ | II | 2 | 3 | No | 4.2 | 3.3 | 1826 | - | Normal |
| 16 | 58 | F | IgG. λ | II | 1 | 2 | yes | 4.2 | 2.1 | 1369 | - | Normal |
ISS, International Staging System; PS, performance score on the ECOG scale; Se, serum; U, urine;
(−) denotes absent serum or urine M-protein; FISH, karyotype assessed by interphase fluorescent in-situ hybridization.
Efficacy
Of the 16 patients, one patient achieved a PR with a 53% reduction in serum M-protein (Patient no. 5). In addition, 5 patients achieved a MR with 32–49% reductions in M-protein. In 6 patients, the disease remained stable for 2 to 15 months while receiving temsirolimus, with patients discontinuing therapy because of toxicity. Disease progression occurred in 4 patients while on therapy. Figure 1 shows the maximum change in serum or urine M-protein compared to baseline for individual patients while on study. The total number of patients who achieved a CR or PR was 6% (95% confidence interval [CI]: 0.16–30.2%). The inclusion of patients who also achieved a MR yields a total response rate of 38% (6 of 16 patients). However, as by study design at least 2 responses defined as CR and/or PR were required at interim analysis to proceed to the second stage of the study, accrual was terminated.
Figure 1. Maximal percentage change in M-protein for individual patients.
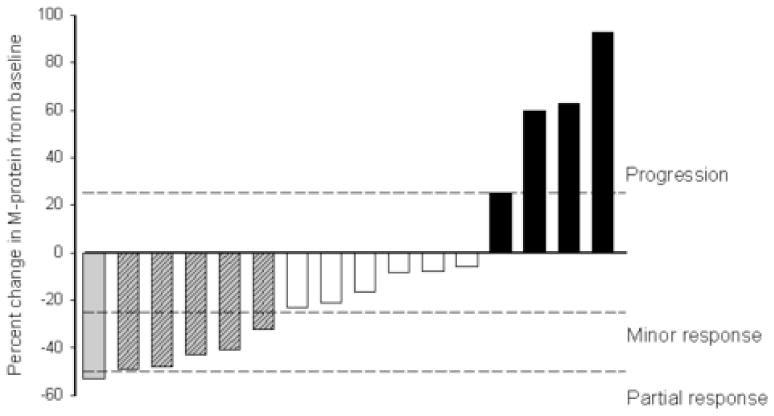
Solid bars represent patients with progression of disease on therapy; white bars, stable disease; gray shaded bars, minor response; gray bars, partial response. See text for definitions of response.
No patient remains on study. Ten patients discontinued therapy because of progressive disease. Other reasons for discontinuation of treatment included grade 3–4 toxicity (n=3), interval illness precluding continuation of treatment (n=1), poor compliance (n=1), and withdrawal of consent (n=1). The median TTP is 138 days (95% CI: 88-not reached) (Figure 2). At the time of reporting, all patients remain alive.
Figure 2.
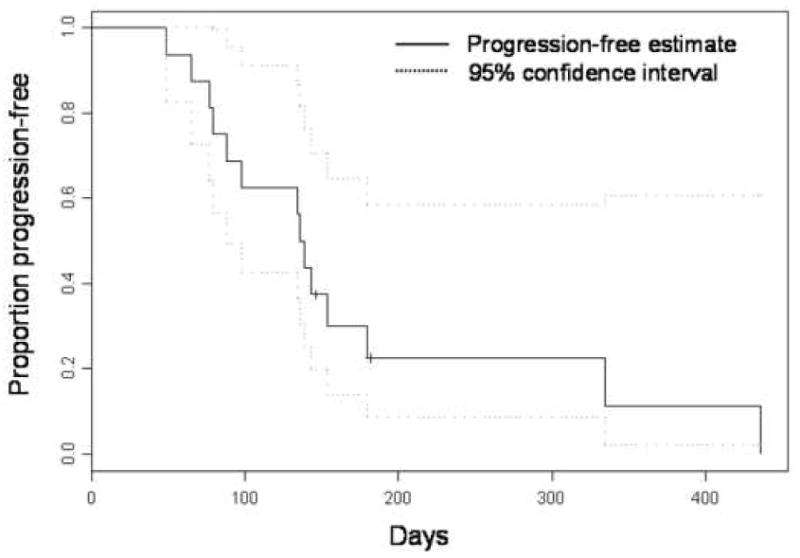
Time to progression for entire study cohort
Toxicity
Table 2 shows the reported treatment-emergent adverse events and their severity. A median of 3.75 (range, 2–14.5) cycles of temsirolimus were administered. The average dose-intensity of temsirolimus, defined as the total dose administered divided by the planned total dose over all cycles given, was 81.2% (range, 58.3–100%). Dose reductions or delays were required because of adverse events in 11 of the 16 patients treated, most commonly due to interval infection or other illness (n=3), fatigue (n=2), neutropenia (n=2), thrombocytopenia (n=2), anemia (n=1), and stomatitis (n=1). Treatment was discontinued in two patients; in one because of grade 3 thrombocytopenia that failed to resolve within 2 weeks, and in another because of interstitial pneumonitis. There were no deaths on study.
Table 2.
Treatment-Emergent Adverse Events for Patients in Any Cycle
| Adverse Event** | Number of patients with adverse event according to grade of severity*
|
|||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Anorexia | 3 | 2 | 0 | 0 |
| Nausea | 6 | 3 | 0 | 0 |
| Vomiting | 2 | 2 | 0 | 0 |
| Diarrhea | 1 | 1 | 1 | 0 |
| Change or loss of taste | 8 | 2 | 0 | 0 |
| Stomatitis | 4 | 2 | 1 | 0 |
| Rash | 4 | 3 | 0 | 0 |
| Hematologic | ||||
| Anemia | 1 | 9 | 3 | 0 |
| Neutropenia | 1 | 3 | 2 | 0 |
| Thrombocytopenia | 0 | 1 | 2 | 0 |
| Fever | 2 | 0 | 0 | 0 |
| Fatigue | 2 | 11 | 2 | 1 |
| Rash | 4 | 3 | 0 | 0 |
| Epistaxis | 7 | 0 | 0 | 0 |
| Alopecia | 0 | 1 | 0 | 0 |
| Elevated transaminases | 1 | 0 | 0 | 0 |
| Dyspnea | 0 | 2 | 1† | 0 |
| Hypertriglyceridemia | 5 | 0 | 0 | 0 |
| Hypercholesterolemia | 6 | 0 | 0 | 0 |
| Muscle pain | 1 | 0 | 0 | 0 |
Grades are according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0. All adverse events were considered at least possibly related to drug.
Each adverse event was counted once (any cycle; highest grade) for each patient.
Dyspnea was associated with bilateral pulmonary infiltrates and impaired gas transfer, consistent with interstitial pneumonitis.
Pharmacokinetics
Temsirolimus plasma concentrations were available for 13 patients who received 25 mg dose on day 1 of the first cycle. In addition, plasma levels were available for all 16 patients following the first dose of cycle 2; 9 who received 25 mg and 7 who received 20 mg of temsirolimus following dose-reduction for toxicity. The plasma concentrations of temsirolimus versus time for the 13 patients who received 25 mg in cycle 1, and for the 7 patients who received 20 mg in cycle 2 are shown in Figures 3A and B, respectively. As shown, most of the plasma concentration versus time profiles clustered closely except for one patient where the profile was higher. On compartmental analysis, the data best fitted a one compartment model. Following a dose of 25 mg temsirolimus, the mean Cmax, AUC, CL, and Vdss were 0.0093 μmol/l (standard deviation [SD], 0.0062 μmol/l), 0.0892 μmol.h/l (SD, 0.0041 μmol.h/l), 319 l/h (SD, 135 l/h), and 3246 l (SD, 1419 l), respectively. There were no significant differences between the PK parameters among patients who received 20 mg and 25 mg temsirolimus in cycles 2 and 1 respectively (P>0.05; data not shown).
Figure 3. Pharmacokinetic parameters of temsirolimus in MM patients.
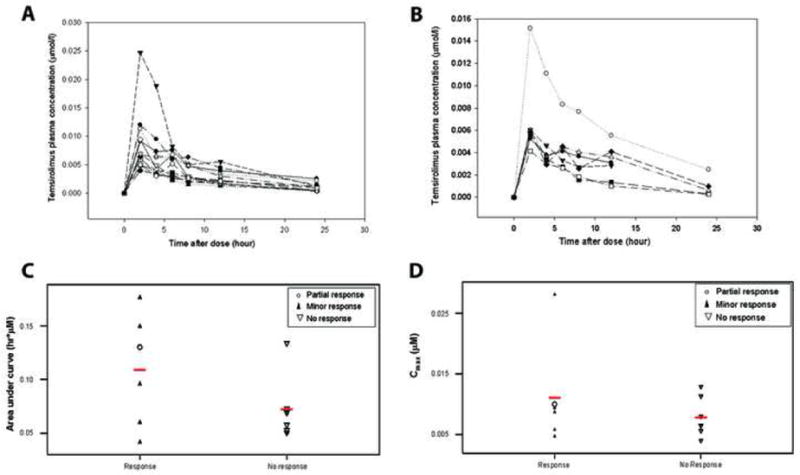
(A) Plasma concentration versus time for 13 patients who received 25 mg dose on day 1 of first cycle, and (B) those who received 20 mg dose on day 1 of cycle 2. AUC (C) and Cmax (D) distributions in patients who achieved at least a minor response (responders) and non-responders.
Figures 3C and D show the distribution of AUC and Cmax, respectively, in patients who achieved a partial or minor response as well as those who did not respond at all. While there was no correlation between Cmax and response, there was a trend toward a higher AUC in patients who achieved at least a MR. Specifically, 4 of 6 patients who had an AUC greater than 0.070 μmol.h/l achieved some response (3 MR, 1 PR) while 6 of 7 patients who did not respond had an AUC less than 0.070 μmol.h/l (χ2 test, P=0.053). The patient with the highest AUC achieved a 32% reduction in serum M-protein after 3 cycles of treatment, but also had significant delays and reduction in dose (dose-intensity, 63.6%) before discontinuing therapy after 5 cycles due to prolonged grade 3 thrombocytopenia.
Inhibition of mTOR in peripheral blood mononuclear cells
We assessed the levels of p-p70S6k and p-4EBP1 in PBMC by Western blots of lysates of PBMC obtained at baseline and at 4, 24, and 48 hours following dosing. Representative blots are shown in Figure 4A and B. Using densitometry, the change in p-p70S6K and p-4EBP1 relative to baseline was quantified and compared at each time point for patients who achieved either at least a MR and those who failed to achieve any significant reduction in M-protein. As shown, patients who achieved partial or minor responses had significant reductions in p-p70S6k (Figure 4C) and p-4EBP1 (Figure 4D) in PBMC, particularly at 48 hours. In contrast, in patients who had stable or progressive disease without response, p-p70S6K and p-4EBP1 either did not change or increased.
Figure 4. Change in levels of p-p70S6k and p-4EBP1 expression in PBMC with temsirolimus in MM patients.

Representative western blots in (A) a patient achieving a PR showing a reduction in p-p70S6k (especially at 48 hours) and p-4EBP1 (at 24 and 48 hours) after a 25 mg temsirolimus dose, compared to (B) a patient who had no reduction in M-protein where no reduction in p-p70S6k and p-4EBP1 is observed. The change in p-p70S6k (C) and p-4EBP1 (D) in PBMC relative to baseline for the entire population in patients who achieved at least a minor response and those who did not is shown in panels (C) and (D) respectively.
DISCUSSION
The results of this phase II trial indicate that temsirolimus given at the dose and schedule used is only modestly active in patients with relapsed and refractory MM. Based on a two-stage design, the trial did not proceed to full accrual because only 1 of 16 patients achieved a PR for a response rate of 6%. This compares unfavorably with the single agent activity of currently approved agents for MM, including thalidomide, bortezomib and lenalidomide in comparable populations [5,18,19]. However, it has recently been recommended to include MR, as defined by the EBMT criteria, in patients with relapsed refractory MM enrolled on phase I/II clinical trials of novel agents to detect a signal of activity [14,15]. Following this recommendation, the additional 5 MRs observed brings the total response rate to 38%. However, these additional minor responses did not translate to an appreciable clinical benefit with a median TTP that was similar to that observed in relapsed or refractory patients treated with dexamethasone alone [3,4].
The toxicity profile was similar to that observed in solid tumors patients receiving temsirolimus, although in the majority of studies significantly higher weekly doses (75–250 mg) were used [20–25]. However, in our population we observed more grade 3/4 hematological toxicity, which occurred in 7 of 16 patients, compared to previously reported experience in solid tumor patients where the incidence has been generally less than 10% [21,22,25]. This may be related to a greater susceptibility of the bone marrow in MM patients where it is a dominant site of disease. Furthermore, temsirolimus was only moderately well tolerated with only 5 of the 16 patients able to receive the drug at the planned dose-intensity.
The temsirolimus PK parameters in our study population were generally similar to those previously reported in solid tumor patients [26,27], although our data best fitted a one-rather than a multi-compartment model as previously reported [22,26]. This may be related to the fact that we measured temsirolimus levels in plasma rather than whole blood where polyexponential and non-linear disposition of the drug has been reported due to specific binding of drug to FK506-binding protein in red cell membranes [22,26,28]. While an association of AUC with the severity of thrombocytopenia and hyperlipidemia has been previously reported in one study of renal cell carcinoma patients [26], this has not been consistent across other studies and was not observed in our study. An association between temsirolimus PK parameters and clinical response, however, has not been previously reported [22]. In contrast, we observed a trend toward a higher AUC in patients who achieved at least a MR, suggesting that alternate dosing schedules that increase temsirolimus AUC may be more efficacious. Our observation, however, requires further investigation in a larger cohort of MM patients before definitive conclusions can be made.
A limitation of our study is the lack of data on the direct effect of temsirolimus on the MM cells. However, we measured the effect of temsirolimus on the phosphorylation of p70S6k and 4EBP1 in PBMC. Importantly, there was a significant association between the reduction in both p-p70S6k and p-4EBP1 and achievement of partial or minor responses. This is consistent with a recent study that showed a similar correlation between in vivo anti-tumor efficacy of another sirolimus derivative, everolimus, with reduction of p-p70S6K and p-4EBP1 in PBMC in a rat pancreatic tumor model [29]. These results suggest that PBMC may be a useful surrogate tissue to analyze for predicting response to temsirolimus and should be further investigated in future trials of mTOR inhibitors.
Several factors may be important in explaining the limited anti-myeloma efficacy of temsirolimus in this study. Although the PI3K/Akt pathway is constitutionally active in MM [6], mTOR is only one of several distal effector molecules in this pathway and therefore its sole inhibition is unlikely to be sufficiently effective. It is known that the mitogen-activated protein (MAP) kinase and Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) pathways are also activated in MM [30,31]. Therefore, it is likely that temsirolimus will be more efficacious in combination with agents that target these pathways. To this end, in vitro synergy has been demonstrated with the combination of sirolimus and lenalidomide [32]. Clinical trials evaluating the combination of mTOR inhibitors with lenalidomide are currently ongoing. Finally, alternative dosing schedules should be explored. Using sirolimus, we observed that inhibition of MM cell lines in vitro occurs only when cells are exposed to drug concentrations between 5–10 nM for more than 72 hours (unpublished data). This is consistent with the association of higher AUCs with anti-myeloma activity seen in the current study. Although we did not measure sirolimus levels, temsirolimus levels were uniformly below 5 nM by 24 hours. The recently reported schedule of daily administration for 5 days every 2 weeks may result in more sustained levels and should be further investigated in MM [33].
In conclusion, our results indicate that temsirolimus at the dose and schedule used has limited clinical efficacy in patients with relapsed and refractory MM. Future studies should aim at exploring alternative dosing schedules and the use of the temsirolimus in combination therapy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Attal M, Harousseau JL, Stoppa AM, Sotto JJ, Fuzibet JG, Rossi JF, Casassus P, Maisonneuve H, Facon T, Ifrah N, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N Engl J Med. 1996;335:91–7. doi: 10.1056/NEJM199607113350204. [DOI] [PubMed] [Google Scholar]
- 2.Child JA, Morgan GJ, Davies FE, Owen RG, Bell SE, Hawkins K, Brown J, Drayson MT, Selby PJ. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. N Engl J Med. 2003;348:1875–83. doi: 10.1056/NEJMoa022340. [DOI] [PubMed] [Google Scholar]
- 3.Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2006;24:431–6. doi: 10.1200/JCO.2005.03.0221. [DOI] [PubMed] [Google Scholar]
- 4.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–98. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 5.Richardson PG, Blood E, Mitsiades CS, Jagannath S, Zeldenrust SR, Alsina M, Schlossman RL, Rajkumar SV, Desikan KR, Hideshima T, et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood. 2006;108:3458–64. doi: 10.1182/blood-2006-04-015909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsu J, Shi Y, Krajewski S, Renner S, Fisher M, Reed JC, Franke TF, Lichtenstein A. The AKT kinase is activated in multiple myeloma tumor cells. Blood. 2001;98:2853–5. doi: 10.1182/blood.v98.9.2853. [DOI] [PubMed] [Google Scholar]
- 7.Pene F, Claessens YE, Muller O, Viguie F, Mayeux P, Dreyfus F, Lacombe C, Bouscary D. Role of the phosphatidylinositol 3-kinase/Akt and mTOR/P70S6-kinase pathways in the proliferation and apoptosis in multiple myeloma. Oncogene. 2002;21:6587–97. doi: 10.1038/sj.onc.1205923. [DOI] [PubMed] [Google Scholar]
- 8.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–62. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 9.Shi Y, Gera J, Hu L, Hsu JH, Bookstein R, Li W, Lichtenstein A. Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res. 2002;62:5027–34. [PubMed] [Google Scholar]
- 10.Frost P, Moatamed F, Hoang B, Shi Y, Gera J, Yan H, Frost P, Gibbons J, Lichtenstein A. In vivo antitumor effects of the mTOR inhibitor CCI-779 against human multiple myeloma cells in a xenograft model. Blood. 2004;104:4181–7. doi: 10.1182/blood-2004-03-1153. [DOI] [PubMed] [Google Scholar]
- 11.Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–57. [PubMed] [Google Scholar]
- 12.Cavo M, Galieni P, Zuffa E, Baccarani M, Gobbi M, Tura S. Prognostic variables and clinical staging in multiple myeloma. Blood. 1989;74:1774–80. [PubMed] [Google Scholar]
- 13.Blade J, Samson D, Reece D, Apperley J, Bjorkstrand B, Gahrton G, Gertz M, Giralt S, Jagannath S, Vesole D. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102:1115–23. doi: 10.1046/j.1365-2141.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- 14.Anderson KC, Kyle RA, Rajkumar SV, Stewart AK, Weber D, Richardson P. Clinically relevant end points and new drug approvals for myeloma. Leukemia. 2008;22:231–9. doi: 10.1038/sj.leu.2405016. [DOI] [PubMed] [Google Scholar]
- 15.Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23:3–9. doi: 10.1038/leu.2008.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan EL, Meier P. Nonparametric estimation from incomoplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 18.Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, Munshi N, Anaissie E, Wilson C, Dhodapkar M, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565–71. doi: 10.1056/NEJM199911183412102. [DOI] [PubMed] [Google Scholar]
- 19.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609–17. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 20.Margolin K, Longmate J, Baratta T, Synold T, Christensen S, Weber J, Gajewski T, Quirt I, Doroshow JH. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–8. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- 21.Duran I, Kortmansky J, Singh D, Hirte H, Kocha W, Goss G, Le L, Oza A, Nicklee T, Ho J, et al. A phase II clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. Br J Cancer. 2006;95:1148–54. doi: 10.1038/sj.bjc.6603419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, Eiermann W, Hess D, Morant R, Semiglazov V, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23:5314–22. doi: 10.1200/JCO.2005.66.130. [DOI] [PubMed] [Google Scholar]
- 23.Chang SM, Wen P, Cloughesy T, Greenberg H, Schiff D, Conrad C, Fink K, Robins HI, De Angelis L, Raizer J, et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357–61. doi: 10.1007/s10637-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 24.Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, Peralba JM, Jenkins RB, Dakhil SR, Morton RF, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294–304. doi: 10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- 25.Atkins MB, Hidalgo M, Stadler WM, Logan TF, Dutcher JP, Hudes GR, Park Y, Liou SH, Marshall B, Boni JP, et al. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol. 2004;22:909–18. doi: 10.1200/JCO.2004.08.185. [DOI] [PubMed] [Google Scholar]
- 26.Boni JP, Leister C, Bender G, Fitzpatrick V, Twine N, Stover J, Dorner A, Immermann F, Burczynski ME. Population pharmacokinetics of CCI-779: correlations to safety and pharmacogenomic responses in patients with advanced renal cancer. Clin Pharmacol Ther. 2005;77:76–89. doi: 10.1016/j.clpt.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 27.Raymond E, Alexandre J, Faivre S, Vera K, Materman E, Boni J, Leister C, Korth-Bradley J, Hanauske A, Armand JP. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol. 2004;22:2336–47. doi: 10.1200/JCO.2004.08.116. [DOI] [PubMed] [Google Scholar]
- 28.Cunningham EB. An inositolphosphate-binding immunophilin, IPBP12. Blood. 1999;94:2778–2789. [PubMed] [Google Scholar]
- 29.Boulay A, Zumstein-Mecker S, Stephan C, Beuvink I, Zilbermann F, Haller R, Tobler S, Heusser C, O’Reilly T, Stolz B, et al. Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative RAD001 correlates with prolonged inactivation of ribosomal protein S6 kinase 1 in peripheral blood mononuclear cells. Cancer Res. 2004;64:252–61. doi: 10.1158/0008-5472.can-3554-2. [DOI] [PubMed] [Google Scholar]
- 30.Lonial S, Waller EK, Simons JW, Heffner LT., Jr Signal transduction and myeloma: new targets, new hope. Cancer Biol Ther. 2003;2:310–9. doi: 10.4161/cbt.2.4.425. [DOI] [PubMed] [Google Scholar]
- 31.Mitsiades CS, Mitsiades N, Munshi NC, Anderson KC. Focus on multiple myeloma. Cancer Cell. 2004;6:439–44. doi: 10.1016/j.ccr.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 32.Raje N, Kumar S, Hideshima T, Ishitsuka K, Chauhan D, Mitsiades C, Podar K, Le Gouill S, Richardson P, Munshi NC, et al. Combination of the mTOR inhibitor rapamycin and CC-5013 has synergistic activity in multiple myeloma. Blood. 2004;104:4188–93. doi: 10.1182/blood-2004-06-2281. [DOI] [PubMed] [Google Scholar]
- 33.Hidalgo M, Buckner JC, Erlichman C, Pollack MS, Boni JP, Dukart G, Marshall B, Speicher L, Moore L, Rowinsky EK. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res. 2006;12:5755–63. doi: 10.1158/1078-0432.CCR-06-0118. [DOI] [PubMed] [Google Scholar]