

Sporadic adrenomedullary hyperplasia may mimic pheochromocytoma. Laparoscopic adrenalectomy is recommended for the management of this entity.
Keywords: Laparoscopic adrenalectomy, Sporadic adrenomedullary hyperplasia, Pheochromocytoma, Diagnosis
Abstract
Background and Objectives:
Sporadic adrenomedullary hyperplasia (AMH) is characterized by a medical history of hypertension, excessive catecholamine excretion, and histomorphometric evidence of increased adrenomedullary tissue relative to the cortex in the absence of multiple endocrine neoplasia. The aim of this study was to perform a retrospective analysis of patients after laparoscopic adrenalectomy for AMH, an early form of sporadic adrenal medulla–related endocrine hypertension, as well as to update our understanding of the clinical features and management of this clinicomorphologic entity.
Methods:
We performed a retrospective review of the medical records of patients operated on between 2007 and 2011 at Reina Sofia University General Hospital, Murcia, Spain, with a diagnosis of AMH. Patient characteristics, diagnostic studies, surgical procedures, and histologic findings were analyzed.
Results:
Seven hypertensive patients with intermittent adrenergic crises were found to have AMH (3 men and 4 women; mean age, 44 years). Catecholamine levels were increased. Radiologic studies included 1 or more of the following: magnetic resonance imaging, computed tomography, positron emission tomography imaging with fluorodeoxyglucose, dihydroxyphenylalanine–positron emission tomography–computed tomography, Octreoscan (Mallinckrodt Pharmaceuticals, St. Louis, MO, USA) and 123I-metaiodobenzylguanidine scintigraphy. Laparoscopic adrenalectomy was performed in all cases. One patient underwent bilateral adrenalectomy because of persistent symptomatology after unilateral adrenalectomy. Surgery was associated with normalization of catecholamine hypersecretion and complete disappearance of symptoms, as well as the reduction or abstention of antihypertensive therapy.
Conclusions:
Sporadic AMH is a clinicomorphologic entity that may mimic pheochromocytoma clinically. Recent advances in diagnostic and surgical methods have changed the management and outcome of this unusual disease. Laparoscopic adrenalectomy may be recommended as the gold standard in the treatment of this entity. Definitive diagnosis is provided by histologic study.
INTRODUCTION
Sporadic adrenomedullary hyperplasia (AMH) is characterized by a clinical history of paroxysmal hypertension, increased serum and/or urinary catecholamine levels, a lack of association with multiple endocrine neoplasia (MEN) syndromes, and histomorphometric evidence of an increased medullary cell mass. It is an extremely infrequent etiology of pheochromocytoma, but both disorders may cause identical clinical symptoms and biochemical findings. Clinical and morphologic diagnosis of sporadic AMH may be difficult; therefore, treatment must be carefully evaluated.1
We present our experience in 7 patients diagnosed with sporadic AMH during a 4-year period. The aim of this study was to corroborate the clinicomorphologic entity of AMH as an early form of sporadic adrenal medulla–related endocrine hypertension and to update our understanding of the characteristics, diagnosis, treatment, and outcomes of these patients after laparoscopic adrenalectomy.
METHODS
Seven patients operated on between 2007 and 2011 at the surgical department of Reina Sofia University General Hospital, Murcia, Spain, were found to have AMH.
The design of the study was a case series. Clinical outcomes were recorded retrospectively from the database and the patients' records.
Routine preoperative laboratory studies included 24-hour urine and serum testing with catecholamine levels—basal or during the adrenergic crises. Biochemical data show the increased parameters and the highest level of catecholamine obtained (Table 1). Radiologic study included a combination of one or more of the following diagnostic methods: magnetic resonance imaging (MRI), thoracoabdominal computed tomography (CT), positron emission tomography (PET) imaging with fluorodeoxyglucose (FDG), dihydroxyphenylalanine-PET-CT, Octreoscan, and 123I-metaiodobenzylguanidine (MIBG) scintigraphy (Table 2).
Table 1.
Epidemiologic, Medical History, Symptomatology, and Biochemical Data
| Patient No./Age (y)/Sex | Medical History | Symptomatology | Biochemical Data |
|---|---|---|---|
| 1/57/M | DM-II, HT, hyperuricemia, overweight, chronic atrial fibrillation, dyslipidemia | HT, adrenergic crises | uTC, 820 μg/24 h; uE, 370 μg/24 h; uNE, 226 μg/24 h; uMN, 4 450 μg/24 h; uNM, 680 μg/24 h; uVMA, 18 mg/24 h |
| 2/36/F | HT | HT, adrenergic crises | uTC, 560 μg/24 h; uE, 433 μg/24 h |
| 3/21/M | Allergic rhinitis, HT | HT | uTC, 247 μg/24 h; uE, 312 μg/24 h; uDA, 644 μg/24 h |
| 4/49/F | β-Lactam antibiotic and metamizol allergy, seborrheic dermatitis, HT | HT, adrenergic crises | Before first surgery: uTC, 385 μg/24 h; uNE, 372 μg/24 h; uVMA, 21 mg/24 h; uDA, 692 μg/24 h Before second surgery: uTC, 141 μg/24 h; uNE, 165 μg/24 h |
| 5/51/F | Fallopian tubes ligation, urinary incontinence surgery, HT | HT, adrenergic crises | uTC, 926 μg/24 h; uNE, 99 μg/24 h; uMN, 148 μg/24 h |
| 6/42/M | HT, thyroidectomy due to Graves-Basedow disease, groin hernioplasty | HT, adrenergic crises | uTC, 193 μg/24 h; uE, 47 μg/24 h; uNE, 103 μg/24 h |
| 7/53/F | Obesity, OSAS, HT, appendectomy | HT, adrenergic crises | uTC, 1 169 μg/24 h; uNE, 183 μg/24 h; uNM, 1 017 μg/24 h; uVMA, 16 mg/24 h |
DM-II = type 2 diabetes mellitus; F = female; HT = arterial hypertension; M = male; OSAS = obstructive sleep apnea syndrome; uDA = urine dopamine; uE = urine epinephrine; uMN = urine metanephrine; uNE = urine normetanephrine; uNM = urine normetanephrine; uTC = urine total catecholamines; uVMA = vanillylmandelic acid.
The normal values for catecholamines are as follows: urine total catecholamines, 14 to 110 μg/24 h; urine metanephrine, 24 to 96 μg/24 h; urine epinephrine, 0.5 to 20 μg/24 h; urine normetanephrine, 75 to 375 μg/24 h; urine norepinephrine, 15 to 80 μg/24 h; serum epinephrine, 0 to 900 pg/mL; urine dopamine, 65 to 400 μg/24 h; serum norepinephrine, 0 to 600 pg/mL; and urine vanillylmandelic acid, 2 to 7 mg/24 h.
Table 2.
Radiologic Study Methods
| Patient No. | CT | MRI | PET | SPECT | 123I-MIBG Scintigraphy | Octreotide Scan | DOPA-PET-CT |
|---|---|---|---|---|---|---|---|
| 1 | N | N | N | Increased left adrenal gland uptake | Increased left adrenal gland uptake | N | N |
| 2 | N | N | — | — | Increased right adrenal gland uptake | — | — |
| 3 | N | Enlarged left adrenal gland | — | Increased left adrenal gland uptake | Increased left adrenal gland uptake | — | — |
| 4 | N | N | N | — | Increased right adrenal gland uptake | N | — |
| — | — | — | — | Increased left adrenal gland uptake | — | — | |
| 5 | N | N | — | Increased left adrenal gland uptake | Increased left adrenal gland uptake | N | — |
| 6 | N | — | — | Increased left adrenal gland uptake | Increased left adrenal gland uptake | — | — |
| 7 | N | — | — | — | Increased right adrenal gland uptake | — | — |
DOPA = dihydroxyphenylalanine; N = normal; SPECT = single photon emission computed tomography.
After complete studies, the patients were thought, clinically and endocrinologically, to have a pheochromocytoma. A familial history of MEN syndrome was ruled out in all patients.
Adrenalectomy was performed by a transperitoneal laparoscopic approach in all 7 cases. Left adrenalectomy was performed with the patient placed in the right lateral decubitus position by use of 3 ports, whereas right adrenalectomy was performed with the patient in the left lateral decubitus position with 4 ports.
RESULTS
The mean age of the patients was 44 years (range, 21–57 years). There were 3 men, with a mean age of 40 years, and 4 women, with a mean age of 47 years.
Epidemiologic, medical history, symptomatology, and biochemical data of the patients are shown in Table 1. Symptoms were adrenergic recurrent episodes including dizziness, flushing, tremor, diaphoresis, headache, palpitations, sweating, and anxiety in 6 cases, as well as resistant hypertension despite antihypertensive therapy in all cases. All patients had increased catecholamine concentrations in urine or serum (or both). Electrocardiograms and chest radiographs showed no lesion.
Six patients had normal-sized adrenal glands on abdominal CT or MRI, and in 1 patient, the adrenal gland appeared enlarged in size. 123I-MIBG showed abnormal unilateral uptake in all patients (Table 2).
All patients underwent lateral transperitoneal laparoscopic unilateral adrenalectomy. In each case surgery showed a diffusely enlarged adrenal gland without a discrete tumor, and the mean weight was 10.65 g (Figure 1). One patient underwent bilateral adrenalectomy because of persistent adrenal crises 1 year after right adrenalectomy, suggesting that the stimulus to AMH may possibly affect other chromaffin tissues. There was no case of conversion to open surgery. The operative and postoperative course was uneventful in all patients. The mean postoperative stay in the hospital was 3.3 days (range, 3–4 days). There were no deaths. The follow-up period for the patients ranged from 15 months to 5 years.
Figure 1.
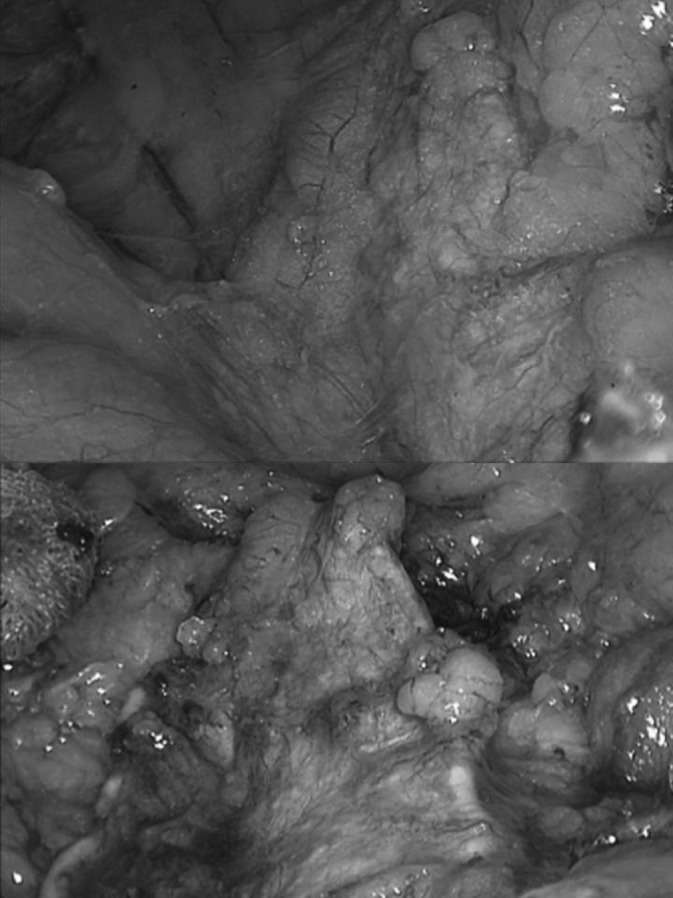
Appearance of the dissection of the left adrenal gland during two different moments of the surgery.
Six patients who underwent unilateral adrenalectomy had improved symptomatology, with partial or complete remission of symptoms and signs and a reduction in antihypertensive therapy. The bilaterally adrenalectomized patient has been free of symptoms for 3 years after the first surgery. Thus it would appear that AMH may occur unilaterally or asynchronously in the two glands. Removal of the hyperplastic adrenal gland resulted in a decrease in the plasma and urinary catecholamine levels and no high uptake on 123I-MIBG scintigraphy.
According to histomorphometry, an increased adrenomedullary cell mass was shown, thus confirming AMH. The removed glands weighed 9.42 g on average (range, 5.6–14 g). Pathologic data of the resected glands are shown in Table 3.
Table 3.
Histopathology Data
| Patient 1 | Patient 2 | Patient 3 | Patient 4 (R) | Patient 4 (L) | Patient 5 | Patient 6 | Patient 7 | |
|---|---|---|---|---|---|---|---|---|
| Pathology findings | DMH | DMH | DMH | NMH | DMH | NMH | DMH | DMH |
| Measurements (cm) | 5 × 2.5 × 1.3 | 5.5 × 3 × 1.2 | 6 × 4 × 3 | 6 × 4 × 1 | 4.5 × 3 × 2 | 6 × 2 × 2 | 4.8 × 2 × 1.5 | 5 × 3.5 × 2 |
| Weight (g) | 6.6 | 9.4 | 9 | 14 | 8.5 | 13.4 | 5.6 | 18.7 |
DMH = diffuse medullary hyperplasia; L = left; NMH = nodular medullary hyperplasia; R = right.
DISCUSSION
Hyperfunction of endocrine glands is generally associated with either hyperplasia or neoplasia of the involved gland. Diseases of the adrenal medulla comprise a small fraction among hypertensive patients (0.1%–0.2%).1 Attempts at proving the existence of AMH as a clinicopathologic entity have appeared since 1933, but perhaps the most convincing example based on clinical and postoperative grounds is that reported by Montalbano et al. in 1962.2,3 This clinicopathologic entity has been widely reported to be associated with familial or type 2 MEN syndrome.2,4 In most cases AMH—usually bilateral—is a component of type 2 MEN together with medullary thyroid cancer and parathyroid hyperplasia.5 Up to now, sporadic AMH has been documented in a relatively small number of cases.1
Whereas pheochromocytomas are defined as adrenal tumors producing catecholamines that typically cause hypertension, AMH is not generally recognized as a clinical entity–causing syndrome that clinically resembles that of adrenomedullary adenoma.1 AMH may represent the morphologic and endocrine counterpart to the adenomatous manifestation.1
The sequence of morphologic events preceding the development of pheochromocytoma is largely unknown.1 Carney et al.2 described the likely sequence of events during AMH. Initially, hypertrophy and hyperplasia of the medullary cells occur, which cause a slight increase in the volume of the medulla without a significant alteration in weight. However, small increases in the volume of the medulla will decrease the corticomedullary ratio. With increasing degrees of hyperplasia, the enlargement of the medulla will become obvious grossly and there will be a measurable increase in the weight of the gland. Eventually, the medulla will become thicker than the cortex. The shape of the gland may be variably altered because, at any stage, zones of increased hyperplastic activity may develop and result in formation of 1 or several nodules; however, in the initial phase, the normal configuration of the gland probably will be maintained.5
It has been proposed that diffuse hyperplasia of the adrenal medulla may be an early manifestation of adrenal pathologic change in this syndrome; this is followed by a stage of “diffuse and multinodular enlargement” (nodular hyperplasia) of the medulla and then by the final adrenal manifestation of the syndrome: pheochromocytoma.5
From clinicomorphologic studies, pheochromocytoma in familial and type 2 MEN–associated adrenomedullary diseases is regarded as the neoplastic end stage of a stepwise development beginning with hypertrophy and hyperplasia of the medullary cells1; however, the frequency and clinical significance of preneoplastic forms of sporadic pheochromocytoma are unknown as yet.1
AMH may produce symptoms similar to those of pheochromocytoma. It is usually bilateral and may be nodular and diffuse,4 as in our series.
Diagnosis is based on clinical and biochemical findings. As many studies suggest, summarizing clinical, laboratory, and imaging results and the indication for operation in suspected AMH requires evidence of the following1:
Hypertension
Increased urinary excretion of epinephrine
Localization of the lesion by 123I-MIBG scintigraphy or, if it is negative, cavovenous blood sampling with selective catheterization of both suprarenal veins
Plasma free metanephrines provide the best test for excluding or confirming pheochromocytomas because they are produced continuously by tumor cells as opposed to episodic secretion of catecholamines.6 Despite the negative 24-hour urine test for catecholamines, a pheochromocytoma may still be present,7 and repeated determinations may be necessary to exclude the diagnosis.8
Having confirmed the diagnosis of pheochromocytoma after biochemical studies, radiologic imaging should be completed.7,8 There are several imaging methods available for tumor localization.8 Morphologic imaging—CT and MRI—in combination with radionuclide imaging procedures—MIBG scintigraphy, PET imaging with FDG, dihydroxyphenylalanine-PET-CT, and Octreoscan—also accurate localize these tumors.7,8
Ultrasonography is able to detect adrenal pheochromocytoma but cannot exclude extra-adrenal or multifocal disease.8 MRI provides excellent anatomic detail, is the morphologic imaging modality of choice in localizing pheochromocytoma and extra-adrenal paraganglioma, and has the advantage of lack of ionizing radiation.8 CT plays a major role in detecting and evaluating adrenal pathology and can detect masses smaller than 0.5 cm in diameter, although it shows reduced specificity.8 CT and MRI should be included in the diagnosis program to detect or exclude major lesions of the adrenal gland, extra-adrenal paraganglia, or other intraperitoneal abnormalities and contribute to planning the surgical technique.1
123I-MIBG scintigraphy is highly specific and seems to be superior to CT scan and ultrasonography in identifying neoplastic and hyperplastic adrenomedullary lesions.1,9 However, approximately 20% of normal adrenals take up some 123I-MIBG.9 Scintigraphy in the form of MIBG scanning is slightly less sensitive but is useful in determining extra-adrenal, multifocal, or recurrent disease.7 For patients with negative 123I-MIBG scintigraphy or patients who may have malignant pheochromocytoma, octreotide scan and FDG-PET are other options for localization.7
Imaging techniques may not confirm the suspicion, however. Therefore it has been proposed by most authors that definitive diagnosis can only be ascertained by quantitative histomorphometric analysis of resected adrenal gland and by the disappearance of the clinical syndrome.1
Because AMH is rare, it is necessary to formulate specific pathologic criteria to ensure its accurate diagnosis when the degree of hyperplasia is mild to moderate.2 Some authors consider the diagnosis of AMH only when there is a significant decrease in the corticomedullary ratio in the head and body of the gland with or without extension of the medulla into the tail.1
In patients with Rearranged during Transfection (RET) mutations, responsible for type 2 MEN syndrome, and with other mutations such as succinate dehydrogenase subunit B, bilateral AMH has been identified. It is hypothesized that AMH is a precursor lesion that can develop into pheochromocytoma.10 Other mutations in genes predisposing to pheochromocytoma are von Hippel–Lindau, neurofibromatosis type 1, and succinate dehydrogenase subunit C or D.11 Furthermore, AMH is often found in the adrenal tissue adjacent to pheochromocytoma.10
Medullary hyperplasia is a proliferation of cells containing normal cellular architecture as opposed to the nests of cytologically atypical polygonal cells that characterize pheochromocytoma.10 On gross examination, as well as radiologic imaging, medulla hyperplasia has no specific nodule, unlike pheochromocytoma, which usually presents as an enlarged adrenal nodule arising from the medulla.10 Morphologically, AMH is characterized by increased medullary mitotic activity and a many-fold increase in medullary volume, as well as total catecholamine content.1 Detection of mitotic activity in the adrenal medulla should arouse suspicion of hyperplasia or neoplasia because mitotic figures are rarely seen in normal medulla.5
To establish the diagnosis of hyperplasia in a medulla that exhibits mitotic activity but is not grossly thickened, it is necessary to demonstrate an increased amount of medullary tissue relative to the cortex. This can be done simply and accurately by planimetric determination of the corticomedullary ratio. The corticomedullary ratio in adult humans has been shown to be about 10:1 and to be slightly greater in women than in men. In the normal adrenal gland, most of the medulla is in the head; there is some in the body and none in the tail.5
The relationship between total adrenal catecholamine content and medullary hyperplasia is unknown, but it is reasonable to expect that the content will be proportional to the medullary mass.5
Rudy et al.2 proposed the following criteria for the diagnosis of AMH:
Clinical history of episodic attacks suggesting pheochromocytoma, generally associated with increased urinary catecholamine levels during attacks.
Diffuse expansion of the adrenal medulla into the tail and the two alae of the gland with or without nodule formation.
A medulla composed of enlarged cells with or without pleomorphism.
An increased medulla-cortex ratio, together with an increased calculated medullary weight.
Diagnosis of AMH in our series was consistent with all criteria; however, increased medullary weight was not calculated. From our point of view, histologic quantitative criteria should not be determinants to establish the diagnosis of AMH because hyperplasia and cellular medullary activity are shown and adrenalectomy results in amelioration of symptoms and signs.
When all studies lead the clinician to suspect a clinical pheochromocytoma, surgical removal of the gland is the optimal treatment choice. Our patients were treated by laparoscopic unilateral adrenalectomy in 6 cases, whereas 1 case required bilateral adrenalectomy. Histologic study showed nodular hyperplasia in 2 cases and diffuse hyperplasia in 5. This minimally invasive procedure is now considered the “gold standard” for the removal of benign functioning and nonfunctioning adrenal tumors.12 The use of a laparoscopic approach for the excision of malignant lesions remains a controversial issue.13
Laparoscopic adrenalectomies have smaller incisions; less intraoperative blood loss; and reduced postoperative wound complications, pain, and infections. Shorter postoperative stays and faster postoperative recovery make this approach a safe and effective alternative to the traditional open adrenalectomy.13 Because of the limitations of minimally invasive surgery, the size of the adrenal tumor could have an influence on the performance of the procedure.12 Transperitoneal laparoscopic adrenalectomy appears to be the procedure of choice for pheochromocytoma and bilateral adrenal tumors, as well as lesions that are larger than 6 cm in diameter.12 A lateral transperitoneal approach offers excellent exposure because of the effects of gravity, and the anatomy is very familiar to abdominal surgeons; it is suitable for removal of tumors up to a maximal diameter of 8 to 15 cm.14 In our series, laparoscopic localization of the adrenal gland seemed to be more difficult than localization in patients presenting with enlarged adrenal glangs or adenoma glands because of the normal size of the adrenal glands.
In the case of surgery for chromaffin tumors, as in our series, transperitoneal access is usually required because of the need for an early closure of the adrenal vein. It allows early occlusion of the vein, avoiding the life-threatening output of catecholamines.12
The rate of conversion to an open approach ranges between 0% and 13%.15 According to many authors, uncontrollable bleeding is the main indication for conversion and the number of conversions may mostly depend on the experience of the surgeon rather than the type of hormonal disorder.12 In our series all cases could be completed by the laparoscopic approach, and no important complications were found during the surgical procedure.
It is recommended that after undergoing successful unilateral adrenalectomy, patients should be followed up because of the metachronous nature of the adrenomedullary lesions.
CONCLUSIONS
Sporadic AMH is an infrequent clinicomorphologic entity. Advances in diagnostic modalities such as 123I-MIBG scintigraphy and surgical techniques have changed the management and outcome of this unusual disease in recent years. The treatment of choice is laparoscopic surgery, which could be offered to such patients after a complete preoperative study, particularly when the clinical spectrum does not improve with medical treatment and there is no other possible cause of the syndrome. Accordingly, a clear set of diagnostic criteria to identify the patients who might benefit from adrenalectomy needs to be established. Definitive diagnosis is provided by histologic study of the resected specimen.
Contributor Information
Miguel Ruiz Marín, General and Digestive Surgery Department, General University “Reina Sofía” Hospital, Murcia, Spain..
Maria Fe Candel Arenas, General and Digestive Surgery Department, General University “Reina Sofía” Hospital, Murcia, Spain.; Surgery, Pediatrics, Gynecology and Obstetrics Department, University of Murcia, Murcia. Spain.
Francisco Miguel González Valverde, General and Digestive Surgery Department, General University “Reina Sofía” Hospital, Murcia, Spain.; Surgery, Pediatrics, Gynecology and Obstetrics Department, University of Murcia, Murcia. Spain.
Emilio Terol Garaulet, General and Digestive Surgery Department, General University “Reina Sofía” Hospital, Murcia, Spain..
María Maestre Maderuelo, General and Digestive Surgery Department, General University “Reina Sofía” Hospital, Murcia, Spain..
Amparo Meoro Avilés, Internal Medicine Department, Endocrinology Section, General University “Reina Sofía” Hospital, Murcia, Spain..
Francisco Pastor Quirante, Pathology Department, General University “Reina Sofía” Hospital, Murcia, Spain.; Pathology Department, University of Murcia, Murcia, Spain.
Antonio Albarracín Marín Blázquez, General and Digestive Surgery Department, General University “Reina Sofía” Hospital, Murcia, Spain.; Surgery, Pediatrics, Gynecology and Obstetrics Department, University of Murcia, Murcia. Spain.
References:
- 1. Dralle H, Schröder S, Gratz KF, Grote R, Padberg B, Hesch RD. Sporadic unilateral adrenomedullary hyperplasia with hypertension cured by adrenalectomy. World J Surg. 1990;14(3):308–315 [DOI] [PubMed] [Google Scholar]
- 2. Kurihara K, Mizuseki K, Kondo T, Ohoka H, Mannami M, Kawai K. Adrenal medullary hyperplasia. Hyperplasia-pheochromocytoma sequence. Acta Pathol Jpn. 1990;40(9):683–686 [DOI] [PubMed] [Google Scholar]
- 3. Rudy FR, Bates RD, Cimorelli AJ, Hill GS, Engelman K. Adrenal medullary hyperplasia: a clinicopathologic study of four cases. Hum Pathol. 1980;11(6):650–657 [DOI] [PubMed] [Google Scholar]
- 4. Carney JA, Sizemore GW, Tyce GM. Bilateral adrenal medullary hyperplasia in multiple endocrine neoplasia, type 2: the precursor of bilateral pheochromocytoma. Mayo Clin Proc. 1975;50(1):3–10 [PubMed] [Google Scholar]
- 5. Montalbano FP, Baronofsky ID, Ball H. Hyperplasia of the adrenal medulla JAMA. 1962;182:264–267 [Google Scholar]
- 6. Lenders JW, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287(11):1427–1434 [DOI] [PubMed] [Google Scholar]
- 7. Ghayee HK, Wyne KL, Yau FS, et al. The many faces of pheochromocytoma. J Endocrinol Invest. 2008;31(5):450–458 [DOI] [PubMed] [Google Scholar]
- 8. Brink I, Hoegerle S, Klisch J, Bley TA. Imaging of pheochromocytoma and paraganglioma. Fam Cancer. 2005;4(1):61–68 [DOI] [PubMed] [Google Scholar]
- 9. Qupty G, Ishay A, Peretz H, et al. Pheochromocytoma due to unilateral adrenal medullary hyperplasia. Clin Endocrinol (Oxf). 1997;47(5):613–617 [DOI] [PubMed] [Google Scholar]
- 10. Grogan RH, Pacak K, Pasche L, Huynht TT, Greco RS. Bilateral adrenal medullary hyperplasia associated with an SDHB mutation. J Clin Oncol 2011;29(8):200–202 [DOI] [PubMed] [Google Scholar]
- 11. Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;9:346(19):1459–1466 [DOI] [PubMed] [Google Scholar]
- 12. Lubikowski J, Kiedrowicz B, Szajko M, et al. Laparoscopic adrenalectomy for functioning and non-functioning adrenal tumours. Endokrynol Pol. 2011;62(6):512–516 [PubMed] [Google Scholar]
- 13. O'Farrell NJ, Collins CG, Stafford AT, Broe PJ. Laparoscopic adrenalectomy: single centre experience. Surgeon. 2011;9(6):300–304 [DOI] [PubMed] [Google Scholar]
- 14. Germain A, Klein M, Brunaud L. Surgical management of adrenal tumors. J Visc Surg. 2011;148(4):250–261 [DOI] [PubMed] [Google Scholar]
- 15. Gumbs AA, Gagner M. Laparoscopic adrenalectomy. Best Pract Res Clin Endocrinol Metab. 2006;20:483–499 [DOI] [PubMed] [Google Scholar]