

Three tyrosine-based sorting signals in the gap junction protein connexin 43 were identified, two of which function cooperatively as adaptor protein complex-2 binding sites. The analyses provide a molecular model for clathrin to efficiently internalize large plasma membrane structures and suggest a mechanism for regulating constitutive versus acute gap junction internalization.
Abstract
Gap junction (GJ) channels that electrically and chemically couple neighboring cells are formed when two hemichannels (connexons) of apposed cells dock head-on in the extracellular space. Remarkably, docked connexons are inseparable under physiological conditions, and we and others have shown that GJs are internalized in whole, utilizing the endocytic clathrin machinery. Endocytosis generates double-membrane vesicles (annular GJs or connexosomes) in the cytoplasm of one of the apposed cells that are degraded by autophagosomal and, potentially, endo/lysosomal pathways. In this study, we investigated the structural motifs that mediate Cx43 GJ endocytosis. We identified three canonical tyrosine-based sorting signals of the type “YXXΦ” in the Cx43 C-terminus, two of which function cooperatively as AP-2 binding sites. We generated a set of green fluorescent protein–tagged and untagged Cx43 mutants that targeted these two sites either individually or together. Mutating both sites completely abolished Cx43-AP-2/Dab2/clathrin interaction and resulted in increased GJ plaque size, longer Cx43 protein half-lives, and impaired GJ internalization. Interestingly, Dab2, an accessory clathrin adaptor found earlier to be important for GJ endocytosis, interacts indirectly with Cx43 via AP-2, permitting the recruitment of up to four clathrin complexes per Cx43 protein. Our analyses provide a mechanistic model for clathrin's efficient internalization of large plasma membrane structures, such as GJs.
INTRODUCTION
Gap junction intercellular communication (GJIC) has been shown to play a crucial role for all aspects of multicellular life, including embryonic development, tissue function, and cellular homeostasis. Connexins (Cxs) are the four-pass transmembrane proteins that form gap junction (GJ) channels. Twenty-one different Cx isoforms have been identified in humans. They are named Cx, followed by the predicted molecular weight of the protein. Cx43 is the most abundant Cx type expressed in most tissues. Six Cx polypeptides oligomerize to form a hemichannel termed a connexon. Complete double membrane–spanning GJ channels are formed when two connexons of apposed cells dock head-on in the extracellular space. Typically, tens to many thousands of GJ channels cluster together to form arrays of channels, termed GJ plaques. GJ channels electrically and chemically couple neighboring cells and mediate intercellular communication by allowing direct transfer of small hydrophilic molecules <1.5 kDa in size (e.g., second messengers, such as cAMP and IP3; ions; metabolites; and potentially small RNA molecules) through the channels via passive diffusion. GJs, based on their tightly sealed double membrane–spanning configuration, are likely to also significantly contribute to physical cell-to-cell adhesion.
Mutations in Cxs have been linked to a number of severe diseases: mutations in the GJB2 gene (encoding Cx26) are a major cause for inherited and sporadic nonsyndromic hearing impairments (Morell et al., 1998; Kenneson et al., 2002); mutations in the GJB1 gene (encoding Cx32) were found in X-linked Charcot-Marie-Tooth syndrome neuropathy patients (Lee et al., 2002); mutations in Cx46 and Cx50 were found to cause cataracts (Gong et al., 1997; White et al., 1998); and mutations in GJA1 (encoding Cx43) lead to serious cardiac diseases, such as hypertrophy, ischemia, and heart failure (Fontes et al., 2012), as well as craniofacial bone defects (oculodentodigital dysplasia, ODDD syndrome) and a number of acute skin disorders (Batra et al., 2012; Xu and Nicholson, 2013). Clearly, all of these examples demonstrate the crucial role of GJIC in multicellular life and illustrate that GJ channel assembly and degradation needs to be regulated precisely to ensure accurate physiological functions.
Elegant studies by Goodenough and Gilula (1974) and Ghoshroy et al. (1995) revealed that, once docked, connexons cannot be separated back into individual connexons under physiological conditions. Yet active, proliferating cells, including embryonic and somatic stem cells, are known to uncouple from their neighbors in order to undergo mitosis (Boassa et al., 2011), and modulation of cellular uncoupling and recoupling plays an important role in many additional physiological and pathological conditions, such as cell migration during development and wound healing, apoptosis, leukocyte extravasation, ischemia, hemorrhage, edema, and cancer metastasis. Furthermore, channels of GJ plaques have been shown to be constitutively endocytosed (Falk et al., 2009), correlating with the short half-life of 1–5 h reported for Cx proteins and GJs (Fallon and Goodenough, 1981; Beardslee et al., 1998; Falk et al., 2009). However, the signals and mechanisms that trigger GJ endocytosis remain unclear. We and others found that cells remove their GJ plaques from the plasma membrane (PM) by an endocytic process that resembles clathrin-mediated endocytosis (CME) and involves the coat protein clathrin itself; the classical PM clathrin adaptor, adaptor protein complex-2 (AP-2); the alternative clathrin adaptor protein Disabled-2 (Dab2); the GTPase dynamin2; the retrograde actin motor myosin-VI; and actin filament assembly (Piehl et al., 2007; Baker et al., 2008; Gilleron et al., 2008, 2011; Gumpert et al., 2008; Johnson et al., 2013). GJ endocytosis generates cytoplasmic double-membrane vesicles termed annular gap junctions (AGJs) or connexosomes (Jordan et al., 2001; Gaietta et al., 2002; Lauf et al., 2002; Piehl et al., 2007; Falk et al., 2009). AGJs subsequent to internalization were found to be degraded by autophagosomal (Hesketh et al., 2010; Lichtenstein et al., 2011; Bejarano et al., 2012; Fong et al., 2012) and, in phorbol ester 12-o-tetradecanoylphorbol-13-acetate (TPA)-treated cells, endo/lysosomal pathways (Leithe et al., 2009; Fykerud et al., 2012). To better understand how clathrin and clathrin adaptors may internalize GJs, we analyzed the Cx43 amino acid sequence for potential AP-2 and Dab2 interaction sites, mutated putative sites by generating a set of amino acid exchange and deletion constructs, and analyzed their impact on GJ internalization. We found that two separate AP-2 binding sites located in the Cx43 C-terminus interact with and recruit AP-2, Dab2, and clathrin to the Cx43 C-terminus, and that deletion or mutation of these sites impaired GJ internalization if only one site was mutated or completely abolished it if both sites were mutated together. Interestingly, our analyses revealed the potential recruitment of up to four clathrin triskelia per Cx43 polypeptide, suggesting a potential molecular mechanism for the efficient CME of large PM structures such as GJs.
RESULTS
Design and construction of Cx43 mutants that interfere with AP-2/clathrin binding
GJ internalization was found to utilize the CME machinery (Piehl et al., 2007; Gumpert et al., 2008; Johnson et al., 2013), correlating with earlier observations of clathrin colocalizing with vesicular, cytoplasmic GJ structures (Larsen et al., 1979). Clathrin typically does not interact directly with its cargo (here the Cx43 protein) but instead requires adaptor proteins to link the cargo to the clathrin coat. As noted above, both clathrin adaptors, AP-2 and Dab2, have been shown previously to be involved in GJ internalization, and knocking down AP-2 and Dab2 protein levels using RNA interference (RNAi) technology significantly reduced GJ internalization (Piehl et al., 2007; Gumpert et al., 2008). AP-2 is a well-characterized CME adaptor protein complex consisting of α-adaptin, β2, σ2, and μ2 subunits that is known to bind to PM-localized cargo proteins via three different binding motifs: two types of tyrosine-based sorting signals with either 1) YXXΦ (in which Y is tyrosine, an essential residue that cannot be substituted, even by structurally related phenylalanine or phospho-tyrosine residues; X, which can be any amino acid; and Φ, which is an amino acid with a bulky hydrophobic side chain, such as F, M, L, I, or V); 2) NPXY sequence (in which N is asparagine, P is proline, X is any amino acid, Y is tyrosine) that bind to the μ2 subunit; or 3) via a dileucine-based sorting signal, [D/E]XXXL[L/I], that can bind to both μ2 and β2 subunits (Bonifacino and Traub, 2003). Dab2 is also known to bind to NPXY sequences (Morris and Cooper, 2001; Mishra et al., 2002; Motley et al., 2003). We did not find any NPXY signals in the Cx43 sequence, suggesting that Dab2 may interact indirectly with Cx43 (see below) and consistent with the observation that NPXY motifs are typically only present in single-spanning membrane proteins (Bonifacino and Traub, 2003). We also did not find any dileucine-based sorting signals in the Cx43 sequence; however, dileucine-based sorting signals are present in the C-terminus of a number of other Cxs (Thévenin et al., 2013). We found three conserved YXXΦ consensus sequences in the Cx43 C-terminus located at positions Y230VFF, Y265AYF, and Y286KLV. These sites were named sorting signals, S1, S2, and S3, respectively (Figure 1A). Endocytic YXXΦ signals normally are located at least 10 amino acid residues away from a transmembrane domain to allow sufficient space for AP-2 access (Collins et al., 2002; Bonifacino and Traub, 2003). Sorting signal S1 (Y230VFF) is located immediately adjacent to transmembrane domain 4 (TM4), which makes it unlikely that S1 would serve as a functional AP-2 binding site. Indeed, we did not gain any evidence that suggests that this site in Cx43 interacts with AP-2 (see results of mutants with sites S2 and S3 [S2+3] mutated/deleted presented in the following section). We generated more than 20 different Cx43 mutants (all either untagged or tagged with green fluorescent protein [GFP] on the C-terminus), including 15 point mutations (Y to H or F to A) and seven small (three to seven amino acids) and two large (36 and 128 amino acids) deletion mutants that targeted the putative S1, S2, and S3 AP-2 binding sites and rendered them nonfunctional. Both deletions and point mutants were constructed and tested to reduce the potential impact of side effects. Selected constructs characterized here are shown in Figure 1A. Note that the sorting signal S3 is a portion of a larger, previously identified regulatory sequence (termed proline-rich region) that consists of the S3 signal and a directly upstream, overlapping PY motif that both were found to influence GJ stability without, however, additive effect (Thomas et al., 2003; see Discussion for further detail). To avoid potential interference of these two signals, we deleted seven amino acid residues, including the PY-motif and the S3 sorting signal, in S3 and S2+3 mutants (mutants 6–8 in Figure 1A). Also note that the Y286 (knocking out both signals) was mutated in the S3 motif, while F268 of the S2 motif was mutated (mutants 2, 4, and 5 in Figure 1A). The Y265 in the S2 motif is a well-known c-Src phosphorylation site (Postma et al., 1998; Solan and Lampe, 2008; reviewed in Thévenin et al., 2013), and mutation of this amino acid residue was expected to generate severe site effects. Mutating either Y or Φ of the canonical YXXΦ motif was found to abolish its function (Bonifacino and Traub, 2003). When transfected into HeLa cells, all constructs produced Cx43 proteins that migrated on SDS–polyacrylamide gels according to their predicted molecular weight (shown for GFP-tagged constructs in Figure 1B). Also, all constructs assembled into GJ plaques in apposing membranes of transfected cells, as shown for selected constructs in Figures 3, 4, 5, and 7, which are discussed later in this article.
FIGURE 1:

Cx43 mutants that interfere with AP-2/clathrin binding. (A) The Cx43 C-terminus harbors three canonical tyrosine-based sorting signals of the type YXXΦ at positions Y230VFF, Y265AYF, and Y286KLV, which we named S1, S2, and S3 (shown in red), that can function as putative binding sites for the classical PM clathrin adaptor, AP-2. To test their role in recruiting AP-2 to Cx43, we generated a set of mutants that abolished the AP-2 binding capacity by mutating known critical residues of the motifs (Y or F) into H or A, or by deleting the amino acid residues of the motifs, either partially (three amino acid residue deletions) or entirely (seven or more amino acid deletions) (depicted with asterisks, see the text for details). Single-, double-, and triple-site mutants (depicted S1, S2, S3, S2+3, and S1+2+3) were constructed. Mutants 2–10 characterized in this work are shown. (B) Immunoblot analysis (using anti-GFP antibodies) of wild type (lane 1) and mutant Cx43-GFP constructs (lanes 2–10). Wild type and mutants produced Cx43 proteins that migrated on SDS–PAGE gels corresponding to their predicted molecular weight. M, marker proteins.
FIGURE 3:

The accessory clathrin adaptor, Dab2, interacts indirectly with Cx43 via AP-2. (A) HeLa cells were transfected with wild-type Cx43-GFP (a), Cx43-ΔL254–290-GFP (b, mutant S2+3), and Cx43-ΔL254–CT-GFP (c, mutant S2+3), and GJ plaques and AGJ vesicles were detected by their emitted fluorescence (green). In-cell protein colocalization assays between Cx43 and Dab2 were performed with mouse anti-Dab2 and rabbit anti-Cx43 antibodies using in situ Duolink technology (red fluorescence). Cx43 wild-type AGJ vesicles colocalized with Dab2, detectable as red/yellow puncta in the magnified views (a). The Cx43-ΔL254–290 mutant is unable to recruit AP-2 and also does not recruit Dab2, as indicated by the negative Duolink signal (b). The Cx43-ΔL254–CT mutant is also unable to interact with the Cx43 antibodies and served as a negative Duolink control (c). Representative images are shown. (B) Quantitative analyses of results described in (A) revealing the impaired ability of AP-2 binding–deficient Cx43 mutants to recruit Dab2. Results are consistent with coimmunoprecipitation results presented in Figure 2.
FIGURE 4:

AP-2 binding–deficient Cx43 mutants have more GJ channels in their apposed PMs. (A) Untagged wild type and S2, S3, and S2+3 Cx43 mutants were cotransfected with mCherry-H2B (red; to better reveal transfected cell pairs) into HeLa cells. Cx43 was stained for immunofluorescence analyses using primary rabbit anti-Cx43 and secondary goat anti-rabbit Alexa Fluor 488 (green) labeled antibodies. Images of transfected cell pairs were acquired using a 60× oil-immersion objective. Three representative images of wild type (a) and selected mutants (b, F268A [S2]; c, Y286H [S3]; d, F268A+Y286H [S2+3]; e, ΔY265AY+ΔP283PGYKLV [S2+3]) are shown. (B) Quantitative analyses of the number of GJ channels present in apposing PMs (performed by measuring PM localized Alexa Fluor 488 fluorescence intensity of wild type and selected S2, S3, and S2+3 mutants) revealed a twofold increase in Cx43-F268A (S2) and Cx43-Y286H (S3) mutants (blue bars) and a threefold increase in Cx43-F268A+Y286H and ΔY265AY+ΔP283PGYKLV (S2+3) mutants (orange/red bars) compared with wild-type Cx43–expressing cells (green bar).
FIGURE 5:

GJ plaques assembled by AP-2 binding–deficient Cx43 mutants are internalization impaired. (A) HeLa cells were transfected with GFP-tagged wild type, S2, S3, and S2+3 mutant constructs and imaged every 2 or 5 min for up to 72 h (phase-contrast and green fluorescence channels). GJ plaque formation, maturation, and endocytosis were captured (depicted by arrows). Selected merged image frames of wild type, F268A (S2), Y286H (S3), and F268A+Y286H-GFP and ΔL254–290-GFP (S2+3) are shown (see also Movies S1–S5). (B) Quantitative analysis of time periods required for GJ plaques to constitutively turn over (form, mature, and be removed) revealed that GJ plaques assembled of wild type Cx43, on average, turned over in ∼5 h (green bar), while GJ plaques assembled from S2 and S3 mutants turned over in 18–19 h (blue bars), and GJ plaques assembled from S2+3 double mutants were removed after ∼30 h (red bar). (See Table S1 for complete data set.)
FIGURE 7:

AP-2 binding–deficient Cx43 mutants become donor cells. (A) HeLa cells were separately transfected with wild-type Cx43-mApple (red) or GFP-tagged wild type, S2, S3, or S2+3 mutant constructs (green) and cocultured. Cell pairs of wild-type Cx43-mApple and mutant/wild-type-GFP–expressing cells assemble heterotypic GJs (yellow in the overlays). Heterotypic AGJs (also appearing yellow) are depicted with arrows. Representative images of wild-type Cx43-GFP (a), F268A-GFP (b, S2), Y286H-GFP (c, S3), and F268A+Y286H-GFP (d, S2+3)–expressing cells paired with wild-type Cx43-mApple–expressing cells of three independent experiments are shown. (B) Heterotypic AGJs were counted, and their cellular location in wild-type Cx43-mApple–expressing cells vs. mutant Cx43-GFP–expressing cells was quantified. Cx43 wild-type/wild-type cell pairs exhibited an unbiased average ratio of 1:1 (bar 1), while Cx43 mutants (S2, S3, or S2+3, bars 2–4) were progressively inefficient in internalizing their plaques. Significance values between cell-pair groups, and between cell types within groups compared with wild-type/wild-type pairs are shown above and within columns, respectively.
Cx43 mutants with both AP-2 binding sites (S2+3) mutated no longer coprecipitate clathrin and clathrin adaptors
To test for protein/protein interactions between Cx43 and CME-components, we performed coimmunoprecipitation assays with both GFP-tagged and untagged Cx43 mutants (Figure 2, A and B). Wild-type Cx43 and Cx43 constructs with sorting signals S2, S3, S2+3, and S1+2+3 mutated were transfected into HeLa cells. Cx43 was immunoprecipitated using anti-GFP (Figure 2A) or anti-Cx43 magnetic beads (Figure 2B). Cx43 binding partners were detected by Western blot analyses using their respective antibodies. Representative immunoprecipitations for selected constructs are shown on the left, and quantitative analyses of three independent experiments each are shown on the right. Results show that wild-type Cx43 coprecipitated AP-2 (using anti–α-adaptin subunit antibodies), Dab2, and clathrin (using anti–clathrin heavy chain [CHC] antibodies; Figure 2, A and B, lane 1), consistent with previously published observations (Piehl et al., 2007; Gumpert et al., 2008). Cx43 constructs with either binding motif S2 or S3 mutated also coprecipitated these endocytic proteins (Figure 2, A and B, lanes 2 and 3), although only to ∼50% of wild type, while mutants with both (S2+3, lanes 4 and 5), or all three binding motifs mutated (S1+2+3, lanes 6 and 7) no longer coprecipitated any, or only minor amounts (≤10 ± 8% compared with wild type), of these endocytic proteins. Thus abolishing Cx43-AP-2 binding also abolished Dab2 and clathrin binding. No detectable differences in coprecipitation efficiencies were found between GFP-tagged and untagged Cx43 constructs or between mutants with all three or only binding sites S2+3 mutated, indicating that the GFP tag did not significantly affect Cx43–endocytic protein interaction and that the binding motif S1 does not bind AP-2 or recruit these endocytic proteins. HeLa cell lysates and immunobeads only were analyzed in control (Figure 2, A and B, lanes 8 and 9), and α-tubulin was analyzed as lysate input and loading control. Note that the amount of precipitated Cx43 proteins increases with one (S2 or S3) or two (S2+3) AP-2 binding motifs mutated (Figure 2, A and B, row 1), correlating with the increased half-life of these mutants (see below and Figure 6 later in the paper). Unexpectedly, in the HeLa cell lysates, a weak protein band was detected using Cx43 antibodies under our experimental conditions (Figure 2B, lane 9, top row), suggesting that small amounts of endogenous Cx43 were present in the CCL-2 HeLa cell clone that we purchased from the American Type Culture Collection for these analyses. Quantitative analyses indicated that the content of endogenous Cx43 is minor, only amounting to ∼1–2%, compared with a 100% efficient exogenous (transient or stable) expression (see Supplemental Figure S1). Taken together, these results identify the tyrosine-based sorting signals S2 (Y265AYF) and S3 (Y286KLV) in the Cx43 C-terminus as direct AP-2 binding sites. Both sites can interact with AP-2 and may work jointly to recruit AP-2 adaptors to the Cx43 C-terminus. Both Dab2 and clathrin neither bind nor interact with Cx43 directly but appear to require the AP-2 complex for their indirect interaction with Cx43.
FIGURE 2:

Cx43 mutants with AP-2 binding site S2 or S3 mutated coprecipitate reduced amounts, while mutants with both (S2+3) or all three (S1+2+3) sites mutated no longer coprecipitate clathrin and clathrin accessory proteins. HeLa cells were transfected with GFP-tagged (A) and untagged (B) Cx43 wild type (lane 1); single F268A (S2), Y286H (S3) (lanes 2 and 3); double F268A+Y286H (S2+3), ΔY265AY+ΔP283PGYKLV (S2+3) (lanes 4 and 5); and triple Y230H+F268A+Y286H (S1+2+3), Y230H+ΔY265AY+ΔP283PGYKLV (S1+2+3) (lanes 6 and 7) mutants. Cells were lysed in SDS-free 1× RIPA buffer, and protein complexes were immunoprecipitated using anti-GFP (A) or anti-Cx43 (B) magnetic beads. Bound and coprecipitated endocytic proteins were detected by Western blot analyses using antibodies specific for GFP (row 1 in A), Cx43 (row 1 in B), α-adaptin (AP-2, row 2), Dab2 (row 3), clathrin heavy chain (CHC, row 4), and α-tubulin (loading control, row 5). Cx43 wild type and single-site mutants S2 and S3 coprecipitated AP-2, Dab2, and clathrin, while double (S2+3) and triple (S1+2+3) mutants did not. HeLa cell lysates and beads only were analyzed in control (lanes 8 and 9). Note that both single mutants S2 and S3 precipitated approximately half the amounts of clathrin and clathrin adaptors compared with wild-type Cx43 (see quantitative graphed analyses on the right). Representative blots are shown on the left, and averaged data of three independent experiments are shown on the right. Note that increasing amounts of Cx43 proteins were immunoprecipitated, correlating with the increasing stability of the mutant proteins (see Figure 6) and that a small amount of endogenous Cx43 was detected in overexposed membranes (row 1, lane 9 in B; quantified in Figure S1). Significant differences (p < 0.05) are shown in this and following figures. Results corroborate that motif S1 is not a functional AP-2 binding site.
FIGURE 6:

AP-2 binding–deficient Cx43 mutant proteins exhibit longer half-lives. HeLa cells were transfected with GFP-tagged (A) or untagged (C) wild type, S2, S3, and S2+3 Cx43 mutant constructs and treated with cycloheximide the following day to block protein biosynthesis. Cells were lysed at indicated time points posttreatment and examined by SDS–PAGE and Western blot analyses using anti-GFP (A) and anti-Cx43 (C) antibodies. All membranes were stripped and reprobed with anti–α-tubulin antibodies as a loading control. Representative Western blot analyses for Cx43 wild type (a), F268A (b, S2), Y286H and ΔP283PGYKLV (panels c and d in A, S3), F268A+Y286H and ΔY265AY+ΔP283PGYKLV (panel d in C and panels e and f in A, S2+3) are shown. (B, D) Normalized quantitative analyses of Cx43 protein that remained after the indicated times in three independent analyses indicates that S2 and S3 mutant proteins have approximately doubled half-lives (∼2 h, blue curves), while double S2+3 mutants again have about doubled half-lives (∼4 h, orange/red curves) when compared with wild-type Cx43 protein (estimated half-life of ∼1 h, green curve).
Cx43/Dab2 interaction is indirect and mediated via AP-2
Dab2, an alternative clathrin adaptor that was initially characterized in AP-2–depleted cells (Morris and Cooper, 2001; Mishra et al., 2002; Motley et al., 2003; Traub, 2003), was also found to play a role in Cx43 GJ internalization (Piehl et al., 2007; Gumpert et al., 2008). As mentioned above, Dab2 interacts with its cargo (such as LDL receptor) via a minimal sequence motif that conforms to tyrosine-based sorting signals of the NPXY type (Morris and Cooper, 2001; Mishra et al., 2002; Bonifacino and Traub, 2003; Traub, 2003, 2005). However, we did not find any NPXY motifs in Cx43, only cryptic “PXY” sequences (two in the rat and one in the human Cx43 sequence; Piehl et al., 2007), making a direct interaction between Dab2 and Cx43 unlikely (reviewed in Thévenin et al., 2013). Yet Dab2 previously was found by immunofluorescence staining to robustly colocalize with Cx43, both in GJ plaques and in AGJs, even in cell lines that express very little amounts of Dab2 (Piehl et al., 2007), and Dab2 was shown by RNAi knockdown to play a considerable role in GJ internalization (Gumpert et al., 2008). In addition, as found in this study and shown in Figure 2 above, Dab2 robustly coimmunoprecipitated with GFP-tagged and untagged Cx43 when expressed in HeLa cells, while its coimmunoprecipitation was reduced or lost when AP-2 binding sites were mutated. Dab2 is known to be able to interact with the α subunit of AP-2 and the C-terminal domain of CHC (reviewed in Bonifacino and Traub, 2003), further suggesting that the interaction between Dab2 and Cx43 is indirect and mediated via AP-2. To substantiate this hypothesis further, we performed Duolink in-cell colocalization assays (Figure 3). The Duolink assay allows for visualization of protein–protein interactions, as long as the two proteins are within 40 nm of each other inside fixed cells (Gullberg et al., 2011). Results obtained with wild-type Cx43 and the S2+3 mutants Cx43ΔL254–290-GFP and Cx43ΔL254–CT-GFP are shown. Cx43 and Dab2 antibodies were utilized for the Duolink colocalization reaction (red fluorescent puncta in wild-type Cx43 and in the Cx43ΔL254–290 construct, Figure 3A, a and b). Note that the used Cx43 antibody is directed against a C-terminally located sequence and therefore does not recognize the Cx43Δ254–CT-GFP mutant protein, thus serving as a negative Duolink control (no red puncta in Figure 3Ac). GJs were visualized by their Cx43-GFP fluorescence. Duolink-based Cx43/Dab2 colocalization was quantified by comparative red-channel pixel-intensity analyses. Results show that the AP-2 binding–deficient Cx43ΔL254–290-GFP (S2+3) mutant, as expected, does not colocalize significantly with Dab2 inside cells when compared with wild-type Cx43 (reduced to 27%, SEM ± 16.4, n = 25, p < 0.0001; Figure 3B). As for results presented in Figures S1, 2, and 7 (later in the paper), a small amount of Duolink signal detected in S2+3 mutant–expressing cells (average ∼ 27%; middle bar in Figure 3B) may be generated by small amounts of endogenous wild-type Cx43 that may have hetero-oligomerized with the mutant Cx43 and thus recruited some AP-2 to these heteromeric connexons. Together data presented here using in situ Duolink colocalization assays and coimmunoprecipitation assays presented above (Figure 2, A and B) firmly support the conclusion that the interaction between Cx43 and Dab2 is indirect and mediated via the classical clathrin adaptor AP-2.
Cells expressing Cx43 with AP-2/clathrin binding sites mutated have more GJ channels in their apposing plasma membranes
Because GJ plaque size has been reported to be regulated by binding of the scaffolding protein ZO-1 (Hunter et al., 2005; Rhett et al., 2011), and GFP-tagged Cxs are unable to bind to ZO-1 (Hunter et al., 2003; Thévenin et al., 2013), untagged Cx43 wild type and mutants were transiently transfected into HeLa cells; this was followed by immunofluorescence staining using primary antibodies against Cx43 and Alexa Fluor 488 (green)-labeled secondary antibodies. mCherry-tagged histone H2B (mCherry-H2B; red) was cotransfected into the cells to identify transfected cell pairs. In each group, more than 30 cell pairs were imaged and analyzed. Representative images are shown in Figure 4A. As fluorescence intensity above the threshold is directly proportional to the number of GJ channels in the PM, the number of channels (more channels correlate with larger GJ plaques) of each group was assessed by quantitatively measuring and comparing Alexa Fluor 488 (GJ) pixel quantity and intensity located in the apposing PMs of transfected cell pairs (see Materials and Methods). Results indicate that cells that expressed S2 and S3 mutants had, on average, twice as many GJ channels in their apposing PMs compared with wild-type Cx43–expressing cells, as indicated by the doubled pixel intensity (p < 0.0001; shown for F268A and Y286H mutants in Figure 4A, a–c, and 4B, blue and green bars), while cells expressing S2+3 double mutants (F268A+Y286H; ΔY265AY+ΔP283PGYKLV) had about three times as many GJ channels in their apposing PMs (p ≤ 0.001; Figure 4A, d and e, and 4B, orange and red bars), indicating a significant internalization impairment of the AP-2 binding–deficient Cx43 mutants. Taken together, these results again suggest that AP-2 is recruited to Cx43 GJs and that AP-2 regulates GJ endocytosis. Because mutating sites S2 and S3 individually caused a less significant increase in PM GJ channel number compared with mutating both AP-2 binding sites S2+3 together (either by mutating critical residues of the signal sequence or by deleting the signal), these results further support the conclusion that AP-2 interacts with both sites, S2 and S3, in Cx43 polypeptides.
GJ plaques formed by AP-2/clathrin binding–deficient Cx43 mutants are internalization impaired
As shown in Figure 4 and described above, cells expressing AP-2/clathrin binding–deficient Cx43 mutants exhibit increased amounts of GJ channels in their apposing PMs. To examine whether this increase is indeed caused by inefficient GJ internalization, we investigated HeLa cells expressing wild type and respective Cx43 mutants for extended time periods (up to 72 h) by live-cell, time-lapse microscopy. Representative image frame sequences are shown in Figure 5A. Results indicate that wild-type Cx43-GFP–expressing cells constitutively internalized/turned over their plaques with a time-constant of ∼5 ± 1.0 h (n = 16) counted from visual onset of GJ plaque formation to completion of GJ internalization (Figure 5A, column 1, and 5B, green bar; Supplemental Table S1; Supplemental Movie S1). Single AP-2 binding site mutants, S2 (F268A-GFP) and S3 (Y286H-GFP and ΔP283PGYKLV-GFP) were still able to internalize their plaques, although at a much slower rate determined to be 18 ± 3.7 h (n = 7) and 19 ± 1.3 h (n = 17) (Figure 5A, columns 2 and 3, shown for F268A-GFP and Y286H-GFP, and 5B, blue bars; Table S1; Movies S2 and S3). S2+3 double mutants (F268A+Y286H-GFP, ΔY265AY+ΔP283PGYKLV-GFP, and ΔL254–290-GFP, Cx43-ΔL254–CT-GFP) were found very inefficient/unable to internalize their plaques, because they were present for 30 ± 2.7 h (n = 9), or even longer in the case of the ΔL254–CT-GFP mutant (Figure 5A, columns 4 and 5, shown for F268A+Y286H-GFP and ΔL254–290-GFP, 5B, red bar; Table S1; Movies S4 and S5). The fact that double-site mutants (S2+3) were observed to remove GJs at all, albeit only after prolonged time periods, again may be explained by small amounts of endogenously expressed Cx43 that may have hetero-oligomerized with the expressed mutant proteins. Alternatively, other yet uncharacterized cellular processes may have accommodated GJ removal under these conditions. Together these results indicate that GJ plaques assembled from AP-2/clathrin binding–deficient Cx43 mutants are internalization impaired.
AP-2/clathrin binding–deficient Cx43 mutant proteins exhibit longer half-lives
To further investigate whether impaired GJ plaque internalization correlates with extended mutant Cx43 protein half-life, we analyzed the half-lives of GFP-tagged and untagged wild-type and mutant Cx43 proteins in HeLa cells in which protein biosynthesis was blocked pharmacologically (Figure 6). Cxs in general are known to exhibit short half-lives of 1–5 h (Fallon and Goodenough, 1981; Beardslee et al., 1998; Falk et al., 2009). Channels in GJ plaques are dynamic and, at steady state, are constitutively internalized from plaque centers, while newly synthesized GJ channels are accrued along the outer edges of plaques (Gaietta et al., 2002; Lauf et al., 2002; Falk et al., 2009). HeLa cells were transfected with GFP-tagged and untagged wild-type Cx43, S2, S3, and double S2+3 mutants. The next day, cells were treated with cycloheximide and lysed at indicated times, and Cx protein levels were determined using GFP antibodies and Western blot analyses. Representative blots for GFP-tagged and untagged wild type and mutant constructs are shown in Figure 6, A and C. Quantitative analyses (by stripping blots and reprobing for α-tubulin) showed that wild-type Cx43-GFP exhibited a short half-life of ∼1 h (Figure 6, A–D, green curve) consistent with previous results (Fallon and Goodenough, 1981; Beardslee et al., 1998; Falk et al., 2009), whereas the single-site mutants S2 and S3 exhibited extended half-lives of ∼2 h (Figure 6, A–D, blue and purple curves), with ∼4 h for the S2+3 double-site mutants (Figure 6, A–D, orange and red curves), respectively. All protein half-life experiments were repeated at least three times. Although no significant differences were observed between GFP-tagged and untagged constructs, untagged Cx43 proteins in general appeared somewhat more stable compared with GFP-tagged constructs. Note that cycloheximide treatment in these experiments may generate considerable cytotoxic side effects that may have caused recorded Cx43 protein half-lives to be shorter compared with experiments performed in untreated cells (shown in Figure 5). Together these results further support our hypothesis that GJs are internalized in an AP-2–dependent, clathrin-driven process.
Cells expressing AP-2/clathrin binding–deficient Cx43 mutants become donor cells when paired with wild-type Cx43–expressing cells
To further support our finding that mutating the AP-2 binding sites in Cx43 generates internalization-impaired GJs, we paired cells expressing wild-type Cx43 with cells expressing AP-2 binding site mutants. We transfected HeLa cells separately with wild-type Cx43-mApple (red fluorescence) and wild-type or mutant Cx43-GFP (green fluorescence) by electroporation, as described in Materials and Methods. Transfected cells were mixed at a 1:1 ratio and seeded and cocultured on glass coverslips overnight. Cells were fixed, analyzed, and quantified for the location of heterotypic wild-type/mutant AGJs. Heterotypic GJs formed between Cx43-mApple/Cx43-GFP–expressing cell pairs were composed of mApple-labeled connexons on one side, and GFP-labeled connexons on the other, resulting in GJ plaques exhibiting yellow fluorescence on the red and green overlay images (Figure 7A). Cytoplasmic AGJ vesicles formed by the internalization of these heterotypic GJ plaques also appeared yellow (labeled with arrows in Figure 7A). Counting the number of heterotypic AGJs and recording their location allow for evaluation of how efficiently the mutant-expressing cells can internalize their plaques. In three independent experiments, when wild-type Cx43-GFP–expressing cells were cocultured with wild-type Cx43-mApple–expressing cells, 46 cell pairs with a total of 249 heterotypic AGJ vesicles were analyzed. No overall bias in directionality of GJ internalization was observed in these cell pairs (50:50 ratio, Figure 7A, a, and 7B, bar 1). In contrast, single (S2, S3) and double (S2+3) AP-2 binding site mutant–expressing cells were found to be significantly less efficient in internalizing the assembled heterotypic GJ plaques, and instead became donor cells (shown for Cx43-F268A-GFP [S2], Cx43-Y286H-GFP [S3], and Cx43-F268A+Y286H-GFP [S2+3] in Figure 7A, b–d, and 7B, bars 2–4). In Cx43-F268A-GFP (S2)/wild-type Cx43-mApple cell-pairing experiments, 72 cell pairs with a total of 475 heterotypic AGJ vesicles were analyzed. Only 130 (28% SEM ± 2.9, p = 0.0004) were found in the S2 mutant–expressing cells. In Cx43-Y286H-GFP (S3)/wild-type Cx43-mApple cell-pairing experiments, 101 cell pairs with a total of 507 heterotypic AGJ vesicles were analyzed. Only 148 (29%, SEM ± 1.5, p < 0.001) were found in the S3 mutant–expressing cells. In Cx43-F268A+Y286H-GFP (S2+3)/Cx43-mApple cell-pairing experiments, 75 cell pairs with a total of 425 AGJs were analyzed, and only 125 (21%, SEM ± 6.4, p = 0.003) were found in the S2+3 mutant–expressing cells (Figure 7B). Note that small amounts of heterotypic AGJs detected in S2+3 mutant–expressing cells (∼20%, Figure 7B, bar 4) may again be generated by small amounts of endogenous wild-type Cx43 (see Figure S1) that may have hetero-oligomerized with mutant Cx43, and/or may have been generated by potential other AP-2–independent internalization processes (see Discussion). The impaired ability of S2, S3, and S2+3 mutant–expressing cells to endocytose these heterotypic GJ plaques further supports the conclusion that both sorting signals, S2 and S3, function in Cx43 GJ internalization, which is consistent with results obtained by other assays described above.
DISCUSSION
In 2003, Thomas and colleagues identified a proline-rich region consisting of amino acid residues P280MSPPGYKLVTG291 in the Cx43 C-terminus that, when mutated, increased the steady-state pool and half-life of Cx43 (Thomas et al., 2003). Since then, it has been assumed that Cx43 GJ internalization and turnover is regulated by this sequence. More recently, Wayakanon et al. (2012), using Cx43 constructs with progressively truncated C-termini, identified critical regions in the Cx43 C-terminus that regulate plaque size (C271–N302) and direction of GJ internalization (T325–Q342). However, while these authors spatially located the region/s important for Cx43 GJ internalization, they did not provide any information on possible mechanisms that may lead to GJ internalization. In this paper, we report the identification of three tyrosine-based sorting signals with the general consensus sequence YXXΦ (Y230VFF, Y265AYF, and Y286KLV) located in the Cx43 C-terminus that we termed sorting signals S1, S2, and S3. YXXΦ signals are known to bind physically to the μ2 subunit of phosphorylation-activated AP-2 complexes (Ohno et al., 1995; Collins et al., 2002). Sorting signal S3 (Y286KLV) is identical to the signal located within the proline-rich region that Thomas et al. identified previously. Sorting signal S1 or S2 at positions Y230VFF and Y265AYF, which we identified in addition, were not revealed by Thomas et al. and were not examined experimentally in their study (Thomas et al., 2003). We found that sorting signals S2 and S3 both recruit the classical PM clathrin adaptor complex AP-2, the alternative clathrin adaptor protein Dab2, and clathrin to mediate GJ endocytosis. A very recent study that examined Cx43/AP-2 binding by applying a yeast two-hybrid screen using the Cx43 C-terminus as bait and the AP-2 μ2 subunit as prey suggests that the Cx43 C-terminus may require specific posttranslational modifications (that did not occur in the yeast host) to render the S2 binding site accessible to AP-2 (Johnson et al., 2013). Sorting signal S1, due to its close juxtaposed TM4 location, and consistent with previously published findings (Collins et al., 2002; Bonifacino and Traub, 2003), was not found to serve as an AP-2 interaction/recruitment site.
We characterized the identified internalization motifs by generating and analyzing a set of Cx43 mutants composed of large and small deletions and critical amino acid exchanges. Cx43 mutants with both sorting signals S2 and S3 (S2+3) mutated no longer coprecipitated AP-2, Dab2, or clathrin. These mutants formed more and/or larger GJ plaques and exhibited longer protein half-lives, and double mutants exhibited severe impairment of GJ plaque internalization. Interestingly, mutants with only one of the two sorting signals mutated (either S2 or S3) were also internalization impaired. However, the internalization of these mutants was affected less severely (by approximately one-half when compared with the double S2+3 mutants; Figures 2 and 4–7). We also found that Dab2 interacts with Cx43 indirectly via AP-2 (Figures 2 and 3). Collectively our results suggest that the Cx43 sorting signals S2 and S3 work together to endocytose Cx43 GJs, and they evoke a possible model for clathrin to achieve the efficient internalization of double membrane–spanning GJs. Compared with other PM receptors, such as transferrin, LDL, receptor tyrosine kinases (epidermal growth factor [EGF], vascular endothelial growth factor [VEGF], platelet-derived growth factor [PDGF]), and G protein–coupled receptors (GPCRs) that are endocytosed by the clathrin machinery, GJs are large PM structures and their endocytosis may pose a greater challenge to the cellular endocytic machinery (reviewed in Pauly and Drubin, 2007). In our model, schemed in Figure 8, AP-2 interacts with the Cx43 sorting signals S2 and S3 via its μ2 subunit as the first step of Cx43 GJ endocytosis. Once Cx43 and AP-2 interaction has been established, AP-2 may either recruit clathrin directly via its β2-subunit appendage and/or clathrin-bound Dab2 via its α-subunit appendage. In this model, the interaction between AP-2 and Dab2 is an important step, as recruitment of Dab2 to GJ plaques may allow multiple (up to four) clathrin triskelia to be recruited to a single Cx43 of a GJ plaque (Figure 8). AP-2, Dab2, and other clathrin adaptors have all been found to be present concurrently in endocytic clathrin-coated vesicles (Keyel et al., 2006), suggesting that they play a cooperative/synergistic role in clathrin and clathrin-machinery component recruitment. Thus recruitment of multiple adaptors and clathrin complexes per Cx43 cargo molecule may facilitate efficient endocytosis of these large PM structures. Furthermore, acute GJ internalization in response to certain stimuli (Baker et al., 2008) might, in addition to AP-2, require extra clathrin adaptors (e.g., Dab2) to achieve highly efficient internalization, while constitutive GJ plaque turnover (Falk et al., 2009) may be achieved by AP-2 alone. This, for example, is known to occur in GPCR endocytosis, in which constitutive endocytosis of unstimulated receptors is achieved via AP-2 binding, while rapid ligand-stimulated GPCR internalization requires AP-2 plus an additional clathrin adaptor, for example, β-arrestin or epsin-1 (Moore et al., 2007; Wolfe and Trejo, 2007; Chen et al., 2011). Finally, recent elegant studies from the Kirchhausen lab indicate that, to successfully initiate coat formation, clathrin triskelia need to interact with two AP-2 complexes simultaneously. These interactions typically are mediated via PM-anchored phosphatidylinositol 4,5-bisphosphate (PIP2) interactions (Cocucci et al., 2012). However, significantly less, or even no, PIP2 is present within GJ plaques, suggesting that Cx43 may have two AP-2 binding sites per Cx43 polypeptide to allow for efficient AP-2 binding/clathrin recruitment to successfully initiate clathrin coat formation and GJ internalization. Future research is needed to further address these exciting possibilities.
FIGURE 8:
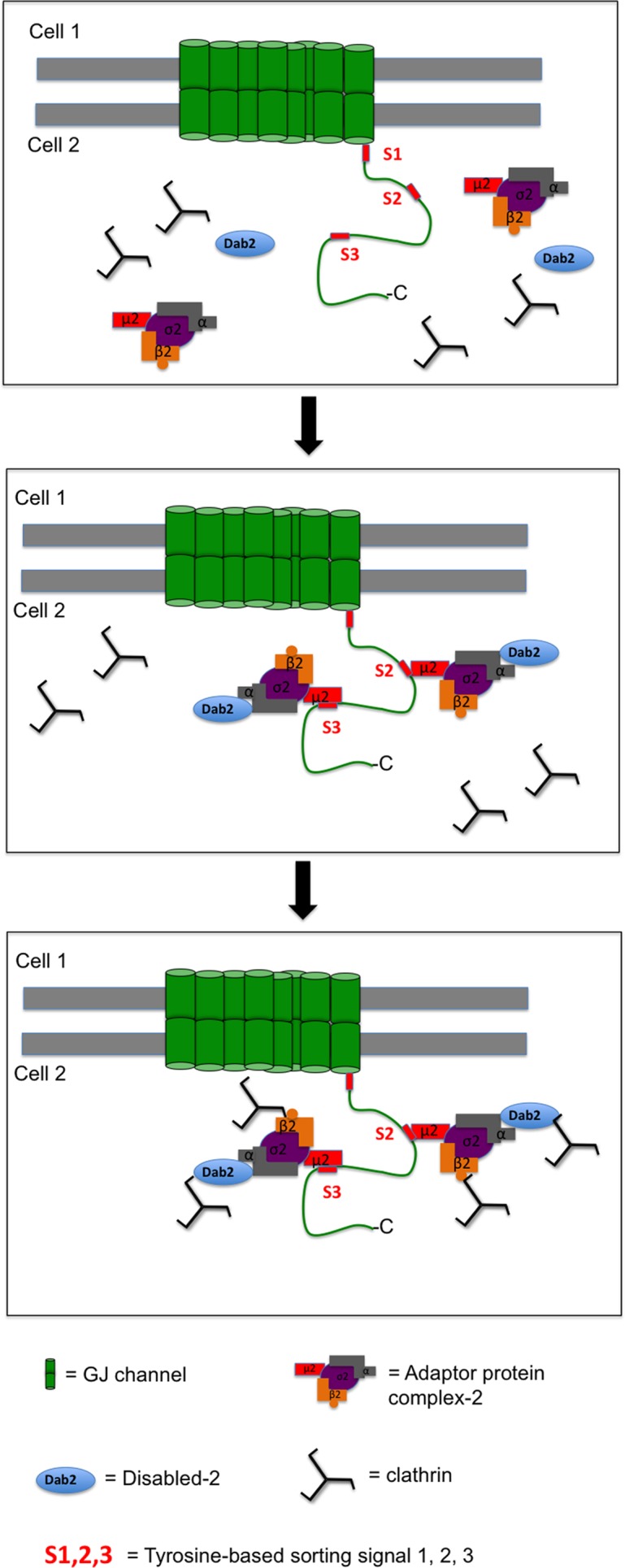
Model of Cx43 recruiting clathrin and endocytic clathrin adaptors. Top, Cx43 encodes three canonical tyrosine-based sorting motifs of the type “YXXΦ” in its C-terminus (S1, S2, and S3). Sorting signals S2 and S3 both can recruit the classical clathrin adaptor AP-2 to Cx43-GJs that are destined for internalization. Middle, internalization-destined Cx43 binds the μ2 subunit of AP-2 via its Y265AYF (S2) and/or its Y286KLV (S3) motif. AP-2 then recruits clathrin either directly via its β2 subunit that binds to the terminal domain of CHC, and/or it interacts via its α subunit with clathrin-bound Dab2. Bottom, as a consequence, one Cx43 polypeptide may recruit up to four clathrin molecules, potentially generating the principal molecular structure required for an efficient endocytosis of GJs.
Interestingly, the sequence SP283PGYKLV289 that Thomas et al. (2003) identified indeed consists of two overlapping signals, a proline-rich PY motif (XP283PXY286) and the YXXΦ tyrosine-rich sorting signal S3 (Y286KLV289), and both may play discrete functions in clathrin-dependent and clathrin-independent GJ internalization mechanisms. For example, in addition to clathrin-dependent internalization, clathrin-independent internalization has been reported for EGF receptors. Clathrin-independent EGF receptor internalization is reliant on ubiquitination and is regulated by the number of stimulated receptors that are present in the PM (Sigismund et al., 2005; reviewed in Aguilar and Wendland, 2005). The proline-rich PY motif (XP283PXY286) in Cx43 has been reported to interact with the HECT E3 ubiquitin ligase Nedd4 via its WW2 domain (Leykauf et al., 2006). Nedd4-mediated ubiquitination of Cx43 allows recruitment of Eps15, a multifunctional ubiquitin-interacting endocytic protein to GJs (Girao et al., 2009; Catarino et al., 2011). Eps15 is a component of clathrin-coated pits and vesicles, and it has been reported to interact directly with AP-2 (Benmerah et al., 1998). However, Eps15 also performs an important function in coupling ubiquitinated cargo to clathrin-independent internalization (Aguilar and Wendland, 2005). Thus ubiquitinated Cx43 may also be recognized by Eps15 through its ubiquitin-interacting motif, possibly resulting in Eps15-mediated clathrin-independent GJ endocytosis. Also, Leithe et al. (2009) identified Cx43 ubiquitination as a potential signal for Cx43 GJ internalization, and Fykerud et al. (2012) recently identified another ubiquitin ligase, Smurf2, that ubiquitinates Cx43 and prepares GJs for internalization and endo/lysosomal degradation in TPA-treated cells. We also found that cells that expressed the double-sorting signal mutants S2+3 were eventually able to remove their GJ plaques (Figure 5 and Movies S1–5), while GJ internalization in the Cx43-ΔL254–CT-GFP mutant was abolished entirely. However, GJ removal in these S2+3 mutants appeared very different and followed a different spatial and temporal pattern, potentially implying a different, as yet uncharacterized cellular mechanism. Together these findings suggest that cells may utilize more than one pathway to remove GJ plaques. Different functional requirements and different upstream cell signaling events leading to different Cx43 modifications (e.g., phosphorylation, ubiquitination, SUMOylation) might dictate which cellular pathway eventually might be utilized (reviewed in Falk et al., 2012; Thévenin et al., 2013). As stated above, the PY motif (P283PXY286) that mediates Nedd4 binding overlaps with the YXXΦ tyrosine-based sorting signal S3 (Y286KLV289), because the Y286 is essential for both signals. Thomas et al. (2003) separately mutated the XP283XXY286 and the Y286XXV289 signals by substituting P283 with leucine (L) and V289 with aspartic acid (D) and found that mutating the latter (YXXΦ) signal resulted in a more significant increase in the steady-state pool of Cx43 (3.5- vs. 1.7-fold, respectively). When both the PXXY and the YXXΦ signals were mutated together by either mutating P283 to L and V289 to D or by mutating Y286 to A, no additive effect was observed. The double mutants behaved similar to the single YXXΦ mutant, suggesting a primarily AP-2–mediated CME-based mechanism for GJ internalization.
Several Cxs, including Cx43, Cx32, Cx36, and Cx46 were reported to interact with caveolin-1 suggesting the possibility that Cx proteins can localize to lipid rafts (Schubert et al., 2002; Lin et al., 2003). However, the size of typical GJ plaques is much larger than the size of lipid rafts (∼50 nm; Yuan et al., 2002; Helms and Zurzolo, 2004), making it unlikely that GJs localize to and internalize utilizing a caveolae-dependent endocytosis mechanism. Caveolae-dependent endocytosis, however, may be utilized for the internalization of PM-localized Cx hemichannels.
In this study, we provide multiple lines of independent evidence that suggest a predominantly AP-2– and clathrin-driven pathway for the internalization of Cx43-based GJs. The alternative clathrin adaptor Dab2 may also be recruited to Cx43 GJs via AP-2 interaction, most likely to enhance the efficacy of this endocytic process. Interestingly, our analyses uncovered two AP-2 binding sites in the Cx43 C-terminus that are both used to convey the binding of up to four clathrin triskelia per Cx43 polypeptide (via the binding of two AP-2 complexes and two Dab2 proteins), suggesting a potential molecular mechanism for clathrin to mediate the internalization of PM structures that can be 50 times larger than typical clathrin-coated vesicles. Future research will need to address which signals regulate access of AP-2 (and of other endocytic adaptors) to Cxs of endocytosis-primed GJs, and how many Cx polypeptides per connexon/GJ channel need to bind to AP-2 to successfully initiate GJ endocytosis. Posttranslational modifications of Cx43, such as phosphorylation and ubiquitination that, for example, are required for GPCR endocytosis (Moore et al., 2007; Wolfe and Trejo, 2007; Chen et al., 2011), binding/release of regulatory proteins as observed for adherens junctions (Nanes et al., 2012), or conformational changes of the Cx43 C-terminus (Solan and Lampe, 2005), are all enticing possibilities.
MATERIALS AND METHODS
cDNA constructs and mutagenesis
GFP-tagged and untagged Cx43 mutants were generated by site-specific mutagenesis. Full-length rat Cx43-GFP previously cloned into pEGFP-N1 vector as described (Falk, 2000) was used as a template for PCR mutagenesis. Site-directed mutagenesis of the Cx43 sequence was performed using an overlap extension PCR mutagenesis procedure developed by Higuchi et al. (1988). PCR mutagenesis reactions were done using proofreading Pfu Ultra II polymerase (cat. no. 600670-51; Stratagene, Santa Clara, CA). Mutated PCR-amplified cDNA fragments were restriction endonuclease digested and ligated into the full-length Cx43-GFP–containing vector to replace the wild-type sequence with the mutated sequence. Untagged Cx43 mutants were generated by QuikChange site-directed mutagenesis. Cx43-GFP mutants were used as templates for PCR amplification. A pair of mutagenic primers reintroducing the authentic Cx43 TAA stop codon behind the last Cx43 amino acid (I382) were used. PCR amplification (using proofreading Pfu Ultra II polymerase, cat. no. 600670-51; Stratagene) generated nicked, circular strands. Template DNA (methylated) was digested using DpnI restriction endonuclease. Constructs were then transformed into DH5α competent Escherichia coli cells. Plasmids were purified using Midi-/Maxiprep plasmid DNA purification kits (cat. nos. 12243 and 12163; Qiagen, Valencia, CA). All constructs were verified via DNA sequence analyses. Plasmids containing histone mCherry-H2B and Cx43-mApple (generous gifts of Michael W. Davidson, National High Magnetic Field Laboratory, Florida State University, Tallahassee, FL) were used to identify Cx43-cotransfected cell pairs (Figure 4) and in Cx43-GFP/Cx43-mApple coculture experiments (Figure 7).
Cell culture and transfections
Newly purchased HeLa cells (cat. no. CCL-2; American Type Culture Collection, Manassas, VA) were maintained under standard conditions in a humidified atmosphere containing 5% CO2 at 37°C in high-glucose DMEM. Media were supplemented for a final concentration of 10% with fetal bovine serum (FBS; cat. no. S11050; Atlanta Biologicals, Norcross, GA), 2 mM l-glutamine (stock 200 mM; Hyclone, cat. no. SH30034.01; Thermo Scientific, Waltham, MA), and 50 IU/ml penicillin and 50 μg/ml streptomycin (cat. no. 30-001-CI; Cellgro, Manassas, VA). Cells were passaged the day before transfection, cultured to 50–75% confluency, and transfected with SuperFect transfection reagent (cat. no. 301307; Qiagen) according to manufacturer's recommendations.
Immunofluorescence microscopy and image analyses
Cells were grown either in glass-bottom dishes or on glass coverslips pretreated with poly-l-lysine (cat. no. P8920; Sigma-Aldrich, St. Louis, MO). Cells were fixed with 3.7% formaldehyde and permeabilized with 0.2% Triton X-100 in phosphate-buffered saline (PBS). Cells were blocked with 10% FBS in PBS at room temperature for 1 h. Cells were incubated with rabbit polyclonal anti-Cx43 primary antibodies (cat. no. 3512; Cell Signaling Technology, Danvers, MA) at 1:500 dilution at 4°C overnight. Secondary antibodies (goat anti-rabbit Alexa Fluor 488, cat. no. A11008; Molecular Probes/Invitrogen, Grand Island, NY) were used at 1:500 dilution at room temperature for 1 h. Cell nuclei were stained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI). Cells were mounted using Fluoromount-G (cat. no. 0100-01; Southern Biotechnology, Birmingham, AL).
Wide-field fluorescence microscopy was performed on a Nikon Eclipse TE 2000E inverted fluorescence microscope equipped with a 60×/1.4 numerical aperture Plan-Apochromat oil-immersion objective (Melville, NY). Images were acquired using MetaVue software version 6.1r5 (Molecular Devices, Sunnyvale, CA). Quantitative analyses were performed using ImageJ version 1.43u (National Institutes of Health [NIH], Bethesda, MD). Adobe Photoshop version 8.0 was used to process images (San Jose, CA).
Live-cell imaging
HeLa cells were transfected with wild-type Cx43-GFP or with Cx43-GFP mutants as described above. Cells were analyzed 24 h posttransfection, and extended live-cell recordings (up to 72 h; phase contrast and fluorescence), as shown in Figure 5, were performed on a Nikon BioStation IM Cell-S1 system using a 40× air objective and glass-bottom dishes. Images were acquired every 2 or 5 min. Time-lapse image sequences were than annotated using NIS Elements AR 3.00.
Western blot analyses
Denatured protein samples derived from Cx43 wild-type and mutant-expressing HeLa cells were analyzed on 10% or 12% SDS–polyacrylamide mini-gels (Bio-Rad, Hercules, CA). Biotinylated protein ladder (cat. no. 7727S; Cell Signaling Technology) was used as a molecular-weight marker. Proteins were transferred onto nitrocellulose membranes (cat. no. 10439396; Whatman, Piscataway, NJ) in an ice bath at 120 V for 1 h before being blocked with 5% nonfat dry milk in Tris-buffered saline (TBS) with 0.1% Tween-20 (TBST) at room temperature for 1 h. Membranes were then incubated with primary antibodies at 4°C overnight. Antibodies used were monoclonal mouse anti-Cx43 (cat. no. 138300; Zymed/Invitrogen) at 1:2000 dilution, polyclonal rabbit anti-Cx43 (cat. no. 3512; Cell Signaling Technology) at 1:2500 dilution, monoclonal mouse anti–α-adaptin (an AP-2 subunit, cat. no. 610501; BD Transduction Laboratories, San Jose, CA) at 1:2000 dilution, monoclonal mouse anti-Dab2 (p96, cat. nos. 610501 and 610464; BD Transduction Laboratories) at 1:2000 dilution, monoclonal mouse anti-CHC (cat. no. 610499; BD Transduction Laboratories) at 1:2000 dilution, monoclonal mouse anti–GFP-horseradish peroxidase (HRP) (cat. no. 130-091-833; Miltenyi Biotec, San Diego, CA) at 1:2500 dilution, and monoclonal mouse anti–α-tubulin (cat. no. T9026; Sigma-Aldrich) at 1:5000 dilution. Membranes were washed three times with TBST; this was followed by incubation with HRP-conjugated secondary antibodies at 1:5000 dilution (cat. no. 81-6520 or 81-6120; Zymed) at room temperature for 1 h. Proteins were detected with ECL plus (cat. no. RPN2132; GE Healthcare, Waukesha, WI). The NIH ImageJ software package was used to quantify the intensity of protein bands.
Coimmunoprecipitation assays
Cx43-transfected HeLa cells were lysed with 1x SDS-free RIPA buffer (cat. no. 9806; Cell Signaling Technology) supplemented with protease and phosphatase inhibitor cocktail (cat. no. 5872; Cell Signaling Technology). Supernatants were incubated either with covalently linked anti-GFP magnetic beads (cat. no. 130-091-288; Miltenyi Biotec) or with rabbit anti-Cx43 antibodies together with Dynabeads protein-G magnetic beads (cat. no. 10003; Invitrogen) at 4°C overnight or at room temperature for 2 h. Beads were washed three times with 1× TBS. Cx43 and bound proteins were eluted from the beads using hot SDS–PAGE sample buffer containing 50 mM dithiothreitol. Samples were analyzed by Western blot as described above.
Cx43 protein half-life analyses
HeLa cells were cultured and transfected with GFP-tagged and untagged wild-type or mutant Cx43 constructs in multiple 3.5-cm dishes as described above. Cells were treated with 50 μg/ml of cycloheximide (cat. no. C-6255; Sigma-Aldrich) the following day to inhibit protein biosynthesis, and cells were lysed and harvested with 500 μl of SDS–PAGE sample buffer at 15 and 30 min and 1, 2, and 4 h posttreatment. Samples were heated at 95°C for 5 min. Cx43 protein levels of each lysate were examined by Western blot analyses using mouse anti–GFP-HRP antibody as described above (15 μl of each protein sample were analyzed). Membranes were stripped with stripping buffer (cat. no. BP-96; Boston Bioproducts); this was followed by a stringent wash in TBST before reprobing with mouse anti–α-tubulin antibody. The protein amount of each band was quantified using NIH ImageJ. Cx43 amounts were corrected for slightly uneven loading by comparison with the amount of α-tubulin.
Transfection and coculture of cells expressing wild-type and mutant Cx43 constructs
HeLa cells were cultured to 70–80% confluency. Cells were dislodged using 0.25% trypsin/EDTA, washed three times with 1× PBS, and pelleted. Cell pellets were resuspended in electroporation buffer (cat. no. ES-003-D; Millipore), and transferred into 4-mm-gap-size electroporation cuvettes. Electroporations were performed with 3 × 106 cells and 20 μg DNA in a total volume of 750 μl at 250 V and 500 μF using a Gene Pulser Xcell Total System (cat. no. 165-2660; Bio-Rad). Cells were rested on ice for 10 min before seeding of wild-type and mutant Cx43 transfected cells at an equal ratio onto poly-l-lysine–coated glass coverslips. Cells were cultured for ∼30 h, fixed, and stained with DAPI as described above before imaging.
Duolink in situ protein colocalization assays
HeLa cells were either transfected with wild-type Cx43-GFP or with double S2+3 mutant-GFP constructs with putative AP-2 binding sites deleted. Cells were fixed, permeabilized, and blocked as described above. Cells were then incubated with mouse anti-Dab2 and rabbit anti-Cx43 antibodies at 1:500 dilution at 4°C overnight. Unbound primary antibodies were removed by being washed three times with 1× PBS. Cells were incubated with secondary antibodies conjugated with PLA-specific oligonucleotide probes (Duolink in situ starter kit, cat. no. 92101; Olink Bioscience, Uppsala, Sweden) at 37°C for 1 h. Unbound antibodies were removed by being washed three times with wash buffer A. Ligase enzyme and ligation buffer were added according to the manufacturer's recommendation in order to hybridize the two PLA probes and to form a closed DNA circle, a reaction that, according to the manufacturer, can occur only if the two PLA probes are located in close proximity to each other (≤40 nm). Samples were then washed three times with wash buffer A. Amplification solution containing deoxyribonucleotides and Texas Red-analogue fluorescent-labeled oligonucleotides (λex. 594 nm, λem. 624 nm) were added, together with DNA polymerase. Amplification reactions were performed at 37°C for 90 min. This will initiate rolling-circle amplification reactions that generate concatemeric DNA strands onto which the fluorescently labeled oligonucleotide detection probes hybridize. Unbound labeled oligonucleotides were removed by washing the samples three times with buffer B before embedding samples in Duolink mounting medium for microscopic observation.
Statistical analyses
Unpaired Student's t tests were performed to analyze the amount of coprecipitated endocytic proteins (Figure 2), percentage of Cx43/Dab2 Duolink colocalization (Figure 3), number of GJ channels in the apposing PMs of coupled cell pairs (Figure 4), average time periods required for GJ plaques to form and internalize (Figure 5), and location and number of heterotypic AGJs/cell type (Figure 7) and to compare the amounts of endogenously versus exogenously expressed Cx43 in HeLa cells (Figure S1) using the GraphPad online software package (GraphPad Software, La Jolla, CA). Data are presented as mean ± SEM. In all analyses, a p value ≤ 0.05 (depicted with *, **, or ***) was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Michael Davidson for mCherry-H2B and Cx43-mApple cDNA constructs and Falk lab members for constructive discussions and critical reading of the manuscript. These works were supported by NIH National Institute of General Medical Science grant GM55725 to M.M.F.
Abbreviations used:
- AGJ
annular gap junction
- AP-2
adaptor protein complex-2
- CME
clathrin-mediated endocytosis
- Cx
connexin
- Dab2
Disabled-2
- GFP
green fluorescent protein
- GJ
gap junction
- GJIC
gap junction intercellular communication
- PM
plasma membrane
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-02-0111) on July 24, 2013.
J.T.F and M.M.F. designed the research, analyzed the data, and wrote the manuscript. J.T.F and R.M.K performed the experiments.
The authors declare that they have no conflict of interest.
REFERENCES
- Aguilar RC, Wendland B. Endocytosis of membrane receptors: two pathways are better than one. Proc Natl Acad Sci USA. 2005;102:2679–2680. doi: 10.1073/pnas.0500213102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SM, Kim N, Gumpert AM, Segretain D, Falk MM. Acute internalization of gap junctions in vascular endothelial cells in response to inflammatory mediator-induced G-protein coupled receptor activation. FEBS Lett. 2008;582:4039–4046. doi: 10.1016/j.febslet.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra N, Kar R, Jiang JX. Gap junctions and hemichannels in signal transmission, function and development of bone. Biochim Biophys Acta. 2012;1818:1909–1918. doi: 10.1016/j.bbamem.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardslee MA, Laing JG, Beyer EC, Saffitz JE. Rapid turnover of connexin43 in the adult rat heart. Circ Res. 1998;83:629–635. doi: 10.1161/01.res.83.6.629. [DOI] [PubMed] [Google Scholar]
- Bejarano E, Girao H, Yuste A, Patel B, Marques C, Spray DC, Pereira P, Cuervo AM. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol Biol Cell. 2012;23:2156–2169. doi: 10.1091/mbc.E11-10-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmerah A, Lamaze C, Begue B, Schmid SL, Dautry-Varsat A, Cerf-Bensussan N. AP-2/Eps15 interaction is required for receptor-mediated endocytosis. J Cell Biol. 1998;140:1055–1062. doi: 10.1083/jcb.140.5.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boassa D, Solan JL, Papas A, Thornton P, Lampe PD, Sosinsky GE. Trafficking and recycling of the connexin43 gap junction protein during mitosis. Traffic. 2011;11:1471–1486. doi: 10.1111/j.1600-0854.2010.01109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Catarino S, Ramalho JS, Marques C, Pereira P, Girao H. Ubiquitin-mediated internalization of connexin43 is independent of the canonical endocytic tyrosine-sorting signal. Biochem J. 2011;437:255–267. doi: 10.1042/BJ20102059. [DOI] [PubMed] [Google Scholar]
- Chen B, Dores MR, Grimsey N, Canto I, Barker BL, Trejo J. Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. J Biol Chem. 2011;286:40760–40770. doi: 10.1074/jbc.M111.299776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocucci E, Aguet F, Boulant S, Kirchhausen T. The first five seconds in the life of a clathrin-coated pit. Cell. 2012;150:495–507. doi: 10.1016/j.cell.2012.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BM, McCoy AJ, Kent HM, Evans PR, Owen DJ. Molecular architecture and functional model of the endocytic AP2 complex. Cell. 2002;109:523–535. doi: 10.1016/s0092-8674(02)00735-3. [DOI] [PubMed] [Google Scholar]
- Falk MM. Connexin-specific distribution within gap junctions revealed in living cells. J Cell Sci. 2000;113:4109–4120. doi: 10.1242/jcs.113.22.4109. [DOI] [PubMed] [Google Scholar]
- Falk MM, Baker SM, Gumpert AM, Segretain D, Buckheit RW., III Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol Biol Cell. 2009;20:3342–3352. doi: 10.1091/mbc.E09-04-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MM, Fong JT, Kells RM, O'Laughlin MC, Kowal TJ, Thevenin AF. Degradation of endocytosed gap junctions by autophagosomal and endo-/lysosomal pathways: a perspective. J Membr Biol. 2012;245:465–476. doi: 10.1007/s00232-012-9464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon RF, Goodenough DA. Five-hour half-life of mouse liver gap-junction protein. J Cell Biol. 1981;90:521–526. doi: 10.1083/jcb.90.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong JT, Kells RM, Gumpert AM, Marzillier JY, Davidson MW, Falk MM. Internalized gap junctions are degraded by autophagy. Autophagy. 2012;8:794–811. doi: 10.4161/auto.19390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontes MS, van Veen TA, de Bakker JM, van Rijen HV. Functional consequences of abnormal Cx43 expression in the heart. Biochim Biophys Acta. 2012;1818:2020–2029. doi: 10.1016/j.bbamem.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Fykerud TA, Kjenseth A, Schink KO, Sirnes S, Bruun J, Omori Y, Brech A, Rivedal E, Leithe E. Smad ubiquitination regulatory factor-2 controls gap junction intercellular communication by modulating endocytosis and degradation of connexin43. J Cell Sci. 2012;125:3966–3976. doi: 10.1242/jcs.093500. [DOI] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- Ghoshroy S, Goodenough DA, Sosinsky GE. Preparation, characterization, and structure of half gap junctional layers split with urea and EGTA. J Membr Biol. 1995;146:15–28. doi: 10.1007/BF00232677. [DOI] [PubMed] [Google Scholar]
- Gilleron J, Carette D, Fiorini C, Dompierre J, Macia E, Denizot JP, Segretain D, Pointis G. The large GTPase dynamin2: a new player in connexin 43 gap junction endocytosis, recycling and degradation. Int J Biochem Cell Biol. 2011;43:1208–1217. doi: 10.1016/j.biocel.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Gilleron J, Fiorini C, Carette D, Avondet C, Falk MM, Segretain D, Pointis G. Molecular reorganization of Cx43, Zo-1 and Src complexes during the endocytosis of gap junction plaques in response to a non-genomic carcinogen. J Cell Sci. 2008;121:4069–4078. doi: 10.1242/jcs.033373. [DOI] [PubMed] [Google Scholar]
- Girao H, Catarino S, Pereira P. Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp Cell Res. 2009;315:3587–3597. doi: 10.1016/j.yexcr.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Gong X, Li E, Klier G, Huang Q, Wu Y, Lei H, Kumar NM, Horwitz J, Gilula NB. Disruption of α3connexin gene leads to proteolysis and cataractogenesis in mice. Cell. 1997;91:833–843. doi: 10.1016/s0092-8674(00)80471-7. [DOI] [PubMed] [Google Scholar]
- Goodenough DA, Gilula NB. The splitting of hepatocyte gap junctions and zonulae occludentes with hypertonic disaccharides. J Cell Biol. 1974;61:575–590. doi: 10.1083/jcb.61.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullberg M, Goransson C, Fredriksson S. Duolink-“In-cell Co-IP” for visualization of protein interactions in situ. Nat Methods. 2011;8 [Google Scholar]
- Gumpert AM, Varco JS, Baker SM, Piehl M, Falk MM. Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett. 2008;582:2887–2892. doi: 10.1016/j.febslet.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms JB, Zurzolo C. Lipids as targeting signals: lipid rafts and intracellular trafficking. Traffic. 2004;5:247–254. doi: 10.1111/j.1600-0854.2004.0181.x. [DOI] [PubMed] [Google Scholar]
- Hesketh GG, Shah MH, Halperin VL, Cooke CA, Akar FG, Yen TE, Kass DA, Machamer CE, Van Eyk JE, Tomaselli GF. Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ Res. 2010;106:1153–1163. doi: 10.1161/CIRCRESAHA.108.182147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16:5686–5698. doi: 10.1091/mbc.E05-08-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter AW, Jourdan J, Gourdie RG. Fusion of GFP to the carboxyl terminus of connexin43 increases gap junction size in HeLa cells. Cell Commun Adhes. 2003;10:211–214. doi: 10.1080/cac.10.4-6.211.214. [DOI] [PubMed] [Google Scholar]
- Johnson KE, Mitra S, Katoch P, Kelsey LS, Johnson KR, Mehta PP. Phosphorylation on serines 279 and 282 of connexin43 regulates endocytosis and gap junction assembly in pancreatic cancer cells. Mol Biol Cell. 2013;24:715–733. doi: 10.1091/mbc.E12-07-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K, Chodock R, Hand AR, Laird DW. The origin of annular junctions: a mechanism of gap junction internalization. J Cell Sci. 2001;114:763–773. doi: 10.1242/jcs.114.4.763. [DOI] [PubMed] [Google Scholar]
- Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med. 2002;4:258–274. doi: 10.1097/00125817-200207000-00004. [DOI] [PubMed] [Google Scholar]
- Keyel PA, Mishra SK, Roth R, Heuser JE, Watkins SC, Traub LM. A single common portal for clathrin-mediated endocytosis of distinct cargo governed by cargo-selective adaptors. Mol Biol Cell. 2006;17:4300–4317. doi: 10.1091/mbc.E06-05-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen WJ, Tung HN, Murray SA, Swenson CA. Evidence for the participation of actin microfilaments and bristle coats in the internalization of gap junction membrane. J Cell Biol. 1979;83:576–587. doi: 10.1083/jcb.83.3.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauf U, Giepmans BN, Lopez P, Braconnot S, Chen SC, Falk MM. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci USA. 2002;99:10446–10451. doi: 10.1073/pnas.162055899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Nelson I, Houlden H, Sweeney MG, Hilton-Jones D, Blake J, Wood NW, Reilly MM. Six novel connexin32 (GJB1) mutations in X-linked Charcot-Marie-Tooth disease. J Neurol Neurosurg Psychiatry. 2002;73:304–306. doi: 10.1136/jnnp.73.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leithe E, Kjenseth A, Sirnes S, Stenmark H, Brech A, Rivedal E. Ubiquitylation of the gap junction protein connexin-43 signals its trafficking from early endosomes to lysosomes in a process mediated by Hrs and Tsg101. J Cell Sci. 2009;122:3883–3893. doi: 10.1242/jcs.053801. [DOI] [PubMed] [Google Scholar]
- Leykauf K, Salek M, Bomke J, Frech M, Lehmann WD, Durst M, Alonso A. Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J Cell Sci. 2006;119:3634–3642. doi: 10.1242/jcs.03149. [DOI] [PubMed] [Google Scholar]
- Lichtenstein A, Minogue PJ, Beyer EC, Berthoud VM. Autophagy: a pathway that contributes to connexin degradation. J Cell Sci. 2011;124:910–920. doi: 10.1242/jcs.073072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Zhou J, Zelenka PS, Takemoto DJ. Protein kinase Cγ regulation of gap junction activity through caveolin-1-containing lipid rafts. Invest Ophthalmol Vis Sci. 2003;44:5259–5268. doi: 10.1167/iovs.03-0296. [DOI] [PubMed] [Google Scholar]
- Mishra SK, Keyel PA, Hawryluk MJ, Agostinelli NR, Watkins SC, Traub LM. Disabled-2 exhibits the properties of a cargo-selective endocytic clathrin adaptor. EMBO J. 2002;21:4915–4926. doi: 10.1093/emboj/cdf487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- Morell RJ, et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med. 1998;339:1500–1505. doi: 10.1056/NEJM199811193392103. [DOI] [PubMed] [Google Scholar]
- Morris SM, Cooper JA. Disabled-2 colocalizes with the LDLR in clathrin-coated pits and interacts with AP-2. Traffic. 2001;2:111–123. doi: 10.1034/j.1600-0854.2001.020206.x. [DOI] [PubMed] [Google Scholar]
- Motley A, Bright NA, Seaman MN, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162:909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanes BA, Chiasson-MacKenzie C, Lowery AM, Ishiyama N, Faundez V, Ikura M, Vincent PA, Kowalczyk AP. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol. 2012;199:365–380. doi: 10.1083/jcb.201205029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, Miyatake S, Saito T, Gallusser A, Kirchhausen T, Bonifacino JS. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science. 1995;269:1872–1875. doi: 10.1126/science.7569928. [DOI] [PubMed] [Google Scholar]
- Pauly BS, Drubin DG. Clathrin: an amazing multifunctional dreamcoat. Cell Host Microbe. 2007;2:288–290. doi: 10.1016/j.chom.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Piehl M, Lehmann C, Gumpert A, Denizot JP, Segretain D, Falk MM. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell. 2007;18:337–347. doi: 10.1091/mbc.E06-06-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma FR, Hengeveld T, Alblas J, Giepmans BN, Zondag GC, Jalink K, Moolenaar WH. Acute loss of cell-cell communication caused by G protein-coupled receptors: a critical role for c-Src. J Cell Biol. 1998;140:1199–1209. doi: 10.1083/jcb.140.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhett JM, Jourdan J, Gourdie RG. Connexin43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell. 2011;22:1516–1528. doi: 10.1091/mbc.E10-06-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert AL, Schubert W, Spray DC, Lisanti MP. Connexin family members target to lipid raft domains and interact with caveolin-1. Biochemistry. 2002;41:5754–5764. doi: 10.1021/bi0121656. [DOI] [PubMed] [Google Scholar]
- Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, Di Fiore PP, Polo S. Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci USA. 2005;102:2760–2765. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711:154–163. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun Adhes. 2008;15:75–84. doi: 10.1080/15419060802014016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenin AF, Kowal TJ, Fong JT, Kells RM, Fisher CG, Falk MM. Proteins and mechanisms regulating gap junction assembly, internalization, and degradation. Physiology (Bethesda) 2013;28:93–116. doi: 10.1152/physiol.00038.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MA, Zosso N, Scerri I, Demaurex N, Chanson M, Staub O. A tyrosine-based sorting signal is involved in connexin43 stability and gap junction turnover. J Cell Sci. 2003;116:2213–2222. doi: 10.1242/jcs.00440. [DOI] [PubMed] [Google Scholar]
- Traub LM. Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J Cell Biol. 2003;163:203–208. doi: 10.1083/jcb.200309175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM. Common principles in clathrin-mediated sorting at the Golgi and the plasma membrane. Biochim Biophys Acta. 2005;1744:415–437. doi: 10.1016/j.bbamcr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Wayakanon P, Bhattacharjee R, Nakahama K, Morita I. The role of the Cx43 C-terminus in GJ plaque formation and internalization. Biochem Biophys Res Commun. 2012;420:456–461. doi: 10.1016/j.bbrc.2012.03.018. [DOI] [PubMed] [Google Scholar]
- White TW, Goodenough DA, Paul DL. Targeted ablation of connexin50 in mice results in microphthalmia and zonular pulverulent cataracts. J Cell Biol. 1998;143:815–825. doi: 10.1083/jcb.143.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe BL, Trejo J. Clathrin-dependent mechanisms of G protein-coupled receptor endocytosis. Traffic. 2007;8:462–470. doi: 10.1111/j.1600-0854.2007.00551.x. [DOI] [PubMed] [Google Scholar]
- Xu J, Nicholson BJ. The role of connexins in ear and skin physiology—functional insights from disease-associated mutations. Biochim Biophys Acta. 2013;1828:167–178. doi: 10.1016/j.bbamem.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C, Furlong J, Burgos P, Johnston LJ. The size of lipid rafts: an atomic force microscopy study of ganglioside GM1 domains in sphingomyelin/DOPC/cholesterol membranes. Biophys J. 2002;82:2526–2535. doi: 10.1016/S0006-3495(02)75596-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.