

Abstract
Purpose
We have previously shown that p90 ribosomal protein S6 kinase 4 (RSK4), an X-linked gene, is highly up-regulated in mammary tumors of MMTV-c-Myc transgenic mice. In this study, we further investigated whether RSK4 inhibits or promotes breast tumor growth and progression.
Experimental Design
Stable overexpression or small interfering RNA – mediated knockdown of RSK4 was done in the MDA-MB-231cell line. Stable clones were tested for cell proliferation, anchorage-independent growth in soft agar, invasive and metastatic ability of these clones in vitro and tumorigenesis, invasive and metastatic ability in vivo in severe combined immunodeficient mice.
Results
Here, we show that exogenous expression of RSK4 resulted in decreased cell proliferation and increased accumulation of cells in G0–G1phase, which paralleled with enhanced expression of tumor suppressor genes: retinoblastoma protein, retinobl astoma-associated 46 kDa protein, and p21 protein. Overexpression of RSK4 resulted in reduced colony formation in soft agar and suppressed invasive and migratory activities of MDA-MB-231cells both in vitro and in vivo. Importantly, RSK4-overexpressing cells showed up-regulation of claudin-2 and down-regulation of CXCR4, both of these play roles in invasion and chemotaxis.
Conclusions
These results indicate that RSK4 expression may limit the oncogenic, invasive, and metastatic potential of breast cancer cells. Anti-invasive and antimetastatic activities of RSK4 may be, in part, due to its regulation of claudin-2. Increased expression of RSK4 in c-Myc-overexpressing cells and a dose-dependent induction of luciferase reporter gene activity suggest that c-Myc may regulate RSK4 expression.
Breast cancer still remains one of the most common malignancies and a major cause of cancer-related deaths among women in the United States. The main cause of cancer-related death is due to the progression of noninvasive solid tumor to invasive and metastatic phenotype, yet the mechanisms underlying the progression of noninvasive to fatal metastatic breast cancer remain unclear. Tumor progression from nonin-vasive to metastatic results from a multitude of genetic events. Identification of molecules or endogenous factor(s) that play a role in the process of cancer progression from noninvasive to invasive and metastatic will allow development of new therapeutic approaches to prevent or treat metastatic breast cancer.
In our search for c-Myc target X-linked genes, we showed previously a significant up-regulation of ribosomal protein S6 kinase 4 (RSK4) mRNA in mammary tumors of MMTV-c-Myc transgenic mice compared with normal mammary glands (1). RSK4, an intracellular serine/threonine kinase, belongs to p90 ribosomal S6 kinase (RSK) family of related kinases that includes both activating and inhibitory isoforms and signals downstream of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase pathway (2–4). RSK family members RSK1, RSK2, and RSK3 are the central mediators of extracellular signal-regulated kinase in the regulation of survival, growth, proliferation, invasion, and metastasis via phosphorylation of numerous intracellular proteins including several transcription factors and coregulators such as CREB, CBP, p300, IκBα, c-fos, and ERα (5–12). RSK4 is constitutively activated in serum-starved cells and shown to suppress cell proliferation as opposed to growth factor–dependent RSK1, RSK2, and RSK3 (13). Depletion of RSK4 bypassed the p53-dependent G1 cell cycle arrest (14) and strongly suppressed mRNA expression of cyclin-dependent kinase inhibitor p21cip1 (p21). In another study, Myers et al. showed that RSK4 expression disrupted mesoderm formation induced by the fibroblast growth factor-Ras-extracellular signal-regulated kinase signaling pathway (15). Together, these studies suggest that RSK4 is distinct from RSK1-3 and appears to have growth-inhibitory function. Interestingly, we observed that RSK4-overexpressing mammary tumors of MMTV-c-Myc transgenic mice are noninvasive and do not metastasize, suggesting that noninvasive and nonmetastatic properties of these tumors may be attributed by RSK4. Moreover, weak to moderate expression of RSK4 protein in human invasive ductal carcinoma and strong positive staining in ductal carcinoma in situ may further suggest its anti-invasive and antimetastatic role in breast cancer (16). Based on our and other previous studies and our observation, we hypothesized that increased expression of RSK4 may restrict tumor growth and suppress mammary tumor invasion and metastasis. Our results indicate that RSK4 can suppress tumorigenic phenotype and may play a critical role in invasion and migration of breast cancer cells.
Materials and Methods
Cell lines, plasmids, and transfection
The human breast cancer cell lines, MDA-MB-231 and T47D, were cultured in DMEM/F-12 or RPMI 1640 (Invitrogen), respectively, with 10% fetal bovine serum (FBS; Invitrogen) at 37°C and 5% CO2. The human MDA-MB-231, a highly invasive and metastatic cell line, was chosen to generate the RSK4-overexpressing stable clones because it showed lower levels of endogenous RSK4. The RSK4 construct (pMT2-HA-RSK4) was kindly provided by Dr. Frodin Morten (Kinase Signaling Laboratory, Biotech Research and Innovation Centre). The RSK4 (GenBank accession no. NM_014496) insert, which encodes the entire cDNA sequence (13), was subcloned in pcDNA3.1/Neo(−) vector for stable transfection. The pBabe-Puro-MycER expression vector contained the mutant hormone-binding domain of the murine estrogen receptor (which retains the ability to bind 4-hydroxytamoxifen but not estrogen) fused at the COOH terminus of the human c-Myc protein (amino acids 1-435) to form c-MycER (14). The pcDNA3.1/Neo-RSK4 or pBabe-Puro-MycER plasmids were used to stably transfect the MDA-MB-231 or T47D cell lines to overexpress the human RSK4 or c-Myc gene. Transfection was done using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Briefly, MDA-MB-231 or T47D cells were cultured in six-well plates at 80% to 90% confluency. Plasmid DNA (1.0 μg/well) was mixed with 10 μL LipofectAMINE 2000 to transfect the MDA-MB-231 or T47D cells. After 48 h, cells were trypsinized and replated into 10-cm culture dishes in the presence of selection drug (800 μg/mL neomycin or 2.5 μg/mL puromycin). Single-cell clones were isolated for clone expansion. Each cell clone was screened by reverse transcription-PCR and Western blot to determine the RSK4 expression at both mRNA and protein levels. Stable clones were cultured and maintained (400 μg/mL neomycin and 1 μg/mL puromycin) at least 15 passages before using them for various experiments.
RNA extraction and reverse transcription-PCR
Total RNA was extracted from transfected cells using RNeasy (Qiagen) spin columns according to the manufacturer’s instruction and stored at −80°C until use. Exogenous RSK4 was identified by reverse transcription-PCR using Taqman RT kit (Applied Biosystems) and Ready PCR Mix (Sigma) in stably transfected clones. Transfected RSK4 was identified using the upstream primer located in the vector (BGH: 5′-TAGAAGGCACAGTCGAGG-3′) and downstream primer located in RSK4 insert (5′-CCCAGTTTCCACCACTCAAA-3′). For the detection of RSK4 mRNA expression, the upstream primer was replaced with 5′-GGAACGGGAGGCTAGTGATA-3′. The vector control clones of pcDNA3.1-neo were identified by using T7 and BGH primer pairs (T7: 5′-TAATACGACTCACTATAGGG-3′ and BGH: 5′-TAGAAGGCA-CAGTCGAGG-3′).
Immunoblotting
For immunoblotting, cells were washed twice with ice-cold PBS and lysed in radioimmunoprecipitation assay buffer (50 mmol/L Tris-HCl, 150 nmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L DTT, 0.1% Tween 20, 1 mmol/L phenylmethylsulfonyl fluoride, 10 mmol/L β-glycerophos-phate, 1 mmol/L NaF, 2 mmol/L Na3VO4, 1–5 μg/mL leupeptin, and 1–5 μg/mL aprotinin). Nuclear extract was prepared by using nuclear extraction buffer as described previously (17). Protein was quantitated using a BCA protein assay (Pierce). Protein aliquots (50 μg/lane) or 25 μL immunoprecipitated samples were separated on SDS-PAGE and transferred onto a nitrocellulose membrane for detection by Western blot and proteins were visualized by chemiluminescence (Pierce). We used rabbit anti-human RSK4 (Abgent), anti-retinoblastoma (Rb)–associated 46 kDa (RbAp46; Abcam) anti-CXCR4 (Santa Cruz), anti-cyclin E (Santa Cruz), anti-cyclin A (Santa Cruz), anti-Snail-2 (Santa Cruz), anti-E-cadherin (Santa Cruz), anti-MMP-2 (Santa Cruz) and anti-MMP-9 (Santa Cruz), p44/42 MAPK (Cell Signaling), phopho-p44/p42 (Thr202/Tyr204) MAPK (Cell Signaling) polyclonal antibodies; anti-p21 (Upstate), anti-pRb (Santa Cruz), anti-c-Myc (EMD), and anti-cyclin D1 (Santa Cruz) monoclonal antibodies; and goat anti-claudin-2 (CLDN2; Santa Cruz) antibody.
Immunostaining
Cells were cultured on coverslips for 24 h and then fixed with 10% formalin for 10 min, washed with PBS, and stored at 4°C. For staining, coverslips were treated with 0.5% Triton X-100 in PBS for 10 min and blocked for nonspecific binding and endogenous peroxidase activity for 1 h at room temperature followed by incubation with primary antibody (anti-RSK4, rabbit polyclonal) at 37°C for 2 h in blocking buffer with 0.05% Triton X-100. After washing three times, cells were incubated with biotin-conjugated anti-rabbit antibody in the blocking buffer for 1 h at 37°C. After three washes, cells were incubated with streptavidin followed by washing three times, staining with 3,3′-diaminobenzidine, and counterstaining with hematoxylin. Images were captured on a Zeiss microscope system.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
Cells were seeded in 96-well plate at 1,500 per 100 μL per well. Cells were grown in culture medium containing 10% FBS for 72, 96, and 120 h. At the end of incubation, 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (40 μL/well of 5 mg/mL 3-(4,5-dime-thylthiazolyl-2)-2,5-diphenyltetrazolium bromide in PBS) was added to each well and plates were incubated in the dark for 3 h at 37°C. After removal of the medium, the dye crystals were dissolved in isopropanol and viable cells were detected by reading the absorption at 595 nm in the Ultra plate reader (Tecan Research). Experiments were repeated three times in quadruplicate wells to ensure the reproducibility of results.
Cell cycle analysis
Synchronized subconfluent vector control and RSK4-overexpressing cells cultured in the presence or absence of 5′-deoxy-5-fluorouridine (5′-dFUR) or low serum/serum deprived were harvested and washed twice with PBS. Pellets were resuspended in 0.5 mL PBS, fixed in 4.5 mL of 70% ethanol, and incubated overnight at −20°C. Cells were collected by centrifugation and washed once with PBS and the pellets were resuspended in 1 mL of 0.2 mg/mL propidium iodide containing 0.1% Triton X-100 (Sigma) and RNase A (1 mg/mL; Sigma). The cell suspension was incubated in the dark for 30 min at room temperature and subsequently analyzed on a Coulter EPICS 753 flow cytometer for DNA content. The percent of cells in different phases of the cell cycle was determined using a ModFit 5.2 computer program.
Small interfering RNA silencing of RSK4
Human RSK4-specific small interfering RNA (siRNA) were purchased from Ambion Biosciences. MDA-MB-231 cells (5 × 105 per well in six-well plate) were transfected with 200 pmol of the siRNA duplex for 48 h using Lipofectamine 2000 (Invitrogen). To assess the RSK4-regulated proteins, RSK4-overexpressing cells were transfected with either a scrambled sequence siRNA, which served as a negative control, or siRNA against RSK4. Cells were harvested after 48 h and cell lysates were prepared to analyze the protein expression of RSK4 target proteins.
In vitro assay for tumorigenesis
Soft agar growth transformation assay
Anchorage-independent growth was assessed by colony formation on soft agar. Briefly, equal volumes of agar (1%, DNA grade) and 2× DMEM (with 20% FBS) were mixed at 40°C to make 0.5% agar in six-well tissue culture plates (Corning) as a base agar. Cells (0.1 mL of 2.0 × 105/mL) were suspended in 3 mL of 2× DMEM (with 20% FBS) and 3 mL of 0.7% agar, and 1.5 mL cell suspension was then added to each well (as 0.35% top agar) with final concentration of 5,000 cells per well. Top agar was covered with culture medium. Plates were incubated at 37°C and 5% CO2 in humidified incubator for 3 to 4 weeks and medium was changed every 3 to 4 days. Colony formation was observed by light phase-contrast microscope and photomicrographed after staining with 0.5 mL of 0.01% crystal violet in PBS for 45 min at room temperature Experiments were repeated twice to ensure the reproducibility of our results.
Matrigel invasion assay
The RSK4-overexpressing or vector control MDA-MB-231 cells were suspended at 1 × 106/mL in medium containing DMEM/F-12 with 0.1% bovine serum albumin and 100 μL cell suspension was added to the upper well of Transwell inserts coated with 1 mg/mL Matrigel (BD Biosciences). In lower wells, 0.6 mL medium was added. The plates were incubated for 24 h at 37°C in 5% CO2. After incubation, the inserts were carefully lifted and cells from the upper surface were gently removed and the remaining cells at the bottom side of the filter were fixed and stained using the Diff-Quick Staining. The results are expressed as the percent of RSK4-overexpressing cells, which invaded the Matrigel and migrated to the bottom side of the Transwell membrane relative to the vector control cells. Each experiment was repeated at least three times in triplicates to ensure the reproducibility of our results.
Chemotaxis assay
Chemotaxis assay was done as described previously (18). Briefly, RSK4-overexpressing or vector control MDA-MB-231 cells were washed twice in serum-free medium (DMEM/F-12) containing 0.1% bovine serum albumin. For chemotaxis assay, 100 μL of 1 × 106 cells/mL were added onto the upper well containing Transwell inserts with 8-mm pore size. In the lower well, either 0.6 mL medium containing CXCL12 (10 nmol/L) or vehicle was added. The plates were incubated for 6 h at 37°C in 5% CO2. After incubation, the cells from the upper surface were carefully removed, cells adhered on the top side of the membrane were scraped, and cells on the lower surface of the membrane were then stained using Diff-Quick staining and counted in at least five different fields. Each experiment was repeated at least three times in triplicates. Percentage chemotaxis in RSK4-overexpressing cells is relative to the number of cells that migrated in the presence of CXCL12 in vector control cells.
In vivo tumorigenesis assay
Six week-old severe combined immunodeficient (SCID) mice were obtained from Taconic Farms. Mice were injected s.c. (n = 5 mice per clone) with 1 × 106 cells resuspended in 0.1 mL DMEM without serum from two RSK4-overexpressing (MR11 and MR12) and two vector control (MN10 and MN11) clones. Tumor growth was monitored twice weekly. At the end of the monitoring period, animals were sacrificed by CO2 asphyxiation, and tumor samples were removed and preserved in formalin. Thin sections were stained with H&E and examined by standard light microscopy.
In vivo experimental metastasis
For experimental lung metastasis, 1 × 107 RSK4-overexpressing cells/mL (MR11 and MR12) or vector control cells (MN10 and MN11) in 100 μL DMEM (n = 5 mice/clone) were injected into the lateral tail veins of 6-week-old female SCID mice. At 10 weeks postinjection, lungs, liver, brain, kidney, and bones were harvested, fixed, and processed for histologic analyses.
RSK4 promoter-luciferase reporter construct and luciferase reporter assay
We cloned a regulatory region of the human RSK4 gene. A 640-bp nucleotide sequence was amplified by PCR using the following primers: 5′-gagtggtaccTCAGCTTCATTGTCAAGGAGT-3′ and 5′-cagtctcgagGGC-TAAGCTTGAAGCAGCTA-3′. Forward primer contained XhoI restriction site and the reverse primer harbored a HindIII restriction site and a few additional nucleotides before the restriction sites. The PCR product was digested with XhoI and HindIII, cloned in TOPO vector (Invitrogen), and subcloned into the reporter vector pGL3-Basic (Promega) to generate a reporter construct. The construct was sequenced to check that no mutations were introduced during this process. For transfection, 200 ng pGL3 or putative RSK4 promoter-luciferase plasmid and combinations of various concentrations (0.5–3.0 μg) of c-Myc expression plasmid were used along with a β-galactosidase expression plasmid for normalization of transfection efficiencies. Luciferase and β-galactosidase activity was determined 48 h post-transfection using a luciferase reporter assay system (Promega). Luciferase units were calculated as luciferase activity/β-galactosidase activity and are presented as the mean ± SE of three individual experiments in triplicates. The fold change was calculated by comparison with the promoterless luciferase vector. HeLa cells were chosen for the transfection because these cells do not express endogenous RSK4.
Statistical analysis
All in vitro experiments were done in triplicate at least three times and in vivo experiments were done at least twice. Values are expressed as mean ± SE. Comparisons between groups were assessed with the Student’s t test. P values <0.05 were considered significant.
Results
Generation of RSK4-overexpressing stable clones
We used MDA-MB-231 cell line to overexpress RSK4 as it showed lower levels of endogenous RSK4 compared with other human breast cancer cell lines, which were screened for the expression of RSK4 and c-Myc (Fig. 1A, row 1). Stable clones of MDA-MB-231 cells overexpressing RSK4 were generated by transfection with a pcDNA3.1/neo-RSK4 plasmid or with pcDNA3.1/neo empty vector to establish control clones. Reverse transcription-PCR showed exogenous RSK4, identified by using the upstream primer located in the vector and downstream primer located in RSK4 insert, in several stable clones, whereas no specific product representing exogenous RSK4 mRNA was seen in the vector control clones (Fig. 1A, row 2, left). Typically, RSK4 clones showed ~2.5- to 3.5-fold higher RSK4 protein expression (Fig. 1A, row 2, right). Immunostaining showed both nuclear and cytoplasmic localizations of RSK4. Nuclear localization could be due to the presence of a nuclear localization signal in a loop within the NH2-terminal kinase domain of RSK4 (13). A representative immunostaining in vector control-MN11 clone, RSK4-overexpressing-MR11 clone, and HeLa cells (negative control) is shown in (Fig. 1A, row 3). To confirm the nuclear localization of RSK4, we probed nuclear and cytoplasmic fractions for RSK4 proteins in vector control and RSK4-overexpressing clones. Representative data are shown in Fig. 1A (row 4).
Fig. 1.

A, stable expression of RSK4. Row 1, human breast cancer cell lines showing relative expression levels of endogenous RSK4 and c-Myc. Row 2, left, MDA-MB-231 clones transfected with pcDNA3.1-RSK4 construct (MR11, MR12, and MR19) or pcDNA3.1-empty vector (MN9, MN10, and MN11) showing exogenous RSK4 mRNA expression; right, RSK4 protein expression in RSK4-overexpressing (MR11and MR12) and empty vector (MN10 and MN11) transfected clones. Row 3, immunostaining showing predominant nuclear localization of RSK4 in MR11clone, whereas MN11clone shows the weak cytoplasmic and nuclear staining. No positive staining was observed in HeLa cells. Row 4, RSK4 expression in nuclear (N) and cytoplasmic (C) extracts. Rb levels served as nuclear protein loading control. B, 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide analysis showing significantly decreased (P < 0.05) cell proliferation in RSK4-overexpressing clones (MR11and MR12) compared with vector control clones (MN10 and MN11) at 120 h. C, left, RSK4-overexpressing clones show increased expression of cyclin D1, p21, RbAp46, and hypophosphorylated pRb proteins compared with vector control clones; right, differential expression pattern of pRb, RbAp46, and cyclin D1after serum starvation and treatment with 20 μmol/L 5′-dFUR at 72 h in RSK4-overexpressing cells. Serum starvation resulted in decreased expression of RbAp46 protein in MR11cells, whereas treatment with 5′-dFUR induced expression of p21, hyperphosphorylated pRb, and RbAp46 proteins. In the vector control clone (MN11), no change in the expression pattern of p21, pRb, and RbAp46 protein was observed. Cyclin D1levels were reduced or lost in 5′-dFUR-treated MR11and MN11cells, respectively. Bottom left, expression of p44/42 MAPK and phospho-p44/p42 (Thr202/Tyr204) MAPK proteins. No difference in their expression was observed between RSK4-overexpressing cells and vector control cells. D, RSK4 silencing using two RSK4-specific siRNA (RSK4-siRNA1and RSK4-siRNA2) or control siRNA (C-siRNA) in two RSK4-overexpressing cells, MR11and MR12, showed clear down-regulation of RbAp46 and moderate decrease in Rb and cyclin D1proteins but no change in phospho-p44/p42 (Thr202/Tyr204) MAPK protein compared with control siRNA-transfected cells after 48 h. All experiments were repeated at least three times and representative data are presented.
RSK4-overexpressing cells show decreased cell proliferation and increased accumulation in G0–G1 phase
We used 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide assay to assess the cell proliferation of the RSK4 or empty vector-transfected stable clones by measuring the number of viable cells at different time points (72, 96, and 120 h). RSK4-expressing clones showed significantly decreased cell proliferation (P < 0.05) at 120 h postseeding compared with vector control clones (Fig. 1B). The cell cycle distribution in RSK4-overexpressing and vector control cells was determined by fluorescence-activated cell sorting analysis under various serum conditions (10%, 5%, 2.5%, 1%, and 0% FBS) as well as after treatment with various concentrations (5, 10, 20, and 30 μmol/L) of 5′-dFUR. Under serum-supplemented conditions, RSK4-overexpressing clones showed a significantly higher (10% FBS, P = 0.038; 5% FBS, P = 0.042) percentage of cells accumulated in the G0–G1 phase compared with vector control clones. Treatment with 5′-dFUR resulted in significantly increased accumulation of cells in S phase with all three concentrations (10 μmol/L, P = 0.037; 20 μmol/L, P = 0.029; and 30 μmol/L, P = 0.022) tested in RSK4-overexpressing clones compared with vector control clones. Representative data at 96 h are shown in Table 1 with 10% or 0% FBS and 20 μmol/L 5′-dFUR in two RSK4-overexpressing clones (MR11 and MR12) and two vector control clones (MN10 and MN11).
Table 1.
Cell cycle distribution of RSK4-overexpressing (MR11 and MR12) and vector control (MN10 and MN11) cells by fluorescence-activated cell sorting analysis
| MR11 | MR12 | MN10 | MN11 | |
|---|---|---|---|---|
| 10% FBS | ||||
| G0–G1 | 80.0 ± 6.0* | 86.0 ± 9.0* | 68.0 ± 7.5 | 65.0 ± 5.0 |
| G2-M | 8.0 ± 5.0 | 5.5 ± 1.5 | 18.0 ± 2.5 | 20.0 ± 2.5 |
| S | 12.0 ± 2.0 | 8.5 ± 2.5 | 14.0 ± 2.2 | 15.0 ± 4.5 |
| 0% FBS | ||||
| G0–G1 | 74.5 ± 4.0 | 84.0 ± 6.0 | 71.0 ± 7.0 | 69.0 ± 7.0 |
| G2-M | 8.0 ± 3.0 | 4.0 ± 2.5 | 16.5 ± 2.0 | 17.5 ± 3.0 |
| S | 17.5 ± 1.5 | 12.0 ± 3.0 | 12.5 ± 2.0 | 13.5 ± 3.0 |
| 5′-dFUR (20 μmol/L) | ||||
| G0–G1 | 31.0 ± 6.0 | 57.0 ± 4.5 | 57.0 ± 4.5 | 52.0 ± 5.0 |
| G2-M | 11.5 ± 3.0 | 0.0 ± 0.0 | 16.0 ± 2.0 | 19.5 ± 2.0 |
| S | 57.5 ± 2.0* | 43.0 ± 6.0* | 27.0 ± 3.0 | 28.5 ± 4.0 |
P < 0.05.
RSK4-overexpressing cells show increased expression of p21, pRb, and RbAp46 proteins
Because RSK4-overexpressing cells showed increased accumulation of cells in G0–G1 phase, we then investigated the molecular targets involved in mediating this effect. We determined the expression pattern of proteins critical for G1–S-phase transition. RSK4-overexpressing clones MR11 and MR12 showed increased levels of p21, hypophos-phorylated pRb, and RbAp46 proteins, but no difference was observed for cyclin E, cyclin A, p44/42 MAPK, and phospho-p44/p42 (Thr202/Tyr204) MAPK proteins (Fig. 1C, left and bottom left). No difference was observed in the expression levels of Cdk2, Cdk4, and Cdk6 and Kip1/p27 between RSK4-overexpressing and vector control clones (data not shown). Surprisingly, MR11 and MR12 clones also showed increased levels of cyclin D1 compared with MN10 and MN11 vector control clones. Next, we determined the effect of serum starvation and treatment with 20 μmol/L 5′-dFUR on the expression of p21, pRb, RbAp46, and cyclin D1. Serum starvation resulted in reduced expression of RbAp46, whereas no changes were evidenced in either the phosphorylation status or the protein levels of p21, pRb, and cyclin D1 in RSK4-overexpressing cells. Treatment with 5′-dFUR induced phos-phorylation of pRb, increased expression of RbAp46 and p21 proteins, and slightly decreased expression of cyclin D1 in RSK4-overexpressing cells (Fig. 1C, right). A previous study has already shown that RSK4 may regulate p21 (14). In this study, we assessed whether RSK4 contributes to the regulation of pRb, RbAp46, cyclin D1, and phospho-p44/p42 MAPK expression. RSK4-specific siRNA was used to silence the expression of RSK4 in two RSK4-overexpressing clones, MR11 and MR12. siRNA-mediated depletion of RSK4 showed reduction in pRb, RbAp46, and cyclin D1 protein levels, but no change was observed in phospho-p44/p42 (Thr202/Tyr204) MAPK protein compared with control siRNA-transfected cells (Fig. 1D).
RSK4 can suppress the malignant phenotype of MDA-MB-231 breast cancer cells
We further investigated whether the RSK4-mediated effect on the cell cycle can influence the malignant phenotype of MDA-MB-231 cells. We first assessed if RSK4 overexpression affects anchorage-independent growth and colony formation of MDA-MB-231 cells in soft agar. A significant reduction (P < 0.0005) in colony formation was observed in RSK4-overexpressing MDA-MB-231 cells manifested by both colony number and colony size compared with vector control clones (Fig. 2A). RSK4-overexpressing cells incubated for another 2 weeks did not show any increase in colony number or colony size. To rule out the possibility that this is not a cell-specific effect, we verified RSK4 mediated inhibition of colony formation in soft agar in RSK4-overexpressing T47D stable clones (TR1 and TR2) and vector control clones (TN1 and TN2). RSK4-overexpressing T47D clones also showed significantly lower colony formation (P < 0.0005) compared with vector control clones. Results are shown in Fig. 2B, which were consistent with RSK4-expressing MDA-MB-231 cells.
Fig. 2.

RSK4 suppresses malignant phenotype of MDA-MB-231breast cancer cells. A, crystal violet staining showing soft agar colony growth of RSK4 (MR11and MR12) or empty vector (MN10 and MN11) transfected MDA-MB-231stable clones. MR11and MR12 clones showed significantly reduced (P < 0.0001) or no growth in soft agar compared with an efficient anchorage-independent growth by vector control MN10 and MN11clones in soft agar. Bottom, growth in soft agar by light phase-contrast micrography (×50 magnification) of MR11, MR12, MN10, and MN11seeded plates. B, significantly reduced (P < 0.0003) soft agar colony growth of RSK4-transfected T47D stable (TR1and TR2) compared with empty vector (TN1and TN2) clones. Bottom, soft agar growth by light phase-contrast micrography (×50 magnification). C, RSK4-transfected and vector control clones subjected to chemotactic assay in the presence of 10 nmol/L concentration of chemokine-CXCL12. MR11and MR12 showed significantly reduced (P < 0.0001) CXCL12-induced chemotaxis compared with vector control MN10 and MN11cells (inset, ×50 magnification). Addition of anti-CXCR4 antibody in the upper wells with MN10 and MN11showed ~50% reduced chemotaxis. D, RSK4-transfected clones analyzed for their chemoinvasive activity in a Matrigel invasion assay showed significantly reduced (P < 0.0001) invasion compared with vector control cells (inset, ×50 magnification). Addition of anti-CXCR4 antibody in the upper wells with MN10 and MN11showed ~40% reduced chemotaxis. All experiments were repeated at least three times and representative data are presented. ***, P < 0.0005.
Because MDA-MB-231 is a highly invasive and metastatic cell line and RSK4 overexpression resulted in the suppression of soft agar colony formation by MDA-MB-231 and T47D cells, we next tested whether RSK4 can affect the invasive and chemotactic ability of MDA-MB-231 cells. RSK4-overexpressing clones were subjected to chemotaxis against CXCL12 and chemoinvasion on Matrigel containing CXCL12 recombinant protein. MR11 and MR12 (RSK4-overexpressing clones) clones showed a significantly reduced (P < 0.0001) chemotaxis against CXCL12 as well as a significantly reduced (P < 0.0001) chemoinvasion compared with vector control MN10 and MN11 clones (Fig. 2C and D).
RSK4 inhibits chemotactic and invasive ability of MDA-MB-231 cells by regulating the expression of CXCR4 and CLDN2 proteins
To further explore the mechanism for the inhibition of invasion and migration, we determined the expression pattern of CXCR4, CLDN2, Snail-2, E-cadherin, MMP-2, and MMP-9 genes, which play roles in invasion and tissue-specific migration. RSK4-overexpressing clones showed reduced expression of CXCR4 and increased expression of CLDN2 proteins (Fig. 3A). No difference was observed in MMP-2 and MMP-9 protein expression levels or their activation status, and no detectable expression of E-cad was found in any of the samples (data not shown). Silencing of RSK4 expression using siRNA resulted in decreased expression of CLDN2 and increased expression of CXCR4 proteins (Fig. 3B). Furthermore, addition of a CXCR4 antibody in the upper wells with vector control clones (MN10 and MN11) showed ~40% to 50% reduction in chemotactic and chemoinvasive activities (Fig. 2C and D). These results indicate that suppression of invasion and directed migration of MDA-MB-231 cells by RSK4 may be mediated through the regulation of CXCR4 and CLDN2 expression.
Fig. 3.
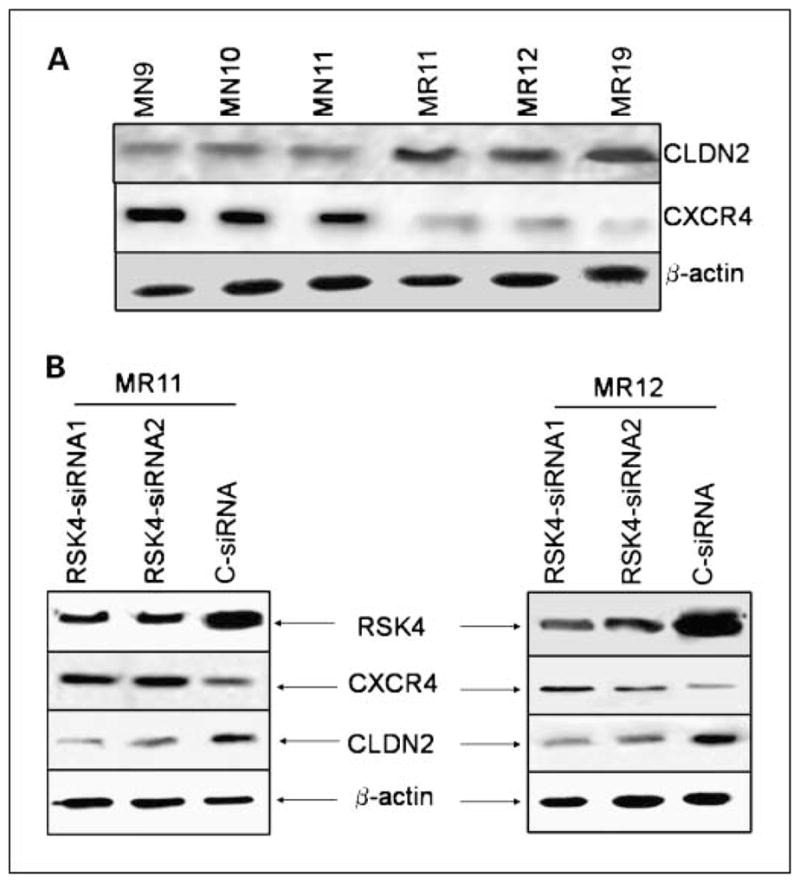
RSK4-regulated suppression of chemotaxis and invasion is mediated by CXCR4 and CLDN2. A, RSK4-overexpressing clones (MR11, MR12, and MR19) show reduced levels of CXCR4 but elevated levels of CLDN2 protein compared with empty vector clones (MN9, MN10, and MN11). B, siRNA silencing of RSK4 in MR11and MR12 clones showed increased levels of CXCR4 and reduced levels of CLDN2 proteins.
In vivo suppression of tumor growth
We next tested whether RSK4-overexpressing clones show difference in vivo tumorigenicity in SCID mice. RSK4-overexpressing (MR11 and MR12) and vector control (MN10 and MN11) cells were injected into the mammary fat pads of five female SCID mice and experiment was repeated at least twice. All mice (10 of 10) injected with MN10 and MN11-vector control cells developed palpable xenograft tumors at both left and right injection sites between 6 and 7 weeks; in MR11- and MR12-injected cells, 6 of 10 and 7 of 10 animals, respectively, formed tumors at 6 to 7 weeks postinjection. Mice injected with RSK4-overexpressing cells showed much smaller tumor size and significantly reduced (P < 0.001) tumor volume and weight compared with mice injected with vector control cells (Fig. 4A, top and middle). Histologic comparison of tumors showed penetrating growth in stromal tissue, indicating the highly invasive ability of tumors derived from vector control cells. On the other hand, tumors developed from RSK4-overexpressing clones showed a pseudocapsule, indicative of noninvasive growth. Representative photomicrographs are shown in Fig. 4A (bottom).
Fig. 4.

A, tumor formation by RSK4-overexpressing or vector control MDA-MB-231cells in SCID mice. SCID mice were inoculated s.c. with 106 cells from MR11, MR12, MN10, and MN11clones; mice (n = 5 × 2) were monitored twice weekly for the evidence of tumor growth. Top, gross appearance of tumors formed by vector control (MN11) or RSK4-overexpressing (MR11) MDA-MB-231cells at 6 to 7 wk postinjection; middle, average ± SE tumor weight and tumor volume from each group; bottom, representative histologic appearance of tumors formed by vector control or RSK4-overexpressing MDA-MB-231cells. **, P < 0.005. B, experimental metastasis. Top left, gross examination of lungs from mice injected with vector control cells (MN10) show numerous surface metastatic lesions (arrows); middle and bottom left, histologic examination revealing multiple metastases (arrows) in vector control cells (MN11) injected mice (middle left, low magnification; bottom left, high magnification); top right, mice injected with RSK4-overexpressing cells (MR11) revealed fewer surface lesions; middle and bottom right, histologic evaluation also showed much fewer metastatic lesions (middle right, low magnification; bottom right, high magnification) in the lungs of mice injected with RSK4-overexpressing cells (MR11).
In vivo suppression of experimental metastasis
We used in vivo experimental model to determine whether RSK4-over-expressing MDA-MB-231 cells can suppress metastatic progression. We injected RSK4-overexpressing cells (MR11 and MR12) and vector control cells (MN10 and MN11) i.v. in SCID mice. In control group, metastasis developed in 80% of mice, whereas in mice injected with RSK4-overexpressing MDA-MB-231 cells metastasis was developed in 40% of mice. The number of gross metastatic lesions was noticeably declined in mice injected with RSK4-overexpressing cells compared with the control group. Representative examples of gross appearance of metastatic lesions in lungs of vector control-MN-10 (top left) and RSK4-overexpressing-MR11 (top right) injected clones are shown in Fig. 4B. Likewise, histologic examination of H&E-stained paraffin sections of lungs revealed numerous metastases in mice injected with vector control cells (Fig. 4B, middle and bottom left), whereas RSK4-overexpressing cells showed markedly decreased metastatic lesions (Fig. 4B, middle and bottom right). No metastatic lesions were detected in bones, liver, brain, or kidney either injected with RSK4-overexpressing cells or vector control cells.
RSK4 may be a transcriptional target of c-Myc
Because high expression of RSK4 was evident in the primary mammary tumors of MMTV-c-Myc transgenic mice by microarray analysis and quantitative reverse transcription-PCR (1), in this study, we further investigated whether c-Myc is likely to play a role in the regulation of RSK4. In agreement with our previous observation, MycER-overexpressing T47D cells showed up-regulation of RSK4 (Fig. 5A) on conditional activation of c-MycER fusion protein (~97 kDa) with the selective estrogen receptor modulator 4-hydroxytamoxifen. Next, we investigated whether c-Myc can regulate RSK4 transcription. To address this question, we cloned a regulatory region of human RSK4 gene to investigate whether this region is regulated by c-Myc. The sequence analysis (using TF Bind software) revealed TATA and CAAT boxes and several other transcription factor binding sites including c-Myc consensus binding sequence (Fig. 5B). This fragment was then cloned into a pGL3 reporter vector. Overexpression of c-Myc in HeLa cells cotransfected with a luciferase reporter gene driven by RSK4 regulatory sequence (a putative RSK4 promoter) showed a dose-dependent induction of luciferase activity (Fig. 5C), thereby suggesting that c-Myc may regulate RSK4 through specific binding to a regulatory region of the RSK4 gene.
Fig. 5.

RSK4 may be a transcriptional target of c-Myc. A, RSK4 mRNA and protein expression after activation of c-Myc by 4-hydroxytamoxifen inT47D cells transfected with pBabe-Puro-MycER construct or vector control pBabe-Puro (VC). B, A 640-bp fragment of human RSK4 gene showing a consensus c-Myc binding site and the relative positions of the binding sites of the other transcription factor. C, pcDNA3.1-c-Myc-transfected HeLa cells cotransfected with a reporter construct (reporter construct, 200 ng/well in six-well plate at 80–85% confluency) show >5-fold increase in luciferase activity. Transient transfection with increasing amounts of c-Myc (0.5–3 μg) expression plasmid show dose-dependent reporter activity. Luciferase units were calculated as luciferase activity/β-galactosidase activity. Mean ± SE of three individual experiments in triplicates. The fold change was calculated by comparison with the promoterless luciferase vector.
Discussion
This study shows that 90-kDa RSK4 can suppress the tumor growth and the invasive and metastatic ability of MDA-MB-231 breast cancer cells. We measured these phenotypic changes in RSK4-overexpressing cells by their ability to (a) grow independently of substrate adhesion, migrate through the transmem-brane to the bottom side of the membrane, and degrade an artificial extracellular matrix (Matrigel) in vitro and (b) grow noninvasive tumors in SCID mice and reduced metastatic lesion in lungs after i.v. injected RSK4-overexpressing cells in SCID mice in vivo. Results of these experiments indicate that increased expression of RSK4 protein can impair the malignant properties of MDA-MB-231 cells. To exclude the possibility that tumor size may contribute to invasion of surrounding tissues, mice injected with RSK4-overexpressing cells were kept for additional 4 weeks to see whether observed tumor size is due to slow-growing properties of these cells and whether increase in size will affect the invasive properties of RSK4-overexpressing cells. Even after 4 weeks, tumors did not grow significantly as well as the tumor morphology and edge was confined (data not shown).
Intriguingly, RSK4-overexpressing cells showed decreased expression of CXCR4 and increased expression of CLDN2 and vice versa after silencing of RSK4. Both of these factors have been shown to play roles in invasion and chemotaxis. Chemotaxis is critical for preferential homing of cancer cells to metastatic sites. In particular, up-regulation of CXCR4 on cancer cells and CXCR4/CXCL12-induced motility have been implicated in the metastasis of breast cancer in lymph nodes, lungs, and bones (19–21). Similarly, CLDN2, a transmem-brane protein, is an essential component of tight junction strands that contribute to the maintenance of epithelial tissue integrity (22–24). Alteration in tight junction barrier function can result in epithelial to mesenchymal transition; therefore, down-regulation of any of the tight junction components consequently can cause cells to adopt a migratory behavior. Loss or reduced expression of claudins has been implicated in epithelial to mesenchymal transition and metastasis (25, 26). Antibody-mediated inhibition of CLDN2 resulted in enhanced invasive activity (data not shown), whereas antibody-mediated inhibition of CXCR4 showed ~50% reduced chemotactic activity. These data suggest that RSK4-regulated CLDN2 and CXCR4, in part, may abrogate the invasive and migratory phenotype of MDA-MB-231 cells.
Overexpression of RSK4 results in decreased cell proliferation and significantly increased accumulation of cells in the G0–G1 phase of the cell cycle, whereas robust S-phase accumulation was observed on treatment with 5′-dFUR. Significantly increased accumulation of cells in the G0–G1 phase is in concurrence with a previous study where depletion of RSK4 abrogated the p53-dependent G1 cell cycle arrest (14). RSK4-overexpressing clones show predominantly hypo-phosphorylated Rb protein and increased expression of p21 protein under normal serum conditions (10% FBS), which may be responsible for the increased accumulation of RSK4-overexpressing cells in the G1 phase. It is known that hypophosphorylation of pRb and increased expression of p21 can arrest cells in the G1 phase (27, 28). The precise molecular mechanism by which RSK4 drives cells into S phase on treatment is unclear. Phosphorylation of pRb is known to be responsible for G1-S-phase progression (29). It is likely that phosphorylation of pRb on treatment in RSK4-overexpressing clones may drive these cells into S phase. Interestingly, RSK4-overexpressing clones also showed marked induction of RbAp46; however, it is yet to be determined whether RbAp46 also contributes to G0–G1 under various serum conditions or S-phase accumulation after treatment with 5′-dFUR. RbAp48/RbAp46 physically associates with E2F1 in the presence of Rb and histone deacetylase (30) to repress transcription of E2F-responsive genes; thus, increased expression of RbAp46 is likely to arrest cells in G1 phase. We also found high expression of cyclin D1 in RSK4-over-expressing cells. Overexpression of cyclin D1 occurs frequently in several types of human cancer and plays diverse roles contingent on its expression level, cell type, and cell context. Various studies have shown that overexpression of cyclin D1 can promote cell proliferation and enhance tumorigenesis (31, 32). On the other hand, many studies have shown that cyclin D1 can inhibit cell cycle progression, arrests cells either in G0–G1 or S phase, and induces apoptosis in response to various stimuli (33–35). It is likely that RSK4-induced increased expression of cyclin D1 contributes to RSK4-mediated cell cycle arrest.
RSK4 maps to chromosome Xq21, a region where deletions have been shown in various cancer patients (36–38). We have also recently shown the loss of expression or reduced expression of RSK4 mRNA in >50% of human breast cancer cases compared with adjacent normal tissues (16). Many studies have reported 14% to 30% loss of Xq region in primary breast carcinomas and infiltrative ductal or lobular carcinomas (38–40). Whether expression patterns of RSK4 can predict the predisposal of breast tumors for invasion and metastasis needs further investigations.
Finally, we show marked induction of endogenous RSK4 and increased luciferase reporter gene activity (−640-bp regulatory region of RSK4-pGL3) in c-Myc-overexpressing cells. These data suggest that RSK4 expression may be regulated by c-Myc. We have shown previously up-regulation of RSK4 in the mammary tumors of MMTV-c-Myc transgenic mice compared with normal mammary glands and mammary tumors of MT-TGFα transgenic mice (1). Consistent with our findings that RSK4 is able to inhibit invasion and metastasis of breast cancer cells, mammary tumors of MMTV-c-Myc trans-genic mice are also noninvasive and nonmetastatic. Based on these data, we speculate that the noninvasive and non-metastatic phenotype of mammary tumors from MMTV-c-Myc transgenic mice, in part, could be due to c-Myc-induced expression of RSK4.
In summary, we have shown that RSK4 may play a critical role in breast cancer invasion. RSK4 being a ribosomal protein kinase may not only play a role in the regulation of gene expression through post-translational modifications but also through transcriptional and post-transcriptional modifications of transcription factors, coregulators, and other unknown target proteins. How RSK4 can suppress transformation, invasion, and metastasis warrants further investigations. Understanding of mechanism(s) of how RSK4 regulates these phenotypes may allow manipulation of RSK4-mediated signaling and to design a new therapeutic strategy.
Acknowledgments
Grant support: NIH grant R01CA100864 (D.J. Liao), Elsa U. Pardee Foundation (A.Thakur), and Susan G. Komen Breast Cancer Foundation grant BCTR02-01648 (D.J. Liao).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Thakur A, Xu H, Wang Y, Bollig A, Biliran H, Liao JD. The role of X-linked genes in breast cancer. Breast Cancer ResTreat. 2005;93:135–43. doi: 10.1007/s10549-005-4516-0. [DOI] [PubMed] [Google Scholar]
- 2.Bjorbaek C, Zhao Y, Moller DE. Divergent functional roles for p90rsk kinase domains. J Biol Chem. 1995;270:18848–52. doi: 10.1074/jbc.270.32.18848. [DOI] [PubMed] [Google Scholar]
- 3.Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 4.Gavin AC, Nebreda AR. A MAP kinase docking site is required for phosphorylation and activation of p90(rsk)/MAPKAP kinase-1. Curr Biol. 1999;9:281–4. doi: 10.1016/s0960-9822(99)80120-1. [DOI] [PubMed] [Google Scholar]
- 5.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–62. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 6.Clark DE, Errington TM, Smith JA, Frierson HF, Jr, Weber MJ, Lannigan DA. The serine/threonine protein kinase, p90 ribosomal S6 kinase, is an important regulator of prostate cancer cell proliferation. Cancer Res. 2005;65:3108–16. doi: 10.1158/0008-5472.CAN-04-3151. [DOI] [PubMed] [Google Scholar]
- 7.Clark DE, Poteet-Smith CE, Smith JA, Lannigan DA. Rsk2 allosterically activates estrogen receptor α by docking to the hormone-binding domain. EMBO J. 2001;20:3484–94. doi: 10.1093/emboj/20.13.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frodin M, Jensen CJ, Merienne K, Gammeltoft S. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 2000;19:2924–34. doi: 10.1093/emboj/19.12.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joel PB, Smith J, Sturgill TW, Fisher TL, Blenis J, Lannigan DA. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol. 1998;18:1978–84. doi: 10.1128/mcb.18.4.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panta GR, Kaur S, Cavin LG, et al. ATM and the catalytic subunit of DNA-dependent protein kinase activate NF-κB through a common MEK/extracellular signal-regulated kinase/p90(rsk) signaling pathway in response to distinct forms of DNA damage. Mol Cell Biol. 2004;24:1823–35. doi: 10.1128/MCB.24.5.1823-1835.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roux PP, Richards SA, Blenis J. Phosphorylation of p90 ribosomal S6 kinase (RSK) regulates extracellular signal-regulated kinase docking and RSK activity. Mol Cell Biol. 2003;23:4796–804. doi: 10.1128/MCB.23.14.4796-4804.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–63. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 13.Dummler BA, Hauge C, Silber J, et al. Functional characterization of human RSK4, a new 90-kDa ribo-somal S6 kinase, reveals constitutive activation in most cell types. J Biol Chem. 2005;280:13304–14. doi: 10.1074/jbc.M408194200. [DOI] [PubMed] [Google Scholar]
- 14.Berns K, Hijmans EM, Mullenders J, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–7. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 15.Myers AP, Corson LB, Rossant J, Baker JC. Characterization of mouse Rsk4 as an inhibitor of fibroblast growth factor-RAS-extracellular signal-regulated kinase signaling. Mol Cell Biol. 2004;24:4255–66. doi: 10.1128/MCB.24.10.4255-4266.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thakur A, Rahman KW, Wu J, et al. Aberrant expression of X-linked genes RbAp46, Rsk4, and Cldn2 in breast cancer. Mol Cancer Res. 2007;5:171–81. doi: 10.1158/1541-7786.MCR-06-0071. [DOI] [PubMed] [Google Scholar]
- 17.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandis AZ, Prasad A, Band H, Klosel R, Ganju RK. Regulation of CXCR4-mediated chemotaxis and chemoinvasion of breast cancer cells. Oncogene. 2004;23:157–67. doi: 10.1038/sj.onc.1206910. [DOI] [PubMed] [Google Scholar]
- 19.Kato M, Kitayama J, Kazama S, Nagawa H. Expression pattern of CXC chemokine receptor-4 is correlated with lymph node metastasis in human invasive ductal carcinoma. Breast Cancer Res. 2003;5:R144–50. doi: 10.1186/bcr627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 21.Murphy PM. Chemokines and the molecular basis of cancer metastasis. N Engl J Med. 2001;345:833–5. doi: 10.1056/NEJM200109133451113. [DOI] [PubMed] [Google Scholar]
- 22.Hoevel T, Macek R, Mundigl O, Swisshelm K, Kubbies M. Expression and targeting of the tight junction protein CLDN1in CLDN1-negative human breast tumor cells. J Cell Physiol. 2002;191:60–8. doi: 10.1002/jcp.10076. [DOI] [PubMed] [Google Scholar]
- 23.Offner S, Hekele A, Teichmann U, et al. Epithelial tight junction proteins as potential antibody targets for pancarcinoma therapy. Cancer Immunol Immun-other. 2005;54:431–45. doi: 10.1007/s00262-004-0613-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soini Y. Expression of claudins 1, 2, 3, 4, 5 and 7 in various types of tumours. Histopathology. 2005;46:551–60. doi: 10.1111/j.1365-2559.2005.02127.x. [DOI] [PubMed] [Google Scholar]
- 25.Morin PJ. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res. 2005;65:9603–6. doi: 10.1158/0008-5472.CAN-05-2782. [DOI] [PubMed] [Google Scholar]
- 26.Soini Y. Claudins 2, 3, 4, and 5 in Paget’s disease and breast carcinoma. Hum Pathol. 2004;35:1531–6. doi: 10.1016/j.humpath.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 27.Choi YH, Choi BT, Lee WH, Rhee SH, Park KY. Doenjang hexane fraction-induced G1 arrest is associated with the inhibition of pRB phosphorylation and induction of Cdk inhibitor p21 in human breast carcinoma MCF-7 cells. Oncol Rep. 2001;8:1091–6. doi: 10.3892/or.8.5.1091. [DOI] [PubMed] [Google Scholar]
- 28.Musgrove EA, Lee CS, Cornish AL, Swarbrick A, Sutherland RL. Antiprogestin inhibition of cell cycle progression inT-47D breast cancer cells is accompanied by induction of the cyclin-dependent kinase inhibitor p21. Mol Endocrinol. 1997;11:54–66. doi: 10.1210/mend.11.1.9869. [DOI] [PubMed] [Google Scholar]
- 29.Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem. 1997;272:10882–94. doi: 10.1074/jbc.272.16.10882. [DOI] [PubMed] [Google Scholar]
- 30.Taylor-Harding B, Binne UK, Korenjak M, Brehm A, Dyson NJ. p55, the Drosophila ortholog of RbAp46/RbAp48, is required for the repression of dE2F2/RBF-regulated genes. Mol Cell Biol. 2004;24:9124–36. doi: 10.1128/MCB.24.20.9124-9136.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. JCellBiochem. 2006;97:261–74. doi: 10.1002/jcb.20690. [DOI] [PubMed] [Google Scholar]
- 32.Kenny FS, Hui R, Musgrove EA, et al. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res. 1999;5:2069–76. [PubMed] [Google Scholar]
- 33.Han EK, Begemann M, Sgambato A, et al. Increased expression of cyclin D1in a murine mammary epithelial cell line induces p27kip1, inhibits growth, and enhances apoptosis. Cell Growth Differ. 1996;7:699–710. [PubMed] [Google Scholar]
- 34.Han EK, Sgambato A, Jiang W, et al. Stable overexpression of cyclin D1 in a human mammary epithelial cell line prolongs the S-phase and inhibits growth. Oncogene. 1995;10:953–61. [PubMed] [Google Scholar]
- 35.Coco Martin JM, Balkenende A, Verschoor T, Lallemand F, Michalides R. Cyclin D1 overexpression enhances radiation-induced apoptosis and radiosensitivity in a breast tumor cell line. Cancer Res. 1999;59:1134–40. [PubMed] [Google Scholar]
- 36.Hogdall EV, Ryan A, Kjaer SK, et al. Loss of heterozygosity on the X chromosome is an independent prognostic factor in ovarian carcinoma: from the Danish “MALOVA” Ovarian Carcinoma Study. Cancer. 2004;100:2387–95. doi: 10.1002/cncr.20213. [DOI] [PubMed] [Google Scholar]
- 37.Kersemaekers AM, Kenter GG, Hermans J, Fleuren GJ, van deVijver MJ. Allelic loss and prognosis in carcinoma of the uterine cervix. Int J Cancer. 1998;79:411–7. doi: 10.1002/(sici)1097-0215(19980821)79:4<411::aid-ijc17>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 38.Nishizaki T, Chew K, Chu L, et al. Genetic alterations in lobular breast cancer by comparative genomic hybridization. Int J Cancer. 1997;74:513–7. doi: 10.1002/(sici)1097-0215(19971021)74:5<513::aid-ijc6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 39.Kuukasjarvi T, Karhu R, Tanner M, et al. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997;57:1597–604. [PubMed] [Google Scholar]
- 40.Tirkkonen M, Tanner M, Karhu R, Kallioniemi A, Isola J, Kallioniemi OP. Molecular cytogenetics of primary breast cancer by CGH. Genes Chromosomes Cancer. 1998;21:177–84. [PubMed] [Google Scholar]