

Abstract
Thrombin-activatable procarboxypeptidase B (proCPB or thrombin-activatable fibrinolysis inhibitor or TAFI) is a plasma procarboxypeptidase that is activated by the thrombin-thrombomodulin complex on the vascular endothelial surface. The activated CPB removes the newly exposed carboxyl terminal lysines in the partially digested fibrin clot, diminishes tissue plasminogen activator and plasminogen binding, and protects the clot from premature lysis. We have recently shown that CPB is catalytically more efficient than plasma CPN, the major plasma anaphylatoxin inhibitor, in inhibiting bradykinin, activated complement C3a, C5a, and thrombin-cleaved osteopontin in vitro. Using a thrombin mutant (E229K) that has minimal procoagulant properties but retains the ability to activate protein C and proCPB in vivo, we showed that infusion of E229K thrombin into wild type mice reduced bradykinin-induced hypotension but it had no effect in proCPB-deficient mice, indicating that the beneficial effect of E229K thrombin is mediated through its activation of proCPB and not protein C. Similarly proCPB-deficient mice displayed enhanced pulmonary inflammation in a C5a-induced alveolitis model and E229K thrombin ameliorated the magnitude of alveolitis in wild type but not proCPB-deficient mice. ProCPB-deficient mice also displayed enhanced arthritis in an inflammatory arthritis model. Thus, our in vitro and in vivo data support the thesis that thrombin-activatable CPB has broad anti-inflammatory properties. By specific cleavage of the carboxyl terminal arginines from C3a, C5a, bradykinin and thrombin-cleaved osteopontin, it inactivates these active inflammatory mediators. Along with the activation of protein C, the activation of proCPB by the endothelial thrombin-thrombomodulin complex represents a homeostatic feedback mechanism in regulating thrombin’s pro-inflammatory functions in vivo.
1. Introduction
Thrombin-activatable procarboxypeptidase B (proCPB or thrombin-activatable fibrinolysis inhibitor or TAFI) is a recently described plasma procarboxypeptidase that plays a role in modulating fibrin clot lysis. In this chapter, we reviewed recent studies and suggest that its physiological function is not limited to controlling fibrinolysis but may serve a much broader anti-inflammatory role in the regulation of thrombin and tissue inflammation.
2. Procoagulant and anticoagulant properties of thrombin
The procoagulant activities of thrombin are well established: it is the final enzyme of the clotting cascade and responsible for forming the fibrin clot; in a positive feedback fashion, it also activates other clotting factors and accelerates the efficiency of the cascade; and it activates platelets via the cleavage of the protease activated receptors (PAR) to form platelet aggregates. Thus thrombin is the major physiological mediator for the formation of platelet-fibrin thrombus at the site of vascular injury (Coughlin, 2005). It is noteworthy that the expression of the PAR receptors is not restricted to platelets and they also exist on endothelial cells, monocytes, fibroblasts, and smooth muscle cells; activation of PARs in these cells generally leads to a proliferative and pro-inflammatory phenotype and contributes to thrombin’s proinflammatory properties.
While the procoagulant properties of thrombin are well recognized, it is less well appreciated that thrombin also has anticoagulant properties (Esmon, 2003). This is mediated by its binding to thrombomodulin (TM), an integral membrane protein found on the vascular endothelial cell surface. Binding of thrombin to TM acts like a molecular switch - thrombin is not inhibited as an enzyme, but its substrate specificity is completely altered. When bound to TM, thrombin no longer clots fibrinogen or activates platelets, but instead acquires the ability to activate protein C, a pro-enzyme (zymogen) in plasma, efficiently (by >1000 fold). Activated protein C cleaves and inactivates activated clotting factors Va and VIIIa, two key cofactors in the clotting cascade, and dampens thrombin generation. It also has intrinsic anti-inflammatory properties and is efficacious in the treatment of severe sepsis (Esmon, 2005; Bernard et al., 2001).
3. Dissociation of thrombin’s procoagulant and anticoagulant properties
It is remarkable that the procoagulant and anticoagulant properties of thrombin can be completely dissociated by a single mutation, with the discovery of E229K thrombin (Leung and Gibbs, 1997). To define the structure-function relationships of thrombin, a library of 53 thrombin mutants was created; based on the crystal structure of thrombin and using the alanine scanning mutagenesis approach, we systematically replaced all the surface exposed hydrophilic amino acid residues on thrombin with alanine. The thrombin mutants were then screened empirically for those that retained protein C activation but had diminished clotting function. Thrombin mutant E229A was identified (Gibbs et al., 1995); glutamic acid 229 is located quite a distance from the active site of thrombin, and from a priori considerations, one would not have guessed that replacement of glutamic acid at this location would have such a profound effect. Saturation mutagenesis at position E229 was performed and replacement of glutamic acid with lysine (E229K) gave the most desirable property of minimal clotting function (<1%) while retaining significant protein C activation in the presence of thrombomodulin (~50% WT activity) (Tsiang et al., 1996). (Subsequent crystal structure analysis revealed that E229K thrombin has an occluded active site - E229 is located at a significant Na+-binding site and disruption of Na+ binding leads to collapse of the active site (Carter et al. 2004). Presumably when E229K binds to thrombomodulin, the conformation of the active site is partially regained). When infused into a monkey, E229K thrombin prolonged the PTT clotting time, and the anticoagulant activity correlated with the activation of protein C. It did not cause any consumption of platelets or fibrinogen. E229K thrombin also functions similarly in mice. Thus E229K is an engineered anticoagulant thrombin.
4. Thrombin-activatable procarboxypeptidase B as a physiological substrate for the thrombin/thrombomodulin complex
Given the biological importance of the thrombin-thrombomodulin protein C activation pathway, does the thrombin-thrombomodulin complex activate any other physiological substrates? Thrombin-activatable procarboxypeptidase (proCPB, also termed thrombin-activatable fibrinolysis inhibitor or TAFI), is a plasma procarboxypeptidase and is now recognized as a second physiological substrate for the thrombin-thrombomodulin complex (its activation is enhanced by ~1200 fold as compared to thrombin alone) (Redlitz et al., 1995; Bajzar et al., 1996; Broze and Higuchi, 1996). When activated, CPB is specific for cleavage of carboxyl terminal arginine and lysine, and functions as a fibrinolysis inhibitor. During clot lysis, tissue type plasminogen activator (tPA) and plasminogen bind to the fibrin clot, plasmin is generated locally and digests the clot. In the process, new carboxyl terminal lysines are exposed in the partially digested clot, which serve as additional binding sites for plasminogen and tPA, and leads to enhanced clot lysis. The activated CPB removes the newly exposed carboxyl terminal lysines in the clot, diminishes additional tPA and plasminogen binding, thus protecting the clot and functioning as a fibrinolysis inhibitor.
From the hemostasis standpoint, activated protein C and CPB can be perceived as playing complementary roles (Fig. 1). At the site of vascular injury, thrombin is generated and forms the fibrin clot to stop bleeding. Thrombin binding to thrombomodulin on the neighboring intact endothelium will activate protein C and the activated protein C dampens the clotting cascade, preventing excessive fibrin clot formation. At the same time, the activation of CPB will stabilize and protect the clot already formed from premature lysis.
Fig. 1.

Complementary roles for aPC and CPB at sites of vascular injury.
5. Thrombin-activatable carboxypeptidase B as an anti-inflammatory molecule
Given the fact that thrombin has well documented pro-inflammatory properties (via its activation of cellular PAR receptors), and activated protein C also has well established anti-inflammatory properties (via its protective effect on endothelial cells), the activity of CPB may not be limited to fibrin protection, and it may have intrinsic anti-inflammatory functions. In contrast to carboxypeptidase N (CPN), the major plasma anaphylatoxin inhibitor that is constitutively active and stable, CPB is thermolabile (plasma t1/2 ~ 15 min) and requires activation by thrombin-thrombomodulin. It may thus function as a stimulus-responsive anti-inflammatory molecule that becomes activated locally at sites of tissue injury. In support of this, we and others have shown that CPB can efficiently inhibit bradykinin (BK), activated complement C3a, C5a, and thrombin-cleaved osteopontin (OPN) in vitro (Shinohara et al., 1994; Campbell et al., 2002; Myles et al., 2003).
OPN is a pro-inflammatory cytokine and immunoregulatory protein found in plasma and the extracellular matrix. It is involved in bone remodeling and wound healing. OPN interacts with many different cell types, including T cells, macrophages and neutrophils, and is upregulated at sites of injury and inflammation. Its cellular interactions are largely mediated by a RGD site that allows it to bind to many different integrins including αvβ3 (vitronectin receptor). However there is also a thrombin-cleavage site within OPN, in close proximity to the RGD sequence, and thrombin cleavage leads to the exposure of a new integrin-binding site (SVVYGLR) at the carboxyl terminus, which is specific for α4β1 and α9β1, a subset of β1 integrins that has a much more restricted cellular expression. We recently showed that thrombin cleavage of OPN requires the engagement of both anion-binding exosites in thrombin and establishes OPN as a bona fide substrate for thrombin (Myles and Leung, 2008). The thrombin-cleaved OPN (OPN-R or OPN-Arg) becomes much more chemotactic for neutrophils and certain T cell subsets. Recombinant OPN-R is ~5-fold more potent than full length OPN in supporting Jurkat cell binding in vitro. Removal of the carboxyl terminus arginine from OPN-R, converting it to OPN-L (or OPN-Lys), renders it unable to bind to α4β1 and reduces its cell adhesion. It supports the notion that sequential thrombin and CPB cleavages of OPN will in turn up-regulate and down-modulate its cell binding activity (Myles et al., 2003).
CPB also inactivates C5a. Using an in vitro neutrophil myeloperoxidase release assay, CPB, at a molar ratio of 1:100 to C5a, effectively inhibited C5a, with complete inhibition of its neutrophil activation activity within 5 min of incubation (Fig. 2).
Fig. 2.

Inhibition of C5a by CPB (TAFIa) in vitro.
A formal comparison of the catalytic efficiencies (as measured by Kcat/km) of CPN and CPB, using a series of synthetic peptides to represent the various substrates, showed that CPB is about 10–25 fold more efficient than CPN in cleaving BK, thrombin-cleaved OPN, and C5a, while they are about the same in cleaving fibrin-based peptides. Thus the in vitro data show that CPB has broad substrate reactivity and may function as an anti-inflammatory molecule, in addition to its role as a fibrinolysis inhibitor.
6. Carboxypeptidase B reduces bradykinin-induced hypotension in vivo
To study the in vivo role of CPB, we used E229K thrombin to activate proCPB in vivo. E229K activates protein C and proCPB when infused intravenously in mice, without concomitant fibrinogen or platelet consumption. Pre-infusion of E229K thrombin into wild type (WT) mice reduced BK-induced hypotension but it had no effect in proCPB-deficient mice, indicating that the beneficial effect of E229K thrombin is mediated through its activation of proCPB and not protein C. It suggests that CPB can supplement CPN in inactivating BK.
7. Carboxypeptidase B ameliorates C5a-induced alveolitis in vivo
To test its role in C5a inhibition in vivo, we utilized a C5a-induced alveolitis mouse model (Nishimura et al., 2007). C5a was instilled intra-tracheally into the mouse; after 6 hrs, bronchoalveolar lavage was performed and the magnitude of pulmonary inflammation measured by cell counts and protein levels in the lavage fluid. In WT mice, C5a induced pulmonary alveolitis in a dose-dependent manner. Inflammation was significantly increased (by 2-fold) in the proCPB-deficient mice. E229K thrombin pre-infusion significantly reduced the pulmonary inflammation in WT but not proCPB-deficient mice (Fig. 3). Histological examination of the pulmonary tissues confirmed the finding of increased inflammation in the proCPB mice as compared to WT mice and its amelioration by E229K thrombin infusion in WT but not proCPB-deficient mice.
Fig. 3.
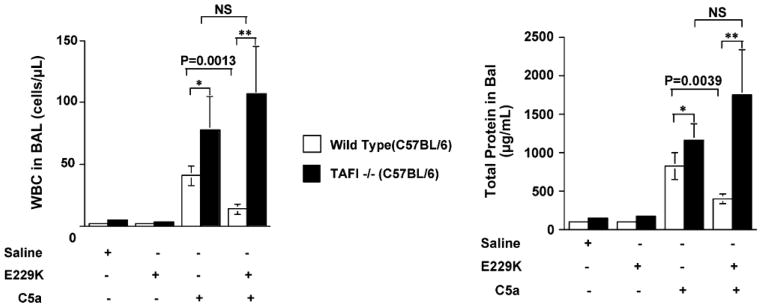
E229K thrombin attenuated C5a-induced alveolitis in WT but not in proCPB (TAFI)-deficient mice in vivo.
Delayed administration of E229K by 2 hours negated its beneficial effect, suggesting that it was mediated by CPB inhibition of C5a, rather than CPB’s effect(s) on some other downstream inflammatory mediators.
8. ProCPB-Deficient mice are predisposed to inflammatory arthritis in vivo
We have tested the proCPB-deficient mice in an anti-collagen antibody-induced inflammatory arthritis model. Both OPN, and specifically thrombin-cleaved OPN, as well as C5a have been reported to play an important role in this process. ProCPB-deficient mice developed enhanced arthritis in this model, with significant swelling in the paws and histology confirmed significant inflammatory cell infiltration in the synovial tissues (Song et al., 2007). C5a plays a key role in this model since C5-deficient mice were protected from arthritis development. The data suggest that in the proCPB-deficient mice, enhanced C5a generation led to more severe arthritis. Whether thrombin-cleaved OPN also plays a role remains to be determined.
9. Regulation of thrombin’s inflammatory properties by thrombin-activatable procarboxypeptidase B
In summary, we have obtained in vitro and in vivo data supporting the thesis that thrombin-activatable CPB has broad anti-inflammatory properties. By specific cleavage of the carboxyl terminal lysines or arginines from C3a, C5a, BK and thrombin-cleaved OPN, it inactivates these active inflammatory mediators. Along with the activation of protein C, the activation of proCPB by the endothelial thrombin-thrombomodulin complex represents a homeostatic feedback mechanism in regulating thrombin’s proinflammatory functions in vivo (Fig. 4). With the recent reports of non-conventional direct thrombin activation of C3 and C5 (Huber-Lang et al., 2006; Clark et al., 2008), the potential interactions between the coagulation and complement pathways offer an exciting new avenue in research in inflammatory diseases.
Fig. 4.
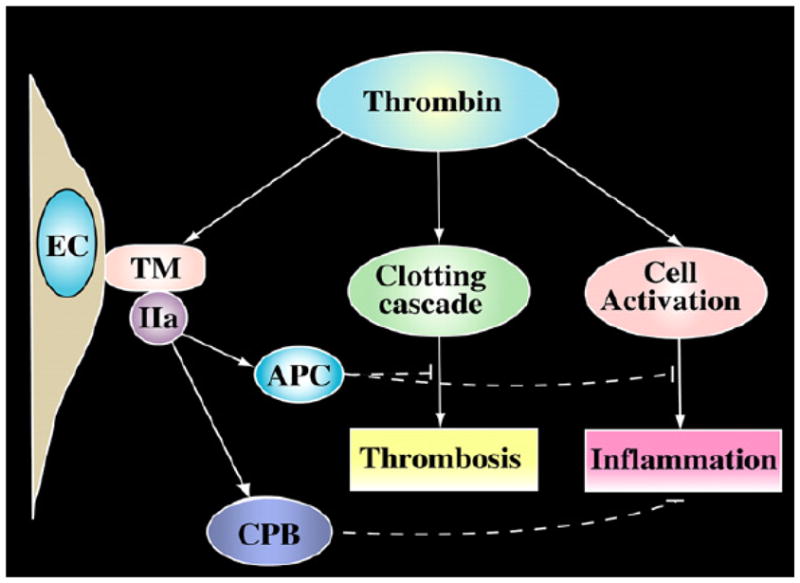
CPB (TAFIa) as an anti-inflammatory molecule.
Acknowledgments
This work was supported by NIH grant RO1 HL57530 (LL).
References
- Bajzar L, Morser J, Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem. 1996;271:16603–16608. doi: 10.1074/jbc.271.28.16603. [DOI] [PubMed] [Google Scholar]
- Bernard GR, Vincent JL, Laterre PF, LaRosa SP, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- Broze G, Higuchi DA. Coagulation-dependent inhibition of fibrinolysis: role of carboxypeptidase-U and the premature lysis of clots from hemophilic plasma. Blood. 1996;88:3815–3823. [PubMed] [Google Scholar]
- Campbell WD, Lazoura E, Okada N, Okada H. Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol Immunol. 2002;46:131–134. doi: 10.1111/j.1348-0421.2002.tb02669.x. [DOI] [PubMed] [Google Scholar]
- Carter WJ, Myles T, Gibbs CS, Leung LL, Huntington JA. Crystal structure of anticoagulant thrombin variant E217K provides insights into thrombin allostery. J Biol Chem. 2004;279:26387–26394. doi: 10.1074/jbc.M402364200. [DOI] [PubMed] [Google Scholar]
- Clark A, Weymann A, Hartman E, Turmelle Y, Carroll M, Thurman JM, Holers VM, Hourcade DE, Rudnick DA. Evidence for non-traditional activation of complement factor C3 during murine liver regeneration. Mol Immunol. 2008;45:3125–3132. doi: 10.1016/j.molimm.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–14. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- Esmon CT. The protein C pathway. Chest. 2003;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131:417–430. doi: 10.1111/j.1365-2141.2005.05753.x. [DOI] [PubMed] [Google Scholar]
- Gibbs CS, Coutre SE, Tsiang M, Li WX, Jain AK, Dunn KE, Law VS, Mao CT, Matsumura SY, Mejsa SJ, Paborsky LR, Leung LLK. Conversion of thrombin into an anticoagulant by protein engineering. Nature. 1995;378:413–416. doi: 10.1038/378413a0. [DOI] [PubMed] [Google Scholar]
- Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- Leung LLK, Gibbs CS. Modulation of thrombin’s procoagulant and anticoagulant properties. Thromb Haemost. 1997;78:577–580. [PubMed] [Google Scholar]
- Myles T, Nishimura T, Yun TH, Nagashima M, Morser J, Patterson AJ, Pearl RG, Leung LLK. Thrombin activatable fibrinolysis inhibitor, a potential regulator of vascular inflammation. J Biol Chem. 2003;278:51059–51067. doi: 10.1074/jbc.M306977200. [DOI] [PubMed] [Google Scholar]
- Myles T, Leung LLK. Thrombin hydrolysis of human osteopontin is dependent on thrombin anion-binding exosites. J Biol Chem. 2008;283:17789–17796. doi: 10.1074/jbc.M708629200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Myles T, Piloponsky P, Kao P, Berry GJ, Leung LLK. Thrombin-activatable procarboxypeptidase B regulates activated complement C5a in vivo. Blood. 2007;109:1992–1997. doi: 10.1182/blood-2006-03-012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redlitz A, Tan AK, Eaton DL, Plow EF. Plasma carboxypeptidases as regulators of the plasminogen system. J Clin Invest. 1995;96:2534–2538. doi: 10.1172/JCI118315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara T, Sakurada C, Suzuki T, Takeuchi O, Campbell W, Ikeda S, Okada N, Okasda H. Pro-carboxypeptidase R cleaves bradykinin following activation. Int Arch Allergy Immunol. 1994;103:400–404. doi: 10.1159/000236661. [DOI] [PubMed] [Google Scholar]
- Song J, Nishimura T, Garcia M, Myles T, Ho P, Du XY, Sharif S, Leung L, Robinson W. Thrombin-activatable carboxypeptidase B prevents anti-collagen antibody induced arthritis. Clinical Immunology. 2007;123(suppl):S98. [Google Scholar]
- Tsiang M, Paborsky LR, Li WX, Jain AK, Mao CT, Dunn KE, Lee DW, Matsumura SY, Matteucci MD, Coutre SE, Leung LLK, Gibbs CS. Protein engineering thrombin for optimal specificity and potency of anticoagulant activity in vivo. Biochemistry. 1996;35:16449–16457. doi: 10.1021/bi9616108. [DOI] [PubMed] [Google Scholar]