

Abstract
CNS glia and neurons express connexins, the proteins that form gap junctions in vertebrates. We review the connexins expressed by oligodendrocytes and astrocytes, and discuss their proposed physiologic roles. Of the 21 members of the human connexin family, mutations in three are associated with significant central nervous system manifestations. For each, we review the phenotype and discuss possible mechanisms of disease. Mutations in GJB1, the gene for connexin 32 (Cx32) cause the second most common form of Charcot-Marie-Tooth disease (CMT1X). Though the only consistent phenotype in CMT1X patients is a peripheral demyelinating neuropathy, CNS signs and symptoms have been found in some patients with CMT1X. Recessive mutations in GJC2, the gene for Cx47, are one cause of Pelizaeus-Merzbacher-like disease (PMLD), which is characterized by nystagmus within the first 6 months of life, cerebellar ataxia by 4 years, and spasticity by 6 years of age. MRI imaging shows abnormal myelination. A different recessive GJC2 mutation causes a form of hereditary spastic paraparesis, which is a milder phenotype than PMLD. Dominant mutations in GJA1, the gene for Cx43, cause oculodentodigital dysplasia (ODDD), a pleitropic disorder characterized by oculo-facial abnormalities including micropthalmia, microcornia and hypoplastic nares, syndactyly of the fourth to fifth fingers and dental abnormalities. Neurologic manifestations, including spasticity and gait difficulties, are often but not universally seen. Recessive GJA1 mutations cause Hallermann-Streiff syndrome, a disorder showing substantial overlap with ODDD.
Keywords: connexin, gap junction, CMT1X, PMLD1, SPG44, ODDD
1. Introduction
CNS glia and neurons express connexins, the proteins that form gap junctions (GJs) in vertebrates. (See Fig. 1 for nomenclature of gap junction channels and hemichannels.) To date 20 members of the connexin family have been identified in mice and 21 have been demonstrated in humans. (Reviewed by Rackauskas et al. [1].) Mutations in at least 10 of these connexin genes cause human disease, [2] including three connexins with significant CNS manifestations; these are the focus of this review. Here we emphasize the clinical phenotypes and current understanding of the pathogenesis of these genetic diseases, which will require a brief overview of connexin expression within CNS glial cells, reviewed in more detail elsewhere. [3, 4]
Figure 1.
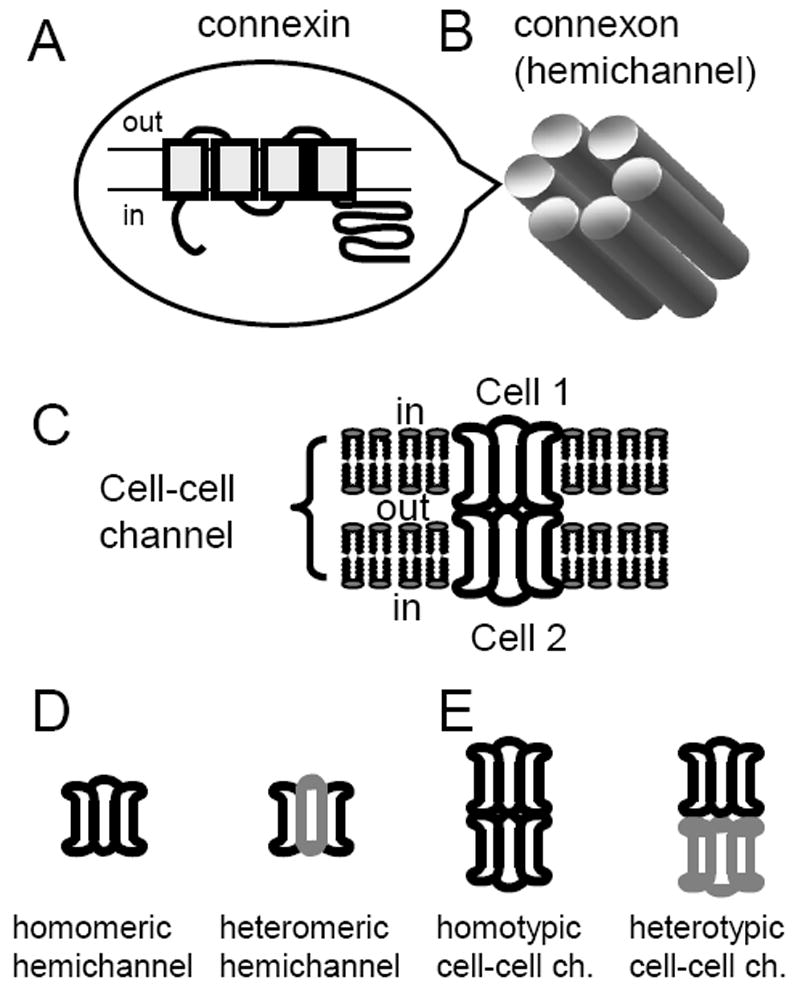
Connexin nomenclature A. Connexins are integral membrane proteins with four transmembrane domains and intracellular N and C termini. B. Each connexon or hemichannel is composed of six subunits called connexins. C. Cell-cell channels are formed by docking of two apposed connexons from apposed cells. D. If all six subunits are identical the connexon is termed homomeric. In cells expressing more than one type of connexin, different connexins may aggregate to form heteromeric connexons. E. Many members of the connexin family can form functional GJs by associating with a like connexon (a homotypic junction) or a different one (a heterotypic junction).
2. Connexins expressed by astrocytes and oligodendrocytes
2.1 Distribution and coupling
Astrocytes (As)1 express Cx30 and Cx43, which can form homotypic (Cx43/Cx43 and Cx30/Cx30) but not heterotypic (Cx43/Cx30) GJs in a model system of transfected cells. [5] Anatomical approaches also provide evidence that A/A GJs are comprised of Cx43/Cx43 and Cx30/Cx30 homotypic channels. [6-9] Cx43-positive GJ plaques are found in both gray and white matter, whereas Cx30-positive GJ plaques are more prominent in gray matter. Although there are contradictory data, [10] some astrocytes may also express Cx26, [7, 9, 11, 12] which can also form homotypic as well as Cx26/Cx30 heterotypic channels. In mice, the deletion of the genes encoding Cx43 (Gja1) and Cx30 (Gjb6) results in the complete loss of A/A coupling. [13] Mutations in GJB2 (encoding Cx26) and GJB6 (encoding Cx30) are not associated with CNS abnormalities in humans or mice.
Oligodendrocytes (Os) express Cx29, Cx32, and Cx47. [9, 14-17] Cx29 is localized to the adaxonal membrane (apposing the axonal membrane) of CNS myelin sheaths, but does not form GJs. [11, 17-19] In transfected cells, Cx29 does not form functional channels, [20] but its human orthologue, Cx31.3, may form hemi-channels. [21] In rodents, Cx47 and Cx32 are found in GJ plaques that are localized on the cell bodies of oligodendrocytes, [17] and Cx32 is also localized within the myelin sheaths themselves, [15] although not as distinctly localized as in Schwann cell myelin sheaths [22]. Though O/O coupling was not seen in a number of brain regions by conventional electron microscopy (EM) or freeze replica immune labeling (FRIL), [8, 23] two groups have recently reported the functional O/O coupling in the mouse corpus callosum. [24, 25] The O/O coupling is mediated by Cx32/Cx32 and Cx47/Cx47 homotypic channels, as it is present in mice lacking Cx32 or Cx47, but is lost in mice that lack both Cx32 and CxCx47. Furthermore, EM demonstrated that oligodendrocytes are directly joined by GJs. [24].
Electron microscopic studies in the 1980s provided anatomical evidence that oligodendrocytes were GJ coupled to astrocytes, but not to themselves [26, 27]. Our work in a model system of transfected cells shows that Cx43/Cx47 and Cx30/Cx32 (but not Cx43/Cx32 or Cx30/Cx47) form morphologic and functional GJs that have distinct electrophysiological properties, [5] but using a different approach, another group [28] showed that Cx30 and Cx47 can form functional channels. Anatomical studies in the CNS [7, 9, 13, 15] make a strong case that A/O GJs consist predominantly of Cx43/Cx47 and Cx30/Cx32 heterotypic channels (a role for Cx26/Cx32 channels remains to be excluded or proved). In the cerebral cortex, A/O coupling is lost in Cx47- (Gjc2), but not in Cx32- (Gjb1) null mice, indicating that Cx43/Cx47 channels (and not Cx30/Cx32 channels) are required for A/O coupling, at least in this region. [24]
2.2 Roles of glial connexins in CNS glia
2.2.1 Lessons from mice with targeted ablation of one or more connexins
The roles of connexins in CNS glia are now beginning to be explicated. Mice with targeted deletion of oligodendrocyte connexins Cx29, Cx32, or Cx47 and mice with targeted deletion of astrocyte connexins Cx30 and Cx43 have been examined. A thorough understanding of the phenotypes of these mice is a critical to understanding of the pathogenesis of disorders associated with mutations in these connexins. Cx32- (Gjb1-) null mice develop a demyelinating peripheral neuropathy after 3 months of age, [29, 30] but only subtle changes in CNS myelin thickness [31, 32] are seen. To date, no behavioral abnormalities have been described in this mouse. Similarly, Cx47- (Gjc2-) null mice show no behavioral abnormalities and only minimal and regionally limited abnormalities in some myelin sheaths. [14, 16] In contrast, mice lacking both Cx32 and Cx47 present with abnormal movements and seizures as well as striking CNS pathology including demyelinated and remyelinated axons, and enlarged extracellular space that separates the axon from its myelin sheath (See Fig. 2); these findings lead to the speculation that this vacuolization might be due to failure of spatial buffering of potassium with increased periaxonal accumulation in the double KO mouse. [16] This idea is supported by data showing that mice with targeted deletion of Kir4.1 [33], an inwardly rectifying K+ channel [34, 35], also develop vacuoles, [33] and that the null allele of Kir4.1 genetically interacts with the null alleles of Cx32 and Cx47. [36]
Figure 2.
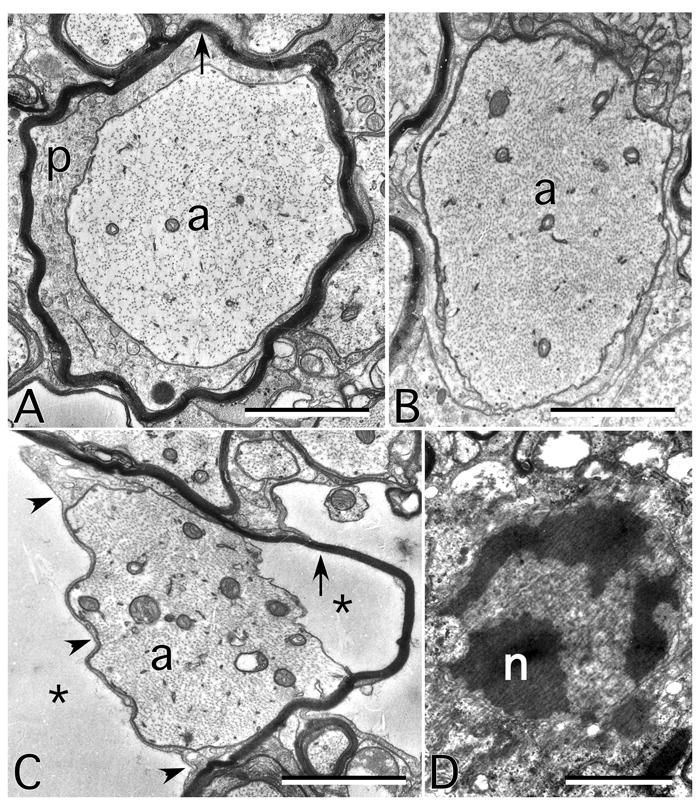
Ultrastructural findings in the CNS myelin of mice lacking expression of Cx32 and Cx47. All are electron micrographs of the ventral funiculus from P31 mice. A. An axon (a) is surrounded by a periaxonal cytoplasmic collar thin myelin sheath (arrow). B. A demyelinated axon. Note that neurofilament packing is tighter than in A. C. An axon still partially surrounded by the adaxonal membrane (arrowheads), but separated from its myelin sheath (arrow) by- extracellular space (asterisks). D. An apoptotic oligodendrocyte nucleus (n). Scale bar, 1μm. From Menichella et al. 2003, [16] used with permission of Society for Neuroscience.
Though the original theory of spatial buffering of potassium referred only to the role of astrocytes (and presumably A/A coupling) in regulating the levels of extracellular potassium [37], work by Menichella et al. [36] supports the hypothesis that oligodendrocyte Cx32 and Cx47 are involved in spatial buffering of K+ during neuronal activity. Optic nerve vacuoles are reduced by suppressing activity of retinal ganglion cell (intraocular injection of tetrodotoxin); conversely, enhanced activity (intraocular injection of cholera toxin) causes increased vacuolization.
Although Cx29 (the mouse ortholog of human Cx31.3) is widely expressed in the CNS during development [38] and in the small myelinated fibers of mature brain and spinal cord [11, 17-19], no CNS physiological or pathological phenotype is seen in mice with targeted ablation of Cx29 [39] though peripheral auditory pathways are affected. [9, 40] Whether mutations in GJE2, [41] the gene encoding Cx31.3, cause hearing loss in humans needs to be confirmed.
Studies of mice with targeted deletion of Cx30 and Cx43, the two astrocyte connexins have also been revealing. Both Wallraff et al. [13] and Theis et al. [42] studied mice with astrocyte specific deletion of Cx43 alone and found approximately a 50% reduction in A/A coupling. Mice expressing no astrocyte Cx43 and no Cx30 in any cell (Cx43Astro-Cx30 dKO) showed no A/A coupling and reduced hippocampal K+ buffering [13] as well as loss of metabolic coupling through astroglial networks. [43] Unlike the Cx32-Cx47 dKO mice, [16] these mice had no gross behavioral abnormalities (but see below in this section) or reduced life expectancy. [13, 44] These findings indicate that loss of O/O coupling has greater impact on CNS function than does loss of A/A coupling, since in both cases it is likely that all A/O coupling is eliminated. Theis et al. [42] found increased locomotor activity and an increase in the velocity of spreading depression in the astrocyte specific Cx43KO mouse. The authors suggest that astrocyte GJ communication may attenuate spreading depression by reducing extracellular K+ or glutamate. This hypothesis is supported by the recent work of Pannasch and collegues [45] showing that astroglial gap junction coupling is required for clearance of extracellular glutamate generated by synaptic activity. Lutz et al. [44] found prominent white matter abnormalities in Cx43Astro-Cx30 dKO mice that are similar to those noted in Cx32-Cx47 dKO and Kir4.1 KO mice (See Fig. 3). Grey matter involvement was restricted to astrocyte swelling in the CA1 region of the hippocampus. Behavioral testing showed decreased performance on testing of hippocampal spatial learning. Detailed behavioral analyses of this mouse as well as the Cx43Astro KO and Cx30 KO mice have been performed and are summarized in Table 1.
Figure 3.
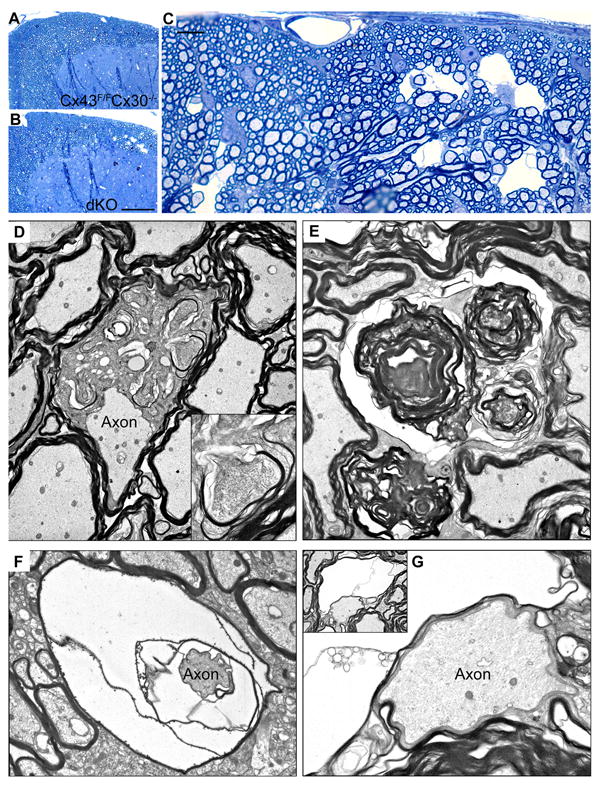
Pathologic findings in the CNS myelin of mice lacking expression of Cx30 globally and Cx43 in astrocytes (Cx30 Cx43astro dKO). A–C. Toluidine blue stained 1μm epoxy sections of the dorsal cuneate of the lumbar spinal cord of non-Cre (Cx43fl/fl Cx30-/- (A) and Cx30 Cx43astro dKO (B, C) mice. D, Electron micrograph showing enlarged oligodendrocyte cytoplasm interpolating between an apparently normal axon and its myelin sheath. Inset depicts ectopic expression of myelin in one of several locations within the oligodendrocyte cytoplasm. E. Myelin whorls, likely representing Wallerian axonal degeneration. F and G. Edema and splitting of myelin sheaths. Scale bar: (A, B) 100μm; (C) 10μm. Magnification: (D) 4000X, inset: 15,000X; (E) 9000X; (F) 8000X; (G) 13,000X, inset: 4000X. From Lutz et al. 2009 [44] used with permission of Society for Neuroscience.
Table 1.
Behavioral phenotypes of astrocyte connexin KO mice. NR not reported a-not analyzed day by day
| phenotype
|
Cx43Astro KO vs. WT [199] | Cx30 KO vs. WT [200] | Cx43Astro-Cx30 vs. WT [44] |
|---|---|---|---|
| Test | |||
|
| |||
| Rotarod | Reduced time on rod day 1. | Trend toward reduced time on rod. | Reduced time on rod.a |
|
| |||
| Balance beam | NR | NR | More falls, longer crossing time. |
|
| |||
| Spatial memory | NR | Reduced contact with displaced object. | Reduced contact with displaced object. |
|
| |||
| Visual memory | NR | NR | No difference. |
|
| |||
| Open field | Increased horizontal activity. | Reduced time in center and increased time in corners of field. | NR |
| Reduced rearings. | |||
|
| |||
| Graded anxiety | No difference. | NR | |
|
| |||
| Water maze | Steeper learning curve. | NR | NR |
|
| |||
| Plus maze | Slight increased entry into closed arms. | NR | NR |
2.2.2 Physiologic roles of glial connexins
The best recognized role of connexins is to form GJs, which provide a pathway for electrical and/or metabolic coupling between cells. [46] In glial cells, GJ coupling is likely to underlie the formation of a glial syncitium. [47] Because glial gap junction coupling is a dynamic process regulated by intra- or extracellular signals (reviewed in Giaume et al. [48]) it has been suggested that coupled glia should be referred to as a network rather than a syncitium. One function of such a syncytium or network is propagation of Ca2+ waves (see [49] for more detail). As noted above (Section 2.1) Cx32 may also provide reflexive coupling between adjacent loops of non-compact myelin. [50] In the last decade, however, other roles are being described for connexins. GJs may be part of a larger complex that contains other junctional elements. Several connexins, including Cx43 and Cx47, have a PDZ-binding domain on their C-terminus, and bind to ZO-1, one of the defining members of PDZ family [19, 51-56] In Gjc2-null mice the association between Cx47 and ZO-1, ZO-1–associated nucleic acid binding proteins (ZONAB), and multi-PDZ domain protein 1 (MUPP1) is lost. [57] Instead of being co-localized with Cx47, ZONAB, a Y-box transcription factor, has increased cytoplasmic and nuclear localization. This redistribution could affect oligodendrocytes, as ZONAB can repress ErbB2, [58] a tyrosine kinase receptor implicated in oligodendrocyte development, differentiation and survival. [59-61] ZONAB also regulates a number of cell cycle regulatory genes. [62, 63] These findings suggest that the appropriate localization of Cx47 in the oligodendrocyte may be important for stabilization/organization of the oligodendrocyte membrane junctional complex, as well as for appropriate regulation of gene expression and control of the cell cycle. Other data suggest that connexins may have roles in regulation of cell growth [64-68] and resistance to both apoptotic and necrotic cell death [69] that are independent of formation of functional GJ channels. Recent work from one of our laboratories has shown that Schwann cell proliferation is regulated by Cx32 in a GJ-independent manner. [68]
Connexin hexamers can form functional membrane hemichannels that may mediate a variety of cellular processes, including ATP release and signaling [70-72], glutamate release [72, 73], glutathione release, [74] and responses to spinal cord injury [75]. Astrocyte Cx43 hemichannels are reportedly modulated in a number of pathogenic states including metabolic inhibition [76] and brain abscesses, [77] and may be upregulated by proinflammatory cytokines secreted from microglia. [78] Cannabinoids may antagonize the effect of proinflammatory cytokines; [79] this may be neuroprotective. [80] However, recent work suggests that at least in cultured astrocytes, pannexin 1 and not Cx43 is responsible for the membrane conductances and permeabilities ascribed to astrocytic hemichannels. [81] Hemichannel activities ascribed to Cx30, [82, 83] Cx47, [84] Cx32, [85-87] and Cx31.3 [21] have all been identified in cultured cells.
3. Disease manifestations of GJB1 mutations
3.1 CMT1X clinical phenotypes MIM ID #302800
Charcot-Marie-Tooth disease (CMT) is the eponym for large number of inherited disorders affecting predominantly or exclusively the peripheral nerves. Symptoms are those of peripheral neuropathy and include weakness and sensory loss of the distal more than proximal extremities. The most common X-linked form of CMT, termed CMT1X, arises in patients with mutations in GJB1, the gene that encodes Cx32, which is expressed in both Schwann cells and oligodendrocytes and a number of other cell types. [88] In spite of its widespread expression, a demyelinating peripheral neuropathy is the only consistent phenotype of the more than 400 mutations that have been reported to date. The CNS signs and symptoms that have been found in some patients with CMT1X are described below, along with their potential mechanisms.
CNS manifestations of CMT1X can be divided into three groups: 1) subclinical abnormalities of visual evoked responses (VERs) and auditory evoked responses (BAERs); 2) overt, generally mild fixed abnormalities on neurological examination and/or CNS imaging and which may or may not be accompanied by clinical manifestations; 3) severe transient CNS dysfunction accompanied by white matter abnormalities on MRI. See Fig. 4 for a schematic of mutations associated with these abnormalities. Although hearing loss has been associated with several mutations including R142Q [89], E186K [90] T191FS, T55R [91] and V38A [92] it remains to be shown that this is truly a phenotypic manifestation of these mutations.
Figure 4.
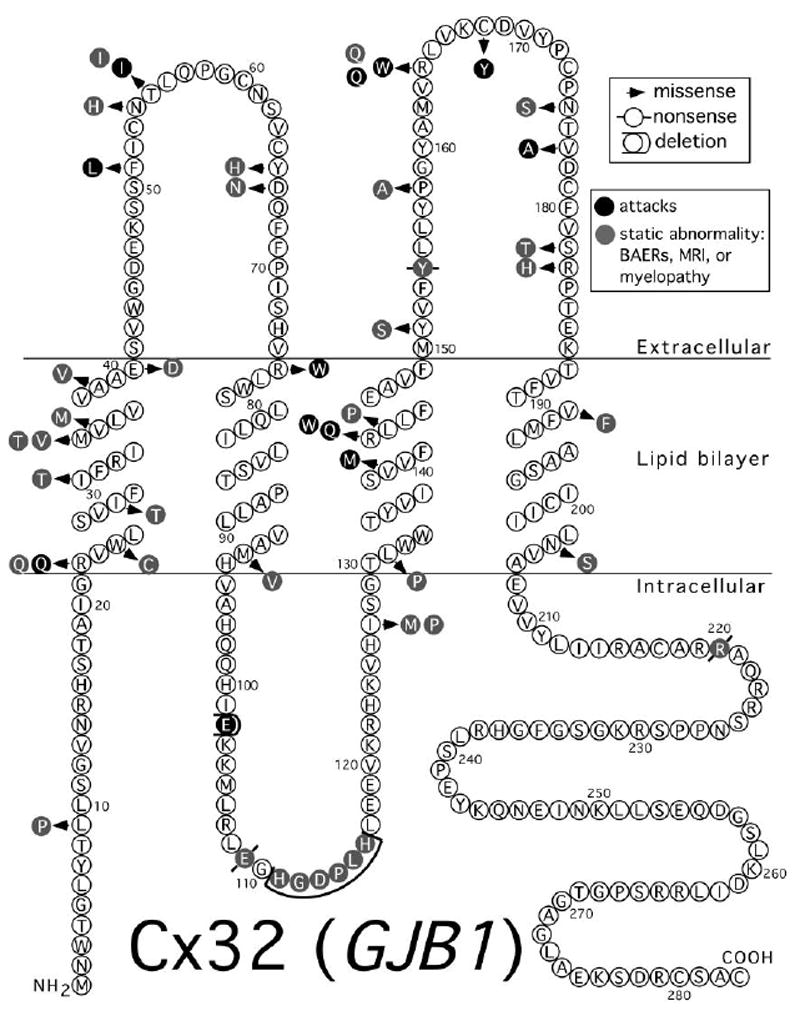
Mutations in Cx32 associated with CNS manifestations. Topology is as depicted in Yeager and Nicholson (1996) [198]
3.1.1 CMT1X subclinical manifestations
Nicholson and collegues [93, 94] were the first to report prolongation of BAERs in many but not all patients with CMT1X. A number of other investigators have confirmed and extended these finding, showing conduction slowing of VERs [89, 91, 95-101] as well as motor [102] and sensory evoked potentials. (SEPs) [103] In a particularly noteworthy study Zambelis et al. [104] measured evoked potentials (blink responses, SEPs, and transcranial magnetic central motor conduction time) in seven affected family members (3 female and 4 male) carrying the Y154STOP mutation. All male and female patients had abnormalities in at least one of these tests; however, males tended to have abnormalities in multiple tests. [104] Nicholson and collegues [94] reported that several mutations in CMT1X patients they evaluated were not associated with abnormalities of BAERs, including L239I [94] and Y151K, V181M, R183C (G Nicholson, personal communication). Interestingly, patients with complete absence of the coding sequence of Cx32 [90, 105] have no abnormalities of VERs or BAERs, suggesting that these CNS findings are due to gain of function.
3.1.2 Subtle clinical manifestations
A few mutations are associated with mild, static clinical abnormalities. For example, a number of patients have been reported with extensor plantar responses [91, 106-108]. In addition, fixed spasticity, hyperreflexia, dysarthria and ataxia have also been reported. [102, 107, 109, 110] A number of studies have reported MRI imaging abnormalities in CMT1X patients. In most cases, these imaging abnormalities are accompanied by clinical findings, [91, 102, 108, 109] while in at least two case no mention is made of accompanying clinical signs or symptoms [111, 112] Interestingly, two reports [102, 109] describe diffuse involvement of the corticospinal/corticobulbar tracts, suggesting that this tract may represent a selectively vulnerable region; the predominance of findings indicative of corticospinal dysfunction in patients with fixed abnormalities, and the predominance of corticospinal findings in patients with florid acute syndromes (see below) also suggest this possibility. A few additional reports describe patients with scattered white matter lesions. [91, 108, 111]
3.1.3 Florid syndromes
A growing number of CMT1X patients who experienced transient neurological dysfunction accompanied by transient white matter abnormalities on MRI have been reported (see Fig. 5). Findings typically include motor weakness and dysarthria [97, 113-119]. In addition, ataxia [113], respiratory distress [114], dysphagia [115, 117] and disorientation [119] have also been described. The duration of symptoms varies, but they usually resolve between a few hours and a few weeks. However, the MRI changes often lag, taking months to return to baseline. [97, 113, 115, 119, 120] Some of these episodes appear to occur without provocation, [97, 116] but many are associated with stressors such as hyperventilation or exertion [97, 115, 120] re-acclimitzation after return from high altitude above 8000 feet, [113] fever, [114, 119] or minor infections. [115, 117]
Figure 5.
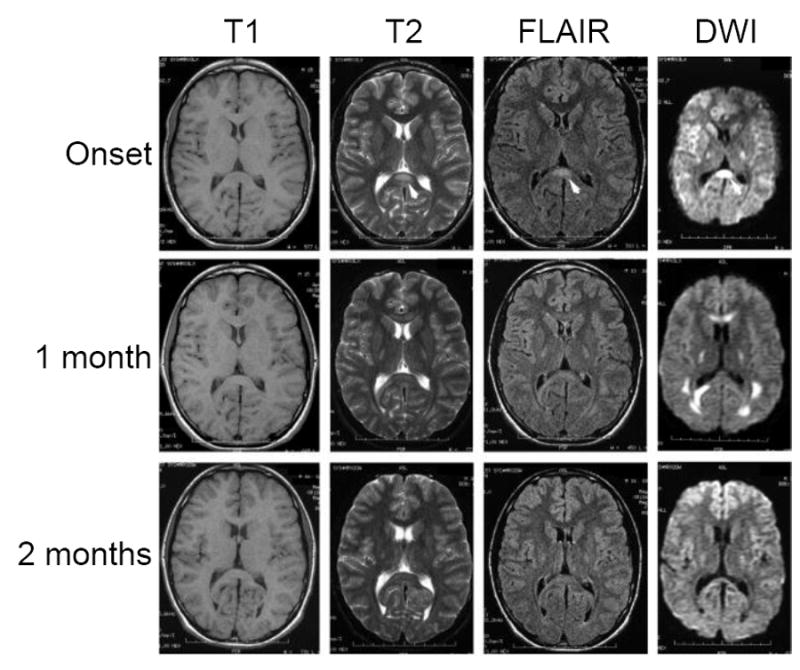
MRI imaging of a CMT1X patient with an acute florid CNS presentation. T1-weighted, T2-weighted, fluid-attenuated inversion recovery (FLAIR), and diffusion-weighted magnetic resonance imaging (DWI) images during onset of an acute CNS syndrome and 1 and 2 months later show splenium of corpus callosum (arrow in the T2, FLAIR and DWI images at onset) and posterior limb of the internal capsule abnormalities at all three time points; signal intensities have almost returned to normal after 2 months. Modified from Paulson et al. (2003) [113], used with permission of Wiley-Davis.
3.2 Pathophysiology of CNS phenotypes in CMT1X patients
The absence of clinical or even subclinical abnormalities of VERs or BAERs in patients with complete absence of the coding sequence of Cx32, [90, 105] and the absence of overt CNS manifestations in most patients with CMT1X and in the Cx32 KO mouse [29-31] suggest that Cx32 is not critical for CNS functioning. [30] Thus, the subclinical and overt CNS manifestations of CMT1X are unlikely to arise through simple loss of function of Cx32, suggesting that Cx32 mutants cause a gain of function. However, the nature of that gain of function is not known. One possibility is that Cx32 mutants interact heteromerically with Cx47 expressed in the same cells, thereby reducing the activity of Cx47. The additive effect would be greater than the loss of Cx32 activity alone, and sufficient to cause the subclinical symptoms or to predispose patients to the acute, florid syndromes described above, just as mice lacking expression of both Cx32 and Cx47 have a much more severe phenotype than mice lacking either connexin alone, as described in Section 2.2.1.
We and others have expressed some of these CMT1XCNS mutants in heterologous cells, and examined their ability to form functional GJs. [86, 107, 121-128] Because many mutants associated with CNS phenoypes are localized intracellularly and do not form GJ plaques, it is tempting to speculate that abnormal subcellular localization predicts abnormal interaction with Cx47 and thereby predicts CNS manifestations. [124] (For example, among mutant showing abnormal subcellular localization [107, 124, 128, 129], R142W, R164W, and R164Q are associated with acute attacks as described in section 3.1.3 and A39V and L143P are associated with clinical and/or MRI findings as described in section 3.1.2.) However, other mutants associated with these same CNS phenotypes can form GJs [124, 127-129] (V139M is associated with acute attacks; V35M, P158A, N205S, R220x, show evoked potential abnormalities but no overt phenotype; and M93V and R183H are associated with clinical and/or MRI findings). Further, some mutants with abnormal subcellular localization have no clinical or subclinical CNS phenotype. [94]
As noted in section 3.1.3, the acute, reversible encephalopathy that occurs in CMT1X has often been associated with stressors such as an acute febrile illness or return to lower altitudes after adapting to higher altitudes. The precise physiological trigger(s) have not been identified, but pro-inflammatory cytokines (for systemic illnesses) and decreases in pH (associated with going from high to low altitude) are strong possibilities. We postulate that Cx32 mutants associated with these transient CNS events have trans-dominant-negative effects on Cx47, thereby reducing O/O and/or O/A coupling. Pro-inflammatory factors such as tumor necrosis factor-α may directly inhibit glial GJs, [130] exacerbating already tenuous GJ coupling and leading to clinical manifestations. Returning to lower altitudes after adapting to higher altitudes decreases the pH of the cerebrospinal fluid; (CSF) [131] lower pH reduces GJ coupling. [132]. At 13 hours after return to normoxia only a small drop in CSF pH is seen (0.03 pH units) [131]; however longer time points were not studied and the full extent of this drop may be greater. In any case a small pH drop could conceivably affect already tenuous O/A and O/O coupling, since reducing the pH in patch electrodes from 7.4 to 7.2 has dramatic effects on Lucifer Yellow transfer from oligodendrocytes [24].
4. Disease manifestations of GJC2 mutations
4.1 Pelizaeus-Merzbacher-like disease 1 (PMLD1) MIM ID #608804
Pelizaeus-Merzbacher disease (PMD) is an X-linked disorder caused by mutations in PLP, the gene for proteolipoprotein (PLP), the major protein of CNS myelin [133]. Pelizaeus-Merzbacher-Like Disease is a group of diseases in patients with a PMD phenotype but lacking PLP mutations. Patients typically develop nystagmus within the first 6 months of life, cerebellar ataxia by 4 years, and spasticity by 6 years of age [134-137]. In 2004 Uhlenberg and collegues reported that mutations in GJC2, the gene encoding Cx47, were found in some PMLD patients [134]. GJC2 mutations are found in only ~8% of PMLD patients. [138] (GJC2 associated PMLD is termed PMLD1 or HLD2 (hereditary leukodystrophy type 2.) To date, 23 different CNS disease associated recessive mutations in the coding region of the gene for Cx47 have been identified. [134-139] All patients with PMLD have extensive white matter disease consistent with abnormal myelination on MRI; relative sparing of the corticospinal tracts has been reported (but is not particularly evident on the images shown in Fig. 7) [134-137] MRI spectroscopy studies in two patients showed normal or near normal choline, N-acetyl aspartate and creatine levels [137] (normal N-acetyl aspartate levels indicate lack of significant axonal damage). Figure 6 shows a schematic of Cx47 mutants associated with human disease.
Figure 7.
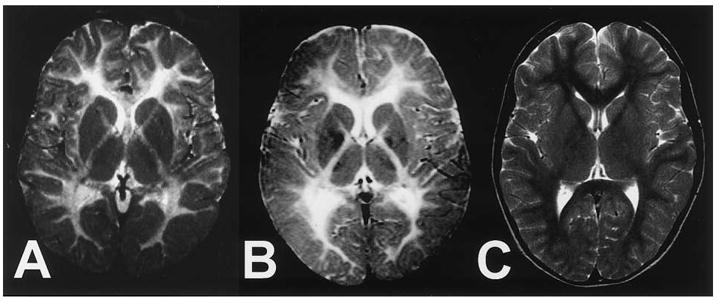
MRI imaging of patients with PMLD1. Axial T2-weighted magnetic resonance images of the brain at the level of the basal ganglia. A. Patient with PMLD at 6 years of age. B. Patient with PMD at 7 years of age. The patients in panels A and B show nearly identical patterns consistent with hypomyelination of central white matter, as indicated by diffusely enhanced signal intensity. C. Low signal of normal myelination in an unaffected child. From Uhlenberg et al. (2004) [134] used with permission of Elsevier.
Figure 6.
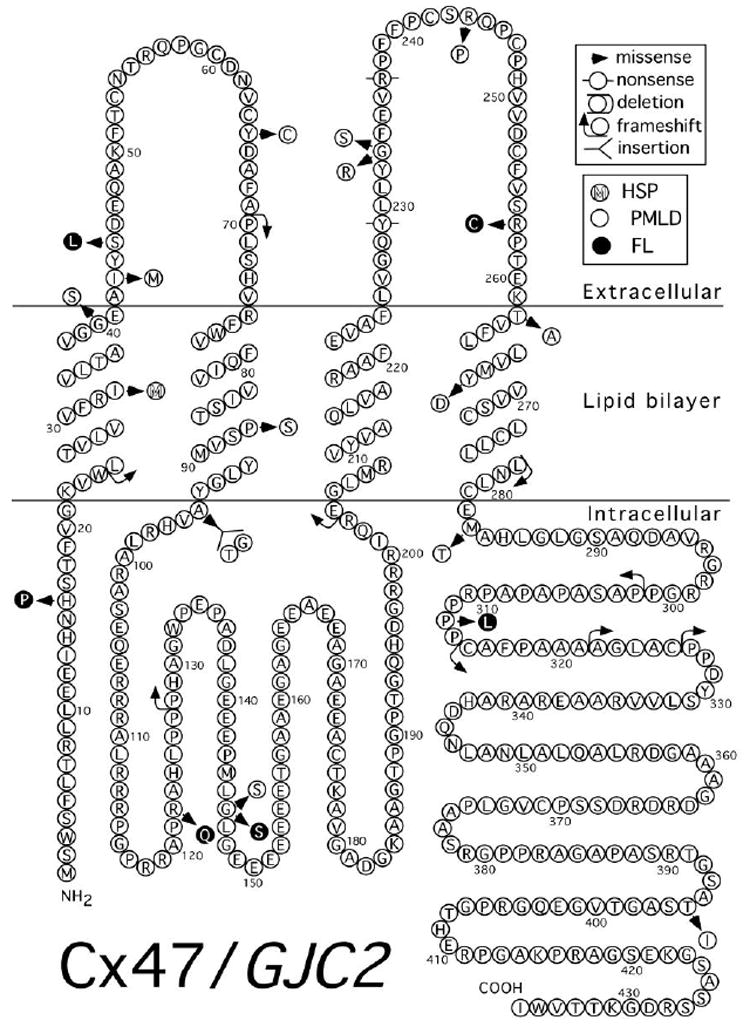
Mutations in Cx47 associated with human disease. Topology is as depicted in Yeager and Nicholson (1996) [198]
4.2 Hereditary Spastic Paresis (SPG44) MIM ID #613206
Orthmann-Murphy et al. [140] described a large family with three affected members carrying a homozygous I33M mutation of GJC2 (Fig. 6). One male patient (age 38) developed moderate walking difficulties, leg stiffness and dysarthria around age 31. His brother (age 36) had minimal motor difficulties since infancy but began to show progressive worsening of his gait and speech after age 20; he walks with a cane. A female cousin (age 55) was always considered dull, had difficulty walking and dysarthria in her teens and has been wheelchair bound since age 30; more recently she has developed urinary incontinence followed by retention. All three patients had spasticity, hyperreflexia, and intention tremor on exam and none had nystagmus. All heterozygous individuals in the family were normal. In summary, although only identified in one family thus far, SPG44 is a clinically much milder disease than is PMLD1 with correspondingly milder changes on the cerebral MRI (Fig. 8). Notably, some PLP1 mutations can also present with a paraperesis phenotype (SPG2). [141] Thus, SPG44 and SPG2 are the only two known causes of hereditary spastic paraparesis associated with mutations in genes that are expressed primarily by oligodendrocytes, and both can also cause a more severe diffuse disorder of myelination. This is particularly interesting because the spastic paraplegia phenotype is thought to represent a length dependent axonopathy, and suggests that mutant forms of Cx47 may directly interfere with interactions between an oligodendrocyte and their associated axons. (The central motor axons to the lumbar segments are the longest central axons in the body and therefore, the most susceptible to length-dependent pathology.)
Figure 8.
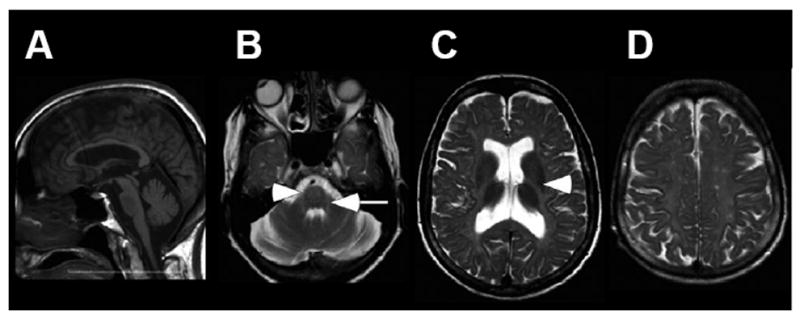
MRI imaging of a patient with SPG44. A. Sagittal T1-weighted imaging shows diffuse thinning of the corpus callosum. B -D. Axial T2-weighted imaging shows symmetric hyperintensity in the region of the corticospinal/corticobulbar tracts at the level of the pons (B, arrows), and the posterior limb of internal capsule (C, arrowhead). In addition, there is diffuse hyperintensity in the subcortical, lobar and periventricular white matter (C and D), and enlarged ventricles (C) Modified from Orthmann-Murphy et al. (2009) [140] Used with Permission of Oxford University Press.
4.3 Hereditary lymphedema 1C MIM ID #613480
A recent report [142] identified six dominant mutations in Cx47 associated with hereditary lymphedema (Fig. 6). However, no neurological histories, examinations, or imaging were provided.
4.4 Pathogenesis of Cx47 associated diseases
One explanation, consistent with the recessive inheritance pattern and relatively uniform phenotype, is that PMLD1 arises due to loss of channel function of Cx47 in oligodendrocytes. The recent finding by Osaka and collegues [143], that a mutation in the SOX10 binding site in the GJC2 promoter causes a mild PMLD phenotype [144], intermediate between that of PMLD1 and SPG44, lends support to the idea that in humans, loss of function of Cx47 is pathogenic and that degree of loss of function may predict severity of phenotype. However, these findings do not rule out gain of function as a contributor to the more severe phenotypes. Data from our laboratories [140] suggest that PMLD1 mutations lead to complete loss of gap junctional coupling, while the SPG44 mutation causes radical alterations in gating, predicted to essentially eliminate Cx47-mediated coupling; thus, it is unlikely that the milder phenotype of SPG44 results from retention of a small degree of Cx47 mediated coupling. The recent finding by Osaka and collegues [143], that a mutation in the SOX10 binding site in the GJC2 promoter causes a mild PMLD phenotype [144], intermediate between that of PMLD1 and SPG44, lends support to the idea that in humans, loss of function of Cx47 is pathogenic and that degree of loss of function may predict severity of phenotype. However, these findings do not rule out gain of function as a contributor to the more severe phenotypes. The work of Diekmannn et al. [84]85] suggests that loss of hemichannel function is a feature of some Cx47 mutants, and may represent an additional component of loss of function of some or all of the Cx47 mutants. The finding that mice with targeted ablation of the gene for Cx47 show minimal [14] CNS pathology or phenotypic abnormalities is also inconsistent with a pure loss-of-function model for PMLD1; however it could simply reflect that mouse oligodendrocytes are less dependent on expression of Cx47 for normal function than are human oligodendrocytes. This notion is supported by the recent demonstration [145] that mice with targeted insertion of the Cx47M282T mutation (corresponding to the human PMLD1 causing Cx47M283T mutation) have a very mild phenotype compared to that seen in humans.
A second possibility is that both the PMLD1 and SPG44 mutants lead to a loss of channel function, but that the PMLD1 mutations lead to the loss of an additional non-channel based function. We found that each of three missense Cx47 mutants associated with PMLD1 accumulate in the endoplasmic reticulum, [146] while the SPG44 mutant I33M forms GJ plaques that are indistinguishable from WT Cx47 [140] (see Fig. 9). Our data [146] suggest that PMLD1 mutants may not fold and/or assemble properly. PLP mutants that cause the most severe phenotypes also accumulate in the ER, and activate the unfolded protein response (UPR), [147-149] a group of cellular signaling pathways that can lead to apoptosis. [150, 151] The three missense Cx47 mutants associated with PMLD1 did not activate CHOP, a downstream effector of the UPR [146] that is activated in PMD. [148] However, a more complete evaluation of the UPR pathway, particularly in oligodendrocytes, is needed before rejecting the hypothesis that the UPR is not involved in PMLD1. Finally, as discussed in section 2.2, the loss of junctional Cx47 leads to mislocalization of ZONAB to the nucleus, which could alter gene expression, and thus contribute to the greater severity of the PMLD1 phenotype.
Figure 9.
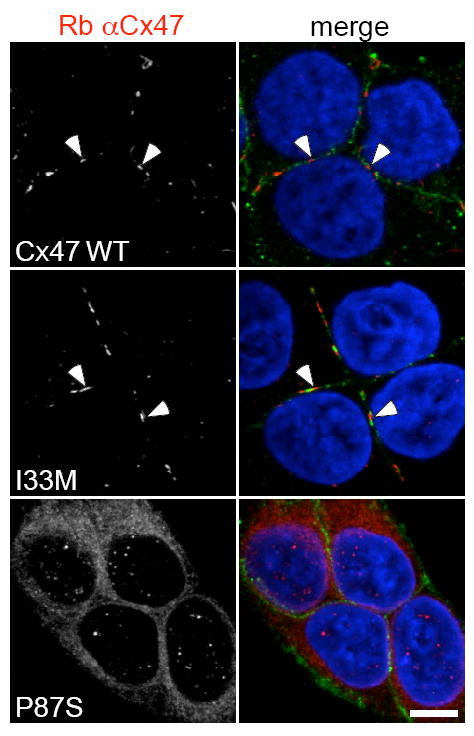
Immunostaining for the I33M and P87S mutants. The SPG44 associated I33M mutant forms GJ plaques while the PMLD1 associated P87S mutant does not. These are confocal images of bulk-selected HeLa cells that express WT Cx47 or the indicated mutants, immunostained with a rabbit antiserum against human Cx47 (red) and a mouse monoclonal antibody against pan-cadherin (green), and counterstained with DAPI. The pan-cadherin staining at cell borders interdigitates with the cell surface staining of Cx47 in cells that express WT Cx47 (arrowheads) or I33M (arrowheads), but surrounds the staining of cells expressing the mutant P87S, which is localized in the endoplasmic reticulum. Scale bar: 10 μm. From Orthmann-Murphy et al. [140] Used with Permission of Oxford University Press.
5. Disease manifestations of GJA1 mutations
5.1 Oculodentodigital dysplasia (ODDD) MIM ID #164200
ODDD is a pleitropic autosomal dominant disorder seen in patients with mutations in GJA1, the gene for Cx43. [152]; these are depicted in Fig. 10. Although a number of patients with this disorder had already been described, [153] the definitive delineation of ODDD dates to the German language publication of “The Microphthalmos Syndrome” by Meyer-Schwickerath in 1957, [154] in which the term ‘dysplasia oculo-dento-digitalis’ was introduced. In 1963, Gorlin et al. [155] reviewed the known cases and defined the core features of syndrome of ODDD as oculo-facial abnormalities including micropthalmia, microcornia and hypoplastic nares, syndactyly of the fourth to fifth fingers and dental abnormalities such as hypoplastic enamel and microdontia. See supplementary tables 3 to 5 in Paznekas et al. 2009 [156] for detailed and comprehensive information on the general phenotypes of patients carrying specific mutations in Cx43. Neurologic manifestations are often but not universally seen, and may not become evident until the second or third decade. The most common are gait difficulties, [153, 157-169] due to either spasticity or ataxia, and urinary incontinence; [166-169] these typically present by the second decade. A wider range of neurologic symptoms have been reported, [153] including cognitive deficiencies, [157, 170] deafness, [171, 172] disorders of extraocular motility, [161, 168, 171] generalized muscle weakness, and seizures. [152] MRI abnormalities include hypointensity of the deep grey matter, which may reflect iron deposition, and changes in the occipital and periventricular white matter [153, 164, 165]. (See Table 2 for further references to neuroimaging.) In some cases where CT imaging was used, the basal ganglia were calcified. [161, 173] Abnormalities in CNS imaging accompany overt CNS findings, but may also be seen in patients without neurological issues, [170] raising the possibility that many mutations reported as showing no neurologic phenotype may still cause subclinical alterations CNS changes. Along this line, an MRI imaging study [165] of a neurologically symptomatic mother and neurologically normal daughter with ODDD both showed identical changes in the white matter. The clinical and subclinical neurological findings in patients with ODDD are summarized in Table 2.
Figure 10.
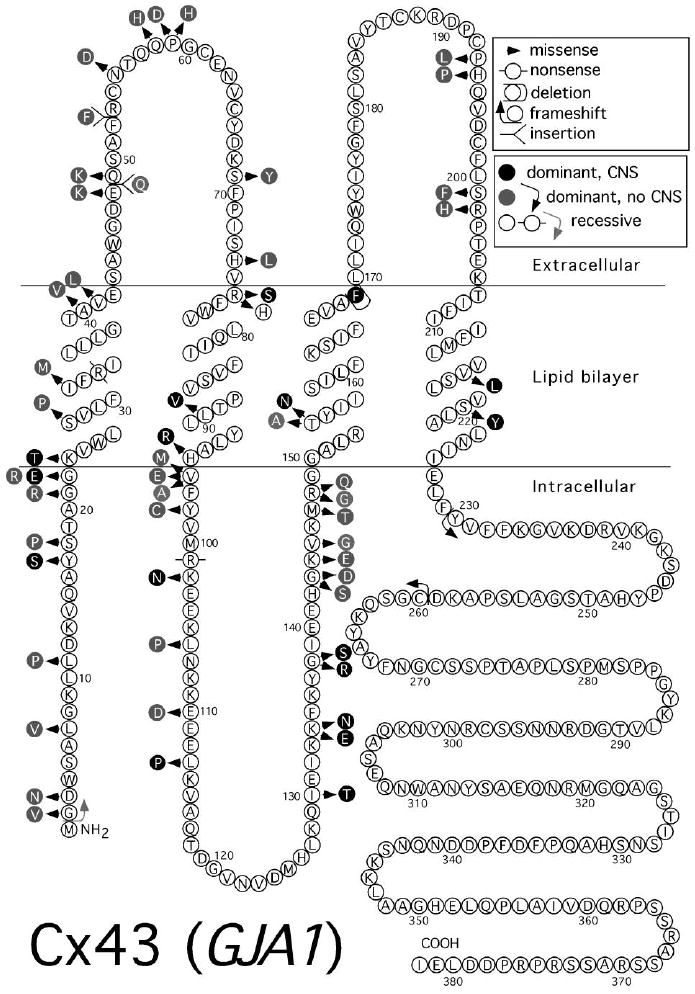
Mutations in Cx43 associated with human disease. Topology is as depicted in Yeager and Nicholson (1996) [198]
Table 2.
Features of mutations in Cx43 associated with ODDD
| Mutation | Functional assay (transfected cell lines unless noted) | Localization in mammalian cells (transfected cell lines unless noted) | Neurologic phenotype | References |
|---|---|---|---|---|
|
| ||||
|
Explicit statement of no neurologic manifestations
| ||||
| S5C | 9 YO “no mental retardation or brain wave abnormality.” [201] | [201] | ||
|
| ||||
| L11P | 13 YO, de novo mutation. | [202] | ||
| No neuro. findings. [202] | ||||
|
| ||||
| S18P | GJ-like plaques. (S. Scherer, unpublished) | No neuro. findings. [152] | [152] originally described in [203, 204]; | |
|
| ||||
| V41A | 11 YO, neuro. exam nl. | [205] | ||
|
| ||||
| Q49K | Low homotypic coupling and minimal dominant negative effect on endogenous Cx43 mediated NRK cell coupling. [185] | GJ-like plaques. [185] (S. Scherer, unpublished) | No neuro. findings. [152] | [152] originally described in [206]; |
|
| ||||
| F52dup | No junctional homotypic coupling. [189] | ER staining pattern [189] Very reduced puncta. [188] Normal localization (basolateral) in MDCK cells. [186] | No neuro. findings. [152] | [152] originally described in [207]; |
| No dye transfer by scrape load or hemichannel activity by dye uptake. [188] | Endosomal. (S. Scherer, unpublished) | |||
|
| ||||
| P59H | No neuro. findings. | [208] | ||
|
| ||||
| R76H | Affected child has homozygous mutation; parents are heterozygotes; no neuro. findings. | [209]. | ||
|
| ||||
| V96E | 12 YO with de novo mutation in; no neuro. findings. | [157] | ||
|
| ||||
| Y98C | GJ-like plaques. (S. Scherer, unpublished) | No neuro. findings. [152] | [152] originally described in [206, 210]; | |
|
| ||||
| H194P | Normal hemichannel activity. No neurobiotin transfer [193] | GJ-like plaques and cytopasmic. [187] | No syndactyly or spastic paraplegia. [156, 211] | [211] |
|
| ||||
| R202H | No homotypic coupling. [189] | GJ-like plaques (S. Scherer, unpublished) | No neuro. findings. [152] | [152, 212, 213] |
| Low coupling and dominant negative effect on endogenous Cx43 mediated NRK coupling. [185] | No plaques, some overlap with ER marker. [189] | |||
| Few plaques partial intracellular retention. [185] | ||||
|
| ||||
| C260fs | Decreased endogenous plaques when expressed in NRK cells; decreased coupling and dominant negative effect on WT Cx43 in transfected cells [192] | ER retained [192] | Neurologically normal [214] | [214] |
|
| ||||
| Y230fs | No neuro. or eye problems. [156, 215] | [215] | ||
|
| ||||
| A253V | Polymorphism | [152] | ||
|
| ||||
|
Neurologic manifestations not mentioned
| ||||
| G2V | Nothing stated. | [216] | ||
|
| ||||
| L7V | Nothing stated. | [156] | ||
|
| ||||
| G22R | Nothing stated. | [156] | ||
|
| ||||
| S27P | Nothing stated. | [212] | ||
|
| ||||
| I31M | Nothing stated. | [212] | ||
|
| ||||
| E48K | Nothing stated. | [217] | ||
| [218] | ||||
|
| ||||
| Q49dup | Nothing stated. | [156] | ||
|
| ||||
| N55D | Nothing stated. | [156] | ||
|
| ||||
| Q58H | Nothing stated. | [156] | ||
|
| ||||
| P59A | Nothing stated. | [156] | ||
|
| ||||
| P59H | Nothing stated. | [219] | ||
|
| ||||
| S69Y | Nothing stated. | [212] | ||
|
| ||||
| H74L | Nothing stated. | [156] | ||
|
| ||||
| V85M | Nothing stated. | [213] | ||
|
| ||||
| V96M | Nothing stated. | [220] | ||
|
| ||||
| E110D | Nothing stated. | [221] | ||
|
| ||||
| I131M | Increased hemichannel activity. | GJ-like plaques and cytopasmic. [187] | Nothing stated. | [212] |
| No neurobiotin transfer.[187] | ||||
|
| ||||
| G138D | Nothing stated. | [213] | ||
|
| ||||
| G138S | Nothing stated. | [156, 213] | ||
|
| ||||
| G143S | Increased hemichannel activity. | GJ-like plaques and cytopasmic. [187] | Nothing stated. | [212] |
| No neurobiotin transfer. [193] | ||||
|
| ||||
| G143D | Nothing stated. | [213] [156] | ||
|
| ||||
| K144E | Nothing stated. | [156] | ||
|
| ||||
| V145G | Nothing stated. | [156] | ||
|
| ||||
| M147T | Sporadic case; nothing stated. | [221] | ||
|
| ||||
| R148Q | Nothing stated. | [212] originslly described in [222] | ||
|
| ||||
| R148G | Nothing stated. | [156] | ||
|
| ||||
| F169del | Nothing stated. | [221] | ||
|
| ||||
| P193L | Nothing stated. | [156] | ||
|
| ||||
| S201F | Nothing stated. | [156] | ||
|
| ||||
| S201Y | Nothing stated. | [213] | ||
|
| ||||
|
Explicit description of neurologic manifestations
| ||||
| G2fs | G2fs+R101x; severe CNS phenotype, psychomotor regression onset age 10, seizures; massive calcifications, hypomyelination, and atrophy on CT/MRI. [173] | [173] | ||
|
| ||||
| D3N | Decreased coupling in patient fibroblasts. [169] | Partial localiztion in Golgi (but also some WT in golgi distribution) in patient fibroblasts. [169] | Neurogenic bladder, UMN. [169] | [156, 213] [169] |
|
| ||||
| Y17S | No junctional coupling. [189] | GJ-like plaques. (reduced nos.) [188] | “Neuro. deficits were prominent.” [206] UMN, ur. inc. [156] | [152] originally described in [206, 223] |
| No dye transfer by scrape load or hemichnnel activity by dye uptake. [188] | GJ-like plaques. (S. Scherer, unpublished) | |||
|
| ||||
| G21R | No homotypic junctional coupling. [189] [190] No dye transfer by scrape load or hemichnnel activity by dye uptake. [188] | GJ-like plaques. (reduced numbers) [189] [188, 190] | No neuro. symptoms but patient is a 2 YO with a sporadic mutation. [152] UMN. [156] | [152] |
| Dominant negative effect on endogenous 43 in NRK cells [190] and Hela and N2a cells. [224] | ||||
|
| ||||
| G22E | GJ-like plaques. (S. Scherer, unpublished) | “Neuro. Symptoms.” [152] | [152] originally described in [225] | |
|
| ||||
| K23T | GJ-like plaques. (S. Scherer, unpublished) | “Neuro. Symptoms.” [152] UMN, MRI WMC, tremor. [156] | [152] originally described in [155] | |
|
| ||||
| R33x | 2 siblings homozygous for this mutation; speech delay, UMN, grossly abnormal grey/white matter differentiation on head CT. [180] | [180, 181] | ||
|
| ||||
| A40V | No homotypic coupling. [189] | GJ-like plaques. (reduced numbers) [188, 189] | No neuro. symptoms but patient is a 2 YO with a sporadic mutation. [152] gait difficulty, ur. urgency [156] | [152, 212, 221] |
| No dye transfer by scrape load or hemichnnel activity by dye uptake.[188] | GJ-like plaques. (S. Scherer, unpublished) | |||
|
| ||||
| Q49P | Ur. inc. | [156] | ||
|
| ||||
| R76S | GJ-like plaques. (S. Scherer, unpublished) | “Neuological deficits were prominent” [206] Epilepsy [152] MRI WMC [156] | [152] originally described in [226] | |
|
| ||||
| L90V | Coupling very low, single channel conductance normal. [189] Low junctional conductance and dominant negative effect on endogenous Cx43 mediated NRK coupling. [185] | GJ-like plaques. (reduced nos.) [188, 189] | “Neuro. deficits were prominent” [206] | [152] originally described in [206, 227, 228] |
| Disrupted localization (partial apical) in MDCK cells. [186 | Epilepsy. [152] | |||
| GJ-like plaques. [185] | Ur. inc. and spastic paraplegia (9/9 patients) [156, 227] | |||
| GJ-like plaques. (S. Scherer, unpublished) | ||||
| No dye transfer by scrape load or hemichnnel activity by dye uptake. [188] | ||||
|
| ||||
| H95R | Ur. inc. and spasticity [166] MRI WMC [156] | [166] | ||
|
| ||||
| V96A | MRI WMC. | [156] originally described in [162] | ||
|
| ||||
| R101x | Compound hetrozygote G2fs+R101x; See G2fs above. | [173] | ||
|
| ||||
| K102N | GJ-like plaques. (S. Scherer, unpublished) | Neurologic symptoms. [152] ur. and bowel inc. [156] | [152] | |
|
| ||||
| L106P | Spasticity, ur. inc. | [156] | ||
|
| ||||
| L113P | Spastic paraparesis. [157] UMN, MRI WMC and low signal in grey matter c/w iron deposition.[153] | [157, 212, 229] originally in [153] | ||
| Others note no neurologic findings [229] | ||||
|
| ||||
| I130T | Markedly decreased coupling; single channel size normal. [189, 191] | GJ-like plaques. [191] | Spastic para- or tetraparesis; epilepsy, ur Inc. MRI cerebellum/midbrain atrophy or hypoplasia [167] | [152, 167] |
| No dye transfer by scrape load or hemichannel activity by dye uptake. [188] | GJ-like plaques (very reduced). [189] | |||
| GJ-like plaques (slightly reduced). [188] | ||||
|
| ||||
| K134E | Markedly decreased or absent coupling; reduced single channel size (54 pS) [191] Reduced coupling. [189] | GJ-like plaques. [191] | “Neurologic symptoms.” [152] UMN, MRI WMC. [156] | [152] |
| GJ-like plaques (very reduced). [189] | ||||
|
| ||||
| K134N | Reduced coupling.[189] | GJ-like plaques (very reduced). [189] | UMN. [156] | [212] probably described previously[230] |
|
| ||||
| G138R | No homotypic,coupling, shifted Gj-Vj relation in hetrotypic with Cx43-EGFP. | GJ-like plaques. [191] | 4 generations 1 and 2: no neurologic findings. 3 and 4: spastic bladder paraparesis, square wave jerks and intention tremor presented 2nd to 4th decade. documented WMC on MRI [168] UMN, ur. inc., MRI WMC. [156] | [152] originally described in [168, 206] |
| Increased hemichannel activity no neurobiotin transfer[193] [187] | GJ-like plaques and cytopasmic. [187] | |||
| No homotypic coupling. [190, 191] Dominant negative effect on endogenous Cx43 in NRK cells [190] and Hela and N2a cells. [224] | ||||
|
| ||||
| T154A | Mild psychomotor delay and WMC on MRI scan. [231] Gait abnl, UMN, ur. inc., MRI WMC. [156] | [156, 231] | ||
|
| ||||
| T154N | Spastic paraparesis, inc., tremor. [157] | [157] | ||
|
| ||||
| V216L | Decreased coupling in patient fibroblasts. [169] | Partial overlap with Golgi marker (but also some WT in Golgi distribution) in patient fibroblasts. [169] | Spasticity, WMC on MRI.[172] | [152, 169] originally described in [172, 206] |
| No conductance and dominant negative effect on endogenous Cx43 mediated NRK coupling. [185] | Few GJ-like plaques partial intracellular retention [185] | Gastrointestinal hypomotility, neurogenic bladder, lost ambulation at age 54; hyperreflexia, spasticity. [169] | ||
| GJ-like plaques. (S. Scherer, unpublished) | UMN, MRI WMC ur. and bowel inc. [156] | |||
|
| ||||
| S220Y | Developmental and language disorder. [157] | [157] | ||
|
| ||||
|
No identified mutation
| ||||
| ? | Spasticity. | [158] | ||
|
| ||||
| ? | Severe spasticity. | [159] | ||
|
| ||||
| ? | Spastic tetraparesis. | [160] | ||
|
| ||||
| ? | Spastic paraparesis, nystagmus. | [161] | ||
|
| ||||
| ? | Spastic paraparesis and decreased signal c/w iron deposition in globus pallidus on MRI. | [232] | ||
|
| ||||
| ? | 21 YO F spastic paraparesis MRI WMC and low signal in grey matter c/w iron deposition. | [164] | ||
|
| ||||
| ? | MRI diffuse WMC, Abnl low signal in globus pallidus, substantia nigra without calcification on CT c/w iron deposition. | [165] | ||
WMC- white matter changes, typically increase signal on T2 weighted imaging. c/w-consistent with. GJ-gap junction. YO-years old. abnl-abnormal. nl-normal. h/o-history of. Gj-Vj – normalized conductance-voltage relation. UMN-spasticity/hyperreflexia. ur inc-urinary incontinence. neuro-neurological.
5.2 Hallermann-Streiff syndrome (HSS); MIM ID 234100
HSS is an autosomal recessive or sporadic syndrome that is at least sometimes associated with mutations in GJA1. The clinical phenotype shows substantial overlap with the ODDD, characterized by brachycephaly with frontal bossing, micrognathia, a beaked nose, microphthalmia, cataracts, dental anomalies, hypotrichosis, skin atrophy, and short stature [174-176] Mental retardation is present in some patients. [177] A homozygous R76H mutation was noted in a boy with HSS; [178, 179] his parents were heterozygous carriers of the mutation and were clinically normal. In another case of HSS, Pizzuti et al. [179] found no GJA1 mutation in the open reading frame, but did not rule out a mutation in the promoter or untranslated region of GJA1. Reports of homozygous [180, 181] or compound heterozygous mutations [173] causing ODDD further demonstrate the overlap between HSS and ODDD. A few case reports of HSS had abnormal neuroimaging, but were not studied for GJA1 mutations. Sigrici and collegues [182] describe a four year old boy with the HSS phenotype, nystagmus, spasticity, optic atrophy and complete agenesis of the corpus callosum. Decreased cerebellar and medullary size on MRI has also been reported. [183] These reports should be viewed cautiously since, as noted above, at least one patient with a “full blown” HSS phenotype had no mutations in the GJA1 coding region. [179]
5.3 Pathogenesis of Cx43 associated diseases
The genetics of ODDD and HSS indicate that ODDD is not the result of haploinsufficiency. The heterozygous parents of patients with the R33X [180] and R76S [179] mutations were normal. The mother of the severely affected girl with a compound G2frameshift and R101X mutations (both of which would likely cause loss of function) was heterozygous for the R101X mutation, and said to be have “mild microphthalmia and hypoplastic alae nasi” [173], although those features are difficult to appreciate in the image provided. In accord, heterozygous Gja1 knockout mice show no overt phenotype in any organ system [184], whereas mice with dominant Gja1 mutations have phenotypes that resemble those in ODDD (see below).
Many ODDD mutants have been expressed by transfection in heterologous cells (Table 2). These results bring up the following points:
Irrespective of known CNS involvement, most, but not all, ODDD mutants studied can form GJ plaques. [169, 185-191] (The number may be reduced for some mutants.) Thus, there is no apparent association between the targeting of the Cx43 mutant to gap junctions and the presence of a CNS phenotype.
Functional analyses show that ODDD mutants lead to partial or complete loss of intercellular GJ mediated coupling, and many have dominant-negative effects on wild type Cx43. [185, 188-190, 192] The degree of this dominant-negative effect varies, and it remains to be determined whether this correlates with the severity of disease, including the presence of CNS manifestations. For example, L90V and V216L (“CNS mutants”) and R202H (a “non-CNS mutant”) reduce Cx43-mediated cell coupling to a similar degree. [185] In addition, it is possible that “trans-dominant-negative” effects of Cx43 mutants on Cx30 contribute to their clinical phenotypes.
Several mutants affect hemichannel properties: Y17S, G21R, A40V, F52dup, L90V and 1130T all lack hemichannel function, I131M, G138R, G143S all show increased hemichannel activity, while the hemichannel activity of H94P is similar to that of WT. [188, 193] It is possible, but by no means proven, that hemi-channel activity contributes to the ODDD phenotype.
I131M, G138R, G143S, and H194P have reduced phosphorylation, a post-translational modification of Cx43 that is associated with increased coupling. [194] Because some (G138R and H194P) but not all (I131M and G143S) of these mutants are associated with neurological findings, the role of phosphorylated Cx43 in this regard remains unproved.
The accumulation of Cx43 mutants in intracellular organelles may have deleterious effects on the cells, possibly by activating the UPR as discussed for PMLD1 in Section 4.3. [147-149]
Reduced Cx43 in GJ plaques may have deleterious effects on localization of other proteins with which it normally interacts, as outlined in Section 2.2.2.
There are several mouse models of ODDD. Jrt mice are heterozygous for a dominant G60S mutation, and show many of the features of ODDD but have no overt CNS findings or brain MRI abnormalities at 52-60 weeks. [195] Immunohistochemistry and immunoblotting of spinal cord and cerebellum from Jrt mice show decreased staining and decreased levels of Cx43, but astrocytes were still well coupled by GJs, both in vitro and in vivo. [196] In transfected cells, G60S formed GJs that did not pass Lucifer Yellow, but a dominant-negative effect on wild type Cx43 could not be demonstrated [196]. Heterozygous mice with a targeted replacement of WT Cx43 by the ODDD I130T [197] or G138R [193] mutations have reduced embryonic viability, cardiac dysrhythmias, and reduced Cx43 levels in heart tissue, but the CNS was not investigated. Because none of the findings seen in the mouse models have been reported in heterozygous Gja1 knockout mice [184], they are dominant effects.
In summary, the analysis of cellular and mouse models of ODDD demonstrate that ODDD mutants have several kinds of dominant effects. Of these, dominant-negative effects on wild type Cx43 are by far the best documented, but it remains to be shown that they are sufficient to account for all of the various attributes of ODDD. The striking phenotypic similarities between patients with ODDD (cause by gain of function mutations) and HSS (caused by loss of function mutations) makes a dominant-negative mechanism the most parsimonious explanation.
6. Conclusion
There are five CNS disorders associated with mutations in three connexin genes. Two disorders, PMLD1 and SPG44, are defined by their CNS manifestations and are associated with recessive GJC2 mutations. Available evidence suggests loss of function of Cx47 is at least partially responsible for these disorders. The difference in severity between the patients with PMLD1 and SPG44 is not well understood, but may reflect either lesser degrees of loss of function for the mutation causing the milder SPG44 phenotype or a component of gain of function for the mutations causing the more severe PMLD1 phenotype. GJB1 mutations cause CMT1X, which always manifests as a peripheral neuropathy, are often associated with subclinical neurological findings, and are rarely associated with a severe but transient syndrome. Though the neuropathy appears related primarily to loss of function of Cx32 in the Schwann cell, CNS manifestations are more likely due to a toxic gain of function in oligodendrocytes. Dominant GJA1 mutations cause ODDD, which is often associated with a CNS phenotype, and recessive GJA1 mutations cause HSS, which strongly resembles ODDD. The genetics of HSS suggest that haploinsufficiency cannot explain the ODDD phenotype. However, a dominant negative effect on wild-type Cx43 has been demonstrated for a number of ODDD mutants; it remains to be demonstrated that such effects will provide an explanation for the full range of phenotypic manifestations of this disorder.
Highlights.
Astrocytes express Cx30 and Cx43 and Oligodendrocytes express Cx29, Cx32, and Cx47.
Mutations in GJB1, cause a form of Charcot-Marie-Tooth disease (CMT1X). Some patients have CNS signs and symptoms.
Recessive mutations in GJC2, the gene for Cx47, are one cause of Pelizaeus-Merzbacher-like disease.
A different recessive GJC2 mutation causes a form of hereditary spastic paraparesis.
Dominant mutations in GJA1, cause oculodentodigital dysplasia (ODDD). Spasticity and gait difficulties may be seen.
Acknowledgments
This work was supported by grants from the NIH (NS067404 and NS050705 to C.K. A. and NS55284 to S.S.S.) and the National Multiple Sclerosis Society (10029126 to S.S.S.).
Footnotes
Non-standard abbreviations. A-astrocyte O-oligodendrocyte GJ- gap junction KO-knockout(null) dKO-double knockout WMC:white matter changes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rackauskas M, Neverauskas V, Skeberdis VA. Diversity and properties of connexin gap junction channels. Medicina. 2010;46:1–12. [PubMed] [Google Scholar]
- 2.Zoidl G, Dermietzel R. Gap junctions in inherited human disease. Pflugers Arch. 2010;460:451–466. doi: 10.1007/s00424-010-0789-1. [DOI] [PubMed] [Google Scholar]
- 3.Orthmann-Murphy JL, Abrams CK, Scherer SS. Gap junctions couple astrocytes and oligodendrocytes. J Mol Neurosci. 2008;35:101–116. doi: 10.1007/s12031-007-9027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abrams CK, Rash JE. Connexins In The Nervous System. In: Harris A, Locke D, editors. Connexins: A Guide. Humana Press; Totowa, N.J: 2009. pp. 323–357. [Google Scholar]
- 5.Orthmann-Murphy JL, Freidin M, Fischer E, Scherer SS, Abrams CK. Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J Neurosci. 2007;27:13949–13957. doi: 10.1523/JNEUROSCI.3395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagy JI, Patel D, Ochalski PA, Stelmack GL. Connexin30 in rodent, cat and human brain: selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience. 1999;88:447–468. doi: 10.1016/s0306-4522(98)00191-2. [DOI] [PubMed] [Google Scholar]
- 7.Nagy JI, Ionescu AV, Lynn BD, Rash JE. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: Implications from normal and connexin32 knockout mice. Glia. 2003;44:205–218. doi: 10.1002/glia.10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rash JE, Yasumura T, Dudek FE, Nagy JI. Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J Neurosci. 2001;21:1983–2000. doi: 10.1523/JNEUROSCI.21-06-01983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altevogt BM, Paul DL. Four classes of intercellular channels between glial cells in the CNS. J Neurosci. 2004;24:4313–4323. doi: 10.1523/JNEUROSCI.3303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filippov MA, Hormuzdi SG, Fuchs EC, Monyer H. A reporter allele for investigating connexin 26 gene expression in the mouse brain. Eur J Neurosci. 2003;18:3183–3192. doi: 10.1111/j.1460-9568.2003.03042.x. [DOI] [PubMed] [Google Scholar]
- 11.Nagy JI, Ionescu AV, Lynn BD, Rash JE. Connexin29 and connexin32 at oligodendrocyte and astrocyte gap junctions and in myelin of the mouse central nervous system. J Comp Neurol. 2003;464:356–370. doi: 10.1002/cne.10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagy JI, Li X, Rempel J, Stelmack G, Patel D, Staines WA, Yasumura T, Rash JE. Connexin26 in adult rodent central nervous system: demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J Comp Neurol. 2001;441:302–323. doi: 10.1002/cne.1414. [DOI] [PubMed] [Google Scholar]
- 13.Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Bussow H, Schilling K, Steinhauser C, Willecke K. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23:4549–4559. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamasawa N, Sik A, Morita M, Yasumura T, Davidson KG, Nagy JI, Rash JE. Connexin-47 and connexin-32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt-Lanterman incisures: implications for ionic homeostasis and potassium siphoning. Neuroscience. 2005;136:65–86. doi: 10.1016/j.neuroscience.2005.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleopa KA, Orthmann JL, Enriquez A, Paul DL, Scherer SS. Unique distributions of the gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia. 2004;47:346–357. doi: 10.1002/glia.20043. [DOI] [PubMed] [Google Scholar]
- 18.Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL. Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci. 2002;22:6458–6470. doi: 10.1523/JNEUROSCI.22-15-06458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Ionescu AV, Lynn BD, Lu S, Kamasawa N, Morita M, Davidson KG, Yasumura T, Rash JE, Nagy JI. Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience. 2004;126:611–630. doi: 10.1016/j.neuroscience.2004.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahn M, Lee J, Gustafsson A, Enriquez A, Lancaster E, Sul JY, Haydon PG, Paul DL, Huang Y, Abrams CK, Scherer SS. Cx29 and Cx32, two connexins expressed by myelinating glia, do not interact and are functionally distinct. J Neurosci. 2008;86:992–1006. doi: 10.1002/jnr.21561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sargiannidou I, Ahn M, Enriquez AD, Peinado A, Reynolds R, Abrams C, Scherer SS, Kleopa KA. Human oligodendrocytes express Cx31.3: function and interactions with Cx32 mutants. Neurobiol Dis. 2008;30:221–233. doi: 10.1016/j.nbd.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scherer SS, Deschenes SM, Xu YT, Grinspan JB, Fischbeck KH, Paul DL. Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci. 1995;15:8281–8294. doi: 10.1523/JNEUROSCI.15-12-08281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rash JE. Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience. 2010;168:982–1008. doi: 10.1016/j.neuroscience.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wasseff SK, Scherer SS. Cx32 and Cx47 mediate oligodendrocyte: Astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiol dis. 2011;42:506–513. doi: 10.1016/j.nbd.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maglione M, Tress O, Haas B, Karram K, Trotter J, Willecke K, Kettenmann H. Oligodendrocytes in mouse corpus callosum are coupled via gap junction channels formed by connexin47 and connexin32. Glia. 2010;58:1104–1117. doi: 10.1002/glia.20991. [DOI] [PubMed] [Google Scholar]
- 26.Massa PT, Mugnaini E. Cell junctions and intramembrane particles of astrocytes and oligodendrocytes: a freeze-fracture study. Neuroscience. 1982;7:523–538. doi: 10.1016/0306-4522(82)90285-8. [DOI] [PubMed] [Google Scholar]
- 27.Massa PT, Mugnaini E. Cell-cell junctional interactions and characteristic plasma membrane features of cultured rat glial cells. Neuroscience. 1985;14:695–709. doi: 10.1016/0306-4522(85)90320-3. [DOI] [PubMed] [Google Scholar]
- 28.Magnotti LM, Goodenough DA, Paul DL. Functional heterotypic interactions between astrocyte and oligodendrocyte connexins. Glia. 2011;59:26–34. doi: 10.1002/glia.21073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anzini P, Neuberg DH, Schachner M, Nelles E, Willecke K, Zielasek J, Toyka KV, Suter U, Martini R. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J Neurosci. 1997;17:4545–4551. doi: 10.1523/JNEUROSCI.17-12-04545.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ. Connexin32-null mice develop demyelinating peripheral neuropathy. Glia. 1998;24:8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8::aid-glia2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 31.Sutor B, Schmolke C, Teubner B, Schirmer C, Willecke K. Myelination defects and neuronal hyperexcitability in the neocortex of connexin 32-deficient mice. Cereb Cortex. 2000;10:684–697. doi: 10.1093/cercor/10.7.684. [DOI] [PubMed] [Google Scholar]
- 32.Sargiannidou I, Vavlitou N, Aristodemou S, Hadjisavvas A, Kyriacou K, Scherer SS, Kleopa KA. Connexin32 mutations cause loss of function in Schwann cells and oligodendrocytes leading to PNS and CNS myelination defects. J Neurosci. 2009;29:4736–4749. doi: 10.1523/JNEUROSCI.0325-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. 2001;21:5429–5438. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–931. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Head V, Timpe LC. Identification of an inward rectifier potassium channel gene expressed in mouse cortical astrocytes. Glia. 2001;33:57–71. doi: 10.1002/1098-1136(20010101)33:1<57::aid-glia1006>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 36.Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci. 2006;26:10984–10991. doi: 10.1523/JNEUROSCI.0304-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- 38.Sohl G, Eiberger J, Jung YT, Kozak CA, Willecke K. The mouse gap junction gene connexin29 is highly expressed in sciatic nerve and regulated during brain development. Biol Chem. 2001;382:973–978. doi: 10.1515/BC.2001.122. [DOI] [PubMed] [Google Scholar]
- 39.Eiberger J, Kibschull M, Strenzke N, Schober A, Bussow H, Wessig C, Djahed S, Reucher H, Koch DA, Lautermann J, Moser T, Winterhager E, Willecke K. Expression pattern and functional characterization of connexin29 in transgenic mice. Glia. 2006;53:601–611. doi: 10.1002/glia.20315. [DOI] [PubMed] [Google Scholar]
- 40.Tang W, Zhang Y, Chang Q, Ahmad S, Dahlke I, Yi H, Chen P, Paul DL, Lin X. Connexin29 is highly expressed in cochlear Schwann cells, and it is required for the normal development and function of the auditory nerve of mice. J Neurosci. 2006;26:1991–1999. doi: 10.1523/JNEUROSCI.5055-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang JJ, Huang SH, Chou KH, Liao PJ, Su CC, Li SY. Identification of mutations in members of the connexin gene family as a cause of nonsyndromic deafness in Taiwan. Audiol Neurootol. 2007;12:198–208. doi: 10.1159/000099024. [DOI] [PubMed] [Google Scholar]
- 42.Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Doring B, Frisch C, Sohl G, Teubner B, Euwens C, Huston J, Steinhauser C, Messing A, Heinemann U, Willecke K. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci. 2003;23:766–776. doi: 10.1523/JNEUROSCI.23-03-00766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–1555. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- 44.Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci. 2009;29:7743–7752. doi: 10.1523/JNEUROSCI.0341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pannasch U, Vargova L, Reingruber J, Ezan P, Holcman D, Giaume C, Sykova E, Rouach N. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci U S A. 2011;108:8467–8472. doi: 10.1073/pnas.1016650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- 47.Mugnaini E. Cell junctions of astrocytes, ependyma, and related cells in the mammalian central nervous system, with emphasis on the hypothesis of a generalized functional syncytium of supporting cells. In: Fedoroff S, Vernadakis A, editors. Astrocytes. I. Academic; New York: 1986. pp. 329–371. [Google Scholar]
- 48.Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N. Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci. 2010;11:87–99. doi: 10.1038/nrn2757. [DOI] [PubMed] [Google Scholar]
- 49.Scemes E, Giaume C. Astrocyte calcium waves: what they are and what they do. Glia. 2006;54:716–725. doi: 10.1002/glia.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balice-Gordon RJ, Bone LJ, Scherer SS. Functional gap junctions in the schwann cell myelin sheath. J Cell Biol. 1998;142:1095–1104. doi: 10.1083/jcb.142.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–934. doi: 10.1016/s0960-9822(07)00375-2. [DOI] [PubMed] [Google Scholar]
- 52.Kausalya PJ, Reichert M, Hunziker W. Connexin45 directly binds to ZO-1 and localizes to the tight junction region in epithelial MDCK cells. FEBS Lett. 2001;505:92–96. doi: 10.1016/s0014-5793(01)02786-7. [DOI] [PubMed] [Google Scholar]
- 53.Nielsen PA, Baruch A, Shestopalov VI, Giepmans BN, Dunia I, Benedetti EL, Kumar NM. Lens connexins alpha3Cx46 and alpha8Cx50 interact with zonula occludens protein-1 (ZO-1) Mol Biol Cell. 2003;14:2470–2481. doi: 10.1091/mbc.E02-10-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X, Olson C, Lu S, Kamasawa N, Yasumura T, Rash JE, Nagy JI. Neuronal connexin36 association with zonula occludens-1 protein (ZO-1) in mouse brain and interaction with the first PDZ domain of ZO-1. Eur J Neurosci. 2004;19:2132–2146. doi: 10.1111/j.l460-9568.2004.03283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Penes MC, Li X, Nagy JI. Expression of zonula occludens-1 ZO-1 and the transcription factor ZO-1-associated nucleic acid-binding protein (ZONAB)-MsY3 in glial cells and colocalization at oligodendrocyte and astrocyte gap junctions in mouse brain. Eur J Neurosci. 2005;22:404–418. doi: 10.1111/j.1460-9568.2005.04225.x. [DOI] [PubMed] [Google Scholar]
- 56.Stout C, Goodenough DA, Paul DL. Connexins: functions without junctions. Curr Opin Cell Biol. 2004;16:507–512. doi: 10.1016/j.ceb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 57.Li X, Penes M, Odermatt B, Willecke K, Nagy JI. Ablation of Cx47 in transgenic mice leads to the loss of MUPP1, ZONAB and multiple connexins at oligodendrocyte-astrocyte gap junctions. Eur J Neurosci. 2008;28:1503–1517. doi: 10.1111/j.1460-9568.2008.06431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Balda MS, Matter K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. Embo J. 2000;19:2024–2033. doi: 10.1093/emboj/19.9.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flores AI, Mallon BS, Matsui T, Ogawa W, Rosenzweig A, Okamoto T, Macklin WB. Akt-mediated survival of oligodendrocytes induced by neuregulins. J Neurosci. 2000;20:7622–7630. doi: 10.1523/JNEUROSCI.20-20-07622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park SK, Miller R, Krane I, Vartanian T. The erbB2 gene is required for the development of terminally differentiated spinal cord oligodendrocytes. J Cell Biol. 2001;154:1245–1258. doi: 10.1083/jcb.200104025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim JY, Sun Q, Oglesbee M, Yoon SO. The role of ErbB2 signaling in the onset of terminal differentiation of oligodendrocytes in vivo. J Neurosci. 2003;23:5561–5571. doi: 10.1523/JNEUROSCI.23-13-05561.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Balda MS, Garrett MD, Matter K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J Cell Biol. 2003;160:423–432. doi: 10.1083/jcb.200210020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sourisseau T, Georgiadis A, Tsapara A, Ali RR, Pestell R, Matter K, Balda MS. Regulation of PCNA and cyclin D1 expression and epithelial morphogenesis by the ZO-1-regulated transcription factor ZONAB/DbpA. Mol Cell Biol. 2006;26:2387–2398. doi: 10.1128/MCB.26.6.2387-2398.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qin H, Shao Q, Curtis H, Galipeau J, Belliveau DJ, Wang T, Alaoui-Jamali MA, Laird DW. Retroviral delivery of connexin genes to human breast tumor cells inhibits in vivo tumor growth by a mechanism that is independent of significant gap junctional intercellular communication. J Biol Chem. 2002;277:29132–29138. doi: 10.1074/jbc.M200797200. [DOI] [PubMed] [Google Scholar]
- 65.Omori Y, Yamasaki H. Mutated connexin43 proteins inhibit rat glioma cell growth suppression mediated by wild-type connexin43 in a dominant-negative manner. Int J Cancer. 1998;78:446–453. doi: 10.1002/(sici)1097-0215(19981109)78:4<446::aid-ijc10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 66.Moorby C, Patel M. Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp Cell Res. 2001;271:238–248. doi: 10.1006/excr.2001.5357. [DOI] [PubMed] [Google Scholar]
- 67.Doble BW, Dang X, Ping P, Fandrich RR, Nickel BE, Jin Y, Cattini PA, Kardami E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J Cell Sci. 2004;117:507–514. doi: 10.1242/jcs.00889. [DOI] [PubMed] [Google Scholar]
- 68.Freidin M, Asche S, Bargiello TA, Bennett MV, Abrams CK. Connexin 32 increases the proliferative response of Schwann cells to neuregulin-1 (Nrg1) Proc Natl Acad Sci U S A. 2009;106:3567–3572. doi: 10.1073/pnas.0813413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin JH, Yang J, Liu S, Takano T, Wang X, Gao Q, Willecke K, Nedergaard M. Connexin mediates gap junction-independent resistance to cellular injury. J Neurosci. 2003;23:430–441. doi: 10.1523/JNEUROSCI.23-02-00430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dale N. Dynamic ATP signalling and neural development. J Physiol. 2008;586:2429–2436. doi: 10.1113/jphysiol.2008.152207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 72.Orellana JA, Froger N, Ezan P, Jiang JX, Bennett MV, Naus CC, Giaume C, Saez JC. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J Neurochem. 2011 doi: 10.1111/j.1471-4159.2011.07210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rana S, Dringen R. Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci Lett. 2007;415:45–48. doi: 10.1016/j.neulet.2006.12.043. [DOI] [PubMed] [Google Scholar]
- 75.O’Carroll SJ, Alkadhi M, Nicholson LF, Green CR. Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell communication & adhesion. 2008;15:27–42. doi: 10.1080/15419060802014164. [DOI] [PubMed] [Google Scholar]
- 76.Contreras JE, Sanchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, Bukauskas FF, Bennett MV, Saez JC. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci U S A. 2002;99:495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Karpuk N, Burkovetskaya M, Fritz T, Angle A, Kielian T. Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J Neurosci. 2011;31:414–425. doi: 10.1523/JNEUROSCI.5247-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Retamal MA, Froger N, Palacios-Prado N, Ezan P, Saez PJ, Saez JC, Giaume C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci. 2007;27:13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Froger N, Orellana JA, Cohen-Salmon M, Ezan P, Amigou E, Saez JC, Giaume C. Cannabinoids prevent the opposite regulation of astroglial connexin43 hemichannels and gap junction channels induced by pro-inflammatory treatments. J Neurochem. 2009;111:1383–1397. doi: 10.1111/j.1471-4159.2009.06407.x. [DOI] [PubMed] [Google Scholar]
- 80.Froger N, Orellana JA, Calvo CF, Amigou E, Kozoriz MG, Naus CC, Saez JC, Giaume C. Inhibition of cytokine-induced connexin43 hemichannel activity in astrocytes is neuroprotective. Mol Cell Neurosci. 2010;45:37–46. doi: 10.1016/j.mcn.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 81.Iglesias R, Dahl G, Qiu F, Spray DC, Scemes E. Pannexin 1: the molecular substrate of astrocyte “hemichannels”. J Neurosci. 2009;29:7092–7097. doi: 10.1523/JNEUROSCI.6062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Valiunas V, Weingart R. Electrical properties of gap junction hemichannels identified in transfected HeLa cells. Pflugers Arch. 2000;440:366–379. doi: 10.1007/s004240000294. [DOI] [PubMed] [Google Scholar]
- 83.Essenfelder GM, Bruzzone R, Lamartine J, Charollais A, Blanchet-Bardon C, Barbe MT, Meda P, Waksman G. Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum Mol Genet. 2004;13:1703–1714. doi: 10.1093/hmg/ddh191. [DOI] [PubMed] [Google Scholar]
- 84.Diekmann S, Henneke M, Burckhardt BC, Gartner J. Pelizaeus-Merzbacher-like disease is caused not only by a loss of connexin47 function but also by a hemichannel dysfunction. Eur J Hum Genet. 2010;18:985–992. doi: 10.1038/ejhg.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Vuyst E, Decrock E, Cabooter L, Dubyak GR, Naus CC, Evans WH, Leybaert L. Intracellular calcium changes trigger connexin 32 hemichannel opening. The EMBO journal. 2006;25:34–44. doi: 10.1038/sj.emboj.7600908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Castro C, Gomez-Hernandez JM, Silander K, Barrio LC. Altered formation of hemichannels and gap junction channels caused by C-terminal connexin-32 mutations. J Neurosci. 1999;19:3752–3760. doi: 10.1523/JNEUROSCI.19-10-03752.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sanchez HA, Orellana JA, Verselis VK, Saez JC. Metabolic inhibition increases activity of connexin-32 hemichannels permeable to Ca2+ in transfected HeLa cells. Am J Physiol Cell Physiol. 2009;297:C665–678. doi: 10.1152/ajpcell.00200.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 89.Stojkovic T, Latour P, Vandenberghe A, Hurtevent JF, Vermersch P. Sensorineural deafness in X-linked Charcot-Marie-Tooth disease with connexin 32 mutation (R142Q) Neurology. 1999;52:1010–1014. doi: 10.1212/wnl.52.5.1010. published erratum appears in Neurology 1999 Jun 10;52(9):1952. [DOI] [PubMed] [Google Scholar]
- 90.Takashima H, Nakagawa M, Umehara F, Hirata K, Suehara M, Mayumi H, Yoshishige K, Matsuyama W, Saito M, Jonosono M, Arimura K, Osame M. Gap junction protein beta 1 (GJB1) mutations and central nervous system symptoms in X-linked Charcot-Marie-Tooth disease. Acta Neurol Scand. 2003;107:31–37. doi: 10.1034/j.1600-0404.2003.01317.x. [DOI] [PubMed] [Google Scholar]
- 91.Lee MJ, Nelson I, Houlden H, Sweeney MG, Hilton-Jones D, Blake J, Wood NW, Reilly MM. Six novel connexin32 (GJB1) mutations in X-linked Charcot-Marie-Tooth disease. J Neurol Neurosurg Psychiatry. 2002;73:304–306. doi: 10.1136/jnnp.73.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karadima G, Panas M, Floroskufi P, Kalfakis N, Vassilopoulos D. A V38A mutation in X-linked Charcot-Marie-Tooth neuropathy with unusual clinical features. J Neurol. 2004;251:222–223. doi: 10.1007/s00415-004-0284-8. [DOI] [PubMed] [Google Scholar]
- 93.Nicholson G, Corbett A. Slowing of central conduction in X-linked Charcot-Marie-Tooth neuropathy shown by brain stem auditory evoked responses. J Neurol Neurosurg Psychiatry. 1996;61:43–46. doi: 10.1136/jnnp.61.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nicholson GA, Yeung L, Corbett A. Efficient neurophysiologic selection of X-linked Charcot-Marie-Tooth families: ten novel mutations. Neurology. 1998;51:1412–1416. doi: 10.1212/wnl.51.5.1412. [DOI] [PubMed] [Google Scholar]
- 95.Bahr M, Andres F, Timmerman V, Nelis ME, Van Broeckhoven C, Dichgans J. Central visual, acoustic, and motor pathway involvement in a Charcot-Marie-Tooth family with an Asn205Ser mutation in the connexin32 geen. J Neurol Neurosurg Psychiatry. 1999;66:202–206. doi: 10.1136/jnnp.66.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Murru MR, Vannelli A, Marrosu G, Cocco E, Corongiu D, Tranquilli S, Cherchi MV, Mura M, Barberini L, Mallarini G, Marrosu MG. A novel Cx32 mutation causes X-linked Charcot-Marie-Tooth disease with brainstem involvement and brain magnetic resonance spectroscopy abnormalities. Neurol Sci. 2006;27:18–23. doi: 10.1007/s10072-006-0560-8. [DOI] [PubMed] [Google Scholar]
- 97.Srinivasan J, Leventer RJ, Kornberg AJ, Dahl HH, Ryan MM. Central Nervous System Signs in X-Linked Charcot-Marie-Tooth Disease After Hyperventilation. Pediatr Neurol. 2008;38:293–295. doi: 10.1016/j.pediatrneurol.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 98.Huang Y, Sirkowski EE, Stickney JT, Scherer SS. Prenylation-defective human connexin32 mutants are normally localized and function equivalently to wild-type connexin32 in myelinating Schwann cells. J Neurosci. 2005;25:7111–7120. doi: 10.1523/JNEUROSCI.1319-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cao F, Eckert R, Elfgang C, Nitsche JM, Snyder SA, DF Hu, Willecke K, Nicholson BJ. A quantitative analysis of connexin-specific permeability differences of gap junctions expressed in HeLa transfectants and Xenopus oocytes. J Cell Sci. 1998;111:31–43. doi: 10.1242/jcs.111.1.31. [DOI] [PubMed] [Google Scholar]
- 100.Panas M, Karadimas C, Avramopoulos D, Vassilopoulos D. Central nervous system involvement in four patients with Charcot-Marie-Tooth disease with connexin 32 extracellular mutations [letter] J Neurol Neurosurg Psychiatry. 1998;65:947–948. doi: 10.1136/jnnp.65.6.947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seeman P, Mazanec R, Ctvrteckova M, Smilkova D. Charcot-Marie-Tooth type X: A novel mutation in the Cx32 gene with central conduction slowing. Int J Mol Med. 2001;8:461–468. [PubMed] [Google Scholar]
- 102.Kassubek J, Bretschneider V, Sperfeld AD. Corticospinal tract MRI hyperintensity in X-linked Charcot-Marie-Tooth Disease. J Clin Neurosci. 2005;12:588–589. doi: 10.1016/j.jocn.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 103.Kawakami H, Inoue K, Sakakihara I, Nakamura S. Novel mutation in X-linked Charcot-Marie-Tooth disease associated with CNS impairment. Neurology. 2002;59:923–926. doi: 10.1212/wnl.59.6.923. [DOI] [PubMed] [Google Scholar]
- 104.Zambelis T, Panas M, Kokotis P, Karadima G, Kararizou E, Karandreas N. Central motor and sensory pathway involvement in an X-linked Charcot-Marie-Tooth family. Acta Neurol Belg. 2008;108:44–47. [PubMed] [Google Scholar]
- 105.Hahn AF, Ainsworth PJ, Naus CC, Mao J, Bolton CF. Clinical and pathological observations in men lacking the gap junction protein connexin 32. Muscle Nerve. 2000;999:S39–S48. [PubMed] [Google Scholar]
- 106.Marques W, Jr, Sweeney JG, Wood NW, Wroe SJ, Marques W. Central nervous system involvement in a novel connexin 32 mutation affecting identical twins. J Neurol Neurosurg Psychiatry. 1999;66:803–804. doi: 10.1136/jnnp.66.6.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kleopa KA, Zamba-Papanicolaou E, Alevra X, Nicolaou P, Georgiou DM, Hadjisavvas A, Kyriakides T, Christodoulou K. Phenotypic and cellular expression of two novel connexin32 mutations causing CMT1X. Neurology. 2006;66:396–402. doi: 10.1212/01.wnl.0000196479.93722.59. [DOI] [PubMed] [Google Scholar]
- 108.Karadima G, Panas M, Floroskufi P, Kalfakis N, Vassilopoulos D. Four novel connexin 32 mutations in X-linked Charcot-Marie-Tooth disease with phenotypic variability. J Neurol. 2006;253:263–264. doi: 10.1007/s00415-005-0955-0. [DOI] [PubMed] [Google Scholar]
- 109.Siskind C, Feely SM, Bernes S, Shy ME, Garbern JY. Persistent CNS dysfunction in a boy with CMT1X. J Neurol Sci. 2009;279:109–113. doi: 10.1016/j.jns.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 110.Hisama FM, Lee HH, Vashlishan A, Tekumalla P, Russell DS, Auld E, Goldstein JM. Clinical and molecular studies in a family with probable X-linked dominant Charcot-Marie-Tooth disease involving the central nervous system. Arch Neurol. 2001;58:1891–1896. doi: 10.1001/archneur.58.11.1891. [DOI] [PubMed] [Google Scholar]
- 111.Basri R, Yabe I, Soma H, Matsushima M, Tsuji S, Sasaki H. X-linked Charcot-Marie-Tooth disease (CMTX) in a severely affected female patient with scattered lesions in cerebral white matter. Intern Med. 2007;46:1023–1027. doi: 10.2169/internalmedicine.46.0047. [DOI] [PubMed] [Google Scholar]
- 112.Bort S, Nelis E, Timmerman V, Sevilla T, Cruz-Martinez A, Martinez F, Millan JM, Arpa J, Vilchez JJ, Prieto F, Van Broeckhoven C, Palau F. Mutational analysis of the MPZ, PMP22 and Cx32 genes in patients of Spanish ancestry with Charcot-Marie-Tooth disease and hereditary neuropathy with liability to pressure palsies. Hum Genet. 1997;99:746–754. doi: 10.1007/s004390050442. [DOI] [PubMed] [Google Scholar]
- 113.Paulson HL, Garbern JY, Hoban TF, Krajewski KM, Lewis RA, Fischbeck KH, Grossman RI, Lenkinski R, Kamholz JA, Shy ME. Transient central nervous system white matter abnormality in X-linked Charcot-Marie-Tooth disease. Ann Neurol. 2002;52:429–434. doi: 10.1002/ana.10305. [DOI] [PubMed] [Google Scholar]
- 114.Schelhaas HJ, Van Engelen BG, Gabreels-Festen AA, Hageman G, Vliegen JH, Van Der Knaap MS, Zwarts MJ. Transient cerebral white matter lesions in a patient with connexin 32 missense mutation. Neurology. 2002;59:2007–2008. doi: 10.1212/01.wnl.0000038390.29853.46. [DOI] [PubMed] [Google Scholar]
- 115.Hanemann CO, Bergmann C, Senderek J, Zerres K, Sperfeld AD. Transient, recurrent, white matter lesions in X-linked Charcot-Marie-Tooth disease with novel connexin 32 mutation. Arch Neurol. 2003;60:605–609. doi: 10.1001/archneur.60.4.605. [DOI] [PubMed] [Google Scholar]
- 116.Halbrich M, Barnes J, Bunge M, Joshi C. A V139M mutation also causes the reversible CNS phenotype in CMTX. Can J Neurol Sci. 2008;35:372–374. doi: 10.1017/s0317167100008994. [DOI] [PubMed] [Google Scholar]
- 117.Anand G, Maheshwari N, Roberts D, Padeniya A, Hamilton-Ayers M, van der Knaap M, Fratter C, Jayawant S. X-linked hereditary motor sensory neuropathy (type 1) presenting with a stroke-like episode. Dev Med Child Neurol. 2010;52:677–679. doi: 10.1111/j.1469-8749.2010.03674.x. [DOI] [PubMed] [Google Scholar]
- 118.Rosser T, Muir J, Panigrahy A, Baldwin EE, Boles RG. Transient leukoencephalopathy associated with X-linked Charcot-Marie-Tooth disease. J Child Neurol. 2010;25:1013–1016. doi: 10.1177/0883073809352378. [DOI] [PubMed] [Google Scholar]
- 119.Fusco C, Frattini D, Pisani F, Spaggiari F, Ferlini A, Della Giustina E. Coexistent central and peripheral nervous system involvement in a Charcot-Marie-Tooth syndrome X-linked patient. J Child Neurol. 2010;25:759–763. doi: 10.1177/0883073809344119. [DOI] [PubMed] [Google Scholar]
- 120.Taylor RA, Simon EM, Marks HG, Scherer SS. The CNS phenotype of X-linked Charcot-Marie-Tooth disease: more than a peripheral problem. Neurology. 2003;61:1475–1478. doi: 10.1212/01.wnl.0000095960.48964.25. [DOI] [PubMed] [Google Scholar]
- 121.Oh S, Ri Y, Bennett MV, Trexler EB, Verselis VK, Bargiello TA. Changes in permeability caused by connexin 32 mutations underlie X- linked Charcot-Marie-Tooth disease. Neuron. 1997;19:927–938. doi: 10.1016/s0896-6273(00)80973-3. [DOI] [PubMed] [Google Scholar]
- 122.Ressot C, Gomes D, Dautigny A, Pham-Dinh D, Bruzzone R. Connexin32 mutations associated with X-linked Charcot-Marie-Tooth disease show two distinct behaviors: loss of function and altered gating properties. J Neurosci. 1998;18:4063–4075. doi: 10.1523/JNEUROSCI.18-11-04063.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Omori Y, Mesnil M, Yamasaki H. Connexin 32 mutations from X-linked Charcot-Marie-Tooth disease patients: functional defects and dominant negative effects. Mol Biol Cell. 1996;7:907–916. doi: 10.1091/mbc.7.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kleopa KA, Yum SW, Scherer SS. Cellular mechanisms of connexin32 mutations associated with CNS manifestations. J Neurosci Res. 2002;68:522–534. doi: 10.1002/jnr.10255. [DOI] [PubMed] [Google Scholar]
- 125.Abrams CK, Freidin M, Bukauskas F, Dobrenis K, Bargiello TA, Verselis VK, Bennett MV, Chen L, Sahenk Z. Pathogenesis of X-linked Charcot-Marie-Tooth disease: differential effects of two mutations in connexin 32. J Neurosci. 2003;23:10548–10558. doi: 10.1523/JNEUROSCI.23-33-10548.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Abrams CK, Freidin MM, Verselis VK, Bennett MV, Bargiello TA. Functional alterations in gap junction channels formed by mutant forms of connexin 32: evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-Tooth disease. Brain Res. 2001;900:9–25. doi: 10.1016/s0006-8993(00)03327-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yum SW, Kleopa KA, Shumas S, Scherer SS. Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiol Dis. 2002;11:43–52. doi: 10.1006/nbdi.2002.0545. [DOI] [PubMed] [Google Scholar]
- 128.Wang HL, Chang WT, Yeh TH, Wu T, Chen MS, Wu CY. Functional analysis of connexin-32 mutants associated with X-linked dominant Charcot-Marie-Tooth disease. Neurobiol Dis. 2004;15:361–370. doi: 10.1016/j.nbd.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 129.Deschenes SM, Walcott JL, Wexler TL, Scherer SS, Fischbeck KH. Altered trafficking of mutant connexin32. J Neurosci. 1997;17:9077–9084. doi: 10.1523/JNEUROSCI.17-23-09077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chandross KJ, Spray DC, Cohen RI, Kumar NM, Kremer M, Dermietzel R, Kessler JA. TNF alpha inhibits Schwann cell proliferation, connexin46 expression, and gap junctional communication. Mol Cell Neurosci. 1996;7:479–500. doi: 10.1006/mcne.1996.0035. [DOI] [PubMed] [Google Scholar]
- 131.Dempsey JA, Forster HV, Bisgard GE, Chosy LW, Hanson PG, Kiorpes AL, Pelligrino DA. Role of cerebrospinal fluid [H+] in ventilatory deacclimatization from chronic hypoxia. J Clin Invest. 1979;64:199–205. doi: 10.1172/JCI109440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys. 2001;34:325–472. doi: 10.1017/s0033583501003705. [DOI] [PubMed] [Google Scholar]
- 133.Garbern JY. Pelizaeus-Merzbacher disease: Genetic and cellular pathogenesis. Cell Mol Life Sci. 2007;64:50–65. doi: 10.1007/s00018-006-6182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Uhlenberg B, Schuelke M, Ruschendorf F, Ruf N, Kaindl AM, Henneke M, Thiele H, Stoltenburg-Didinger G, Aksu F, Topaloglu H, Nurnberg P, Hubner C, Weschke B, Gartner J. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004;75:251–260. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Salviati L, Trevisson E, Baldoin MC, Toldo I, Sartori S, Calderone M, Tenconi R, Laverda A. A novel deletion in the GJA12 gene causes Pelizaeus-Merzbacher-like disease. Neurogenetics. 2007;8:57–60. doi: 10.1007/s10048-006-0065-x. [DOI] [PubMed] [Google Scholar]
- 136.Wolf NI, Cundall M, Rutland P, Rosser E, Surtees R, Benton S, Chong WK, Malcolm S, Ebinger F, Bitner-Glindzicz M, Woodward KJ. Frameshift mutation in GJA12 leading to nystagmus, spastic ataxia and CNS dys-/demyelination. Neurogenetics. 2007;8:39–44. doi: 10.1007/s10048-006-0062-0. [DOI] [PubMed] [Google Scholar]
- 137.Bugiani M, Al Shahwan S, Lamantea E, Bizzi A, Bakhsh E, Moroni I, Balestrini MR, Uziel G, Zeviani M. GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy. Neurology. 2006;67:273–279. doi: 10.1212/01.wnl.0000223832.66286.e4. [DOI] [PubMed] [Google Scholar]
- 138.Henneke M, Combes P, Diekmann S, Bertini E, Brockmann K, Burlina AP, Kaiser J, Ohlenbusch A, Plecko B, Rodriguez D, Boespflug-Tanguy O, Gartner J. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology. 2008;70:748–754. doi: 10.1212/01.wnl.0000284828.84464.35. [DOI] [PubMed] [Google Scholar]
- 139.Wang J, Wang H, Wang Y, Chen T, Wu X, Jiang Y. Two novel gap junction protein alpha 12 gene mutations in two Chinese patients with Pelizaeus-Merzbacher-like disease. Brain & development. 2010;32:236–243. doi: 10.1016/j.braindev.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 140.Orthmann-Murphy JL, Salsano E, Abrams CK, Bizzi A, Uziel G, Freidin MM, Lamantea E, Zeviani M, Scherer SS, Pareyson D. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain. 2009;132:426–438. doi: 10.1093/brain/awn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Inoue K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics. 2005;6:1–16. doi: 10.1007/s10048-004-0207-y. [DOI] [PubMed] [Google Scholar]
- 142.Ferrell RE, Baty CJ, Kimak MA, Karlsson JM, Lawrence EC, Franke-Snyder M, Meriney SD, Feingold E, Finegold DN. GJC2 missense mutations cause human lymphedema. Am J Hum Genet. 2010;86:943–948. doi: 10.1016/j.ajhg.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Osaka H, Hamanoue H, Yamamoto R, Nezu A, Sasaki M, Saitsu H, Kurosawa K, Shimbo H, Matsumoto N, Inoue K. Disrupted SOX10 regulation of GJC2 transcription causes Pelizaeus-Merzbacher-like disease. Annals of neurology. 2010;68:250–254. doi: 10.1002/ana.22022. [DOI] [PubMed] [Google Scholar]
- 144.Nezu A, Kimura S, Uehara S, Osaka H, Kobayashi T, Haraguchi M, Inoue K, Kawanishi C. Pelizaeus-Merzbacher-like disease: female case report. Brain & development. 1996;18:114–118. doi: 10.1016/0387-7604(95)00078-x. [DOI] [PubMed] [Google Scholar]
- 145.Tress O, Maglione M, Zlomuzica A, May D, Dicke N, Degen J, Dere E, Kettenmann H, Hartmann D, Willecke K. Pathologic and phenotypic alterations in a mouse expressing a connexin47 missense mutation that causes pelizaeus-merzbacher-like disease in humans. PLoS genetics. 2011;7:e1002146. doi: 10.1371/journal.pgen.1002146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Orthmann-Murphy JL, Enriquez AD, Abrams CK, Scherer SS. Loss-of-function GJA12/Connexin47 mutations cause Pelizaeus-Merzbacher-like disease. Mol Cell Neurosci. 2007;34:629–641. doi: 10.1016/j.mcn.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gow A, Lazzarini RA. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat Genet. 1996;13:422–428. doi: 10.1038/ng0896-422. [DOI] [PubMed] [Google Scholar]
- 148.Southwood CM, Garbern J, Jiang W, Gow A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron. 2002;36:585–596. doi: 10.1016/s0896-6273(02)01045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gow A, Southwood CM, Lazzarini RA. Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J Cell Biol. 1998;140:925–934. doi: 10.1083/jcb.140.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang K, Kaufman RJ. Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb Exp Pharmacol. 2006:69–91. doi: 10.1007/3-540-29717-0_3. [DOI] [PubMed] [Google Scholar]
- 151.Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 152.Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, Jabs EW. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Amer J Hum Genet. 2003;72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Loddenkemper T, Grote K, Evers S, Oelerich M, Stogbauer F. Neurological manifestations of the oculodentodigital dysplasia syndrome. J Neurol. 2002;249:584–595. doi: 10.1007/s004150200068. [DOI] [PubMed] [Google Scholar]
- 154.Meyer-Schwickerath G, Gruterich E, Weyers H. The microphthalmos syndrome. Klinische Monatsblatter fur Augenheilkunde und fur augenarztliche Fortbildung. 1957;131:18–30. [PubMed] [Google Scholar]
- 155.Gorlin RJ, Meskin LH, Geme JW. Oculodentodigital dysplasia. J Ped. 1963;63:69–75. doi: 10.1016/s0022-3476(63)80304-2. [DOI] [PubMed] [Google Scholar]
- 156.Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, Laurence F, Koivisto PA, Van Maldergem L, Boyadjiev SA, Bodurtha JN, Jabs EW. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat. 2009;30:724–733. doi: 10.1002/humu.20958. [DOI] [PubMed] [Google Scholar]
- 157.Wiest T, Herrmann O, Stogbauer F, Grasshoff U, Enders H, Koch MJ, GrondGinsbach C, Schwaninger M. Clinical and genetic variability of oculodentodigital dysplasia. Clin Genet. 2006;70:71–72. doi: 10.1111/j.1399-0004.2006.00631.x. [DOI] [PubMed] [Google Scholar]
- 158.Reisner SH, Kott E, Bornstein B, Salinger H, Kaplan I, Gorlin RJ. Oculodendtodigital dysplasia. Amer J Dis Child. 1969;118:600–607. doi: 10.1001/archpedi.1969.02100040602013. [DOI] [PubMed] [Google Scholar]
- 159.Cox DR, DiSalvo M, Hall BD. Neurologic abnormalities in oculodentodigital dysplasia: a new finding. Clin Res. 1978;26:193A. [Google Scholar]
- 160.Audry D, Dumas R, Nivelon A, Audry F. Manifestations neurologiques de la dysplasie oculo-dento-osseuse. Rev Otoneuroophtalmol. 1981;53:209–214. [PubMed] [Google Scholar]
- 161.Barnard A, Hamersma H, De Villiers JC, Beighton P. Intracranial calcification in oculodento-osseous dysplasia. S Afr Med J. 1981;59:758–762. [PubMed] [Google Scholar]
- 162.Nivelon-Chevallier A, Audry D, Audry F, Dumas R. Dysplasie oculo-dento-digital. A propos d’un cas avec paraplegie spasmodique. J Genet Hum. 1981;29:171–179. [PubMed] [Google Scholar]
- 163.Schrander-Stumpel CTRM, Franke CL. Central nervous system abnormalities in oculodentodigital dysplasia. Genet Counseling. 1996;7:233–235. [PubMed] [Google Scholar]
- 164.Gutmann DH, Zackai EH, McDonald-McGinn DM, Fischbeck KH, Kamholz J. Oculodentodigital dysplasia syndrome associated with abnormal cerebral white matter. Am J Med Genet A. 1991;41:18–20. doi: 10.1002/ajmg.1320410106. [DOI] [PubMed] [Google Scholar]
- 165.Ginsberg LE, Jewett T, Grub R, McLean WT. Oculodental digital dysplasia: neuroimaging in a kindred. Neuroradiology. 1996;38:84–86. doi: 10.1007/BF00593230. [DOI] [PubMed] [Google Scholar]
- 166.Honkaniemi J, Kalkkila J, Koivisto P, Kahara V, Latvala T, Simola K. Novel GJA1 mutation in oculodentodigital dysplasia. Am J Med Genet A. 2005;139:48–49. doi: 10.1002/ajmg.a.30925. [DOI] [PubMed] [Google Scholar]
- 167.Amador C, Mathews AM, Del Carmen Montoya M, Laughridge ME, Everman DB, Holden KR. Expanding the neurologic phenotype of oculodentodigital dysplasia in a 4-generation Hispanic family. J Child Neurol. 2008;23:901–905. doi: 10.1177/0883073808317730. [DOI] [PubMed] [Google Scholar]
- 168.Shapiro RE, Griffin JW, Stine OC. Evidence for genetic anticipation in the oculodentodigital syndrome. Am J Med Genet. 1997;71:36–41. [PubMed] [Google Scholar]
- 169.Churko JM, Shao Q, Gong XQ, Swoboda KJ, Bai D, Sampson J, Laird DW. Human dermal fibroblasts derived from oculodentodigital dysplasia patients suggest that patients may have wound-healing defects. Hum Mutat. 2011;32:456–466. doi: 10.1002/humu.21472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Alao MJ, Bonneau D, Holder-Espinasse M, Goizet C, Manouvrier-Hanu S, Mezel A, Petit F, Subtil D, Magdelaine C, Lacombe D. Oculo-dento-digital dysplasia: lack of genotype-phenotype correlation for GJA1 mutations and usefulness of neuro-imaging. Eur J Med Genet. 2010;53:19–22. doi: 10.1016/j.ejmg.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 171.Debeer P, Van Esch H, Huysmans C, Pijkels E, De Smet L, Van de Ven W, Devriendt K, Fryns JP. Novel GJA1 mutations in patients with oculo-dento-digital dysplasia (ODDD) Eur J Med Genet. 2005;48:377–387. doi: 10.1016/j.ejmg.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 172.Norton KK, Carey JC, Gutmann DH. Oculodentodigital dysplasia with cerebral white matter abnormalities in a two-generation family. Am J Med Genet A. 1995;57:458–461. doi: 10.1002/ajmg.1320570320. [DOI] [PubMed] [Google Scholar]
- 173.Jamsheer A, Badura-Stronka M, Sowinska A, Debicki S, Kiryluk K, Latos-Bielenska A. A severe progressive oculodentodigital dysplasia due to compound heterozygous GJA1 mutation. Clin Genet. 2010;78:94–97. doi: 10.1111/j.1399-0004.2010.01412.x. [DOI] [PubMed] [Google Scholar]
- 174.Hallermann W. Vogelgesicht und Cataracta congenita. Klin Monatsbl Augenheilkd. 1948;113:315–318. [Google Scholar]
- 175.Francois J. A new syndrome: dyscephalia with bird face and dental anomalies, nanism, hypotrichosis, cutaneous atrophy, microphthalmia, and congenital cataract. Arch Ophthal. 1958;60:842–862. [PubMed] [Google Scholar]
- 176.Streiff EB. Dysmorphie mandibulo-faciale (tete d’oiseau) et alterations oculaires. Ophthalmologica. 1950;120:79–83. doi: 10.1159/000300873. [DOI] [PubMed] [Google Scholar]
- 177.Gorlin R, Cohen M, Levin S. Syndromes of the Head and Neck. 3. Oxford University Press; New York: 1990. Hallermann-Streiff syndrome; pp. 306–308. [Google Scholar]
- 178.Damiano Salpietro C, Briuglia S, Valeria Merlino M, Piraino B, Valenzise M, Dallapiccola B. Hallerman-Streiff syndrome: patient with decreased GH and insulin-like growth factor-1. Am J Med Genet A. 2004;125A:216–218. doi: 10.1002/ajmg.a.20399. [DOI] [PubMed] [Google Scholar]
- 179.Pizzuti A, Flex E, Mingarelli R, Salpietro C, Zelante L, Dallapiccola B. A homozygous GJA1 gene mutation causes a Hallermann-Streiff/ODDD spectrum phenotype. Hum Mutat. 2004;23:286. doi: 10.1002/humu.9220. [DOI] [PubMed] [Google Scholar]
- 180.Joss SK, Ghazawy S, Tomkins S, Ahmed M, Bradbury J, Sheridan E. Variable expression of neurological phenotype in autosomal recessive oculodentodigital dysplasia of two sibs and review of the literature. Eur J Pediatr. 2008;167:341–345. doi: 10.1007/s00431-007-0468-1. [DOI] [PubMed] [Google Scholar]
- 181.Richardson RJ, Joss S, Tomkin S, Ahmed M, Sheridan E, Dixon MJ. A nonsense mutation in the first transmembrane domain of connexin 43 underlies autosomal recessive oculodentodigital syndrome - art. no. e37. J Med Genet. 2006;43:E37. doi: 10.1136/jmg.2005.037655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sigirci A, Alkan A, Bicak U, Yakinci C. Hallermann-Streiff syndrome associated with complete agenesis of the corpus callosum. J Child Neurol. 2005;20:691–693. doi: 10.1177/08830738050200081101. [DOI] [PubMed] [Google Scholar]
- 183.Hou JW. Hallermann-Streiff syndrome associated with small cerebellum, endocrinopathy and increased chromosomal breakage. Acta paediatrica. 2003;92:869–871. doi: 10.1080/08035250310003686. [DOI] [PubMed] [Google Scholar]
- 184.Reaume AG, De Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 185.McLachlan E, Manias JL, Gong XQ, Lounsbury CS, Shao Q, Bernier SM, Bai D, Laird DW. Functional characterization of oculodentodigital dysplasia-associated Cx43 mutants. Cell Commun Adhes. 2005;12:279–292. doi: 10.1080/15419060500514143. [DOI] [PubMed] [Google Scholar]
- 186.Chtchetinin J, Gifford WD, Li S, Paznekas WA, Jabs EW, Lai A. Tyrosine-dependent basolateral targeting of human connexin43-eYFP in Madin-Darby canine kidney cells can be disrupted by the oculodentodigital dysplasia mutation L90V. The FEBS journal. 2009;276:6992–7005. doi: 10.1111/j.1742-4658.2009.07407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Dobrowolski R, Sommershof A, Willecke K. Some oculodentodigital dysplasia-associated Cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J Membr Biol. 2007;219:9–17. doi: 10.1007/s00232-007-9055-7. [DOI] [PubMed] [Google Scholar]
- 188.Lai A, Le DN, Paznekas WA, Gifford WD, Jabs EW, Charles AC. Oculodentodigital dysplasia connexin43 mutations result in non-functional connexin hemichannels and gap junctions in C6 glioma cells. J Cell Sci. 2006;119:532–541. doi: 10.1242/jcs.02770. [DOI] [PubMed] [Google Scholar]
- 189.Shibayama J, Paznekas W, Seki A, Taffet S, Jabs EW, Delmar M, Musa H. Functional characterization of connexin43 mutations found in patients with oculodentodigital dysplasia. Circ Res. 2005;96:e83–91. doi: 10.1161/01.RES.0000168369.79972.d2. [DOI] [PubMed] [Google Scholar]
- 190.Roscoe W, Veitch GI, Gong XQ, Pellegrino E, Bai D, McLachlan E, Shao Q, Kidder GM, Laird DW. Oculodentodigital dysplasia-causing connexin43 mutants are non-functional and exhibit dominant effects on wild-type connexin43. J Biol Chem. 2005;280:11458–11466. doi: 10.1074/jbc.M409564200. [DOI] [PubMed] [Google Scholar]
- 191.Seki A, Coombs W, Taffet SM, Delmar M. Loss of electrical communication, but not plaque formation, after mutations in the cytoplasmic loop of connexin43. Heart Rhythm. 2004;1:227–233. doi: 10.1016/j.hrthm.2004.03.066. [DOI] [PubMed] [Google Scholar]
- 192.Gong XQ, Shao Q, Lounsbury CS, Bai D, Laird DW. Functional characterization of a GJA1 frameshift mutation causing oculodentodigital dysplasia and palmoplantar keratoderma. J Biol Chem. 2006;281:31801–31811. doi: 10.1074/jbc.M605961200. [DOI] [PubMed] [Google Scholar]
- 193.Dobrowolski R, Sasse P, Schrickel JW, Watkins M, Kim JS, Rackauskas M, Troatz C, Ghanem A, Tiemann K, Degen J, Bukauskas FF, Civitelli R, Lewalter T, Fleischmann BK, Willecke K. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum Mol Genet. 2008;17:539–554. doi: 10.1093/hmg/ddm329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Oh SY, Dupont E, Madhukar BV, Briand JP, Chang CC, Beyer E, Trosko JE. Characterization of gap junctional communication-deficient mutants of a rat liver epithelial cell line. Eur J Cell Biol. 1993;60:250–255. [PubMed] [Google Scholar]
- 195.Flenniken AM, Osborne LR, Anderson N, Ciliberti N, Fleming C, Gittens JE, Gong XQ, Kelsey LB, Lounsbury C, Moreno L, Nieman BJ, Peterson K, Qu D, Roscoe W, Shao Q, Tong D, Veitch GI, Voronina I, Vukobradovic I, Wood GA, Zhu Y, Zirngibl RA, Aubin JE, Bai D, Bruneau BG, Grynpas M, Henderson JE, Henkelman RM, McKerlie C, Sled JG, Stanford WL, Laird DW, Kidder GM, Adamson SL, Rossant J. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development. 2005;132:4375–4386. doi: 10.1242/dev.02011. [DOI] [PubMed] [Google Scholar]
- 196.Wasseff S, Abrams CK, Scherer SS. A dominant connexin43 mutant does not have dominant effects on gap junction coupling in astrocytes. Neuron Glia Biol. 2011:1–11. doi: 10.1017/S1740925X11000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kalcheva N, Qu J, Sandeep N, Garcia L, Zhang J, Wang Z, Lampe PD, Suadicani SO, Spray DC, Fishman GI. Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc Natl Acad Sci U S A. 2007;104:20512–20516. doi: 10.1073/pnas.0705472105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yeager M, Nicholson BJ. Structure of gap junction intercellular channels. Curr Opin Struct Biol. 1996;6:183–192. doi: 10.1016/s0959-440x(96)80073-x. [DOI] [PubMed] [Google Scholar]
- 199.Frisch C, Theis M, De Souza Silva MA, Dere E, Sohl G, Teubner B, Namestkova K, Willecke K, Huston JP. Mice with astrocyte-directed inactivation of connexin43 exhibit increased exploratory behaviour impaired motor capacities and changes in brain acetylcholine levels. Eur J Neurosci. 2003;18:2313–2318. doi: 10.1046/j.1460-9568.2003.02971.x. [DOI] [PubMed] [Google Scholar]
- 200.Dere E, De Souza-Silva MA, Frisch C, Teubner B, Sohl G, Willecke K, Huston JP. Connexin30-deficient mice show increased emotionality and decreased rearing activity in the open-field along with neurochemical changes. Eur J Neurosci. 2003;18:629–638. doi: 10.1046/j.1460-9568.2003.02784.x. [DOI] [PubMed] [Google Scholar]
- 201.Himi M, Fujimaki T, Yokoyama T, Fujiki K, Takizawa T, Murakami A. A case of oculodentodigital dysplasia syndrome with novel GJA1 gene mutation. Jpn J Ophthalmol. 2009;53:541–545. doi: 10.1007/s10384-009-0711-6. [DOI] [PubMed] [Google Scholar]
- 202.Kelly SC, Ratajczak P, Keller M, Purcell SM, Griffin T, Richard G. A novel GJA 1 mutation in oculo-dento-digital dysplasia with curly hair and hyperkeratosis. Eur J Dermatol. 2006;16:241–245. [PubMed] [Google Scholar]
- 203.Zach GA. Ocuolodento-osseus dysplasia syndrome. Oral Surg. 1975;40:122–125. doi: 10.1016/0030-4220(75)90354-0. [DOI] [PubMed] [Google Scholar]
- 204.Judisch GF, Mrtin-Casals A, Hanson JW, Olin WH. Oculodentodigital dysplasia - four new reports and a literature review. Arch Ophthalmol. 1979;97:878–884. doi: 10.1001/archopht.1979.01020010436007. [DOI] [PubMed] [Google Scholar]
- 205.Kellermayer R, Keller M, Ratajczak P, Richardson E, Harangi F, Merei E, Melegh B, Kosztolanyi G, Richard G. Bigenic connexin mutations in a patient with hidrotic ectodermal dysplasia. Eur J Dermatol. 2005;15:75–79. [PubMed] [Google Scholar]
- 206.Boyadjiev SA, Jabs EW, LaBuda M, Jamal JE, Torbergsen T, Ptacek LJI, Rogers RC, Nyberg-Hansen R, Opjordsmoen S, Zeller CB, Stine OC, Stalker HJ, Zori RT, Shapiro RE. Linkage analysis narrows the critical region for oculodentodigital dysplasia to chromosome 6q22-q23. Genomics. 1999;58:34–40. doi: 10.1006/geno.1999.5814. [DOI] [PubMed] [Google Scholar]
- 207.Gellis SS, Feingold M. Picture of the month. Am J Dis Child. 1974;128:81–82. doi: 10.1001/archpedi.1974.02110260083015. [DOI] [PubMed] [Google Scholar]
- 208.Vasconcellos JPC, Melo MB, Schimiti RB, Bressanim NC, Costa FF, Costa VP. A novel mutation in the GJA1 gene in a family with oculodentodigital dysplasia. Arch Ophthalmol. 2005;123:1422–1426. doi: 10.1001/archopht.123.10.1422. [DOI] [PubMed] [Google Scholar]
- 209.Pizzuti A, Flex E, Mingarelli R, Salpietro C, Zelante L, Dallapiccola B. a homozygous GJA1 gene mutation causes a Hallermann-Streiff/ODDD spectrum phentoype. Hum Mutation on line. 2004:691. doi: 10.1002/humu.9220. [DOI] [PubMed] [Google Scholar]
- 210.Woolridge WE, Anthony dD, Olson ER, Bates GP, Sammon TJ. Oculodentodigital dysplasia. Mol Med. 1977;74:379–383. [PubMed] [Google Scholar]
- 211.Vitiello C, D’Adamo P, Gentile F, Vingolo EM, Gasparini P, Banfi S. A novel GJA1 mutation causes oculodentodigital dysplasia without syndactyly. Am J Med Genet. 2005;133A:58–60. doi: 10.1002/ajmg.a.30554. [DOI] [PubMed] [Google Scholar]
- 212.Richardson RR, Donnai D, Meire F, Dixon MJ. Expression of Gja1 correlates with the phenotype observed in oculodentodigital syndrome/type III syndactyly. J Med Genet. 2004;41:60–67. doi: 10.1136/jmg.2003.012005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fenwick A, Richardson RJ, Butterworth J, Barron MJ, Dixon MJ. Novel mutations in GJA1 cause oculodentodigital syndrome. Journal of dental research. 2008;87:1021–1026. doi: 10.1177/154405910808701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.van Steensel MAM, Spruijt L, van der Burgt I, Bladergroen RS, Vermeer M, Steijlen PM, van Geel M. A 2-bp deletion in the GJA1 gene is associated with oculo-dento-digital dysplasia with palmoplnatar keratoderma. Am J Med Genet. 2005;132A:171–174. doi: 10.1002/ajmg.a.30412. [DOI] [PubMed] [Google Scholar]
- 215.Vreeburg M, deZwartStorm EA, Schouten MI, Nellen RGL, MarcusSoekarman D, Devies M, vanGeel M, vanSteensel MAM. Skin changes in oculo-dento-digital dysplasia are correlated with C-terminal truncations of connexin 43. Am J Med Genet. 2007;143A:360–363. doi: 10.1002/ajmg.a.31558. [DOI] [PubMed] [Google Scholar]
- 216.de la Parra DR, Zenteno JC. A new GJA1 (connexin 43) mutation causing oculodentodigital dysplasia associated to uncommon features. Ophthalmic Genet. 2007;28:198–202. doi: 10.1080/13816810701538620. [DOI] [PubMed] [Google Scholar]
- 217.Di WL, Gu Y, Common JE, Aasen T, O’Toole E A, Kelsell DP, Zicha D. Connexin interaction patterns in keratinocytes revealed morphologically and by FRET analysis. J Cell Sci. 2005;118:1505–1514. doi: 10.1242/jcs.01733. [DOI] [PubMed] [Google Scholar]
- 218.Feller L, Wood NH, Sluiter MD, Noffke C, Raubenheimer EJ, Lemmer J, van Rensburg EJ. Report of a black South African child with oculodentodigital dysplasia and a novel GJA1 gene mutation. Am J Med Genet A. 2008;146A:1350–1353. doi: 10.1002/ajmg.a.32272. [DOI] [PubMed] [Google Scholar]
- 219.Vasconcellos JP, Melo MB, Schimiti RB, Bressanim NC, Costa FF, Costa VP. A novel mutation in the GJA1 gene in a family with oculodentodigital dysplasia. Arch Ophthalmol. 2005;123:1422–1426. doi: 10.1001/archopht.123.10.1422. [DOI] [PubMed] [Google Scholar]
- 220.Kjaer AW, Hansen L, Eiberg H, Leicht P, Opitz JM, Tommerup N. Novel connexin 43 (GJA1) mutation causes oculo-dento-digital dysplasia with curly hair. Am J Med Genet. 2004;127A:152–157. doi: 10.1002/ajmg.a.20614. [DOI] [PubMed] [Google Scholar]
- 221.Debeer P, VanEsch H, Huysmans C, Pijkels E, DeSmet L, VandeVen W, Devriendt K, Fryns JP. Novel GJA1 mutations in patients with oculo-dento-digital dysplasia (ODDD) Eur J Med Genet. 2005;48:377–387. doi: 10.1016/j.ejmg.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 222.Schrander-Stumpel CT, De Groot-Wijnands JB, De Die-Smulders C, Fryns JP. Type III syndactyly and oculodentodigital dysplasia: a clinical spectrum. Genetic counseling. 1993;4:271–276. [PubMed] [Google Scholar]
- 223.Rajic DS, de Veber LL. Hereditary oculodentoosseous dysplasia. Ann Radiol. 1966;9:224–231. [Google Scholar]
- 224.Gong XQ, Shao Q, Langlois S, Bai D, Laird DW. Differential potency of dominant negative connexin43 mutants in oculodentodigital dysplasia. J Biol Chem. 2007;282:19190–19202. doi: 10.1074/jbc.M609653200. [DOI] [PubMed] [Google Scholar]
- 225.Traboulsi EI, Parks MM. Glaucoma in oculo-dento-osseous dysplasia. Am Journal of Ophthalmol. 1990;109:310–313. doi: 10.1016/s0002-9394(14)74556-8. [DOI] [PubMed] [Google Scholar]
- 226.Stanislaw CL, Narvaez C, Roger RG, Woodard CS. Oculodentodigital dysplasia with cerebral white matter abnormalies: an additional case. Proc Greenwood Genet Center. 1998;17:20–24. [Google Scholar]
- 227.Opjordsmoen S, Nyberg-Hansen R. Hereditary spastic paraplegia with neurogenic bladder disturbances and syndactylia. Acta Neurol Scand. 1980;61:35–41. doi: 10.1111/j.1600-0404.1980.tb02993.x. [DOI] [PubMed] [Google Scholar]
- 228.Mohr OL. Dominant acrocephalosyndactyly in a Norwegian family. Hereditas. 1939;25:193–203. [Google Scholar]
- 229.Musa FU, Ratajczak P, Sahu J, Pentlicky S, Fryer A, Richard G, Willoughby CE. Ocular manifestations in oculodentodigital dysplasia resulting from a heterozygous missense mutation (L113P) in GJA1 (connexin 43) Eye. 2009;23:549–555. doi: 10.1038/eye.2008.77. [DOI] [PubMed] [Google Scholar]
- 230.Gladwin A, Donnai D, Metcalfe K, Schrander-Stumpel C, Brueton L, Verloes A, Aylsworth A, Toriello H, Winter R, Dixon M. Localization of a gene for oculodentodigital syndrome to human chromosome 6q22-q24. Hum Mol Genet. 1997;6:123–127. doi: 10.1093/hmg/6.1.123. [DOI] [PubMed] [Google Scholar]
- 231.van Es RJ, Wittebol-Post D, Beemer FA. Oculodentodigital dysplasia with mandibular retrognathism and absence of syndactyly: a case report with a novel mutation in the connexin 43 gene. International journal of oral and maxillofacial surgery. 2007;36:858–860. doi: 10.1016/j.ijom.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 232.Schrander-Stumpel CT, Franke CL. Central nervous system abnormalities in oculodentodigital dysplasia. Genetic counseling. 1996;7:233–235. [PubMed] [Google Scholar]