

Abstract
Purpose.
To study safety and efficacy of subretinal adeno-associated virus (AAV) vector AAV-Bbs1 injection for treatment of a mouse model of Bardet-Biedl syndrome type 1 (BBS1).
Methods.
Constructs containing a wild-type (WT) Bbs1 gene with and without a FLAG tag in AAV2/5 vectors were generated. Viral genomes were delivered by subretinal injection to right eyes and sham injections to left eyes at postnatal day 30 (P30) to P60. Transgene expression and BBSome reconstitution were evaluated by immunohistochemistry and Western blotting following sucrose gradient ultracentrifugation. Retinal function was analyzed by electroretinogram (ERG) and structure by optical coherence tomography (OCT). Histology and immunohistochemistry were performed on selected eyes.
Results.
Expression of FLAG-tagged Bbs1 was demonstrated in photoreceptor cells using antibody directed against the FLAG tag. Coinjection of AAV-GFP demonstrated transduction of 24% to 32% of the retina. Western blotting demonstrated BBS1 protein expression and reconstitution of the BBSome. ERG dark-adapted bright flash b-wave amplitudes were higher in AAV-Bbs1–injected eyes than in sham-injected fellow eyes in more than 50% of 19 animals. Anti-rhodopsin staining demonstrated improved localization of rhodopsin in AAV-Bbs1–treated eyes. WT retinas injected with AAV-Bbs1 with or without a FLAG tag showed outer retinal degeneration on ERG, OCT, and histology.
Conclusions.
In a knock-in model of BBS1, subretinal delivery of AAV-Bbs1 rescues BBSome formation and rhodopsin localization, and shows a trend toward improved ERG. BBS is challenging to treat with gene therapy due to the stoichiometry of the BBSome protein complex and overexpression toxicity.
Keywords: retinal degeneration, Bardet Biedl syndrome, AAV vector, mouse model, gene therapy
Subretinal gene therapy with an AAV vector can rescue formation of the BBSome and slightly improve retinal function in mice with BBS type 1. However, overexpression toxicity may present challenges for therapy.
Introduction
Bardet-Biedl syndrome (BBS) refers to a group of disorders characterized by obesity, early-onset severe retinal degeneration, polydactyly, renal and gonadal anomalies, anosmia, and cognitive disabilities.1 The protein products of at least 17 different genes associated with BBS interact to affect the structure and/or function of cilia.2 Many of these gene products interact in multisubunit complexes. For example, seven BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, and BBS9), along with another protein called BBIP10, form a complex called the BBSome.3 The BBSome is believed to mediate protein trafficking to the primary cilium and the photoreceptor outer segment. Another complex, the BBS/CCT chaperonin complex, facilitates the BBSome assembly and is composed of three BBS proteins (BBS6, BBS10, and BBS12) and six CCT chaperonin proteins.4 Because the protein products of BBS genes physically interact to perform a common function, mutations of many different genes cause the same unusual combination of phenotypic findings.
The retinal degeneration in BBS is rapidly progressive and devastating to vision, usually causing legal blindness by the age of 20 years. BBS can also present as night blindness and nystagmus in very young children, overlapping the presentation of Leber congenital amaurosis (LCA), the most common retinal degeneration of childhood. In fact, a homozygous nonsense mutation in the gene responsible for the largest fraction of LCA, CEP290, has been reported in a BBS patient, highlighting the interconnectedness of the cilia-related genes.5,6
Recessive, loss-of-function diseases of the retina are potentially treatable with gene replacement therapy. For example, RPE65-associated LCA has been shown to be highly amenable to treatment with subretinal gene therapy in mice, dogs, and humans.7–11 The most common form of BBS in humans is caused by homozygous M390R mutations in BBS1, and knock-in mice homozygous for this mutation closely recapitulate the human retinal disease as well as other associated BBS phenotypes.12 We hypothesized that subretinal delivery of normal copies of the Bbs1 gene might rescue the retinal phenotype in these mice. One potential challenge of gene replacement therapy for a gene whose product is a member of a large multiprotein complex is that if the abundance of one member is altered significantly, the stoichiometry of the complex may be disrupted. Therefore we also entertained the alternative hypothesis that overexpression of wild-type (WT) BBS1 protein might be toxic or might not restore normal function.
Materials and Methods
AAV Vectors
Recombinant AAV-Bbs1 and AAV-GFP vectors were prepared at the University of Iowa Gene Transfer Vector Core using an AAV2-based proviral plasmid (Fig. 1). Chicken β-actin promoter, N-terminal 3× FLAG tag, mouse Bbs1 open reading frame (ORF), and bovine growth hormone (BGH) poly A signal sequences were inserted between the two AAV2 inverted terminal repeats (ITRs) using standard molecular biology techniques (Fig. 1).
Figure 1. .

Structure of the FLAG-tagged and non-FLAG–tagged Bbs1 inserts. The 4.45-kb insert, composed of the chicken β-actin promoter, 3× FLAG sequences in reading frame with the mouse Bbs1 gene, and a poly A tail sequence, is flanked by two inverted terminal repeat sequences (ITR). In the second construct, the insert is 4.35 kb and lacks the FLAG tag.
The recombinant vector was cross-packaged into AAV serotype-5 capsids in Sf9 insect cells. Viruses were initially purified using an iodixanol gradient (15%–60% w/v) and subjected to additional purification via ion exchange using MustangQ Acrodisc membranes (Pall Corporation, East Hills, NY). The viral titers (viral genomes per milliliter) were determined by quantitative polymerase chain reaction (QPCR; see Supplementary Data). The procedures for this vector system have been described in detail elsewhere.13,14
Animals and Subretinal Injections
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Iowa and conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All animals were maintained in 12-hour light–dark cycles and fed standard mouse chow ad libitum.
Homozygous Bbs1M390R/M390R mice on a 129SVEV background were generated by homologous recombination as described previously12 and were selected by genotyping litters from heterozygous crosses. These mice exhibit abnormal outer segments by P21, and the b-wave amplitudes of the dark-adapted bright flash ERG (standard combined response, or SCR) are reduced by approximately 40% at P60 compared to those in WT or heterozygous mice of the same background.12 By 6 to 8 months of age the ERG is essentially nonrecordable.
Transscleral subretinal injections were performed by anesthetizing the mice with a ketamine/xylazine mix (0.1 mL/20 g weight at a concentration of 17.5 mg/mL ketamine and 2.5 mg/mL xylazine), then applying topical povidone iodine 10% solution, tropicamide 1%, and proparacaine 1% (one drop each). A limbal conjunctival peritomy was made superotemporally using 0.12 forceps and Vannas scissors (Bausch and Lomb/Storz Ophthalmics, Rochester, NY). A sclerotomy was made just posterior to the limbus using a 30-gauge half-inch needle viewed through the operating microscope. The injection was then given through the scleral opening by inserting a 33-gauge blunt needle on a Hamilton syringe (Hamilton Company, Reno, Nevada) under the retina and slowly depressing the plunger. The needle was left in place for several seconds after the fluid was delivered. The resulting retinal bleb could be visualized through the operating microscope. If no bleb formed, or if extensive hemorrhage was seen, the eye was excluded.
Vectors delivered to the subretinal space included AAV-Bbs1 with a FLAG tag, AAV-Bbs1 without a FLAG tag, empty AAV capsids, or AAV-GFP as sham injections, all using AAV2/5, at varying dilutions starting with 1 × 1010 viral genomes (VG). Sterile saline or empty AAV capsids were used as a sham injection in some cases, as noted in the figure legends.
Retinal Histology
Mice were euthanized at predetermined time points by CO2 asphyxiation followed by cervical dislocation. The eyes were immersed in cold fixative (4% paraformaldehyde) for >4 hours or one-half strength Karnovsky fixative overnight15 while maintaining orientation, and then stored in PBS for later processing. The eyes were dissected, and the anterior segment and the lens were removed. Samples were embedded in acrylamide solution16 and frozen in optimal cutting temperature (OCT) compound. Posterior poles were oriented in order to obtain superior-to-inferior cross sections of the eye cup. Tissue was sectioned at 10 μm on a cryostat (MICROM GmbH, Walldorf, Germany). Sections were processed for hematoxylin and eosin (H&E) histochemistry and were analyzed using light microscopy (Olympus BX41; Olympus, Center Valley, PA).
Overlapping images of the entire section were acquired using a 20× objective lens. Commercial software (Photoshop; Adobe Systems, San Jose, CA) was used to make montages of the eye cups. Measurements of outer nuclear layer (ONL) thickness were performed adjacent to the needle entry site and equidistant from the ciliary body on the opposite side of the eye cup from the injection site. Five measurements were made on each side of the eye 100 μm apart.
For treated and control eyes, two histologic sections per eye from the same retinal locations were photographed. Beginning 0.5 mm on either side of the optic nerve (ON), five measurements of photoreceptor ONL thickness were made across nonoverlapping 0.5-mm lengths using the caliper on ImageJ (available in the public domain at http://rsbweb.nih.gov/ij/), and averaged as the representative value for that area as described previously.17
Flat mounts of treated and control eyes were prepared by fixing selected eyes in 4% paraformaldehyde for 2 hours, then removing the lens and cornea. Four radial cuts equidistant from the ON were made through the eye cup with scissors. A well was prepared on a glass slide using 2% agarose gel; the eye was suspended in PBS in this well and photographed under 4× magnification (Olympus BX41).
In some eyes after treatment with AAV-Bbs1 injection, histologic sections were prepared as above, followed by labeling with anti-FLAG antibody (Sigma) and an Alexa Fluor 488–conjugated secondary antibody (Invitrogen, Carlsbad, CA), or with anti-rhodopsin antibody (clone RETP1; Santa Cruz Biotechnology, Santa Cruz, CA) and a secondary goat anti-mouse Alexa Fluor 568 antibody (Invitrogen). Immunohistochemistry was performed as described previously.18
Sucrose Gradient Ultracentrifugation and Western Blotting
Sucrose gradient ultracentrifugation and Western blotting were conducted as previously described19 with minor modifications. Briefly, AAV vector–injected eyes were enucleated using forceps and homogenized in ice-cold lysis buffer (PBS with 0.6% Triton X-100). After clearance by centrifugation, the protein extract was concentrated with a Microcon centrifugal filter device (30,000 MWCO; Millipore), loaded on a 4-mL 10% to 40% sucrose linear gradient in PBS with 0.04% Triton X-100, and spun at 166,400gavg for 15 hours. A Gel Filtration HMW Calibration Kit (GE Healthcare) and bovine catalase (Sigma) were used as sedimentation coefficient standards and were run simultaneously on an identical gradient. Fractions (∼210 μL) were collected from the bottom of the tube using a 26-gauge needle. Equal volumes of each fraction were mixed with LDS sample loading buffer (Invitrogen) and loaded onto an SDS-PAGE gel. SDS-PAGE and Western blotting were performed following standard protocols. All BBS antibodies have been previously characterized and described.3,19 IFT88 antibody was obtained from ProteinTech Group.
Electroretinogram (ERG) Recordings
Full-field ERG was obtained using the Espion V5 Diagnosys system (Diagnosys LLC, Lowell, MA). After overnight dark adaptation, mice were anesthetized with an intraperitoneal injection of ketamine (87.5 mg/kg) and xylazine (2.5 mg/kg). ERGs were recorded simultaneously from the corneal surface of each eye after pupil dilation (1% tropicamide) using gold ring electrodes (Diagnosys) referenced to a needle electrode (Roland Consult, Brandenburg an der Havel, Germany; LKC Technologies Inc., Gaithersburg, MD) placed on the back of the head. Another needle electrode placed in the tail served as the ground. A drop of methylcellulose (2.5%) was placed on the corneal surface to ensure electrical contact and to maintain corneal integrity. Body temperature was maintained at a constant temperature of 38°C using a regulated heating pad. All stimuli were presented in a ColorDome (Diagnosys) ganzfeld bowl, and the mouse head and electrode positions were monitored on the camera attached to the system. Dim red light was used for room illumination until dark adapted testing was completed. A modified International Society for Clinical Electrophysiology of Vision (ISCEV) protocol17,20 was used, including a dark-adapted dim flash of 0.01 cd.s/m2, maximal combined response (standard combined response or SCR) to bright flash of 3 cd.s/m2, light-adapted bright flash of 3 cd.s/m2, and 5-Hz flicker stimuli at 3 cd.s/m2.
The a-wave was measured from the baseline to the trough of the first negative wave. The b-wave was measured from the trough of the a-wave to the peak of the first positive wave, or from the baseline to the peak of the first positive wave if no a-wave was present.
Optical Coherence Tomography (OCT)
OCT (Bioptigen, Morrisville, NC) was performed after placing the animals under ketamine/xylazine anesthesia as described above. Tropicamide 1% was used to dilate the pupils, and the retinas were scanned one eye at a time. Methylcellulose lubricant was placed on the corneas, and the noncontact probe was positioned near the cornea until the retinal image could be seen on the screen. This was then focused and oriented with the ON in the middle of the scan as a landmark. Retinal OCT was performed using rectangular volume scan (volumetric acquisition made up of a series of B-Scans) with a length of 1.40 mm at a width of 1.40 mm at a rate of 1000 A-Scan/B-Scan. An average of three repeated B-Scans (of the same region) centered on the ON was used for analysis of retinal thickness. The probe was then rotated to obtain images of the retinal periphery. The total retinal thickness was measured on the OCT image at two locations, each 0.3 mm from the edge of the ON, on either side using the measuring tool provided (Bioptigen). Eyes were excluded from further analysis if retinal disruption more than four times the area of a typical injection site due to hemorrhage or chronic retinal detachment (greater than 1 week after injection) was noted on OCT.
Statistical Analysis
The paired t-test and Wilcoxon signed rank test were used to compare results of the ERG amplitudes between pairs of eyes where the right eye was treated and left eye was sham treated. The paired t-test was used to compare ONL thickness (on OCT or by histology) between treated and control groups.
Results
Expression of AAV-Bbs1 in the Retina
Fluorescence of GFP was evaluated using whole mounts of four eyes in which AAV-GFP had been coinjected with AAV-FLAG-Bbs1. At least 24% of the area of the retina was transduced in each eye (Fig. 2). A similar percentage of retina was transduced whether one or two injections through the same incision in different directions were given per eye. Sections of AAV-FLAG-Bbs1–injected eyes from WT mice were collected and labeled with anti-FLAG antibody or anti-Bbs1 antibody (Fig. 3). Intense anti-FLAG labeling was observed in the outer retina, the ONL, and inner/outer segments as well as the outer plexiform layer.
Figure 2. .

Whole mount of a retina that received 1/10 AAV-GFP mixed with the AAV-FLAG-Bbs1 as a subretinal injection. Subretinal injection was performed in a 1-month-old WT animal. The animal was killed and the whole mount prepared 2 weeks after injection. Note the substantial degree of retinal transduction.
Figure 3. .

Expression of FLAG-BBS1 in photoreceptor cells. (A) Wild-type retina was transduced with the AAV-FLAG-Bbs1 (without AAV-GFP) and labeled with anti-FLAG antibody. Animals received subretinal injection of AAV-FLAG-Bbs1 vector at 1 month of age and were killed 2 weeks after injection. Left: Anti-FLAG immunofluorescence is seen in the retina which was over the injection site. Note the extensive labeling of the FLAG fusion protein in the outer plexiform layer, the inner segments, and to a lesser extent the outer nuclear layer. Right: A field from the same retina remote from the injection site. Scale bar: 50 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; IS/OS, inner and outer segments. (B) Localization of native BBS1 protein. Left: An area of retina from a 6-week-old WT mouse that had received a subretinal injection of AAV-FLAG-Bbs1 2 weeks before. This section was stained with anti-BBS1 antibody (not anti-FLAG antibody). Right: A different section of the same retina remote from the injection site (i.e., not exposed to the vector) labeled with anti-BBS1 antibody. The anti-BBS1 antibody is labeled with Alexa Fluor 488. The amount of immunoreactive BBS1 protein in the injected area is much greater than in the uninjected area. BBS1 protein can be detected without the anti-FLAG stain; however, this antibody does not bind as avidly as the anti-FLAG antibody.
Reconstitution of the BBSome With Recombinant BBS1
To determine whether the virally delivered FLAG-Bbs1 produces recombinant protein and supports BBSome formation, we transduced HEK293T cells with AAV-FLAG-Bbs1 and conducted immunoprecipitation using anti-FLAG antibodies. In this experiment, FLAG-BBS1 pulled down all BBSome components tested (BBS2, BBS4, BBS7, and BBS9), indicating that FLAG-BBS1 does form the BBSome with endogenous BBS proteins (Fig. 4A).
Figure 4. .

AAV-Bbs1 and AAV-FLAG-Bbs1 partly restore the BBSome assembly. (A) FLAG-BBS1 forms the BBSome. HEK293T cells were transduced with AAV-FLAG-Bbs1, and cell lysate was subjected to immunoprecipitation (IP) using anti-FLAG antibody and Western blotting against indicated BBS proteins. Normal HEK293T cells were used as a control. (B) Disruption of the BBSome in Bbs1M390R/M390R mutant eyes. Protein extracts from WT and Bbs1M390R/M390R mutant eyes were fractionated on a 10% to 40% sucrose linear gradient. Collected fractions were subjected to SDS-PAGE and Western blotting using the appropriate antibodies. Fraction numbers are shown at the top. Black and red arrowheads indicate the migration of sedimentation coefficient standards and the BBSome, respectively. In the WT gel, BBS1, -2, -4, -5, -7, and -9 migrate together, indicating a complex. In the Bbs1 gel, only BBBS2, -7, and -9 migrate together at a slightly lower fraction, indicating that the abnormal Bbs1M390R/M390R proteinis not incorporated into the BBSome and disrupts the complex. (C) Virally delivered FLAG-Bbs1 partly rescues the BBSome assembly. Protein extracts from treated animals were analyzed by sucrose gradient ultracentrifugation as in (B). (D) Virally delivered WT Bbs1 (without the FLAG) partly rescued the BBSome assembly just as the FLAG-tagged version did. Note the reappearance of BBS4 in fractions 11 and 12 of the treated eye.
We next compared the status of the BBSome in normal and Bbs1M390R/M390R mutant mouse eyes using 10% to 40% sucrose density gradient ultracentrifugation (Fig. 4B). Consistent with previous results on formation of the BBSome,3,19 all BBSome components from WT mouse eyes were fractionated together with a sedimentation coefficient of ∼14S. In contrast, the elution profiles of BBSome subunits (BBS2, BBS5, BBS7, and BBS9) were shifted toward lower molecular weight fractions in Bbs1M390R/M390R mouse eyes, indicating that at least some of the BBSome subunits were missing. Indeed, BBS1 (M390R mutant protein), BBS4, and BBS8 were hardly detectable in this experiment, suggesting that these BBS proteins may be unstable in the absence of normal BBS1. IFT88, a component of the IFT-B complex, was not associated with the BBSome and did not show changes in its migration rate, serving as a fractionation control.
We examined whether the introduction of FLAG-Bbs1 can restore the BBSome assembly in Bbs1M390R/M390R eyes (Fig. 4C). In AAV-FLAG-Bbs1–injected WT eyes, FLAG-BBS1 was found in the same fractions as the endogenous BBS proteins, indicating that FLAG-BBS1 is integrated into the BBSome in the eye. However, FLAG-BBS1 was also found in multiple additional fractions where endogenous BBS proteins are not found. This result suggests that overexpressed FLAG-Bbs1 forms multiple, heterogeneous protein complexes. In Bbs1M390R/M390R eyes, FLAG-BBS1 slightly shifted the elution profiles of BBS7 and BBS9 toward that of the normal BBSome (or “holo-BBSome”). In addition, injection of AAV-FLAG-Bbs1 partly restored the expression of BBS4, which is barely detectable in sham or uninjected Bbs1M390R/M390R eyes. BBS4 in these animals was found in the same fraction as that of holo-BBSome, suggesting that these BBS4-containing BBSomes are fully assembled. As in WT eyes, FLAG-BBS1 was also found in additional fractions with various migration rates. Since the viral transduction efficiency ranged from 24% to 32% of the entire retina, endogenous BBSome subunits found in lower molecular weight fractions are presumably from nontransduced cells. When the experiment was repeated using a non-FLAG–tagged vector, as shown in Figure 4D, the partial reconstitution of the BBSome was the same. In particular, Bbs4 could be detected in the same fraction as other BBSome components with a normal BBSome sedimentation coefficient in the treated eyes. Elution profiles of BBS5, BBS7, and BBS9 were also shifted closer to those of WT. Taken together, our data indicate that virally delivered FLAG-BBS1 and nontagged BBS1 at least partly restore BBSome assembly in Bbs1M390R/M390R eyes.
Partial ERG Rescue in Mice With BBS1 Gene Therapy
Control eyes receiving subretinal injection of AAV-GFP, empty AAV capsids, or saline had similar-amplitude, slightly reduced ERG b-wave amplitudes compared to uninjected eyes (data not shown). Although this was not statistically significant, we chose to use a sham injection for the contralateral eyes to control for the small diminution in ERG response that occurred due to the injection alone. The ERG was markedly reduced in WT eyes that received AAV-FLAG-Bbs1 or AAV-Bbs1 vectors compared to the contralateral eyes that received sham injections (Figs. 5A, 5B). In 7 of 13 Bbs1M390R/M390R eyes the ERG b-wave amplitudes were slightly higher in treated than control eyes. However, the average amplitudes were not higher, and the difference was not statistically significant (Fig. 5C). When the 1 × 1010 VG solution was diluted by 1/100 or 1/1000 and the same 2- to 3-μL volumes were delivered, the detrimental effect on ERG amplitude in the WT mice was less, but the beneficial effect in the Bbs1 mice was also reduced (Figs. 5D, 5E). Representative ERG tracings of the SCR can be seen in Figure 6.
Figure 5. .

Electroretinogram amplitudes of wild-type (WT) and Bbs1M390R/M390R mice injected with therapeutic and control AAV vectors. The x-axes show right and left eye b-wave and a-wave amplitudes for the dark-adapted bright flash standard combined response (SCR) ERG and the light-adapted 5-Hz flicker ERG. The y-axis is the amplitude of the ERG responses in microvolts. (A) Comparison of ERG amplitudes in 11 WT mice whose right eyes (OD) were injected with 1 × 1010 viral genomes (VG) AAV-FLAG-Bbs1 in 2 μL versus left eyes (OS) injected with 1 × 1010 VG AAV-GFP in 2 μL. Note that the WT right eyes injected with therapeutic vector had significantly worse b-wave amplitudes than control left eyes. (B) Comparison of ERG amplitudes in seven WT mice whose right eyes (OD) were injected with 1 × 1010 VG AAV-Bbs1 without a FLAG tag in 2 μL versus left eyes (OS) injected with 1 × 1010 VG AAV-GFP in 2 μL. Note that the right eyes injected with therapeutic vector had significantly worse b-wave amplitudes than control left eyes, just as seen in the FLAG-tagged vector–injected eyes. (C) Comparison of ERG amplitudes in five Bbs1M390R/M390R mice whose right eyes were injected with 1 × 1010 VG AAV-FLAG-Bbs1 in a volume of 2 μL versus left eyes (OS) injected with sterile saline solution 2 μL. The right eyes injected with therapeutic vector did not have improved ERG compared to control, but neither did they have a worse ERG as in the WT eyes. (D) Comparison of ERG amplitudes in five WT mice whose eyes were injected with a 1/1000 dilution of 1 × 1010 VG AAV-FLAG-Bbs1 in 2 μL, right eyes (OD), versus left eyes (OS) injected with 1/1000 dilution of empty AAV viral capsids in 2 μL. Diluting the therapeutic vector mitigated its toxicity in WT eyes. (E) Comparison of ERG amplitudes in six Bbs1M390R/M390R mice whose right eyes were injected with a 1/1000 dilution of 1 × 1010 VG AAV-FLAG-Bbs1 in a volume of 2 μL, right eyes (OD), versus left eyes (OS) injected with 1/1000 dilution of empty AAV capsids in 2 μL. Diluting the therapeutic vector showed a trend toward less effect in the Bbs1 eyes. Data shown are mean ± standard error of the mean.
Figure 6. .

Representative ERG SCR waveforms in the experimental groups shown graphically in Figure 5. The y-axis is SCR ERG b-wave amplitude in microvolts. The x-axis is in milliseconds. OD, right eye; OS, left eye. In all experiments, animals received subretinal injection at 4 to 6 weeks of age with therapeutic AAV vector in right eyes and control AAV-GFP in the left eyes. 1 × 1010 VG per 2 μL was the stock vector solution. (A) WT mouse; right eye 2 μL AAV-FLAG-Bbs1, left eye 2 μL AAV-GFP. (B) WT mouse; right eye 2 μL AAV-Bbs1 without FLAG, left eye 2 μL AAV-GFP. (C) Bbs1M390R/M390R mouse; right eye 2 μL AAV-FLAG-Bbs1, left eye 2 μL AAV-GFP. (D) WT mouse; right eye 2 μL 1/1000 dilution of AAV-FLAG-Bbs1, left eye 2 μL 1/1000 dilution of AAV-GFP. (E) Bbs1M390R/M390R mouse; right eye 2 μL 1/1000 dilution of AAV-FLAG-Bbs1, left eye 2 μL 1/1000 dilution AAV-GFP.
Giving 1 × 1010 VG in a lower-volume injection (1 μL) either as a single injection, or as two injections of 1 μL each through the same sclerotomy in different directions, resulted in better SCR ERG b-wave amplitudes at 4 weeks postinjection in the therapeutic vector–treated eyes than in the sham-treated eyes (n = 15 eyes, P = 0.02, Wilcoxon) (Fig. 7). These eyes continued to have higher ERG values at three time points out to 3 months postinjection, which were statistically significant at 3 weeks and 3 months. Of note, the rate of decline of the ERG amplitude over time was the same for the therapeutically treated and sham-treated eyes, although the amplitude was higher in the AAV-Bbs1–treated eyes at all time points (Fig. 8).
Figure 7. .
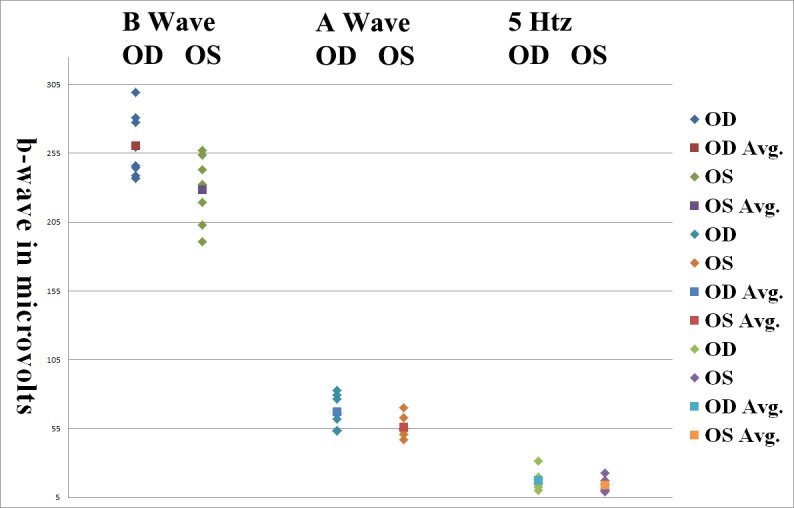
SCR ERG b-wave amplitudes in Bbs1M390R/M390R mice treated with 1-μL injections. N = 15 eyes. Right eyes were treated with 1-μL injections containing 1 × 1010 AAV-FLAG-Bbs1 versus left eyes treated with the same volumes of AAV-GFP. Eyes were injected when mice were 5 weeks old, and ERG was performed 3 weeks later. The b-wave amplitudes are statistically significantly different (P = 0.02, Wilcoxon).
Figure 8. .
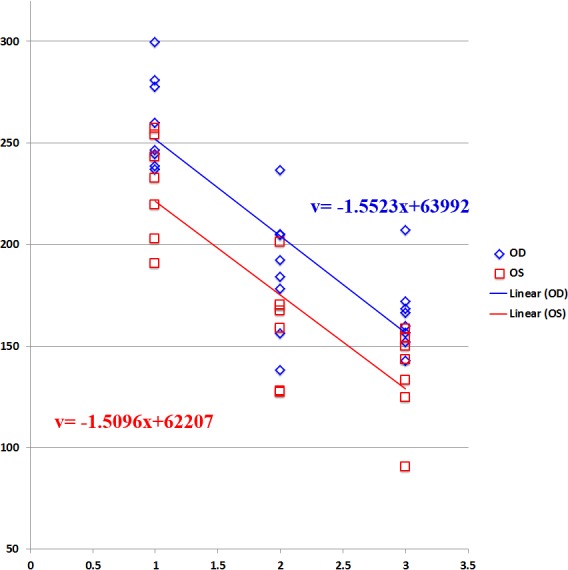
Rate of decline of SCR ERG b-wave amplitude over time in AAV-FLAG-Bbs1–treated versus AAV-GFP–treated Bbs1M390R/M390R mice. The therapeutically treated right eyes (OD) showed better ERG amplitudes at all time points than the sham-treated left eyes (OS); however, the rate of decline remained the same. N = 15 eyes.
Overexpression Toxicity of BBS1 in Wild-Type Retinas
With use of OCT at 1 week after injection, the needle entry point could be detected in 71 of 177 eyes receiving either therapeutic or sham subretinal injections in both WT and Bbs1M390R/M390R eyes (Fig. 9). Of 177 eyes, 33 were excluded from further study due to extensive hemorrhage or chronic retinal detachment on OCT. Without postinjection screening using OCT, these eyes would likely have been included in the study, which could have introduced bias since trauma affected their outcomes.
Figure 9. .
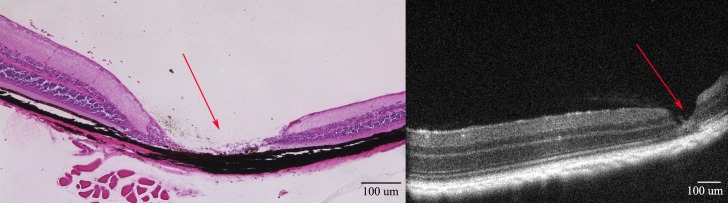
Needle entry site demonstrated in the retinal periphery. Retinal section of a 3-month-old mouse retina 1 month after subretinal injection on the left; on the right, OCT of a living mouse at the same age after the same procedure. Note the normal retina on either side of the needle entry site. In most cases, subretinal injection causes minimal or no damage to the retina detectable on OCT or histology.
In 7 of 11 WT right eyes injected with AAV-FLAG-Bbs1, an area of outer retinal toxicity could be seen on OCT corresponding to the area of retina over the injection bleb (Fig. 10A). The external limiting membrane and inner/outer segment junction or ellipsoid line was no longer visible in the treated areas, and the ONL was disorganized. Clumping and irregular borders of the normal lamination developed. The inner retina was preserved. This appearance was not seen in any of the contralateral left eyes injected with the same volume (2–3 μL) of AAV-GFP (Fig. 10B). Among Bbs1M390R/M390R eyes injected with 2 to 3 μL of the same vector, only 3 of 8 right eyes showed mild outer retinal toxicity, with 5 of 8 appearing identical to the AAV-GFP–injected left eyes (Figs. 10C, 10D). When 1-μL injections were given in Bbs1 eyes, 0 of 9 eyes receiving AAV-Bbs1 demonstrated the toxicity. No significant differences in retinal thickness measurements on OCT could be detected between treated and control groups.
Figure 10. .

OCT demonstration of retinal toxicity due to subretinal injection of AAV-FLAG-Bbs1 in WT mice retinas versus Bbs1 retinas. (A, B) The outer retinal layers to the right of the optic nerve in the photos of the right eyes (OD) from two different WT mice demonstrate disruption of the ELM and IS/OS junction and ellipsoid region in the area where the subretinal injection was delivered (A). In corresponding areas of the left eyes (OS) of the same two WT mice (B), which received subretinal AAV-GFP, the retinal lamination is retained and ONL appears normal. The right and left eye pairs in (A) and (B) are from two different animals at the same age after the same treatment. (C) In Bbs1M390R/M390R eyes, the AAV-FLAG-Bbs1–injected right eye (OD) looks similar to the AAV-GFP–injected left eye (OS), shown in (D). All OCTs were performed 1 month after injections given at 4 to 8 weeks old.
H&E staining of WT retinas treated with AAV-FLAG-Bbs1 revealed destruction of the ONL in the retina, which had been over the bleb. This observation corresponded to the findings on OCT. Contralateral WT eyes treated with AAV-GFP did not show this effect (Fig. 11). ONL thickness measured on a representative section was 26.2 μm in the AAV-FLAG-Bbs1–injected eye versus 49.6 μm in the control.
Figure 11. .

Histology of WT mouse retinas following subretinal injection of AAV-FLAG-Bbs1 (A) or AAV-GFP (B). The retina to the left of the optic nerves was under the injection bleb. ONL measured 26.8 μm in the right eye in the area of the therapeutic bleb, versus 49.6 μm in the left eye in the area of the control bleb. Histology was performed 1 month after injection of 4- to 8-week-old mice.
ONL Thickness and Rho Localization in Bbs1 Mutant Mice
Selected eyes of the treated and control groups were sectioned and stained with H&E. Among Bbs1M390R/M390R eyes, as noted above, only 3 of 17 eyes injected with the AAV-FLAG-Bbs1 vector showed outer retina toxicity compared to sham–injected eyes on OCT (Figs. 10C, 10D). H&E staining of selected right and left eyes with similar OCT appearance confirmed that there was no significant difference in the ONL thickness (14.2 vs. 14.5 μm) (Fig. 12).
Figure 12. .

Histology of Bbs1 mice retinas following subretinal injection. (A) Histology of Bbs1M390R/M390R right eye, after injection of AAV-FLAG-Bbs1, compared to (B) left eye injected with AAV-GFP. There is no sign of disruption of outer retina, but neither is there a dramatic improvement in the AAV-Bbs1–injected eye, with the ONL thickness measuring 14.2 μm in the right eye and 14.5 μm in the left eye. The retina to the left of the optic nerve was under the injection bleb. Histology was performed 1 month after injection of 4- to 8-week-old mice.
With use of a montage of histologic images to examine the eye cup in treated areas (adjacent to the needle entry site) and remote areas (on the opposite side of the ON at an equal distance from the ciliary body) in Bbs1M390R/M390R eyes, it could be seen that the transduced retina was thicker than untransduced retina in two of three Bbs1M390R/M390R eyes (Fig. 13).
Figure 13. .

Montage of eye cup of Bbs1M390R/M390R eyes treated with AAV-FLAG-Bbs1. Histology was done at P90; injection was performed at age 5 weeks. Note the thicker retina around the needle site (arrow) compared to the contralateral side of the eye cup. The area around the needle entry site is the site of the retinal bleb, under which transduction occurs. Two of three eyes examined in this way demonstrated the difference.
Rhodopsin has been shown to be mislocalized in Bbs1M390R/M390R eyes.12 However, in Bbs1M390R/M390R eyes that received subretinal AAV-FLAG-Bbs1, rhodopsin localization was qualitatively corrected compared to sham-treated eyes (Fig. 14).
Figure 14. .

Rhodopsin mislocalization is improved in Bbs1 mice treated with subretinal AAV-FLAG-Bbs1. (A) Anti-rhodopsin stain (red) and DAPI (blue) of Bbs1M390R/M390R right eye treated with subretinal AAV-FLAG-Bbs1. (B) Only the rhodopsin stain is shown. Note that there is very little rhodopsin mislocalized to the outer nuclear layer in this treated eye. (C) 4′,6-diamidino-2-phenylindole (DAPI) (blue) and anti-rhodopsin (red) stains of retina of the left untreated eye of the same Bbs1M390R/M390R mouse. There are multiple cells in the outer nuclear layer with mislocalized rhodopsin (arrows). (D) Only the anti-rhodopsin stain in this untreated Bbs1M390R/M390R eye is shown.
Discussion
Ciliated cells have complex internal mechanisms that have only recently been intensively studied. The recognition that disorders of cilia share overlapping phenotypes (including polydactyly; retinal degeneration; hearing loss; anosmia; gonadal, renal, and intellectual dysfunction; and in some cases situs inversus) has helped to crystallize the importance of understanding disease mechanisms with the aim of treating these individually rare but collectively clinically important entities.21–23 BBS is a fairly common ciliopathy, and may be even more common than we have believed. We now know that incomplete forms of the disorder occur and may be misdiagnosed. For example, patients have been reported who have only retinitis pigmentosa (RP), even in pedigrees in which others with the same mutations have the entire BBS phenotype.24 RP patients without family members exhibiting signs of BBS would not usually be suspected of having mutations in a BBS gene. Partial expression of syndromic disease is not uncommon; it also occurs in Usher syndrome25 and Senior-Loken syndrome.26
Simons et al. reported rescue of photoreceptors with subretinal gene delivery in another type of BBS, the BBS4 null mouse.27 They were able to transduce approximately 4% of the retina and to demonstrate rescue by showing that retinal thickness and ONL were increased in the area of retina adjacent to the thinned injection entry site. In our hands, it was difficult to identify the injection entry site in many eyes, even with OCT, since the trauma is minimal. However, in two of three eyes in which it could be detected, we found thicker retina adjacent to the site of injection. This may be an area of rescue similar to that described by Simons et al. Whether this would translate to a clinical rescue is uncertain.
Administration of subretinal AAV-Bbs1 to WT mice, either with or without a FLAG tag, is toxic to the outer retina, and is only mildly therapeutic in affected Bbs1 mice based on full-field ERG, OCT, and histologic examination. This is in concordance with data observed in zebrafish suggesting that overexpression of Bbs1 causes a phenotype similar to that produced by knockdown of the gene (Bay LM, Sheffield V, Slusarksi DC, unpublished data, 2012). In Usher syndrome mice, rescue of the Usher protein complex can be demonstrated even though the ocular phenotype is unchanged.28 This is relevant because Usher syndrome also involves disrupted protein complexes that are formed from the products of multiple genes.
Estrada-Cuzcano et al. summarized the known mechanisms of retinal ciliopathies and discussed possible roadblocks to treatment.2 Our data support their conclusion that gene replacement is more complex in this group of disorders. The most striking finding in our studies is the toxicity of excess BBS1 protein in the normal, WT murine retina. This toxicity was apparent as early as 2 weeks after injection and persisted for the lifetime of the mouse. The control eyes of the WT mice did not show toxicity; these eyes were injected with an equal volume of an AAV vector carrying a GFP construct; therefore neither the trauma of injection, injection technique, AAV vector, or expression of a transgene can be implicated in the toxicity. The toxicity was seen with both FLAG and non-FLAG versions of the vector. In mice lacking a normally functioning Bbs1 gene, overexpression of WT BBS1 was not as toxic, especially when delivered in lower volumes. The lack of robust rescue of ERG may have been due to two opposing forces at work: the therapeutic effect of reconstitution of the BBSome in one population of cells, allowing for more normal rhodopsin transport, while cells of a second population were overwhelmed by an excess of BBS1, resulting in abnormal protein complexes and damage to the cells.
ERG amplitudes were higher at every time point for the therapeutically treated eyes when 1-μL volumes of vector were delivered. The rate of decline of the ERG over time was the same for AAV-FLAG-Bbs1–treated eyes as it was for sham-treated eyes. This may indicate that although providing WT BBS1 protein makes living photoreceptors function better, it does not stop cell death. Alternatively it may halt cell death in a small population of transduced cells, but not in surrounding cells, so the mass response detected by the full-field ERG is not rescued. A similar finding was recently reported in adult humans in an RPE65 subretinal gene therapy trial.29 Taken together, these data suggest that subretinal AAV-Bbs1 reconstitutes the BBSome, improves rhodopsin trafficking, and slightly improves ERG in Bbs1M390R/M390R mice.
One of our AAV-Bbs1 vector constructs carries a FLAG tag, which has the potential to influence our results. However, FLAG tags have been used extensively in gene therapy studies without showing toxicity30; and if the FLAG tag were causing the toxicity, we would expect it to occur in the Bbs1M390R/M390R eyes as well as the controls. Numerous experiments in which a non-FLAG–tagged vector was used yielded results identical to those with the FLAG vector. Using smaller volumes of vector also correlated with better therapeutic effect, but not larger volumes of more dilute vector. Thus, the overexpression of Bbs1 protein itself appears to be toxic to the retina. This may be because excess protein cannot incorporate into the normal protein complexes and is free in the cell, interacting with other proteins and hindering their normal function or their normal trafficking. Excess of one protein component may disrupt normal BBSome assembly. It is of interest that transgenic mice with extra copies of the WT rhodopsin gene also show retinal degeneration.31 Rhodopsin requires transporter complexes with Arf4 and ASAP1 in order to be moved into the outer segment and has interactions with Rab8 and Rab11, which interact with the BBSome through Rabin8.3,32
If gene therapy alone improves function but does not stop death of photoreceptors, it could be combined with an antiapoptotic therapy such as systemic Tauroursodeoxycholic acid (TUDCA), which we have previously shown to slow retinal degeneration in Bbs1M390R/M390R mice.17 It is also possible that gene therapy must be delivered at a very young age to both improve function and slow degeneration. It is important to note that effective gene therapy for all BBS genes may not meet with the difficulties observed in this study of BBS1. Subretinal gene therapy in a mouse model of BBS4 showed promise as reported by Simons et al.27 In addition, Chamling et al. recently developed transgenic mice expressing various levels of BBS4. The transgene was able to rescue BBS phenotypes, including the retinopathy, in Bbs4−/− mice despite the fact that the transgene was expressed at vastly different levels in different tissues.33
In conclusion, we report here our experience with subretinal gene therapy in Bbs1 mice. We generated AAV vectors containing the Bbs1 gene, utilized this virus to treat Bbs1 mutant mice by subretinal injection, and established that the virus transduces the outer retina and that Bbs1 protein is expressed. We then characterized the response of WT and Bbs1 mutant mice to AAV transduction using Western blots, ERG, histology, and OCT. We showed that the BBSome is restored and the b-wave of the ERG slightly improved in Bbs1 mutant mice receiving the therapeutic Bbs1 vector (compared to sham-injected controls) and that this improvement is statistically significant in mice receiving the smallest volume (1 μL) under the retina. We also found qualitative improvement of rhodopsin mislocalization in Bbs1 mutant mice receiving the therapeutic vector. Most of our experiments were performed with both FLAG-tagged and untagged vectors, and we found no difference in outcomes. However, since the FLAG tag may conceivably affect some functions of the protein, it is possible that this tag had an influence on our data. In summary, we showed that in WT mice, delivery of the same viral vector load that is tolerated in a mouse retina lacking normal BBS1 protein results in degeneration of the photoreceptor cells, indicating that gene therapy trials in humans will need to carefully consider the impact of overexpression of BBS1 protein on retinal health.
Supplementary Material
Acknowledgments
The authors thank Al Maguire and Dan Chung for training in the subretinal injection technique, and Steve Russell and Megan Riker for contributions to this project.
Supported by a Marjorie Carr Adams Career Development award from the Foundation Fighting Blindness (AVD) National Institutes of Health (NIH) Grant R01-EY022616 (SS), and the Howard Hughes Medical Institute (EMS and VS).
Disclosure: S. Seo, None; R.F. Mullins, None; A.V. Dumitrescu, None; S. Bhattarai, None; D. Gratie, None; K. Wang, None; E.M. Stone, None; V. Sheffield, None; A.V. Drack, None
References
- 1. Drack AV, Mullins RF, Seo S. RP syndromes: Bardet Biedl. In: Hartnett ME. Pediatric Retina. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2012. [Google Scholar]
- 2. Estrada-Cuzcano A, Roepman R, Cremers FP, den Hollander AI, Mans DA. Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum Mol Genet. 2012; 21(R1): R111–R124 [DOI] [PubMed] [Google Scholar]
- 3. Nachury MV, Loktev AV, Zhang Q, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007; 129: 1201–1213 [DOI] [PubMed] [Google Scholar]
- 4. Seo S, Baye LM, Schultz NP, et al. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc Natl Acad Sci U S A. 2010; 107: 1488–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coppieters F, Lefever S, Leroy BP, De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat. 2010; 31: 1097–1108 [DOI] [PubMed] [Google Scholar]
- 6. Leitch CC, Zaghloul NA, Davis EE, et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008; 40: 443–448 [DOI] [PubMed] [Google Scholar]
- 7. Acland GM, Aquirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001; 28: 92–95 [DOI] [PubMed] [Google Scholar]
- 8. Acland GM, Aquirre GD, Bennett J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005; 12: 1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008; 358: 2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008; 358: 2231–2239 [DOI] [PubMed] [Google Scholar]
- 11. Cideciyan AV, Aleman TS, Boye SL, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A. 2008; 105: 15112–15117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davis RE, Swiderski RE, Rahmouni K, et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc Natl Acad Sci U S A. 2007; 104: 19422–19427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther. 2002; 13: 1935–1943 [DOI] [PubMed] [Google Scholar]
- 14. Smith RH, Levy JR, Kotin RM. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol Ther. 2009; 17: 1888–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karnovsky MJ. A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. J Cell Biol. 1965; 27: 137A–138A [Google Scholar]
- 16. Johnson LV, Blanks JC. Application of acrylamide as an embedding medium in studies of lectin and antibody binding in the vertebrate retina. Curr Eye Res. 1984; 3: 969–974 [DOI] [PubMed] [Google Scholar]
- 17. Drack AV, Dumitrescu AV, Bhattarai S, et al. TUDCA slows retinal degeneration in two different mouse models of retinitis pigmentosa and prevents obesity in Bardet-Biedl syndrome type 1 mice. Invest Ophthalmol Vis Sci. 2012; 53: 100–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishimura DY, Fath M, Mullins RF, et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A. 2004; 101: 16588–16593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seo S, Zhang Q, Bugge K, et al. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS Genet. 2011; 7: e1002358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marmor MF, Fulton AB, Holder GE, et al. ISCEV standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009; 118: 69–77 [DOI] [PubMed] [Google Scholar]
- 21. Estrada-Cuzcano A, Neveling K, Kohl S, et al. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am J Hum Genet. 2012; 90: 102–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pretorius PR, Aldahmesh MA, Alkuraya FS, Sheffield VC, Slusarski DC. Functional analysis of BBS3 A89V that results in non-syndromic retinal degeneration. Hum Mol Genet. 2011; 20: 1625–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pretorius PR, Baye LM, Nishimura DY, et al. Identification and functional analysis of the vision-specific BBS3 (ARL6) long isoform. PLoS Genet. 2010; 6: e1000884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abu-Safieh L, Al-Anazi S, Al-Abi L, et al. In search of triallelism in Bardet-Biedl syndrome. Eur J Hum Genet. 2012; 20: 420–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000; 66: 1975–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stone EM, Cideciyan AV, Aleman TS, et al. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch Ophthalmol. 2011; 129: 81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simons DL, Boyle SL, Hauswirth WW, Wu SM. Gene therapy prevents photoreceptor death and preserves retinal function in a Bardet-Biedl syndrome mouse model. Proc Natl Acad Sci U S A. 2011; 108: 6276–6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zou J, Luo L, Shen Z, et al. Whirlin replacement restores the formation of the USH2 protein complex in whirlin knockout photoreceptors. Invest Ophthalmol Vis Sci. 2011; 52: 2343–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cideciyan AV, Jacobson SG, Beltran WA, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci U S A. 2013; 110: E517–E525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kachi S, Binley K, Yokoi K, et al. Equine infectious anemia viral vector-mediated codelivery of endostatin and angiostatin driven by retinal pigmented epithelium-specific VMD2 promoter inhibits choroidal neovascularization. Hum Gene Ther. 2009; 20: 31–39 [DOI] [PubMed] [Google Scholar]
- 31. Olsson JE, Gordon JW, Pawlyk BS, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992; 9: 815–830 [DOI] [PubMed] [Google Scholar]
- 32. Deretic D, Wang J. Molecular assemblies that control rhodopsin transport to the cilia. Vision Res. 2012; 75: 5–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chamling X, Seo S, Bugge K, et al. Ectopic expression of human BBS4 can rescue Bardet-Biedl syndrome phenotypes in Bbs4 null mice. PLoS One. 2013; 8: e59101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.