

Acute suppression of Arp2/3 complex activity in lamellipodia demonstrates its essential role in actin network treadmilling and filament organization and geometry. Arp2/3 complex activity also defines the recruitment of crucial independent factors, including capping protein and cofilin, and is essential for lamellipodia-based keratocyte migration.
Abstract
Lamellipodia are sheet-like protrusions formed during migration or phagocytosis and comprise a network of actin filaments. Filament formation in this network is initiated by nucleation/branching through the actin-related protein 2/3 (Arp2/3) complex downstream of its activator, suppressor of cAMP receptor/WASP-family verprolin homologous (Scar/WAVE), but the relative relevance of Arp2/3-mediated branching versus actin filament elongation is unknown. Here we use instantaneous interference with Arp2/3 complex function in live fibroblasts with established lamellipodia. This allows direct examination of both the fate of elongating filaments upon instantaneous suppression of Arp2/3 complex activity and the consequences of this treatment on the dynamics of other lamellipodial regulators. We show that Arp2/3 complex is an essential organizer of treadmilling actin filament arrays but has little effect on the net rate of actin filament turnover at the cell periphery. In addition, Arp2/3 complex serves as key upstream factor for the recruitment of modulators of lamellipodia formation such as capping protein or cofilin. Arp2/3 complex is thus decisive for filament organization and geometry within the network not only by generating branches and novel filament ends, but also by directing capping or severing activities to the lamellipodium. Arp2/3 complex is also crucial to lamellipodia-based migration of keratocytes.
INTRODUCTION
The actin cytoskeleton is fundamental for establishment and maintenance of forces in both individual cells and cell sheets or tissues and organizes into various structural arrays optimized for exerting specific functions. Migration is commonly initiated by the protrusion of sheets of cytoplasm, so-called lamellipodia, which are filled with networks of actin filaments, the structure, dynamics, and turnover of which have been extensively studied over decades (Pollard and Borisy, 2003; Ridley, 2011; Rottner and Stradal, 2011; Svitkina, 2013). Lamellipodia and the structurally related membrane ruffles are common to a variety of migrating cell types, ranging from epithelial cells to neurons, but are also used, for example, as structures mediating the engulfment of extracellular material as in professional phagocytes (Hall, 2012).
Recent progress indicates that actin filaments that build lamellipodial networks are mostly generated through nucleation or branching effected by actin-related protein 2/3 (Arp2/3) complex (Steffen et al., 2006; Nicholson-Dykstra and Higgs, 2008; Suraneni et al., 2012; Wu et al., 2012), but other potential actin filament nucleators, such as formins or WH2-domain–containing proteins, have also been implicated in regulating different aspects of protrusion (Ahuja et al., 2007; Yang et al., 2007; Zuchero et al., 2009).
Arp2/3 complex activation in lamellipodia is believed to be mediated by pentameric WASP-family verprolin homologous (WAVE) complex, harboring interaction surfaces for both ras-related C3 botulinum toxin substrate (Rac; Sra-1/PIR121) and Arp2/3 complex. Consistent with this view, RNA interference (RNAi)–mediated knockdown of individual WAVE complex subunits suppressed lamellipodia formation and membrane ruffling in various systems, including mammalian and Drosophila cells (Kunda et al., 2003; Rogers et al., 2003; Innocenti et al., 2004; Steffen et al., 2004). The critical function of WAVE complex in lamellipodia formation downstream of Rac also fits the lack of phenotype in lamellipodia formation upon genetic removal of the prominent Arp2/3 activator and Cdc42 effector neural Wiskott–Aldrich syndrome protein (N-WASP; Lommel et al., 2001; Snapper et al., 2001) or the association of related Arp2/3 activators, WHAMM and WASH complex, with other subcellular structures (Campellone et al., 2008; Rottner et al., 2010; Carnell et al., 2011; Duleh and Welch, 2012; Gomez et al., 2012). That Dictyostelium WASP can compensate for suppressor of cAMP receptor (Scar)/WAVE loss of function might indicate less strict functional separation in this distant eukaryote between Scar/WAVE and WASP proteins (Veltman et al., 2012).
Although all these data speak for a linear pathway of lamellipodia formation, at least in mammalian cells, from activation of Rac to Arp2/3-mediated branching via WAVE complex, it has been unclear whether WAVE complex contributes to lamellipodia protrusion solely through WAVE-mediated Arp2/3 activation or through potential additional mechanisms, due—last but not least—to the lack of a genetic model system to eliminate the entire WAVE complex in mammalian tissue culture cells. Thus the recently described Arp2/3-knockout phenotype cannot currently be compared with cells lacking WAVE complex (Suraneni et al., 2012).
Furthermore, dissection of the relative contributions of all of these factors to lamellipodium protrusion is even more challenging if we distinguish between their roles in initiation versus maintenance of lamellipodia (Rottner and Stradal, 2011). For instance, we are just beginning to understand the precise consequences for actin filament generation and turnover of abrupt Arp2/3 complex inhibition in lamellipodia: in neuronal growth cones, an exciting study showed that treatment with the small-molecule inhibitor CK666 (Nolen et al., 2009) causes increased rearward flow rates that depended on myosin II (Yang et al., 2012). It remained unclear, however, whether the inverse relationship between myosin II and Arp2/3 activities in protrusion is specific to growth cones or is a more common phenomenon in actin-based protrusion.
Here we use various experimental protocols to dissect Arp2/3 complex function in fibroblasts harboring established lamellipodia, allowing direct analysis of changes in actin dynamics independent of lamellipodia initiation by Rac and thus uncoupled from upstream regulatory inputs, for instance, through growth factor or integrin receptor signaling. We also explore the consequences of interference with Arp2/3 complex function for targeting and kinetics of other lamellipodial regulators and during the lamellipodia-mediated migration of melanoma cells and keratocytes.
RESULTS
Injection of C -terminus of WAVE proteins harboring WH2, connector, and acidic domains causes instantaneous lamellipodia disruption and Arp2/3 complex delocalization
Lamellipodia formation was previously suppressed by transient (Steffen et al., 2006; Nicholson-Dykstra and Higgs, 2008) or stable (Wu et al., 2012) knockdown of Arp2/3 complex subunits or Arp2/3 sequestration in the cytosol expressing C-terminal Arp2/3-interacting fragments of WAVE1 (formerly called Scar1; Machesky and Insall, 1998). In addition, analysis of fibroblasts lacking the Arp2/3 complex subunit ArpC3 has recently proved the crucial function of the complex in lamellipodia formation (Suraneni et al., 2012). These studies, however, could not determine whether Arp2/3 complex is required solely for lamellipodia initiation or also their maintenance. Moreover, such long-term treatments might cause changes in expression patterns or signaling status of treated cells that could also indirectly affect protrusion, exemplified by increased RhoA levels observed upon Arp2/3 complex knockdown in neuronal cells (Korobova and Svitkina, 2008).
To directly probe the potential function of Arp2/3 complex in continuous protrusion of established lamellipodia in fibroblasts, we performed video microscopy of cells expressing constitutively active Rac1, before and after microinjection with Rac1 alone or mixtures of Rac1 and the C-terminus of WAVE proteins harboring WH2, connector, and acidic domains (WCA) of WAVE1 (see experimental strategy in Figure 1A). Based on long-term expression of the same fragment upon transfection (Machesky and Insall, 1998), it was expected to potently sequester Arp2/3 complex in the cytoplasm and thus cause its instantaneous, competitive removal from its physiological sites of action. Coinjection of constitutively active Rac1 was used to ensure continuous signaling required for lamellipodia protrusion (Figure 1B) and exclude loss of peripheral protrusions due to Rac signaling defects or nonspecific injection artifacts.
FIGURE 1:

Microinjection of WCA/Rac causes Arp2/3 complex sequestration and a shift from lamellipodia to filopodia formation. (A) The strategy used in this study. 1) Expression of constitutively active Rac1 leads to lamellipodia formation, presumably via activation of WAVE- and Arp2/3 complexes. 2) On microinjection, WCA of WAVE1 binds and sequesters the Arp2/3 complex in the cytoplasm. (B) Frames of phase contrast microscopy videos of NIH3T3 cells expressing constitutively active Rac1 and microinjected with active Rac1 as control (top) or a mixture of WCA and active Rac1 (bottom). Time is given in minutes and seconds. Bars, 5 μm. See also Supplemental Movie S1. (C) Fluorescence (left) and phase contrast (right) video frames of NIH3T3 cells transfected with constitutively active Rac1 and pEGFP-p16B before and after Rac1 (top) or WCA/Rac1 (bottom) microinjection (5 min, 29 s, and 5 min, 32 s, for Rac1 and WCA/Rac1, respectively). Bar, 3 μm. Right, graphs showing line scans of fluorescence intensities in regions as indicated. (D) Frames of phase contrast movie of NIH3T3 cells coexpressing constitutively active Rac1 and EGFP-actin (see also Figure 7D later in the paper) before and after microinjection with a mixture of CA and active Rac1. Time in minutes and seconds. Bar, 5 μm.
WCA/Rac injections robustly and reproducibly transformed peripheral lamellipodia into multiple, filopodia-like protrusions within minutes (Figure 1B; see also Supplemental Movie S1), and loss of lamellipodia was stable in these cells up to several hours (data not shown). These data confirm that Arp2/3 complex activity is required for lamellipodia maintenance, as indicated previously by its continuous incorporation from the lamellipodium tip (Iwasa and Mullins, 2007; Lai et al., 2008). To confirm that WCA/Rac affected Arp2/3 complex accumulation at the cell periphery, we injected cells coexpressing enhanced green fluorescent protein (EGFP)–tagged ArpC5, also known as p16B (Millard et al., 2003). WCA/Rac diminished Arp2/3 complex accumulation at the cell periphery, whereas control injections with Rac alone did not (Figure 1C). To test whether WCA injections might cause less specific interference with actin assembly than originally intended, for instance, through actin monomer sequestration effected by the W domain present in the WCA fragment, we used ectopic actin polymerization activated on the surface of microtubules, allowing comparison of the actin assembly capabilities of distinct nucleation mechanisms (Oelkers et al., 2011). WCA injections interfered with Arp2/3-dependent actin accumulation on microtubules but not with actin assembly induced by the nucleation domain of Spir (Spir-NT; Supplemental Figure S1).
To exclude that phenotypes observed were solely due to ectopic Arp2/3 activation instead of sequestration, we explored the effect of the C-terminus lacking the WH2 domain (CA), as the WH2 domain is considered essential for delivering actin monomer to Arp2 and Arp3 during nucleation (Campellone and Welch, 2010). Although one study detected a weak, additional actin-binding activity in the C domain of Scar1 (Kelly et al., 2006), previous data indicated negligible activity concerning Arp2/3 activation of CA fragments using pyrene-actin assays (Hufner et al., 2001; Marchand et al., 2001), as confirmed in our experiments (see later discussion of Figure 7A). When injecting mixtures of Rac with the CA fragment of WAVE1, we observed a similar transformation of the cell periphery from lamellipodia to filopodia (Figure 1D). CA was less potent than WCA, however, and required higher protein concentrations, fully consistent with the partial and concentration-dependent competition with WCA-induced actin assembly in pyrene assays (see later discussion of Figure 7B). It is conceivable that establishment of multiple interactions with Arp2/3 complex, through CA and indirectly through interaction of W with actin, will increase the potency of sequestration by WCA, so for further experiments, we focused on the entire C-terminus (WCA).
FIGURE 7:

CA partially inhibits Arp2/3 complex function in vitro and in vivo. (A) CA does not stimulate Arp2/3 complex–mediated actin polymerization. A total of 2 μM G-actin (10% pyrene labeled) was polymerized in 1× KMEI buffer in the presence of 1 nM Arp2/3 complex and CA at the concentrations indicated. Note that even a 1000-fold molar excess of CA compared with Arp2/3 complex did not affect Arp2/3-dependent actin polymerization. (B) WCA-stimulated activation of Arp2/3 complex–mediated actin polymerization is suppressed, but not fully inhibited, by CA in a concentration-dependent manner. A total of 2 μM G-actin (10% pyrene labeled) was polymerized in 1× KMEI buffer in the presence of 1 nM Arp2/3 complex, WCA, and CA at concentrations as indicated. (C–F) Fluorescence (top) and phase contrast (bottom) video frames of NIH3T3 cells expressing EGFP-tagged p16B (C), actin (D), cofilin (E), and capping protein (F) before and 5 min after injection with Rac/CA. Bar, 3 μm. (G) Line scans of fluorescence intensities as indicated in C–F.
Arp2/3 complex sequestration does not eliminate actin assembly but reduces rearward flow
To explore the consequences of Arp2/3 complex sequestration on dynamics of the peripheral actin cytoskeleton, we performed live-cell imaging of cells transiently coexpressing active Rac and Lifeact-EGFP (Figure 2 and Supplemental Movie S2). As shown, the bright rim of lamellipodial actin network rapidly vanished upon Rac/WCA injection and was replaced by filopodial bundles embedded in actin meshworks of lower intensity. Kymograph analysis allowed determination of flow rates of actin networks before and after injection of respective protein mixture, as indicated (Figure 2B). Flow rates in the absence of net protrusion can be equated with actin assembly rates in lamellipodial networks (Watanabe and Mitchison, 2002), but it is unclear to what extent actin branching by Arp2/3 complex affects actin assembly and thus flow rates of the network in fibroblast lamellipodia. Of interest, injection of Rac alone slightly reduced rearward flow of the actin network, for reasons that remain to be established, but upon WCA/Rac injections, flow rates were diminished down to roughly one-fourth of the levels before injections. Of note, these rates were identical to those seen in lamella regions proximal to the lamellipodium of noninjected cells (no mi lamella, Figure 2C). These data emphasize the relative importance of Arp2/3-mediated actin branching for maintaining rapid actin network flow in the lamellipodium.
FIGURE 2:

Sequestration of Arp2/3 complex diminishes lamellipodial actin networks and reduces peripheral actin flow to lamella levels. (A) NIH3T3 cell transfected with constitutively active Rac1 and EGFP-Lifeact to visualize actin before and after microinjection of WCA/Rac1. Bar, 3 μm. See also Supplemental Movie S2. (B) Examples of kymograph measurements of peripheral regions of NIH3T3 cells transfected with constitutively active Rac1 and EGFP-tagged Lifeact or actin before and after injection of Rac1 (top) or WCA/Rac1 (bottom). Numbered black lines are examples of individual measurements; arrows indicate time points of injection. (C) Results of kymograph analysis of rearward flow at the cell edge or lamella of uninjected (no mi lamella), Rac1-injected, and WCA/Rac1-injected cells. Error bars, SEM. n, number of individual measurements from 12 (no mi), 7 (+ Rac), 13 (+WCA/Rac), and 8 (no mi lamella) independent cells. Flow rates in WCA/Rac-injected cells (cell edge) were confirmed not to be statistically different from those in the lamella of noninjected cells (no mi lamella; p < 0.57).
Arp2/3 complex is essential for actin treadmilling at the cell periphery
In spite of a reduction of flow rate and loss of canonical lamellipodia upon Rac/WCA injection, the actin network seemed to be fueled by continuous polymerization from the membrane, suggesting that actin turnover is still governed by treadmilling from the tip, as seen in lamellipodia networks (Wang, 1985; Iwasa and Mullins, 2007; Lai et al., 2008).
To test this directly, we performed fluorescence recovery after photobleaching (FRAP) analysis upon injections in cells coexpressing EGFP-actin (Figure 3). Rac-alone control–injected cells displayed actin assembly from their tips in a treadmilling regime, as previously seen in fibroblasts coexpressing active Rac1 and EGFP-actin (Lai et al., 2009). Unexpectedly, and in spite of continuous flow from the front, network treadmilling was lost upon Arp2/3 complex sequestration (Figure 3 and Supplemental Movie S3). We previously showed (Lai et al., 2008) that fluorescence recovery times in control cells are typically doubled in the rear part of the lamellipodium compared with the front (Figure 3B). This feature was virtually lost upon Arp2/3 complex inhibition (Figure 3C), and fluorescence recovery times in both front and rear regions of the zone previously occupied by a lamellipodium were similar to those of the front half of control lamellipodia (Figure 3, B and C). This indicated that the average actin polymerization rate (assumed to be key determinant of actin fluorescence recovery in these experiments) was mostly unchanged in the region corresponding to the lamellipodium front before injection and even increased (roughly by a factor of two) in that corresponding to the lamellipodium rear. In other words, net actin polymerization appeared not reduced, in spite of inhibition of a decisive actin filament nucleator at the cell periphery. These data indicate that Arp2/3 complex activity at the tip membrane of established, continuously protruding lamellipodia is less relevant for maintaining the number of growing filament ends than previously believed but is essential instead for biasing filament growth to the front. This activity can be equalized to inhibition of nucleation and polymerization in rear parts of canonical lamellipodia, constituting an essential prerequisite of lamellipodial actin network treadmilling (Figure 3).
FIGURE 3:

Arp2/3 complex is essential for actin treadmilling. (A) Examples of FRAP experiments of EGFP-actin at the cell periphery of NIH3T3 cells expressing constitutively active Rac1 and injected with Rac1 (top) or WCA/Rac1 (bottom). White rectangle indicates bleach region; blue and red rectangles indicate front and rear regions, respectively, as used for treadmilling factor (TMF) analysis. Time in seconds; bars, 5 μm. See also Supplemental Movie S3. (B, C) Graphs displaying curve fits of normalized mean recovery of fluorescence after photobleaching in front (blue) and rear (red) regions in Rac1-injected (B) or WCA/Rac1-injected (C) cells, which were used for TMF analysis. n, number of cells used for analysis.
Our data also suggest that Arp2/3 complex inhibition is sufficient to transform lamellipodia into lamella-like networks, supported by the notion, for instance, that microspike bundles turn over actin filaments in a nontreadmilling regime as soon as they are uncoupled from the lamellipodium tip and enter the lamella (Koestler et al., 2008). This view is also consistent with identical flow rates observed in the lamella of noninjected cells and peripheral actin networks after WCA/Rac injection (Figure 2C).
Acute Arp2/3 complex inhibition reduces filament and branching density
The presence of actin filament branches in lamellipodia was first described using metal shadowing electron microscopy (Svitkina et al., 1997; Svitkina and Borisy, 1999). Electron tomography of negatively stained lamellipodia has more recently shown that branches connect subsets of actin filaments within the three-dimensional network, with variable distances between branches and an average branching frequency—at least in the front part of 3T3 cell lamellipodia—of approximately one branch/0.8 μm of actin filament (Vinzenz et al., 2012). Branching frequency, however, had not been directly correlated with Arp2/3 complex activity. We injected cells with Rac alone or WCA/Rac during video microscopy (not shown) and fixed and subjected them to electron tomography. Figure 4A shows a representative example (right) of the actin network reorganization caused by Arp2/3 complex inhibition as compared to control (left) (see also Supplemental Movie S4). Most apparent at first glance is a strong increase in filaments running approximately parallel to the cell edge at the expense of more-diagonal filaments, a distribution roughly inverse to the control cell situation (left; insets in Figure 4A). Manual as well as automated tracking of the trajectories of individual filaments (Winkler et al., 2012) in these tomograms indicated a modest decrease in average filament density (Figure 4A and data not shown). More important, branch quantitation in tomograms (red dots in tomogram projections in Figure 4A) upon Rac/WCA injection revealed branch numbers per lamellipodial area to be reduced to almost one-third of the numbers obtained in controls (Figure 4B). Considering the decrease in rearward flow rates by a factor of three to four (Figure 2C), we estimate a residual branching frequency in these conditions at the lamellipodium tip of ∼10% of that in control cells. We also sought to explain the absence of actin network treadmilling upon Arp2/3 inhibition (Figure 3), which in theory could be caused by increased nucleation throughout the lamellipodium or loss of filament polarization to the front routinely observed in control lamellipodia (Small et al., 1978; Narita et al., 2012). Owing to detailed structural information available for branch junctions, for instance, obtained by electron microscopy both in vitro (Mullins et al., 1998; Rouiller et al., 2008) and in vivo (Svitkina and Borisy, 1999; Vinzenz et al., 2012), it is possible to determine the polarities of both daughter and mother filaments from the angular orientation of the branch junction (Narita et al., 2012). Of note, filaments oriented parallel to the cell front after Rac/WCA injection exhibited branch junctions with opposite polarities (Figure 4C), uncovering potential reorientation into antiparallel arrays (see also later discussion), as in the lamella (Koestler et al., 2008). In addition, some mother filaments were apparently oriented in the wrong direction (Figure 4C), with their barbed ends pointing away from the membrane, unlike controls. These data suggest that the loss of network treadmilling in these conditions results from the loss of orientation of rapidly polymerizing barbed ends toward the front.
FIGURE 4:

Arp2/3 inhibition reduces branching frequency and causes network rearrangements resulting in antiparallel actin filament arrays. (A) Electron tomograms of negatively stained NIH3T3 cells expressing constitutively active Rac1 and injected with Rac1 (left) or WCA/Rac1 (right). Projections of 20 central slices of 0.746-nm thickness. Identified branches are marked with red dots. Insets display the angular distribution of filaments in the tomogram to the cell edge. Bars, 200 nm. See also Supplemental Movie S4. (B) Number of branches/μm2 in tomograms upon Rac1 and WCA/Rac1 injection, as indicated. Bars correspond to arithmetic means (n = 5) and SEM. Differences were confirmed to be statistically significant (p ≤ 0.0049). (C) Tomogram of the edge of a WCA/Rac-injected NIH3T3 cell transfected with constitutively active Rac1. Red circles indicate branch points of daughter filaments pointing in opposite directions. Cell edge is oriented toward the top of the image. Bar, 200 nm.
Arp2/3 complex inhibition triggers myosin II incorporation
Aside from incorporation of nascent spots of myosin II into actin arcs initiated in the lamellipodium (Burnette et al., 2011) or into minor foci in the large lamellipodia of keratocytes (Svitkina et al., 1997), the majority of myosin II remains excluded from lamellipodial networks (McKenna et al., 1989), due presumably to the lack of bundles of opposite polarity, as observed in contractile arrays such as stress fibers (Langanger et al., 1986). A recent study established filaments with opposite polarities to be sufficient for spatial and specific recruitment of myosin motors in vitro (Reymann et al., 2012), but whether filaments of opposite polarity constitute a prerequisite of myosin II incorporation at the cell periphery is unclear. Because Arp2/3 inhibition induced alterations of filament angle distributions and polarities (Figure 4), we tested whether these features coincided with changes in myosin II dynamics. Indeed, before microinjection with WCA/Rac, EGFP-tagged myosin light chain accumulated exclusively in patches along stress fibers and in the lamella but not in the lamellipodium (Figure 5A and Supplemental Movie S5). Soon after Arp2/3 complex sequestration, however, nascent myosin patches started to form at the cell edge, distal to the lamellipodium–lamella boundary before injection (Figure 5B), indicating that the ultrastructural rearrangements of the actin network by this treatment was sufficient to allow myosin incorporation. Quantifications revealed a >10-fold increase in the presence of myosin spots upon Arp2/3 complex inhibition in regions classified as lamellipodia before injections (Figure 5C). These data, together with recently published work (Reymann et al., 2012), suggest that Arp2/3 complex might counteract myosin II function, as established recently for neuronal growth cones (Yang et al., 2012), through generation of actin networks lacking filaments of opposite polarity.
FIGURE 5:

Arp2/3 sequestration attracts myosin II incorporation at the cell edge. (A) Fluorescence (top) and merge of fluorescence (green) and phase contrast (red) overview images of NIH3T3 cell coexpressing constitutively active Rac1 and EGFP-tagged myosin light chain. (B) Magnification of the region depicted in A before and after injection of WCA/Rac1, as indicated. Time, minutes and seconds. White lines in fluorescence images indicate cell edge as deduced from phase contrast images. See also Supplemental Movie S5. (C) Numbers of myosin light chain spots/μm2 before and after injection of WCA/Rac, as indicated. Data are arithmetic means ± SEM determined from 21 lamellipodial regions (11 cells).
Protrusion regulators show specific and distinct dependence on Arp2/3 complex activity
We next explored the molecular consequences of Arp2/3 complex loss of function for a panel of lamellipodial or filopodial regulators (Figure 6 and Supplemental Movie S6). As expected, we found a strong association with filopodia of the actin-bundling protein fascin both before and after interference with Arp2/3 complex function, confirming that the bundled protrusions induced upon Arp2/3 complex sequestration were filopodia and consistent with the view that Arp2/3 complex can be functionally separated from filopodia formation (Steffen et al., 2006; Nicholson-Dykstra and Higgs, 2008; Suraneni et al., 2012; Wu et al., 2012).
FIGURE 6:

Differential requirement of Arp2/3 complex for incorporation of various protrusion regulators. (A) Fluorescence images of cells transfected with constitutively active Rac1 and EGPF-tagged fusion proteins as indicated before (top) and after injection of WCA/Rac1 (bottom). Bar, 3 μm. See also Supplemental Movie S6 and Supplemental Figure S2A. (B) Line scans of fluorescence intensities as indicated. (C) Quantification of lamellipodial tip intensities of respective components after Rac/WCA injections expressed in percentage before injections. Error bars, SEM. Components confirmed to be statistically different from 100%: cofilin (n = 6), capping protein (CP; n = 7), cortactin (Cttn; n = 6), each p < 0.00001, VASP (n = 13, p < 0.0001). WAVE (n = 7, p < 0.74) and FMNL2∆DAD (n = 8, p < 0.53) were not statistically different from 100%.
In contrast, the lamellipodial marker protein cortactin rapidly vanished after Arp2/3 complex sequestration, consistent with the idea that lamellipodial targeting required both its actin filament and Arp2/3 complex–binding activities (Weed et al., 2000) and with recruitment of cortactin to ectopic actin filament assemblies nucleated by Arp2/3 complex (Oelkers et al., 2011). Of interest, Arp2/3 complex inhibition also rapidly removed cofilin from the cell periphery, providing the first experimental evidence that cofilin specifically requires Arp2/3 to associate with the lamellipodium (Figure 6). The underlying mechanistic reasons are unclear (see also later discussion), but based on these data it is tempting to hypothesize that the absence of cofilin from the lamella or stress fibers is caused by independence of their generation of Arp2/3 complex. The fact that the lamellipodial network components cortactin and cofilin displayed similar and relatively rapid turnover rates within the lamellipodium, with half-time of recovery rates of ∼6 s (Lai et al., 2008), explains their comparable kinetics of disappearance upon Arp2/3 complex inhibition (Supplemental Movie S6).
As opposed to cortactin, the loss of cofilin upon Arp2/3 inhibition is puzzling. A potential explanation includes competitive displacement from the lamellipodium by an actin-binding protein actively recruited upon Arp2/3 inhibition. Indeed, tropomyosin (Tm) was previously shown to antagonize actin filament severing by members of the ADF/cofilin family (Bernstein and Bamburg, 1982; Ono and Ono, 2002). Moreover, suppression of lamellipodia formation by microinjection of skeletal muscle Tm was reported to coincide with a decrease at the cell periphery not only of Arp2/3 complex, but also of cofilin (Gupton et al., 2005). Because Tm was previously also shown to inhibit Arp2/3-mediated branching in vitro, Tm members are thus believed to antagonize lamellipodia formation by counteracting Arp2/3 and cofilin activities (DesMarais et al., 2002; Bugyi et al., 2010). To test whether cofilin delocalization upon Arp2/3 complex sequestration is mediated by a potential Tm member, we first assessed expression of different tropomyosins in our NIH3T3 fibroblasts (Schevzov et al., 2005). Western blotting revealed expression of the high–molecular weight isoforms 1–3, as well as the low–molecular weight isoforms Tm4 and Tm5NM1/2 (Supplemental Figure S2). EGFP- or enhanced yellow fluorescent protein (EYFP)–tagged variants of these isoforms could also be readily expressed, and none of these convincingly localized to the lamellipodium when coexpressed with constitutively active Rac1 (Supplemental Figures S3 and S4 and data not shown). EGFP-Tm3 nicely accumulated in stress fibers and actin filament arrays in the lamella but stayed excluded from the cell periphery upon Arp2/3 complex sequestration. Furthermore, the low–molecular weight isoform Tm5NM1 also failed to target to the cell periphery upon Rac/WCA injection (Supplemental Figure S3). Similar results were obtained with EGFP-Tm2 (data not shown), which appeared more abundant endogenously than Tm3 (Supplemental Figure S2). Finally, coexpression of EGFP-Tm2 with mCherry-cofilin confirmed that Arp2/3 inhibition–mediated cofilin displacement was not accompanied by Tm2 targeting to the cell periphery (Supplemental Figure S4). We conclude that loss of cofilin from the cell periphery cannot be explained by tropomyosin-mediated displacement.
We then turned to heterodimeric capping protein, specific interactor of the rapidly growing barbed ends of actin filaments and considered essential for both lamellipodia formation (Mejillano et al., 2004) and reconstitution of Arp2/3-dependent actin assembly in vitro (Loisel et al., 1999). Capping protein accumulation in lamellipodia is frequently more restricted to their tips (Svitkina et al., 2003; Mejillano et al., 2004; Lai et al., 2008), presumably consistent with the accumulation of barbed ends at the membrane (Lai et al., 2008; Vinzenz et al., 2012). Although capping protein is capable of promoting Arp2/3-dependent filament generation in vitro (Akin and Mullins, 2008), such functional coupling remains to be revealed in vivo. Capping protein is usually considered to stochastically block barbed-end assembly at the lamellipodium tip, but the reason for its specificity for lamellipodial barbed ends versus those located in filopodia or the lamella has not been firmly established. Surprisingly, our data show that capping protein accumulation in the lamellipodium is downstream and not independent of Arp2/3 complex activity (Figure 6 and Supplemental Movie S6), so functional coupling between the two in vivo is more direct than previously appreciated.
We also explored the consequences of Arp2/3 inhibition on fate and dynamics of its upstream regulatory complex (WAVE complex, exemplified by EGFP-tagged WAVE2), as well as actin filament polymerases like VASP and the novel lamellipodial tip component FMNL2 (Block et al., 2012). In spite of loss of bias of actin polymerization to the tip (Figure 3), caused at least in part by network reorganizations or reorientations of filament polarities (Figure 4), all these tip components remained associated with the interface of plasma membrane and actin network (Figure 6, A and B, and Supplemental Movie S6), although VASP appeared slightly more sensitive to WCA/Rac injections on average than WAVE2 and FMNL2 (Figure 6C).
WAVE complex should operate upstream and thus be independent of Arp2/3 incorporation into the lamellipodium. Its turnover in the lamellipodium tip upon Arp2/3 complex inhibition, however, has never been explored. Indeed, a potential feedback of regulation of WAVE turnover by Arp2/3 complex could in theory be deduced from vaccinia actin tail formation, in which N-WASP operates as essential Arp2/3 activator at the viral surface (Snapper et al., 2001). This is because failure of Arp2/3 activation by N-WASP completely abolishes its exchange at the viral surface (Weisswange et al., 2009), indicating functional coupling between Arp2/3 activation and turnover of its activators. To test whether this conclusion would hold for lamellipodial WAVE subunits, we compared WAVE2 turnover at the lamellipodium tip by FRAP in cells injected with Rac alone or WCA/Rac. Although WAVE turnover was slower on average in these Rac-expressing fibroblasts compared with nontreated melanoma cells migrating on laminin (Lai et al., 2008), no significant difference was observed after Arp2/3 inhibition, with half-times of recovery of 14.5 and 16.6 s for Rac and WCA/Rac-injected cells, respectively (Supplemental Figure S2A). These data are also consistent with unchanged WAVE2 turnover upon freezing of actin network turnover by an inhibitor cocktail (Millius et al., 2012). Distinct from observations with the vaccinia virus system (Weisswange et al., 2009), positioning and turnover of lamellipodial WAVE can thus be uncoupled from Arp2/3 complex activation.
Cofilin and capping protein preferentially incorporate into Arp2/3-dependent actin structures
Delocalization of cofilin and capping protein caused by WCA injection could have also been effected formally by WCA-mediated activation of Arp2/3 complex in the cytosol by competing away cofilin and capping protein into the cytoplasm instead of specific loss of fluorescence at the cell periphery due to inhibition of Arp2/3 complex incorporation. To address this experimentally, we studied Arp2/3 complex, cofilin, and capping protein dynamics before and after injection with the mixture of Rac and the truncated CA domain shown in Figure 1D. Although the CA fragment is inactive concerning Arp2/3 activation (Figure 7A) or Arp2/3-independent actin assembly (Supplemental Figure S5B), its injection caused a partial but significant loss not only of Arp2/3 incorporation (Figure 7, C and G), but also of cofilin and capping protein association with the cell periphery (Figure 7, E–G). Actin filament intensities at the cell periphery upon Rac/CA injections were also reduced (Figure 7, D and G), comparable to effects seen after WCA/Rac injections (Figures 2 and 3). Incomplete, CA-mediated Arp2/3 inhibition in vivo perfectly fits the competitive interference with WCA-induced Arp2/3 activation in vitro (Figure 7B). These results demonstrate that loss of cofilin and capping protein accumulation at the cell periphery are a result of Arp2/3 complex delocalization but not ectopic activation of the latter. In complementary experiments, we used the small-molecule inhibitor CK666, which at a concentration >420 μM apparently precipitated in our growth medium. Of interest, although CK666 at 420 μM failed to delocalize Arp2/3 complex in our fibroblasts expressing constitutively active Rac1, it markedly reduced network flow compared with the inactive compound CK689 (data not shown). B16-F1 melanoma cells spontaneously migrating on laminin in a lamellipodia-dependent manner, however, were more sensitive to CK666. Treatment at a concentration of 210 μM allowed acute and reversible inhibition of lamellipodia formation and exploration of the consequences of CK666 treatment for Arp2/3 complex, cofilin, and capping protein accumulation. Of interest, CK666 caused a significant and reversible reduction of Arp2/3 complex, cofilin, or capping protein accumulation. Consistently Arp2/3 complex maintenance at the cell periphery was more sensitive to CK666 treatment than that of actin (Supplemental Figure S6), and cofilin and capping protein behaved in a manner comparable to Arp2/3 complex (Supplemental Figure S7). Collectively these data strongly suggest that cofilin and capping protein targeting to lamellipodia is sensitive to the activity of Arp2/3 complex, irrespective of the method of inhibition.
Because all of these data suggested that the targeting of cofilin and capping protein to a given actin structure is determined, at least in part, by the mechanism of its generation, we sought to test this conclusion using an additional, gain-of-function approach, complementary to the loss-of-function experiments described earlier. We used in vivo actin assembly induced by distinct, ectopic nucleators on the surface of microtubules (Oelkers et al., 2011). Of interest, the C-terminus of N-WASP harboring two WH2 domains followed by connector and acidic domains (WWCA) fused to the microtubule-binding domain of MAP4 known to induce strong actin polymerization on the surface of microtubules (Oelkers et al., 2011) caused a marked accumulation of both cofilin and capping protein at these sites (Figure 8). In contrast, a corresponding construct harboring the N-terminus of the independent actin nucleator Spir and equally potent in actin filament accumulation on microtubules failed to recruit either Arp2/3 complex (Oelkers et al., 2011) or cofilin (Figure 8). Although recruited with low frequency to Spir-induced actin filaments, capping protein also showed a strong preference for Arp2/3-dependent actin filaments polymerized on microtubules (Figure 8). Pull-down experiments using purified Arp2/3 complex, cofilin, and capping protein indicated the absence of direct interaction surfaces for cofilin or capping protein on Arp2/3 complex (Supplemental Figure S5C).
FIGURE 8:

Preferential targeting of cofilin and capping protein to Arp2/3-stimulated actin structures. Fluorescence images of B16-F1 cells coexpressing mCherry-tagged cofilin (A) or capping protein (B) each with EGFP-tagged microtubule binding domain (MBD)–WWCA (top) or MBD-Spir-NT (bottom). Merged images show cofilin or capping protein in red and the respective MBD-fusion protein in green. Bar, 10 μm. (C) Quantification of cofilin and capping protein accumulation on microtubules in cells coexpressing MBD-fusion proteins as indicated. Data are means ± SEM from three independent experiments. n, total number of cells analyzed. Specificity of cofilin and capping protein recruitment to Arp2/3-induced actin structures was confirmed to be statistically significant (cofilin, p < 0.0004; capping protein, p < 0.029).
Together these data suggest that both cofilin and capping protein are targeted indirectly to Arp2/3-dependent actin structures such as the lamellipodium in vivo, likely contributing to the specificity of function exerted by each factor at the cell periphery.
Arp2/3 complex inhibition arrests migration of epidermal fish keratocytes
Stable, RNAi-mediated knockdown of Arp2/3 complex expression in fibroblasts was recently found to reduce but not abolish migration (Wu et al., 2012). In contrast, genetic removal of an Arp2/3 complex subunit in embryonic stem cell–derived fibroblastoid cells established defects in directionality of migration rather than rate (Suraneni et al., 2012). Although the reason for this discrepancy is unclear, it might indicate differential capabilities of different cell types to adapt to Arp2/3 removal and switch to lamellipodia-independent migration. To test this hypothesis, we explored the consequences of our treatment in cells expressing prominent lamellipodia. Fish epidermal keratocytes are very efficient movers, which form a fan-shaped lamellipodium covering a large area at the cell front and dragging forward a rolling cell body (Anderson et al., 1996). Owing to the small size of these cells compared with fibroblasts, successful injection is more challenging and was documented by coinjection of an inert fluorescent marker (insets in Figure 9). Injection of Rac alone had little effect on migration rates besides occasional, modest extension of lamellipodial protrusions to cell flanks and rear (Figure 9 and Supplemental Movie S7). In contrast, WCA/Rac injections not only transformed the front edge of the protruding lamellipodium from a smooth into a jagged appearance, but they also arrested migration. This phenotype again coincided with strong transformation of the ultrastructural organization of WCA/Rac-injected cells versus controls (Supplemental Figure S8) in a manner highly reminiscent of the data obtained with injected fibroblasts (Figure 4). We conclude that continuous Arp2/3 complex activity is essential not just for the initiation but also for the maintenance of protrusion in cells, like keratocytes, the migration of which relies on lamellipodia.
FIGURE 9:

Sequestration of Arp2/3 complex leads to migration arrest of fish keratocytes. Phase contrast images of video sequences of fish keratocytes injected with Rac1 (top) or WCA/Rac1 (bottom). Insets show fluorescence of Texas red–labeled dextran indicating successful injection. Red lines mark position of the protruding cell front in consecutive frames. Time after injection, minutes and seconds; bars, 10 μm. See also Supplemental Movie S7 and Supplemental Figure S5.
DISCUSSION
There has been major progress in recent years concerning the molecular regulation of lamellipodia protrusion derived from the precise determination of biochemical activities of key regulators in vitro, including the discovery of Arp2/3 complex and characterization of its nucleation and branching activity (reviewed in Campellone and Welch, 2010), recognition of Scar/WAVE proteins as its activators at the lamellipodium tip (Stradal et al., 2004), and conformational changes accompanying WAVE complex activation through coincident signals including phospholipids and Rac (Lebensohn and Kirschner, 2009; Chen et al., 2010).
All components of the pathway have been probed for their relevance by RNA interference, and recent highlights include the development of cells genetically deficient for the first nonredundant component of the pathway, the Arp2/3 complex subunit ArpC3 (Suraneni et al., 2012). Such permanent genetic deletions in combination with reintroduction of the missing complex subunit could help to define the earliest phases of lamellipodia protrusion, but here we used the opposite approach, allowing acute functional interference with Arp2/3 complex and thus maintenance of actin structures requiring its continuous activity. The latter is essential for the unidirectional assembly of the lamellipodial actin network from the tip and thus treadmilling during lamellipodia formation.
Our approach also enabled us to dissect the ultrastructural modifications accompanying suppression of Arp2/3 complex activity at the single-cell level and discern components and thus associated biochemical activities to be positioned upstream or downstream of Arp2/3 complex. For example, cofilin localization depends on how peripheral actin networks are nucleated and maintained; otherwise it would target to the cell periphery irrespective of Arp2/3 complex activity. What can we learn from this? Cofilin family members bind and sever actin filaments in vitro (Bernstein and Bamburg, 2010). In addition, they are relevant for lamellipodia protrusion (Hotulainen et al., 2005) and are believed to accumulate in these structures by directly interacting with actin filaments, but how specificity is established is usually not addressed. Surprisingly, loss of cofilin upon Arp2/3 inhibition did not coincide with accumulation of its potential antagonist tropomyosin, so how specificity for the lamellipodium is achieved remains elusive. Notwithstanding this, our data demonstrate that subcellular cofilin targeting is highly sensitive to Arp2/3 complex activity and cannot be solely mediated by interaction with actin filaments. Of interest, similar considerations apply to heterodimeric capping protein. Given that we failed to detect direct interaction of cofilin or capping protein with Arp2/3 complex, their observed preference for Arp2/3-dependent actin structures must derive from additional factors operating downstream of Arp2/3. Future work will aim at identifying these factors and/or mechanisms.
The lamellipodium was recently proposed to spatially overlap with the lamella. Although the latter had originally been defined as the region proximal to the lamellipodium (Small et al., 2002), more recent studies, for instance, using speckle fluorescence microscopy suggested that lamella filaments polymerize up to the front edge of the cell (for review see Chhabra and Higgs, 2007), thereby contributing to protrusion, or even form a separable layer below the lamellipodium (Giannone et al., 2007). Because actin assembly rates and flow in this model are considered to be distinct between lamellipodial and lamella filaments (Ponti et al., 2004), with lamella filaments moving much more slowly than lamellipodial filaments, this would imply that actin regulators at the protrusion tip membrane control polymerization of different filament “categories” at different rates. Such a scenario, however, is hardly compatible with the homogeneous lamellipodial treadmilling observed by FRAP (Wang, 1985; Lai et al., 2008). Moreover, actin bundles in the lamella turn over in a nontreadmilling manner (Koestler et al., 2008), a feature not normally seen in lamellipodial protrusions. Our experiments help to resolve this controversy, as interference with Arp2/3 complex function was sufficient to transform lamellipodia into a network sharing many features with the lamella, such as speed of rearward flow, loss of network treadmilling, and attraction of myosin II. Although our data are compatible with the view that the lamellipodium spatiotemporally evolves into the lamella (Small and Resch, 2005; Burnette et al., 2011) as opposed to significant spatial overlaps of the two structures, it is evident that they do not depend on each other. In other words, lamellipodial filaments likely do not constitute essential prerequisites for filament generation in the lamella. If they did, lamella network intensities, as well as lamella flow rates, should significantly go down upon suppression of lamellipodia formation, but no evidence for this was found. Aside from the residual actin-branching activity seen upon WCA-mediated Arp2/3 complex sequestration, the mechanism of actin filament generation and maintenance at the cell periphery upon Arp2/3 inhibition remains to be established. Whatever the outcome, these mechanisms will likely share features with the ones operating in the lamella. Of interest, filopodia were previously shown to translate from the cell periphery into the lamella (Nemethova et al., 2008), and their formation appears enhanced upon Arp2/3 complex inhibition (the present study). Further investigations will thus also have to establish the relative contribution of filopodia to the maintenance of actin networks both in the lamella and after Arp2/3 complex inhibition.
In conclusion, the approach used here enabled us to confirm that the Arp2/3 complex is crucial not only to lamellipodia initiation, but also to their maintenance. Our experiments showed for the first time that Arp2/3 activity in fibroblast lamellipodia correlates with the rate of actin network flow, as well as with filament geometries and branching frequency. The observed reduction of branching frequency below a certain threshold level, however, had dramatic consequences not only on network organization and ultrastructure, but also on the dynamics of other lamellipodial regulators. In addition, Arp2/3 inhibition instantaneously arrested the motility of fish keratocytes, which is largely dependent on a lamellipodium.
Considering the results together, we suggest that the Arp2/3 complex, aside from amplifying filament numbers via branching, orchestrates the formation of lamellipodial actin networks by at least two additional mechanisms: 1) by focusing actin polymerization to the lamellipodium tip, essential for actin network treadmilling, and 2) by determining the recruitment of additional lamellipodial regulators, including cortactin, cofilin, and capping protein.
MATERIALS AND METHODS
Protein purification, pull downs, and Western blotting
Recombinant Rac1L61 was purified and dialyzed into microinjection buffer essentially as described (Rottner et al., 1999). Additional, histidine- or glutathione S-transferase (GST)–tagged fusion proteins were purified from bacterial lysates using standard procedures. For cofilin-1 purification, GST was removed by proteolytic cleavage and size exclusion chromatography as described (Breitsprecher et al., 2011). Arp2/3 complex was purified from pig brain as described (Block et al., 2012). Ca2+-ATP–actin was purified from rabbit skeletal muscle according to the method of Spudich and Watt (1971), stored in G buffer (2 mM Tris-HCl, pH 8.0, 0.2 mM CaCl2, 0.5 mM dithiothreitol [DTT], 0.2 mM ATP), and prepared for pyrene assays as described (Block et al., 2012). Protein concentrations were determined by absorption spectroscopy using extinction coefficients predicted from the amino acid sequences using Vector NTI software (Invitrogen, Carlsbad, CA).
For the pull down shown in Supplemental Figure S2B, GST-WCA was bound to glutathione Sepharose–coated beads by incubation of 100 μl of bead slurry with 1 ml GST-WCA (2 μM) in incubation buffer (20 mM Tris, pH 7.5, 50 mM NaCl, 2 mM DTT, 0.1 mM ATP, 2 mM MgCl2, 5 mM benzamidine, 5 mM phenylmethylsulfonyl fluoride) for 1 h. The beads were subsequently washed twice with incubation buffer and incubated for another hour with 1 ml of Arp2/3 complex (1 μM) in the presence or absence of 1 ml of capping protein (5 μM) or cofilin-1 (5 μM). Bead incubation with capping protein and cofilin-1 alone served as additional control. Beads were then washed four times with 1 ml of incubation buffer. Bound proteins were eluted from the beads with 3× SDS sample buffer and resolved using 15% SDS–PAGE, followed by Coomassie blue staining.
Western blotting was done according to standard procedures and using primary tropomyosin antibodies as follows: mouse monoclonal α/9d recognizing Tm6, 1, 2, 3, 5a, and 5b (Schevzov et al., 2011); rabbit polyclonal δ/9d recognizing mainly Tm4 but also Tm1, 2, and 3 and mouse monoclonal γ/9d recognizing mainly Tm5NM1 and TM5NM2 (Schevzov et al., 2005). Monoclonal GFP antibody clone 101G4 was purchased from Synaptic Systems (Göttingen, Germany).
Pyrene-actin assays
Proteins to be assayed were diluted in 2× KMEI buffer (100 mM KCl, 2 mM MgCl2, 2 mM ethylene glycol tetraacetic acid, 20 mM imidazole, pH 7.0) to reach a final concentration of 1.18× KMEI. Antifoaming solution Extran AP33 (Merck, Darmstadt, Germany) was added to the mixture to reach a final concentration of 0.05%. We placed 170-μl aliquots of the protein mixtures in nontreated 96-well microtiter plates (Nalge Nunc International, Rochester, NY). The assembly reaction was initiated by injection of 30 μl of a 13.33 μM solution of 10% pyrene-labeled G-actin in G buffer (2 mM Tris-HCl, pH 8.0, 0.2 mM ATP, 0.2 mM CaCl2, 0.5 mM DTT) into the protein mixture to reach a final concentration of 2 μM actin in 1× KMEI by the automated dispenser of a Synergy 4 fluorescence microplate reader (BioTek, Winooski, VT). After a 2-s mixing step, actin polymerization was monitored by measuring the increase of fluorescence of pyrene-actin at 364-nm excitation and 407-nm emission wavelengths for 90 min. The kinetic data were normalized.
Cell culture, transfection, and constructs
NIH3T3 murine embryonic fibroblasts (CRL-1658; American Type Culture Collection, Manassas, VA) were cultured in high-glucose (4.5 g/l) DMEM with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), 2 mM l-glutamine, 1 mM sodium pyruvate, and 1% nonessential amino acids at 37°C in the presence of 5 or 7% CO2. Transfections were carried out with FuGENE HD (Roche, Mannheim, Germany) according to manufacturer's protocols. Cells were plated on glass coverslips coated with 25 μg/ml fibronectin (Roche). B16-F1 mouse melanoma cells were cultured and transfected as described (Lai et al., 2008). Fish keratocytes were prepared from freshly killed brook trout (Salvelinus fontinalis) as previously described (Urban et al., 2010).
pGEX-GST-WCA for protein purification was constructed by cloning WCA of WAVE1 (Machesky and Insall, 1998) into pGEX-6P1 (GE Healthcare, Piscataway, NJ). The WAVE1-CA fragment encoding residues 514–559 and a flexible GS-rich linker at its N-terminus was amplified from human brain cDNA (Invitrogen) using primers 5′- CGCGGATCCGGAGGTTCTGGTTCATCTGTAGAAGAGCAGCGTGAACAGCGCGGATCCCCTGTAATCAGTGATGCCAGGAGT-3′ and 5′-CGCGTCGACTTACTCCAACCAATCTACTTCATC-3′ and ligated into the BamHI and SalI sites of pGEX-6-P1 (GE Healthcare). The constructs for recombinant expression of human heterodimeric capping protein (CapZ), murine cofilin-1 (n-cofilin), and human WAVE1-WCA fragment encoding residues 491–559 were used as previously described (Breitsprecher et al., 2008; Lai et al., 2008; Block et al., 2012). Plasmids expressed in mammalian cells were previously described: mCherry-actin and EGFP-tagged actin, p16B, VASP, WAVE2, capping protein β2, and cofilin (Lai et al., 2008); EGFP-Lifeact (Riedl et al., 2008); EGFP-fascin (Adams and Schwartz, 2000); EGFP-tagged cortactin, MBD-WWCA, and MBD-Spir-NT (Oelkers et al., 2011); and pRK5mycRac1L61 and FMNL2ΔDAD (Block et al., 2012). Constructs that drive expression of EYFP-tagged Tm1 and Tm4 as well as EGFP-Tm2 were described in Tojkander et al. (2011), and EYFP-Tm5NM1 was as in Percival et al. (2004). To create EGFP-Tm3, rat Tm3 cDNA was subcloned into BspEI and BamHI sites of pEGFP-C1 (Clontech, Mountain View, CA). mCherry-cofilin was generated by replacing EGFP in an EGFP-cofilin construct (Mannherz et al., 2005) for mCherry. EGFP-tagged myosin light chain was kindly provided by Rex Chisholm (Northwestern University, Chicago, IL).
Video microscopy, FRAP, and microinjection
Cells were observed in an open, heated chamber (Warner Instruments, Reading, United Kingdom) at 37°C (NIH3T3 and B16-F1) or room temperature (fish keratocytes) on inverted microscopes (Axioscope or Axio Observer) equipped with either a DG-4 epifluorescence illumination system (Sutter Instruments, Novato, CA) and VIS-LED illumination for phase-contrast microscopy or with halogen lamps for both epifluorescence and phase contrast imaging and using 63× or 100×/1.4 numerical aperture (NA) plan-apochromatic objectives or a 100×/1.3 NA Neofluar lens (Carl Zeiss, Jena, Germany). Time-lapse images were acquired using back-illuminated, cooled charge-coupled-device cameras (Roper Micromax or CoolsnapHQ2; Photometrics, Tucson, AZ). B16-F1 cells were imaged on coverslips coated with 25 μg/ml laminin (Sigma-Aldrich) and using Ham's F12–containing microscopy medium (Sigma-Aldrich) supplemented with or without the small-molecule Arp2/3-complex inhibitor CK666. The inhibitor was purchased from Calbiochem (La Jolla, CA), prepared as stock solution as recommended by the manufacturer, and used at a final concentration of 210 μM.
For FRAP, fluorescent molecules in selected regions were bleached with the 2D-VisiFRAP Realtime Scanner (Visitron Systems, Puchheim, Germany) using a 120-mW, 405-nm diode laser. Microscopes were controlled with VisiView (Visitron, Puchheim, Germany) or MetaMorph software (Molecular Devices, Sunnyvale, CA). Image analysis was carried out on MetaMorph.
Microinjections were performed with sterile Femtotips (Eppendorf, Hamburg, Germany) held in a micromanipulator (Narishige [Nikon, Tokyo, Japan] or Leitz [Leica Microsystems, Vienna, Austria]) with a pressure supply from an Eppendorf microinjector 5242 or Femtojet (Eppendorf). Cells were injected with 1 mg/ml L61Rac or 1 mg/ml L61Rac mixed with WCA (1.7 mg/ml) or 1 mg/ml L61Rac mixed with 17.3 mg/ml CA supplemented with 0.25 mg/ml Texas red–labeled dextran (70 kDa; Molecular Probes, Eugene, OR) in microinjection buffer (15 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2), with a continuous outflow mode from the needle under a constant pressure of between 20 and 80 hPa (Rottner et al., 1999).
Kymography, quantitation of myosin accumulation, and fluorescence intensity measurements
For kymography (Figure 2), line regions with a length of 6 μm, corresponding to the y-axis of kymographs, were drawn from inside the lamella across the lamellipodium beyond the cell edge. Regions from each time point of a time-lapse series were pasted next to each other along the x-axis. Lines in kymographs were used to measure rearward flow rates in peripheral regions and the lamella.
Quantitation of accumulation of myosin spots upon Arp2/3 inhibition (Figure 5B) was done as follows. Regions classified as lamellipodia in phase contrast images before injection were outlined and the presence of myosin spots in these regions counted manually before and upon Rac/WCA injection over time (up to 20 min after injection).
Fluorescence intensities of lamellipodial components in Figures 1, 6, and 7 and Supplemental Figures S6 and S7 were measured by drawing a 1- to 5-μm-wide line in an ∼90° angle across the cell edge. In injected fibroblasts, average intensities across the width of the line were measured along the length of the line before and 5 min after injection (between 5 and 50 min for VASP to correct for time-dependent variations) using ImageJ (National Institutes of Health, Bethesda, MD) or MetaMorph. Measurements in B16-F1 cells (Supplemental Figures S6 and S7) were carried out for time points before and after inhibitor treatments as indicated. Background intensities outside the cell were subtracted, and values were normalized to the lamellipodium tip before injection or inhibitor treatment and plotted over the entire line distance.
FRAP analysis
FRAP data were analyzed essentially as described (Rabut and Ellenberg, 2005). Intensity values in bleached regions were measured over time and background levels outside the cell subtracted. Acquisition photobleaching was corrected as recommended (Rabut and Ellenberg, 2005) and resulting values normalized to mean values of the last three frames before photobleaching using Excel 2010 (Microsoft, Redmond, WA). Curves were fitted with SigmaPlot 12.0 (Scientific Solutions SA, Pully-Lausanne, Switzerland) using dynamic curve fits for exponential rise to maximum (f(x) = y0 + a(1 − e−bx)) except for rear lamellipodia regions of Rac-injected cells, which best fitted sigmoidal curves 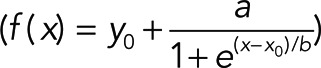 , and the means from different cells were calculated for each time point. Half-times of recovery were calculated from the parameters describing the fitted mean curve using t1/2 = −(1/b) ln 0.5 for exponential and
, and the means from different cells were calculated for each time point. Half-times of recovery were calculated from the parameters describing the fitted mean curve using t1/2 = −(1/b) ln 0.5 for exponential and 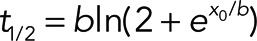 for sigmoidal curves. The treadmilling factor (TMF) describes the difference in fluorescence recovery for front and rear halves of the lamellipodium, as described (Lai et al., 2008), or for corresponding regions upon WCA injections. TMFs were determined as described (Lai et al., 2008), except that the front region corresponded to the region within the first 1 μm measured from tip membrane and the rear one to the region between 1 and 2 μm from the edge. In addition, the gradient in F-actin intensity from lamellipodium front to rear (Watanabe and Mitchison, 2002; Vinzenz et al., 2012) was corrected for by normalization of front and rear regions to their respective maxima.
for sigmoidal curves. The treadmilling factor (TMF) describes the difference in fluorescence recovery for front and rear halves of the lamellipodium, as described (Lai et al., 2008), or for corresponding regions upon WCA injections. TMFs were determined as described (Lai et al., 2008), except that the front region corresponded to the region within the first 1 μm measured from tip membrane and the rear one to the region between 1 and 2 μm from the edge. In addition, the gradient in F-actin intensity from lamellipodium front to rear (Watanabe and Mitchison, 2002; Vinzenz et al., 2012) was corrected for by normalization of front and rear regions to their respective maxima.
Statistics
All data sets were confirmed to be normally distributed using the Anderson–Darling test, allowing data sets to be compared with each other using two-sided, two-sample t tests. Data sets in Figure 6 and cofilin recruitment to WWCA-induced actin filaments on microtubules shown in Figure 8 were tested by one-sample t test to be statistically different from 100 and 0%, respectively.
Correlated live-cell imaging, electron tomography, and analysis
Correlated live-cell imaging, electron tomography, and analysis of tomograms were performed essentially as described (Vinzenz et al., 2012). To correlate responses after injection with structure in the electron microscope, cells were cultured on Formvar-coated coverslips embossed with a grid pattern in gold for cell relocalization (Auinger and Small, 2008).
Tilt series of negatively stained cytoskeletons on Formvar-coated Cu/Pd grids (Maxtaform) were acquired on a FEI Tecnai F30 (Polara; FEI, Hillsboro, OR) microscope, operated at 300 kV and cooled to ∼80 K. Tomograms were generated from two tilt series obtained around orthogonal axes, and images were recorded on a Gatan UltraScan 4000 CCD camera. The primary on-screen magnifications used for image acquisition were 27,500×.
Tracking of filaments in the tomograms was performed manually using IMOD (Kremer et al., 1996), essentially as described (Urban et al., 2010). Orientation analysis and actin branch identification and labeling were performed as reported (Vinzenz et al., 2012). For quantitation, branches from five tomograms for each experimental group were counted manually, followed by statistical analysis.
Supplementary Material
Acknowledgments
This work was supported in part by Deutsche Forschungsgemeinschaft Grants RO2414/3-1 (to K.R.) and FA330/6-1 (to J.F.), Austrian Science Fund Projects FWF 1516-B09 and FWF P21292-B09 (to J.V.S.), the Vienna Science and Technology Fund (WWTF, to J.V.S. and C.S.), and Australian National Health and Medical Research Council Grant APP1004175 (to P.W.G.). We thank J. Adams, R. Chisholm, A. Hall, L. Machesky, H. G. Mannherz, D. Schafer, and R. Wedlich-Söldner for expression constructs and B. Denker, P. Hagendorff, and G. Landsberg for technical assistance.
Abbreviations used:
- Arp2/3
actin-related protein 2/3
- FRAP
fluorescence recovery after photobleaching
- N-WASP
neural Wiskott–Aldrich syndrome protein
- Rac
ras-related C3 botulinum toxin substrate
- RNAi
RNA interference
- Scar
suppressor of cAMP receptor
- Tm
tropomyosin
- WAVE
WASP-family verprolin homologous
- WCA
C-terminus of WAVE proteins harboring WH2, connector, and acidic domains
- WWCA
C-terminus of N-WASP harboring two WH2 domains followed by connector and acidic domains
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-12-0857) on July 24, 2013.
*These authors contributed equally to this work.
REFERENCES
- Adams JC, Schwartz MA. Stimulation of fascin spikes by thrombospondin-1 is mediated by the GTPases Rac and Cdc42. J Cell Biol. 2000;150:807–822. doi: 10.1083/jcb.150.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja R, Pinyol R, Reichenbach N, Custer L, Klingensmith J, Kessels MM, Qualmann B. Cordon-bleu is an actin nucleation factor and controls neuronal morphology. Cell. 2007;131:337–350. doi: 10.1016/j.cell.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akin O, Mullins RD. Capping protein increases the rate of actin-based motility by promoting filament nucleation by the Arp2/3 complex. Cell. 2008;133:841–851. doi: 10.1016/j.cell.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KI, Wang YL, Small JV. Coordination of protrusion and translocation of the keratocyte involves rolling of the cell body. J Cell Biol. 1996;134:1209–1218. doi: 10.1083/jcb.134.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auinger S, Small JV. Correlated light and electron microscopy of the cytoskeleton. Methods Cell Biol. 2008;88:257–272. doi: 10.1016/S0091-679X(08)00414-7. [DOI] [PubMed] [Google Scholar]
- Bernstein BW, Bamburg JR. Tropomyosin binding to F-actin protects the F-actin from disassembly by brain actin-depolymerizing factor (ADF) Cell Motil. 1982;2:1–8. doi: 10.1002/cm.970020102. [DOI] [PubMed] [Google Scholar]
- Bernstein BW, Bamburg JR. ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 2010;20:187–195. doi: 10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block J, et al. FMNL2 drives actin-based protrusion and migration downstream of Cdc42. Curr Biol. 2012;22:1005–1012. doi: 10.1016/j.cub.2012.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher D, Kiesewetter AK, Linkner J, Urbanke C, Resch GP, Small JV, Faix J. Clustering of VASP actively drives processive, WH2 domain-mediated actin filament elongation. EMBO J. 2008;27:2943–2954. doi: 10.1038/emboj.2008.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher D, Koestler SA, Chizhov I, Nemethova M, Mueller J, Goode BL, Small JV, Rottner K, Faix J. Cofilin cooperates with fascin to disassemble filopodial actin filaments. J Cell Sci. 2011;124:3305–3318. doi: 10.1242/jcs.086934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugyi B, Didry D, Carlier MF. How tropomyosin regulates lamellipodial actin-based motility: a combined biochemical and reconstituted motility approach. EMBO J. 2010;29:14–26. doi: 10.1038/emboj.2009.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette DT, Manley S, Sengupta P, Sougrat R, Davidson MW, Kachar B, Lippincott-Schwartz J. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 2011;13:371–382. doi: 10.1038/ncb2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campellone KG, Webb NJ, Znameroski EA, Welch MD. WHAMM is an Arp2/3 complex activator that binds microtubules and functions in ER to Golgi transport. Cell. 2008;134:148–161. doi: 10.1016/j.cell.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol. 2010;11:237–251. doi: 10.1038/nrm2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnell M, Zech T, Calaminus SD, Ura S, Hagedorn M, Johnston SA, May RC, Soldati T, Machesky LM, Insall RH. Actin polymerization driven by WASH causes V-ATPase retrieval and vesicle neutralization before exocytosis. J Cell Biol. 2011;193:831–839. doi: 10.1083/jcb.201009119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, Ismail AM, Umetani J, Billadeau DD, Otwinowski Z, Rosen MK. Structure and control of the actin regulatory WAVE complex. Nature. 2010;468:533–538. doi: 10.1038/nature09623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra ES, Higgs HN. The many faces of actin: matching assembly factors with cellular structures. Nat Cell Biol. 2007;9:1110–1121. doi: 10.1038/ncb1007-1110. [DOI] [PubMed] [Google Scholar]
- DesMarais V, Ichetovkin I, Condeelis J, Hitchcock-DeGregori SE. Spatial regulation of actin dynamics: a tropomyosin-free, actin-rich compartment at the leading edge. J Cell Sci. 2002;115:4649–4660. doi: 10.1242/jcs.00147. [DOI] [PubMed] [Google Scholar]
- Duleh SN, Welch MD. Regulation of integrin trafficking, cell adhesion, and cell migration by WASH and the Arp2/3 complex. Cytoskeleton (Hoboken) 2012;69:1047–1058. doi: 10.1002/cm.21069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone G, et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell. 2007;128:561–575. doi: 10.1016/j.cell.2006.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez TS, Gorman JA, de Narvajas AA, Koenig AO, Billadeau DD. Trafficking defects in WASH-knockout fibroblasts originate from collapsed endosomal and lysosomal networks. Mol Biol Cell. 2012;23:3215–3228. doi: 10.1091/mbc.E12-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton SL, et al. Cell migration without a lamellipodium: translation of actin dynamics into cell movement mediated by tropomyosin. J Cell Biol. 2005;168:619–631. doi: 10.1083/jcb.200406063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho family GTPases. Biochem Soc Trans. 2012;40:1378–1382. doi: 10.1042/BST20120103. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Paunola E, Vartiainen MK, Lappalainen P. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol Biol Cell. 2005;16:649–664. doi: 10.1091/mbc.E04-07-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufner K, Higgs HN, Pollard TD, Jacobi C, Aepfelbacher M, Linder S. The verprolin-like central (vc) region of Wiskott-Aldrich syndrome protein induces Arp2/3 complex-dependent actin nucleation. J Biol Chem. 2001;276:35761–35767. doi: 10.1074/jbc.M106520200. [DOI] [PubMed] [Google Scholar]
- Innocenti M, Zucconi A, Disanza A, Frittoli E, Areces LB, Steffen A, Stradal TE, Di Fiore PP, Carlier MF, Scita G. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6:319–327. doi: 10.1038/ncb1105. [DOI] [PubMed] [Google Scholar]
- Iwasa JH, Mullins RD. Spatial and temporal relationships between actin-filament nucleation, capping, and disassembly. Curr Biol. 2007;17:395–406. doi: 10.1016/j.cub.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AE, Kranitz H, Dotsch V, Mullins RD. Actin binding to the central domain of WASP/Scar proteins plays a critical role in the activation of the Arp2/3 complex. J Biol Chem. 2006;281:10589–10597. doi: 10.1074/jbc.M507470200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koestler SA, Auinger S, Vinzenz M, Rottner K, Small JV. Differentially oriented populations of actin filaments generated in lamellipodia collaborate in pushing and pausing at the cell front. Nat Cell Biol. 2008;10:306–313. doi: 10.1038/ncb1692. [DOI] [PubMed] [Google Scholar]
- Korobova F, Svitkina T. Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol Biol Cell. 2008;19:1561–1574. doi: 10.1091/mbc.E07-09-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Kunda P, Craig G, Dominguez V, Baum B. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr Biol. 2003;13:1867–1875. doi: 10.1016/j.cub.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Lai FP, Szczodrak M, Block J, Faix J, Breitsprecher D, Mannherz HG, Stradal TE, Dunn GA, Small JV, Rottner K. Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 2008;27:982–992. doi: 10.1038/emboj.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai FP, et al. Cortactin promotes migration and platelet-derived growth factor-induced actin reorganization by signaling to Rho-GTPases. Mol Biol Cell. 2009;20:3209–3223. doi: 10.1091/mbc.E08-12-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langanger G, Moeremans M, Daneels G, Sobieszek A, De Brabander M, De Mey J. The molecular organization of myosin in stress fibers of cultured cells. J Cell Biol. 1986;102:200–209. doi: 10.1083/jcb.102.1.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009;36:512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- Lommel S, Benesch S, Rottner K, Franz T, Wehland J, Kuhn R. Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep. 2001;2:850–857. doi: 10.1093/embo-reports/kve197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM, Insall RH. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr Biol. 1998;8:1347–1356. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- Mannherz HG, Gonsior SM, Gremm D, Wu X, Pope BJ, Weeds AG. Activated cofilin colocalises with Arp2/3 complex in apoptotic blebs during programmed cell death. Eur J Cell Biol. 2005;84:503–515. doi: 10.1016/j.ejcb.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Marchand JB, Kaiser DA, Pollard TD, Higgs HN. Interaction of WASP/Scar proteins with actin and vertebrate Arp2/3 complex. Nat Cell Biol. 2001;3:76–82. doi: 10.1038/35050590. [DOI] [PubMed] [Google Scholar]
- McKenna NM, Wang YL, Konkel ME. Formation and movement of myosin-containing structures in living fibroblasts. J Cell Biol. 1989;109:1163–1172. doi: 10.1083/jcb.109.3.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejillano MR, Kojima S, Applewhite DA, Gertler FB, Svitkina TM, Borisy GG. Lamellipodial versus filopodial mode of the actin nanomachinery: pivotal role of the filament barbed end. Cell. 2004;118:363–373. doi: 10.1016/j.cell.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Millard TH, Behrendt B, Launay S, Futterer K, Machesky LM. Identification and characterisation of a novel human isoform of Arp2/3 complex subunit p16-ARC/ARPC5. Cell Motil Cytoskeleton. 2003;54:81–90. doi: 10.1002/cm.10087. [DOI] [PubMed] [Google Scholar]
- Millius A, Watanabe N, Weiner OD. Diffusion, capture and recycling of SCAR/WAVE and Arp2/3 complexes observed in cells by single-molecule imaging. J Cell Sci. 2012;125:1165–1176. doi: 10.1242/jcs.091157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita A, Mueller J, Urban E, Vinzenz M, Small JV, Maeda Y. Direct determination of actin polarity in the cell. J Mol Biol. 2012;419:359–368. doi: 10.1016/j.jmb.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemethova M, Auinger S, Small JV. Building the actin cytoskeleton: filopodia contribute to the construction of contractile bundles in the lamella. J Cell Biol. 2008;180:1233–1244. doi: 10.1083/jcb.200709134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson-Dykstra SM, Higgs HN. Arp2 depletion inhibits sheet-like protrusions but not linear protrusions of fibroblasts and lymphocytes. Cell Motil Cytoskeleton. 2008;65:904–922. doi: 10.1002/cm.20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature. 2009;460:1031–1034. doi: 10.1038/nature08231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelkers JM, et al. Microtubules as platforms for assaying actin polymerization in vivo. PLoS One. 2011;6:e19931. doi: 10.1371/journal.pone.0019931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S, Ono K. Tropomyosin inhibits ADF/cofilin-dependent actin filament dynamics. J Cell Biol. 2002;156:1065–1076. doi: 10.1083/jcb.200110013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival JM, Hughes JA, Brown DL, Schevzov G, Heimann K, Vrhovski B, Bryce N, Stow JL, Gunning PW. Targeting of a tropomyosin isoform to short microfilaments associated with the Golgi complex. Mol Biol Cell. 2004;15:268–280. doi: 10.1091/mbc.E03-03-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. Two distinct actin networks drive the protrusion of migrating cells. Science. 2004;305:1782–1786. doi: 10.1126/science.1100533. [DOI] [PubMed] [Google Scholar]
- Rabut G, Ellenberg J. Photobleaching techniques to study mobility and molecular dynamics of proteins in live cells: FRAP, iFRAP, and FLIP. In: Goldman RD, Spector DL, editors. Live Cell Imaging, A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 2005. pp. 101–126. [Google Scholar]
- Reymann AC, Boujemaa-Paterski R, Martiel JL, Guerin C, Cao W, Chin HF, De La Cruz EM, Thery M, Blanchoin L. Actin network architecture can determine myosin motor activity. Science. 2012;336:1310–1314. doi: 10.1126/science.1221708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Life at the leading edge. Cell. 2011;145:1012–1022. doi: 10.1016/j.cell.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Riedl J, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SL, Wiedemann U, Stuurman N, Vale RD. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J Cell Biol. 2003;162:1079–1088. doi: 10.1083/jcb.200303023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottner K, Hall A, Small JV. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr Biol. 1999;9:640–648. doi: 10.1016/s0960-9822(99)80286-3. [DOI] [PubMed] [Google Scholar]
- Rottner K, Hanisch J, Campellone KG. WASH, WHAMM and JMY: regulation of Arp2/3 complex and beyond. Trends Cell Biol. 2010;20:650–661. doi: 10.1016/j.tcb.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Rottner K, Stradal TE. Actin dynamics and turnover in cell motility. Curr Opin Cell Biol. 2011;23:569–578. doi: 10.1016/j.ceb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Rouiller I, Xu XP, Amann KJ, Egile C, Nickell S, Nicastro D, Li R, Pollard TD, Volkmann N, Hanein D. The structural basis of actin filament branching by the Arp2/3 complex. J Cell Biol. 2008;180:887–895. doi: 10.1083/jcb.200709092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schevzov G, Vrhovski B, Bryce NS, Elmir S, Qiu MR, O'Neill GM, Yang N, Verrills NM, Kavallaris M, Gunning PW. Tissue-specific tropomyosin isoform composition. J Histochem Cytochem. 2005;53:557–570. doi: 10.1369/jhc.4A6505.2005. [DOI] [PubMed] [Google Scholar]
- Schevzov G, Whittaker SP, Fath T, Lin JJ, Gunning PW. Tropomyosin isoforms and reagents. Bioarchitecture. 2011;1:135–164. doi: 10.4161/bioa.1.4.17897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small JV, Isenberg G, Celis JE. Polarity of actin at the leading edge of cultured cells. Nature. 1978;272:638–639. doi: 10.1038/272638a0. [DOI] [PubMed] [Google Scholar]
- Small JV, Resch GP. The comings and goings of actin: coupling protrusion and retraction in cell motility. Curr Opin Cell Biol. 2005;17:517–523. doi: 10.1016/j.ceb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Small JV, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol. 2002;12:112–120. doi: 10.1016/s0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- Snapper SB, et al. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat Cell Biol. 2001;3:897–904. doi: 10.1038/ncb1001-897. [DOI] [PubMed] [Google Scholar]
- Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- Steffen A, Faix J, Resch GP, Linkner J, Wehland J, Small JV, Rottner K, Stradal TE. Filopodia formation in the absence of functional WAVE- and Arp2/3-complexes. Mol Biol Cell. 2006;17:2581–2591. doi: 10.1091/mbc.E05-11-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen A, Rottner K, Ehinger J, Innocenti M, Scita G, Wehland J, Stradal TE. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. 2004;23:749–759. doi: 10.1038/sj.emboj.7600084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stradal TE, Rottner K, Disanza A, Confalonieri S, Innocenti M, Scita G. Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol. 2004;14:303–311. doi: 10.1016/j.tcb.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Suraneni P, Rubinstein B, Unruh JR, Durnin M, Hanein D, Li R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol. 2012;197:239–251. doi: 10.1083/jcb.201112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM. Ultrastructure of protrusive actin filament arrays. Curr Opin Cell Biol. 2013;25:574–581. doi: 10.1016/j.ceb.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–1026. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Bulanova EA, Chaga OY, Vignjevic DM, Kojima S, Vasiliev JM, Borisy GG. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 2003;160:409–421. doi: 10.1083/jcb.200210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, McQuade KM, Borisy GG. Analysis of the actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J Cell Biol. 1997;139:397–415. doi: 10.1083/jcb.139.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojkander S, Gateva G, Schevzov G, Hotulainen P, Naumanen P, Martin C, Gunning PW, Lappalainen P. A molecular pathway for myosin II recruitment to stress fibers. Curr Biol. 2011;21:539–550. doi: 10.1016/j.cub.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Urban E, Jacob S, Nemethova M, Resch GP, Small JV. Electron tomography reveals unbranched networks of actin filaments in lamellipodia. Nat Cell Biol. 2010;12:429–435. doi: 10.1038/ncb2044. [DOI] [PubMed] [Google Scholar]
- Veltman DM, King JS, Machesky LM, Insall RH. SCAR knockouts in Dictyostelium: WASP assumes SCAR's position and upstream regulators in pseudopods. J Cell Biol. 2012;198:501–508. doi: 10.1083/jcb.201205058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinzenz M, et al. Actin branching in the initiation and maintenance of lamellipodia. J Cell Sci. 2012;125:2775–2785. doi: 10.1242/jcs.107623. [DOI] [PubMed] [Google Scholar]
- Wang YL. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J Cell Biol. 1985;101:597–602. doi: 10.1083/jcb.101.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Mitchison TJ. Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science. 2002;295:1083–1086. doi: 10.1126/science.1067470. [DOI] [PubMed] [Google Scholar]
- Weed SA, Karginov AV, Schafer DA, Weaver AM, Kinley AW, Cooper JA, Parsons JT. Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J Cell Biol. 2000;151:29–40. doi: 10.1083/jcb.151.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisswange I, Newsome TP, Schleich S, Way M. The rate of N-WASP exchange limits the extent of ARP2/3-complex-dependent actin-based motility. Nature. 2009;458:87–91. doi: 10.1038/nature07773. [DOI] [PubMed] [Google Scholar]
- Winkler C, Vinzenz M, Small JV, Schmeiser C. Actin filament tracking in electron tomograms of negatively stained lamellipodia using the localized radon transform. J Struct Biol. 2012;178:19–28. doi: 10.1016/j.jsb.2012.02.011. [DOI] [PubMed] [Google Scholar]
- Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Czech L, Gerboth S, Kojima S, Scita G, Svitkina T. Novel roles of formin mDia2 in lamellipodia and filopodia formation in motile cells. PLoS Biol. 2007;5:e317. doi: 10.1371/journal.pbio.0050317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Zhang XF, Pollard TD, Forscher P. Arp2/3 complex-dependent actin networks constrain myosin II function in driving retrograde actin flow. J Cell Biol. 2012;197:939–956. doi: 10.1083/jcb.201111052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchero JB, Coutts AS, Quinlan ME, Thangue NB, Mullins RD. p53-cofactor JMY is a multifunctional actin nucleation factor. Nat Cell Biol. 2009;11:451–459. doi: 10.1038/ncb1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.