

Abstract
It should be noted that 3-phosphoinositide-dependent protein kinase-1 (PDK1) is a protein encoded by the PDPK1 gene, which plays a key role in the signaling pathways activated by several growth factors and hormones. PDK1 is a crucial kinase that functions downstream of phosphoinositide 3-kinase activation and activates members of the AGC family of protein kinases, such as protein kinase B (Akt), protein kinase C (PKC), p70 ribosomal protein S6 kinases, and serum glucocorticoid-dependent kinase, by phosphorylating serine/threonine residues in the activation loop. AGC kinases are known to play crucial roles in regulating physiological processes relevant to metabolism, growth, proliferation, and survival. Changes in the expression and activity of PDK1 and several AGC kinases have been linked to human diseases including cancer. Recent data have revealed that the alteration of PDK1 is a critical component of oncogenic phosphoinositide 3-kinase signaling in breast cancer, suggesting that inhibition of PDK1 can inhibit breast cancer progression. Indeed, PDK1 is highly expressed in a majority of human breast cancer cell lines and both PDK1 protein and messenger ribonucleic acid are overexpressed in a majority of human breast cancers. Furthermore, overexpression of PDK1 is sufficient to transform mammary epithelial cells. PDK1 plays an essential role in regulating cell migration, especially in the context of phosphatase and tensin homologue deficiency. More importantly, downregulation of PDK1 levels inhibits migration and experimental metastasis of human breast cancer cells. Thus, targeting PDK1 may be a valuable anticancer strategy that may improve the efficacy of chemotherapeutic strategies in breast cancer patients. In this review, we summarize the evidence that has been reported to support the idea that PDK1 may be a key target in breast cancer management.
Keywords: 3-phosphoinositide-dependent protein kinase-1, phosphoinositide 3-kinase, AGC protein kinases, oncogenic kinase, cell signaling, breast cancer therapy
Introduction
Breast cancer is the second most common cancer worldwide and, although treatments have improved greatly over the last decade, this cancer remains the leading cause of cancer-related death in women.1 Therefore, there is a huge need to identify novel molecular targets in breast cancer. 3-Phosphoinositide-dependent kinase 1 (PDK1), encoded by the gene PDPK1, is a molecular kinase belonging to the phosphoinositide-3-kinase (PI3K) signaling pathway. Since their discovery over 20 years ago, the enzymes PI3Ks have been established as major signaling molecules implicated in a variety of different cellular functions such as glucose metabolism,2–4 cellular proliferation, cellular survival, and angiogenesis.5–8 Their discovery was also paralleled by the observation that genetic lesions to the PI3K pathway are frequently observed in cancer; abnormal PI3K signaling has been estimated to occur in as many as 50% of all human malignancies.9 The PI3K family contains eight mammalian isoforms grouped into three classes. Class 1A consists of a catalytic subunit and a regulatory subunit. The catalytic subunits include p110α, p110β, and p110δ, while the regulatory subunit consists of p85α, p85β, and p55γ. Class 1B consists of only a p110γ catalytic unit and two regulatory units, p87 and p101.8 Class 2 PI3Ks are monomeric proteins that comprise three isoforms, PI3KC2α, PI3KC2β, and PI3KC2γ,9 whilst class 3 comprises only one isoform (hVps34). PI3Ks catalyze the phosphorylation of position 3 of the inositol head group of phosphoinositides. The best characterized PI3K product is the phospholipid phosphatidylinositol-3,4,5-trisphosphate (PtdIns[3,4,5]P3) derived from the phosphorylation of the 3′ position of phosphatidylinositol-4,5-bisphosphate (PtdIns[4,5] P2).8 PtdIns(3,4,5)P3 is synthesized by class 1 PI3K in response to either receptor tyrosine kinases, or G-protein-coupled receptor activation, and it acts as a second messenger by inducing the translocation of proteins to the membrane and subsequent activation.
The discovery of modular domains, such as the pleckstrin homology (PH) domains, able to interact with 3′-phosphorylated lipids marked a significant step forward in understanding the PI3K pathway10–12 The most significant example of PtdIns(3,4,5)P3-dependent protein activation is that of serine/threonine-specific protein kinase B (Akt). Akt binds to PtdIns(3,4,5)P3 via its PH domain, and it is subsequently phosphorylated at its residue threonine (Thr)308 and activated by PDK1, which itself is also associated to the membrane via PH domain-dependent binding to PtdIns(3,4,5)P3. A second Akt residue is phosphorylated by distinct kinases, mainly the mammalian target of rapamycin (mTOR) complex 2.13–14 In its fully active phosphorylation state, Akt phosphorylates several downstream targets, including glucose synthase kinase, transcription factors such as Foxo-1 and cyclic adenosine monophosphate (cAMP) response element-binding (CREB).16 This pathway is negatively regulated by the phosphatase and tensin homologue (PTEN), which directly dephosphorylates PtdIns(3,4,5)P3 at the 3′ position, hence attenuating the PI3K signal.17
Indeed the oncogenicity of abnormal PI3K signaling is emphasized by the observation that deactivating mutations to the gene encoding PTEN are among the most frequently occurring in human malignancy17 Similarly, activating mutations are also frequently seen in the class 1 p110α subunit including ~8% of breast cancers.18–23 Due to the importance of this pathway in cancer cell signaling, it is of little surprise that molecular inhibitors of the PI3K pathway are often investigated for their potential therapeutic benefit in human malignancy. There are currently vast arrays of PI3K-targeted molecular inhibitors in production, some of which are pan inhibitors designed to target all isoforms, and others which are isoform specific. Whilst the former of the two have raised concerns regarding toxicity, there is growing momentum in the research to suggest that targeting specific isoforms may be sufficient to attenuate the PI3K signal in specific cancer settings.23–25 For example, CAL- 101 a molecular inhibitor specific for the class 1 catalytic subunit p110δ, is currently entering Phase III trials for the treatment of chronic lymphocytic leukemia.26–30
While strategies aimed at attenuating the PI3K signal have been mainly focused on inhibiting the PI3K catalytic subunits, there is currently growing evidence suggesting that PDK1 itself may be a viable target. Indeed evidence is beginning to accumulate suggesting that in particular cancer settings, PDK1 is overexpressed and it activates cancer cell growth and survival in a mechanism that is independent of Akt signaling.31,32 This, therefore, detracts from the conventional notion that PDK1 is merely an Akt-activating stepping stone, but rather it highlights the notion that PDK1 may be an important oncogenic regulator and a potential therapeutic target in cancer.33 In this review, we will discuss the role of PI3K/PDK1 signaling in cancer and more specifically assess the role of PDK1 in breast cancer.
PDK1, mechanisms of activation, and physiological functions
PDK1 belongs to the family of AGC kinases (serine and threonine kinases) that show a sequence homology in their catalytic domain to cAMP-dependent protein kinase 1, cyclic guanosine monophosphate-dependent protein kinase, and protein kinase C (PKC).34 The amino-terminal small lobe and the carboxy-terminal large lobe “sandwich” one adenosine triphosphate molecule essential for the subsequent substrate phosphorylation.34 Many AGC kinases possess two phosphorylation sites that regulate their activation: one in the activation loop, which is located within the kinase domain; and another in the hydrophobic motif, which is located in a region adjacent to the catalytic domain.13 Phosphorylation of these sites increases kinase activity and leads to enzymatic full activation. PDK1 was originally discovered in 1997 as the kinase responsible for the phosphorylation of the Akt activation loop, at residue Thr308, which is essential for enzyme activation.13 Furthermore Akt phosphorylation at Thr308 was dependent on PtdIns(3,4,5)P3 concentration in vitro, linking PDK1 to the upstream activation of PI3K. PDK1 kinase is a protein of 556 amino acids that possesses an N-terminal catalytic domain, a C-terminal PH domain, and a nuclear export sequence (Figure 1). The nuclear export sequence is a short sequence of four amino acids that are essential for exporting PDK1 from the cell nucleus to the cytoplasm through the nuclear pore complex using nuclear transport.
Figure 1.
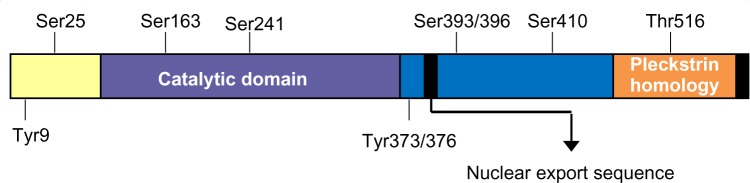
Schematic representation of PDK1 structure.
Abbreviations: PDK1, 3-phosphoinositide-dependent protein kinase-1; Thr, threonine; Tyr, tyrosine; Ser, serine.
Similar to other AGC kinases, PDK1 possesses a phosphorylation site within the activation loop (S241), which is phosphorylated in resting cells and is not affected by growth factor stimulation. The phosphorylation of PDK1 in S241 is catalyzed by an autophosphorylation reaction in trans.35 PDK1 kinase activity is therefore constitutively active, and the regulation of PDK1-activated signaling involves different mechanisms. The first mechanism was discovered investigating the steps involved in Akt T-loop phosphorylation in living cells. PDK1 is localized at the plasma membrane due to the interaction of its PH domain with the phosphoinositides PtdIns(3,4,5)P3, phosphatidylinositol-3,4-bisphosphate [PtdIns(3,4)P2 ], and PtdIns(4,5)P2, with the highest affinity toward the PI3K lipid products.33,36 Although PDK1 membrane localization has been largely investigated and the affinity of the PDK1-PH domain for the PI3K products suggested a potential PI3K-dependent PDK1 membrane translocation, this localization is still controversial. Indeed, the jury is still out on whether PDK1 translocates to the plasma membrane following growth factor stimulation, or it is constitutively localized to the plasma membrane. Nevertheless, it is well established that PDK1 membrane localization is essential for Akt phosphorylation in Thr308 (Figure 2). PDK1 is constitutively associated in a homodimeric complex through PH domain interaction of two PDK1 monomers, and this interaction is important in the regulation of Akt phosphorylation.37
Figure 2.

Activated receptor provides a docking site for PI3K, once bound to the receptor PI3K becomes active and phosphorylates PIP2, forming PIP3. PIP3 acts as a membranous second messenger, providing a docking site for downstream proteins such as Akt and PDK1. Upon binding PIP3, Akt undergoes a conformational change, facilitating PDK1-dependent phosphorylation of Akt at threonine 308. Once phosphorylated, Akt becomes active and dissociates from the membrane and phosphorylates a variety of downstream targets involved in growth and survival pathways, such as mTOR, MDM2 and BAD. PDK1 is also known to phosphorylate a variety of other downstream proteins, which have also been implicated in cancer cell signaling, such as SGK and YAP. There is also evidence to suggest PI3K-independent PDK1-dependent activation of mTOR.
Abbreviations: PDK1, 3-phosphoinositide-dependent protein kinase-1; PI3K, phosphoinositide-3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; SGK, serum glucocorticoid-dependent kinase; PKC, protein kinase C; YAP, Yes-associated protein kinase; Akt, protein kinase B; mTOR, mammalian target of rapamycin; MDM2, mouse double minute 2 homologue; BAD, Bcl-2-associated death promoter; GSK, glycogen synthase inase.
Many other kinases are known to be downstream of PDK1 and are attracting an increasing interest. Among these, serum glucocorticoid-dependent kinase (SGK), p70 ribosomal protein S6 kinases (S6K), p90 ribosomal protein S6 kinase (RSK), and atypical PKC isoforms are known to be direct targets of PDK1, which phosphorylates specific serine/threonine residues of their activation loop (Figure 2).38 The mechanism of activation of these kinases differs from the Akt activation mechanism. PDK1 possesses a hydrophobic pocket, which is termed PDK1 interacting fragment pocket, and is essential for PDK1 interaction with the hydrophobic motif of the targeted protein kinases. Mutations within the PDK1 interacting fragment pocket abolish the binding of their subsequent phosphorylation and activation of PDK1 to PKC, S6K, and SGK1. The physiological role of PDK1 has been investigated in vivo in yeast, Drosophila melanogaster, and mice. These studies have shown that deletion of PDK1 is lethal, indicating that PDK1 is required for normal embryo development. PDK1–/– mice lack branchial arches, and have problems in neural crest specification and forebrain development, as well as several disruptions in the development of a functional circulatory system, which eventually causes death at the E9.5 embryonic stage. In order to study the role of PDK1 in development, hypomorphic mice for PDK1 were generated, in which the neomycin resistance gene is inserted between exons 2 and 3 of the PDK1 gene in order to reduce the expression of PDK1 by 80%–90% in all tissues.
These mice showed a decreased body size of 40%–50% compared to the wild type littermate, but no significant differences in the activation of AKT, S6K, and RSK were induced by insulin. Analysis of organs revealed that the difference in size is due to a decreased cell size rather than a reduction in cell number.
Specific function of PI3K/PDK1 in cancer
The PI3K pathway is one of the most frequently deregulated pathways in human malignancy; indeed, there are a variety of genetic abnormalities observed in this pathway in cancer, including activating and deactivating mutations, copy number changes, and posttranscriptional epigenetic irregularities. Among these, deactivating mutations in the gene encoding the tumor suppressor, PTEN, are among the most common. Evidence for the role of PTEN as a tumor suppressor was first suggested in 1997, where it was seen that PTEN was frequently mutated in patients with the cancer predisposition syndrome, Cowden disease.39,40 Studies in PTEN knockout mice further confirmed PTEN as an important tumor suppressor in a number of tissue types including 38% of endometrial, 14% of prostate, and 20% of central nervous system cancers.41,42 This, together with reports of homozygous deletion of the PTEN allele in cancer and suggestions of epigenetic and micro ribonucleic acid-based mechanism of regulation, have resulted in PTEN being considered one of the most frequently deregulated tumor suppressors in human malignancy.17 Similarly important, PIK3CA – the gene encoding the p110α catalytic subunit – is frequently mutated in human cancer. Indeed, the PI3K sequencing of human tumor tissues samples revealed PIK3CA somatic mutations in different cancer types such as lung (4%), breast (8%), gastric (25%), brain (27%), and colon (32%).19,21 Over 90 different somatic mutations have been identified in PIK3CA,9 with 47% of these found in the helical domain and 33% found in the kinase domain, indicating that these mutations increase p110α lipid kinase activity. Statistical analysis has since revealed three “hot spot” PIK3CA mutations: H1047R, E542K, and E545K. To date, PIK3CA-activating mutations have been observed in a variety of other cancers such as endometrial cancer, head and neck cancer, ovarian cancer, bladder cancer, and skin cancer.18,43 In addition, p110α is not the only isoform known to contain mutations; mutations have been found in all PI3K isoforms, although their prevalence and functional relevance in disease is considered limited. In particular, PIK3CD was seen to be mutated in 6.5% of human neuroblastoma tumor samples compared to 0% in normal tissue, and significant recurrent mutations are also seen in both PIK3CG (9.7%) and PIK3C2B (12.9%) in lung cancer.44,45 There is also evidence to suggest that mutations to the PI3K class 1 regulatory subunit p85α may play a key role in tumorigenicity; mutations have been found in the p85α SH2 domain, which diminish regulatory function of p85α on p110α, thereby increasing p110α activity.
While less frequently mutated, evidence is beginning to accumulate to suggest a role for the other class 1 PI3K isoforms in cancer. In particular, we reported that p110γ is overexpressed in both human pancreatic ductal adenocarcinoma and human hepatocellular carcinoma, where it regulates cell proliferation.25,46 In addition, more recent data indicated that upregulated p110γ conveys the metastatic signal initiated by G-protein coupled receptors in breast cancer cells.47 Expression of physiological levels of p110β, p110δ, and p110γ isoforms in chicken embryo fibroblasts was sufficient to induce oncogenic transformation, whereas the induction of physiological levels of p110α did not induce transformation.48 These data in particular indicate that a variety of class 1 isoforms may be involved in tumorigenesis and tumor maintenance in a cancer-specific setting.
The first major indication that PDK1 itself may be a viable target in cancer came in 2005 when Bayascas et al49 generated transgenic mice that were hypomorphic for PDK1. These mice were crossed with tumorigenic heterozygous PTEN+/− mice, and data showed that the prevalence of tumor development was reduced in mice with deficient PDK1 levels.49 Since then, various groups have investigated the role of PDK1 in a variety of different cancers, where in particular PDK1 is seen to play a key role in the development of breast cancer, as will be discussed in more detail. Increased levels of PDK1 expression have been reported in 45% of patients with acute myeloid leukemia, and PDK1 has also been suggested as a viable target in head and neck cancer, multiple myeloma, pancreatic cancer, and colorectal cancer.50–54 With regards to the latter, Tan et al54 observed that PDK1 induced resistance to rapamycin inhibition via Myc activation; this pathway was seen to be negatively regulated by the B55β-associated protein phosphatase 2A complex. While the mechanism of Myc activation was seen to be PDK1-dependent, it was also suggested to be PI3K/Akt independent, thereby adding to the evidence that in a cancer-specific manner, PDK1 may be a viable target that can act independently of Akt.54 Similarly, Vasudevan et al32 observed that a subset of PIK3CA mutated breast cancer cell lines displayed reduced dependence on Akt for tumorigenicity, but rather relied on PDK1-dependent activation of another AGC kinase and PDK1 target, SGK-3.
While there has been some doubt regarding the effectiveness of PDK1-directed inhibition on in vitro two-dimensional proliferation, both genetic ablation and pharmacological inhibition of PDK1 was sufficient to reduce soft agar colony formation of a panel of cancer cell lines, suggesting a role for PDK1 in anchorage-independent cell growth.55,56 Indeed, perhaps the most convincing evidence to suggest PDK1 as a potential therapeutic target in cancer has come from the investigation of the role of PDK1 in cell migration, invasion, and metastasis. In particular, PDK1 has been shown to play an essential role in the regulation of cellular migration in the context of PTEN deficiency.57 The mechanistic role of PDK1 in the motility of cancer cells was investigated in 2008, when Pinner and Sahai58 reported a PDK1-regulated Rho-associated, coiled-coil containing protein kinase 1 (ROCK1)-dependent contraction of actin–myosin. Interestingly, ROCK1 activation was not dependent on PDK1 kinase activity, but activation involved the direct binding of PDK1 to ROCK1 at the plasma membrane.58 This result was observed both on deformable gels and also in vivo, and further suggests that PDK1 may have important regulatory functions in cancer, which are distinct from Akt activation. This particular observation also suggests that PDK1 may have regulatory functions that are completely distinct from its kinase activity, relying rather on allosteric interactions.58 In this respect, we recently demonstrated that PDK1 plays a role in phospholipase C (PLC)γ1 activation in a mechanism which requires association of the two proteins.59 This novel PDK1-PLCγ1 pathway was shown to regulate the invasion of breast and melanoma cancer cell lines.59
Overview of the evidence suggesting PDK1 as a potential target in breast cancer
As previously mentioned, perhaps the most compelling evidence to suggest PDK1 as a molecular target in human malignancy comes from investigations into the role of PDK1 in breast cancer. The PDPK1 gene maps on chromosome 16 at 16p13.3. The 16p13.3 gain was found to be associated with poor survival of breast cancer patients.60 A similar copy number gain of chromosome 16p13.3 was found in prostate cancer and lung cancer.61 Mapping of the focal 16p13.3 genomic gain has identified PDPK1 as the driver of the gain. Indeed, phosphorylation of PDK1 at serine (SER)241 is frequently elevated in breast cancer, with concomitantly increased phosphorylation of downstream kinases, including Akt mTOR, p70S6K, S6, and signal transducer and activator of transcription (Stat)3.55 Moderate to high levels of PDK1 phosphorylation were found in 86% of high-grade metastasized breast tumors. In addition, PDK1 protein and messenger ribonucleic acid were found to be overexpressed in a majority of human breast cancers, with 21% of tumors having five or more copies of the PDK1 encoding gene PDPK1.55 This copy number variation was seen to correlate with upstream lesions in the PI3K pathway such as PIK3CA mutation, ERBB2 amplification, and PTEN loss, and increased PDK1 expression was suggested to increase AKT activation.55 Several groups have investigated the role of PDK1 in breast cancer in vitro, and both genetic ablation and pharmacological inhibition of PDK1 have proved to be critical for anchorage-independent growth, cellular proliferation, migration, and invasion.56,63,64 In addition to these in vitro findings, the role of PDK1 in vivo has also been explored, in most instances using severe combined immunodeficient mice.65 In these instances, genetic ablation of PDK1 reduced lung colonization of human breast cancer cells and also reduced growth of xenograft tumors. In addition, PDK1 overexpressing mammary epithelial cells readily formed invasive tumors when injected into the inferior mammary fat pad of severe combined immunodeficient mice.55 Notably PDK1 overexpression has been found oncogenic only in the COMMA-1D murine mammary cell model. On the contrary, in breast-derived cell lines, PDK1 overexpression is not tumorigenic, but it is able to potentiate the oncogenic effects of upstream lesions.55
The mechanistic function of PDK1 in breast cancer has also been investigated. PDK1 is integral to the PI3K/AKT pathway, as it phosphorylates the activating segment of AKT, a potent proto-oncogene involved in a variety of cellular functions such as proliferation and survival. With this, the role of PDK1-dependent AKT activation in breast cancer has been explored, where indeed both genetic ablation and pharmacological inhibition of PDK1 in breast cancer cells have been shown to reduce AKT Thr308 phosphorylation.64,65 Similarly, overexpression of PDK1 in the human breast epithelial cell line MCF10A potentiates AKT signaling, indeed suggesting that PDK1 does have important AKT-dependent activity in breast cancer.55 However, as previously mentioned, while PDK1 is integral to the PI3K/AKT pathway, there is a growing indication that in specific cancer settings, PDK1 may have important functions that are AKT independent.63 Results obtained with cancer cell lines and the involvement of PDK1 in resistance mechanisms to several anticancer drugs suggest that PDK1 regulates different oncogenic signaling pathways.71,73 Other suggested downstream targets of PDK1 in breast cancer include SGK-3 (which was required for tumorigenicity in a subset of PIK3CA mutated breast cancer cells), p70 ribosomal kinase (an AGC kinase that can be directly phosphorylated by PDK1), and PLCγ1 (which directly binds to PDK1 and regulates cellular invasion in a PDK1-dependent manner.32,59,64
Recently, it has been shown that PDK1 regulates anchorage-independent growth, resistance to and tumor formation in breast cancer cells not only harboring PIK3CA mutations, but also in the absence of these genetic alterations.55 The effect of PDK1 inhibition using short hairpin ribonucleic acid or chemical inhibitors when apoptosis is induced by absence of anchorage, involves antiapoptotic signaling rather than mitogenic signaling.63 This is in agreement with other studies reporting a specific role for PDK1 in cell migration and invasion, but not in proliferation.57 More recently, the PI3K/PDK1 pathway was also shown to regulate the Hippo pathway in the mammary epithelial cell line MCF10A, and as previously discussed, PDK1 was shown to regulate cancer cell motility by antagonizing inhibition of ROCK1 in highly invasive breast adenocarcinoma cells.58,66 Interestingly, a specific role for PDK1 downstream of mutant KRAS is emerging. It has been found that somatic cell knockdown of both KRAS G12V and oncogenic PIK3CA mutations in human breast epithelial cells results in cooperative activation of the PI3K and mitogen-activated protein (MAP) kinase pathways in vitro and in vivo.67 This oncogene cooperativity seen with concurrent mutations in KRAS and PIK3CA is mediated by Ras/p110α interaction and signaling through PDK1 that directly mediates p90RSK activity. Further evidence that PDK1 is a downstream effector of mutant KRAS has been recently found in models of pancreatic cancer.53 Interestingly, MDA-MB-231 cells, carrying KRAS and p53 mutations, are more sensitive to PDK1 inhibitors than breast cancer cells, such as T47D, harboring PIK3CA mutation.63 Consistently, Akt inhibitors are not able to inhibit anchorage-independent growth of MDA-MB-231 cells, whereas they are highly effective in blocking T47D cell growth.
Interestingly, PDK1 seems to also be involved in the molecular mechanisms by which diurnal and circadian rhythms regulate cell proliferation in human breast cancer xenografts.68 Indeed, tumor growth in nude rats bearing MCF7 breast tumors can be significantly accelerated by exposing the rats to light at night through a PDK1-dependent mechanism.
Molecular targeting of PDK1 in breast cancer
In order to evaluate the clinical potential of PDK1-directed therapy in breast cancer, it is important to determine whether pharmaceutical targeting of PDK1 can be achieved in a safe and specific manner. Indeed with the emerging role of PDK1 in cancer, there has been a plethora of PDK1 targeted molecular inhibitors entering production with varying specificity and structure.69 The first generation of PDK1 inhibitors, reviewed in 2008 by Peifer and Alessi,70 lacked high selectivity for PDK1. In recent years, a few highly selective PDK1 inhibitors have been described.69,70 The most notable include compounds able to bind the PDK1 active site disclosed by GlaxoSmithKline (Benford, UK), Pfizer (Pfizer, Inc, New York, NY, USA), Sunesis (Sunesis Pharmaceuticals, Inc., San Francisco, CA, USA), and Biogen Idec (Biogen Idec Inc., Weston, MA, USA). GlaxoSmithKline and Pfizer compounds bind to the active form of the PDK1 kinase, whereas the Sunesis compound, perhaps the most selective PDK1 kinase inhibitor, binds to the inactive form of PDK1.69 We have developed a selective compound (2-O-Bn-InsP5, a derivative of the natural compound inositol 1,3,4,5,6-pentakisphosphate [InsP5]), which possesses enhanced proapoptotic and antitumor activity compared to the parent molecule, InsP5.71 Kinase profiling assays on almost 60 different kinases revealed a remarkable specificity of this compound for PDK1, inhibiting PDK1-dependent phosphorylation of Akt Thr308 in vitro and in vivo. These results are particularly important considering that 2-O-Bn-InsP5 is among the first compounds to be confirmed as highly specific and also be well tolerated in vivo.71 Similarly, the GlaxoSmithKline compound GSK2334470 has recently been characterized as a highly specific adenosine triphosphate-competitive PDK1 inhibitor.72 Notably, this compound inhibited S6K and SGK more potently than AKT.72 While these compounds do show promise for potential therapeutic activity, there is currently a lack of in vivo characterization, which will be required for progression of these compounds into preclinical testing. The availability of these potent and selective PDK1 inhibitors provides the opportunity to test the pharmacological consequences of PDK1 inhibition. Growing evidence suggests that PDK1 is required for experimental tumor formation in a mechanism that could be PI3K- and Akt-independent. This suggests that the choice of tumor to be treated with a PDK1 inhibitor should not be based solely on the basis of PI3K pathway deregulation. Taking into consideration the data suggesting that PDK1 plays a specific role in cell migration and invasion in breast cancer cells, the best models to test the efficacy of PDK1 inhibitors are certainly the metastatic models.
Combination therapy using PDK1 inhibitors with chemotherapy and other targeted therapies
Acquired drug resistance represents the major problem in the clinical success of targeted therapy. There are indications that PDK1 inhibition represents a valuable tool for tackling drug resistance in breast cancer. We have recently showed that 2-O-Bn-InsP5 increases the effect of tamoxifen and paclitaxel in MCF7 and MDA-MB-468 human breast cancer cell lines, respectively.71 This is consistent with the reported role of PDK1 inhibition in tamoxifen sensitization.73 Many lines of evidence indicate that the components of the PI3K pathway may decisively contribute to the resistant phenotype, and therefore it is a major target for anticancer drug development.74 Crosstalk between mTOR and PI3K/Akt signaling pathways has been recently identified in clinical settings.75 This has prompted the development of a novel targeted strategy aiming at the combination of mTOR and PI3K inhibitors. Interestingly, mTOR inhibitors, such as everolimus and temsirolimus, have recently attracted strong interest due to a reported clinical success combined with endocrine therapies in patients with metastatic breast cancer.76 In addition, several clinical trials are currently evaluating the activity of mTOR, PI3K, Akt, and dual PI3K/mTOR inhibitors in combination with various targeted agents.75,76 Despite some early promising results, toxicity associated with these targeted therapies, especially mTOR inhibitors, represents a major problem for breast cancer patients. Therefore, novel combinations and agents are being actively investigated. Recent data from Najafov et al77 demonstrate that Akt Ser(473) phosphorylation using mTOR inhibitors sensitizes Akt to PDK1 inhibitors.77 Consequently, a combination of PDK1 and mTOR inhibitors reduced Akt activation to below basal levels and markedly inhibited proliferation of the different cell lines being tested, including breast cancer cells. This result marks the utility of combining PDK1 and mTOR inhibitors as a therapeutic strategy for the treatment of cancers that harbor mutations that elevate Akt activity. Interestingly, even though 2-O-Bn-InsP5 is highly selective for PDK1 (inhibitory concentration 50 = 26 nM) it possesses weaker but promising activity toward mTOR (inhibitory concentration 50 = 1.3 micro-M). Therefore, 2-O-Bn-InsP5 may represent the lead compound in developing novel mTOR/PDK1 dual inhibitors. Studies in transgenic mice have also shown that PDK1 and other kinases in the PI3K pathway, such as p110α, Akt, and mTOR, are important for protecting the heart from ischemia reperfusion, and from aortic stenosis-induced cardiac dysfunction.78 This may suggest that cancer therapies targeting this pathway will inevitably have a significant impact on the heart. Nevertheless, it should be noted that these results are confined to mouse models and not to clinical trials. Finally, it is worth stating that the existence of PDK1 pathways that can be PI3K- and Akt-independent is emerging and therefore targeting PDK1 has this unique feature when compared to PI3K/Akt targeting.32,79 This conclusion is further supported by the ability of PDK1 inhibition, but not PI3K and Akt, to sensitize therapeutic response of rapamycin in colorectal cancer cells, as discussed above.54
Conclusion
Emerging evidence indicates that PDK1 plays a critical role in the context of PI3K activation. Indeed, PDK1 has been found to be overexpressed and hyperactivated in several cancers including breast carcinomas. The recently discovered potent and selective, small molecule PDK1 inhibitors provide a unique opportunity to exploit the consequences of PDK1 inhibition in cancer therapy. Key issues that remain to be addressed before proceeding down the long road that leads to clinical testing include the advantage of PDK1 targeting over specific PI3K inhibition, and which particular clinical settings will benefit from specific PDK1 inhibition. Accumulating evidence suggests that PDK1 plays a specific and distinct role from the canonical PI3K/Akt pathway, and that PDK1 may specifically activate signal propagation in tumor progression, as well as in cell migration and invasion. Therefore, breast cancer progression and metastasis represents a major challenge for the future development of PDK1 inhibitors.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Hutchinson L. Breast cancer: Challenges, controversies, breakthroughs. Nature Reviews Clinical Oncology. 2010;7:669–670. doi: 10.1038/nrclinonc.2010.192. [DOI] [PubMed] [Google Scholar]
- 2.Hara K, Yonezawa K, Sakaue H, et al. 1-Phosphatidylinositol 3-kinase activity is required for insulin-stimulated glucose transport but not for RAS activation in CHO cells. Proc Natl Acad Sci U S A. 1994;91(16):7415–7419. doi: 10.1073/pnas.91.16.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wennström S, Hawkins P, Cooke F, et al. Activation of phosphoinositide 3-kinase is required for PDGF-stimulated membrane ruffling. Curr Biol. 1994;4(5):385–393. doi: 10.1016/s0960-9822(00)00087-7. [DOI] [PubMed] [Google Scholar]
- 4.Okada T, Kawano Y, Sakakibara T, Hazeki O, Ui M. Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J Biol Chem. 1994;269(5):3568–3573. [PubMed] [Google Scholar]
- 5.Dudek H, Datta SR, Franke TF, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275(5300):661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 6.Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385(6616):544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 7.Ahmed NN, Grimes HL, Bellacosa A, Chan TO, Tsichlis PN. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci U S A. 1997;94(8):3627–3632. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 9.Fyffe C, Buus R, Falasca M. Genetic and epigenetic regulation of phosphoinositide 3-kinase isoforms. Curr Pharm Des. 2013;19(4):680–686. [PubMed] [Google Scholar]
- 10.Haslam RJ, Koide HB, Hemmings BA. Pleckstrin domain homology. Nature. 1993;363(6427):309–310. doi: 10.1038/363309b0. [DOI] [PubMed] [Google Scholar]
- 11.Harlan JE, Hajduk PJ, Yoon HS, Fesik SW. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature. 1994;371(6493):168–170. doi: 10.1038/371168a0. [DOI] [PubMed] [Google Scholar]
- 12.Lemmon MA, Falasca M, Schlessinger J, Ferguson K. Regulatory recruitment of signalling molecules to the cell membrane by pleckstrinhomology domains. Trends Cell Biol. 1997;7(6):237–242. doi: 10.1016/S0962-8924(97)01065-9. [DOI] [PubMed] [Google Scholar]
- 13.Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 14.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376(6541):599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 15.Stephens L, Anderson K, Stokoe D, et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279(5351):710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- 16.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 18.Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65(23):10669–10673. doi: 10.1158/0008-5472.CAN-05-2620. [DOI] [PubMed] [Google Scholar]
- 19.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 20.Samuels Y, Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol. 2010;347:21–41. doi: 10.1007/82_2010_68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102(3):802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bachman KE, Argani P, Samuels Y, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3(8):772–775. doi: 10.4161/cbt.3.8.994. [DOI] [PubMed] [Google Scholar]
- 23.Falasca M, Maffucci T. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem J. 2012;443(3):587–601. doi: 10.1042/BJ20120008. [DOI] [PubMed] [Google Scholar]
- 24.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 25.Edling CE, Selvaggi F, Buus R, et al. Key role of phosphoinositide 3-kinase class IB in pancreatic cancer. Clin Cancer Res. 2010;16(20):4928–4937. doi: 10.1158/1078-0432.CCR-10-1210. [DOI] [PubMed] [Google Scholar]
- 26.Meadows SA, Vega F, Kashishian A, et al. PI3Kδ inhibitor, GS-1101 (CAL-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of Hodgkin lymphoma. Blood. 2012;119(8):1897–1900. doi: 10.1182/blood-2011-10-386763. [DOI] [PubMed] [Google Scholar]
- 27.Castillo JJ, Furman M, Winer ES. CAL-101: a phosphatidylinositol-3-kinase p110-delta inhibitor for the treatment of lymphoid malignancies. Expert Opin Investig Drugs. 2012;21(1):15–22. doi: 10.1517/13543784.2012.640318. [DOI] [PubMed] [Google Scholar]
- 28.Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603–3612. doi: 10.1182/blood-2011-05-352492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herman SE, Gordon AL, Wagner AJ, et al. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116(12):2078–2088. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin HJ, Hsieh FC, Song H, Lin J. Elevated phosphorylation and activation of PDK-1/AKT pathway in human breast cancer. Br J Cancer. 2005;93(12):1372–1381. doi: 10.1038/sj.bjc.6602862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vasudevan KM, Barbie DA, Davies MA, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16(1):21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raimondi C, Falasca M. Targeting PDK1 in cancer. Curr Med Chem. 2011;18(18):2763–2769. doi: 10.2174/092986711796011238. [DOI] [PubMed] [Google Scholar]
- 34.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11(1):9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 35.Wick MJ, Ramos FJ, Chen H, Quon MJ, Dong LQ, Liu F. Mouse 3-phosphoinositide-dependent protein kinase-1 undergoes dimerization and trans-phosphorylation in the activation loop. J Biol Chem. 2003;278(44):42913–42919. doi: 10.1074/jbc.M304172200. [DOI] [PubMed] [Google Scholar]
- 36.Currie RA, Walker KS, Gray A, et al. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J. 1999;337(Pt 3):575–583. [PMC free article] [PubMed] [Google Scholar]
- 37.Masters TA, Calleja V, Armoogum DA, et al. Regulation of 3-phosphoinositide-dependent protein kinase 1 activity by homodimerization in live cells. Sci Signal. 2010;3(145):ra78. doi: 10.1126/scisignal.2000738. [DOI] [PubMed] [Google Scholar]
- 38.Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15(2):161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 39.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15(4):356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Ye n C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 41.Podsypanina K, Ellenson LH, Nemes A, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96(4):1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19(4):348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 43.Qiu W, Schönleben F, Li X, et al. PIK3CA mutations in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12(5):1441–1446. doi: 10.1158/1078-0432.CCR-05-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carén H, Fransson S, Ejeskär K, Kogner P, Martinsson T. Genetic and epigenetic changes in the common 1p36 deletion in neuroblastoma tumours. Br J Cancer. 2007;97(10):1416–1424. doi: 10.1038/sj.bjc.6604032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu P, Morrison C, Wang L, et al. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis. 2012;33(7):1270–1276. doi: 10.1093/carcin/bgs148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dituri F, Mazzocca A, Lupo L, et al. PI3K class IB controls the cell cycle checkpoint promoting cell proliferation in hepatocellular carcinoma. Int J Cancer. 2012;130(11):2505–2513. doi: 10.1002/ijc.26319. [DOI] [PubMed] [Google Scholar]
- 47.Xie Y, Abel PW, Kirui JK, et al. Identification of upregulated phosphoinositide 3-kinase γ as a target to suppress breast cancer cell migration and invasion. Biochem Pharmacol. 2013;85(10):1454–1462. doi: 10.1016/j.bcp.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang S, Denley A, Vanhaesebroeck B, Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2006;103(5):1289–1294. doi: 10.1073/pnas.0510772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bayascas JR, Leslie NR, Parsons R, Fleming S, Alessi DR. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN(+/−) mice. Curr Biol. 2005;15(20):1839–1846. doi: 10.1016/j.cub.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 50.Pearn L, Fisher J, Burnett AK, Darley RL. The role of PKC and PDK1 in monocyte lineage specification by Ras. Blood. 2007;109(10):4461–4469. doi: 10.1182/blood-2006-09-047217. [DOI] [PubMed] [Google Scholar]
- 51.Bhola NE, Freilino ML, Joyce SC, et al. Antitumor mechanisms of targeting the PDK1 pathway in head and neck cancer. Mol Cancer Ther. 2012;11(6):1236–1246. doi: 10.1158/1535-7163.MCT-11-0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujiwara S, Kawano Y, Yuki H, et al. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br J Cancer. 2013;108(1):170–178. doi: 10.1038/bjc.2012.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eser S, Reiff N, Messer M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23(3):406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 54.Tan J, Lee PL, Li Z, et al. B55β-associated PP2A complex controls PDK1-directed myc signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer Cell. 2010;18(5):459–471. doi: 10.1016/j.ccr.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 55.Maurer M, Su T, Saal LH, et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009;69(15):6299–6306. doi: 10.1158/0008-5472.CAN-09-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagashima K, Shumway SD, Sathyanarayanan S, et al. Genetic and pharmacological inhibition of PDK1 in cancer cells: characterization of a selective allosteric kinase inhibitor. J Biol Chem. 2011;286(8):6433–6448. doi: 10.1074/jbc.M110.156463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finlay DK, Sinclair LV, Feijoo C, et al. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. J Exp Med. 2009;206(11):2441–2454. doi: 10.1084/jem.20090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pinner S, Sahai E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol. 2008;10(2):127–137. doi: 10.1038/ncb1675. [DOI] [PubMed] [Google Scholar]
- 59.Raimondi C, Chikh A, Wheeler AP, Maffucci T, Falasca M. A novel regulatory mechanism links PLCγ1 to PDK1. J Cell Sci. 2012;125(Pt 13):3153–3163. doi: 10.1242/jcs.100511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muranen TA, Greco D, Fagerholm R, Kilpivaara O, Kämpjärvi K, Aittomäki K, Blomqvist C, Heikkilä P, Borg A, Nevanlinna H. Breast tumors from CHEK2 1100delC-mutation carriers: genomic landscape and clinical implications. Breast Cancer Res. 2011;13(5):R90. doi: 10.1186/bcr3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choucair KA, Guérard KP, Ejdelman J, et al. The 16p13.3 (PDPK1) Genomic gain in prostate cancer: a potential role in disease progression. Transl Oncol. 2012;5(6):453–460. doi: 10.1593/tlo.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie Z, Yuan H, Yin Y, Zeng X, Bai R, Glazer RI. 3-phosphoinositide-dependent protein kinase-1 (PDK1) promotes invasion and activation of matrix metalloproteinases. BMC Cancer. 2006;6:77. doi: 10.1186/1471-2407-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gagliardi PA, di Blasio L, Orso F, et al. 3-phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia. 2012;14(8):719–731. doi: 10.1593/neo.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baxi SM, Tan W, Murphy ST, Smeal T, Yin MJ. Targeting 3-phosphoinoside-dependent kinase-1 to inhibit insulin-like growth factor-I induced AKT and p70 S6 kinase activation in breast cancer cells. PLoS One. 2012;7(10):e48402. doi: 10.1371/journal.pone.0048402. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Liu Y, Wang J, Wu M, et al. Down-regulation of 3-phosphoinositide-dependent protein kinase-1 levels inhibits migration and experimental metastasis of human breast cancer cells. Mol Cancer Res. 2009;7(6):944–954. doi: 10.1158/1541-7786.MCR-08-0368. [DOI] [PubMed] [Google Scholar]
- 66.Fan R, Kim NG, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A. 2013;110(7):2569–2574. doi: 10.1073/pnas.1216462110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang GM, Wong HY, Konishi H, et al. Single copies of mutant KRAS and mutant PIK3CA cooperate in immortalized human epithelial cells to induce tumor formation. Cancer Res. 2013;73(11):3248–3261. doi: 10.1158/0008-5472.CAN-12-1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu J, Dauchy RT, Tirrell PC, et al. Light at night activates IGF-1R/PDK1 signaling and accelerates tumor growth in human breast cancer xenografts. Cancer Res. 2011;71(7):2622–2631. doi: 10.1158/0008-5472.CAN-10-3837. [DOI] [PubMed] [Google Scholar]
- 69.Medina JR. Selective 3-phosphoinositide-dependent kinase 1 (PDK1) inhibitors: dissecting the function and pharmacology of PDK1. J Med Chem. 2013;56(7):2726–2737. doi: 10.1021/jm4000227. [DOI] [PubMed] [Google Scholar]
- 70.Peifer C, Alessi DR. Small-molecule inhibitors of PDK1. Chem Med Chem. 2008;3(12):1810–1838. doi: 10.1002/cmdc.200800195. [DOI] [PubMed] [Google Scholar]
- 71.Falasca M, Chiozzotto D, Godage HY, et al. A novel inhibitor of the PI3K/Akt pathway based on the structure of inositol 1,3,4,5,6-pentakisphosphate. Br J Cancer. 2010;102(1):104–114. doi: 10.1038/sj.bjc.6605408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Najafov A, Sommer EM, Axten JM, Deyoung MP, Alessi DR. Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem J. 2011;433(2):357–369. doi: 10.1042/BJ20101732. [DOI] [PubMed] [Google Scholar]
- 73.Iorns E, Lord CJ, Ashworth A. Parallel RNAi and compound screens identify the PDK1 pathway as a target for tamoxifen sensitization. Biochem J. 2009;417(1):361–370. doi: 10.1042/BJ20081682. [DOI] [PubMed] [Google Scholar]
- 74.Falasca M. PI3K/Akt signalling pathway specific inhibitors: a novel strategy to sensitize cancer cells to anti-cancer drugs. Curr Pharm Des. 2010;16(12):1410–1416. doi: 10.2174/138161210791033950. [DOI] [PubMed] [Google Scholar]
- 75.Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw. 2013;11(6):670–678. doi: 10.6004/jnccn.2013.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yardley DA. Combining mTOR inhibitors with chemotherapy and other targeted therapies in advanced breast cancer: rationale, clinical experience, and future directions. Breast Cancer (Auckl) 2013;7:7–22. doi: 10.4137/BCBCR.S10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Najafov A, Shpiro N, Alessi DR. Akt is efficiently activated by PIF-pocket-and PtdIns(3,4,5)P3-dependent mechanisms leading to resistance to PDK1 inhibitors. Biochem J. 2012;448(2):285–295. doi: 10.1042/BJ20121287. [DOI] [PubMed] [Google Scholar]
- 78.Klement GL, Goukassian D, Hlatky L, Carrozza J, Morgan JP, Yan X. Cancer therapy targeting the HER2-PI3K pathway: potential impact on the heart. Front Pharmacol. 2012;3:113. doi: 10.3389/fphar.2012.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Finlay DK, Rosenzweig E, Sinclair LV, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209(13):2441–2453. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]