

Abstract
The Drosophila embryonic gonad is assembled from two distinct cell types, the Primordial Germ Cells (PGCs) and the Somatic Gonadal Precursor cells (SGPs). The PGCs form at the posterior of blastoderm stage embryos and are subsequently carried inside the embryo during gastrulation. To reach the SGPs, the PGCs must traverse the midgut wall and then migrate through the mesoderm. A combination of local repulsive cues and attractive signals emanating from the SGPs guide migration. We have investigated the role of the hedgehog (hh) pathway gene shifted (shf) in directing PGC migration. shf encodes a secreted protein that facilitates the long distance transmission of Hh through the proteoglycan matrix after it is released from basolateral membranes of Hh expressing cells in the wing imaginal disc. shf is expressed in the gonadal mesoderm, and loss- and gain-of-function experiments demonstrate that it is required for PGC migration. Previous studies have established that the hmgcr-dependent isoprenoid biosynthetic pathway plays a pivotal role in generating the PGC attractant both by the SGPs and by other tissues when hmgcr is ectopically expressed. We show that production of this PGC attractant depends upon shf as well as a second hh pathway gene gγ1. Further linking the PGC attractant to Hh, we present evidence indicating that ectopic expression of hmgcr in the nervous system promotes the release/transmission of the Hh ligand from these cells into and through the underlying mesodermal cell layer, where Hh can contact migrating PGCs. Finally, potentiation of Hh by hmgcr appears to depend upon cholesterol modification.
Author Summary
The molecular mechanisms underlying directed cell migration have been studied extensively in different biological contexts. Germ cell migration provides an effective model to study the dynamics of in vivo cell migration. The process of germ cell migration in Drosophila melanogaster results in embryonic gonad formation consisting of primordial germ cells (PGCs) and somatic gonadal precursor cells (SGPs). Moreover, it likely involves a complex series of attractive and repulsive cues. Molecular and genetic analysis has been performed to elucidate the nature of the attractive cue(s) and components that guide germ cells to the SGPs in the mesoderm. One current model proposes that 3-Hydroxy-3-Methylglutaryl Coenzyme A reductase (Hmgcr), synthesized in the SGPs, potentiates signaling downstream of Hedgehog (Hh) ligand also emanating from the SGPs. The model pivots on the novel activity of an established morphogen, Hedgehog, to function as a chemoattractant for the migrating germ cells. A variety of ‘loss-’ and ‘gain-of-function’ strategies manipulating different components of this signaling pathway have been successfully employed in support of the proposed model.
Introduction
The hedgehog (hh) signaling pathway plays a crucial role in patterning in a wide range of multicellular eukaryotes [1]–[4]. In addition to its role in morphogenesis, hh has also been implicated in cell migration, axonal guidance and in shaping polarized cellular extensions [5]–[9]. In most of these contexts, Hh must be able to signal not only to nearby cells but also to cells located at a distance. Recent studies have uncovered an unusual mechanism for long distance hh signaling.
The Hh ligand in Drosophila is synthesized as a precursor polypeptide, which is subject to a series of processing steps [10],[11]. It is initially targeted to the secretory pathway by an N-terminal signal sequence. After removal of the signal sequence, an internal autoproteolytic cleavage coupled with cholesterol addition generates the active 19 kD, Hh-Np (processed) ligand. This peptide is then palmitoylated before it is released from the apical surface of polarized epithelial cells. Although Hh-Np can signal to neighboring cells, its movement along the apical surface is constrained by the lipid modifications, in part through interactions with the glypican Dally, and the transmembrane protein Brother of Ihog [12]–[14]. By contrast, a C-terminal truncation, Hh-N, that lacks the cholesterol modification, diffuses much farther along the apical surface than Hh-Np [15]. Additionally, movement of the unmodified Hh-N ligand is independent of factors like tout-velu (ttv) and dispatched (disp) that are critical for the spreading of the fully modified Hh-Np protein [16]–[19].
Recent studies by Callejo et al. [20] have suggested that long distance hh signaling is mediated by an unusual multistep pathway in which Hh-Np is transmitted from the basolateral membranes of the sending cells. In this model, most of the Hh-Np that reaches the apical surface is recaptured and internalized by an endocytic pathway that depends upon dynamin and Rab5. Instead of being targeted for lysosomal degradation, the Hh-Np endosomes are recycled by a Rab8 dependent mechanism into exocytic vesicles that are targeted to the basolateral membrane of the hh expressing cells. It is possible that Disp, with its sterol sensing domain, plays a role in selecting Hh-Np containing endocytic vesicles for recycling to the basolateral membrane. Disp is found co-localized with Hh-Np in endocytic vesicles near the apical surface and along the basolateral membrane. It is also in an immunoprecipitable complex with Hh-Np. Two proteins previously implicated in facilitating the response of cells receiving the Hh signal, the Dally-like protein (Dlp) and Interference Hh (Ihog), also seem to participate in the targeting and/or release of Hh-Np from the basolateral membrane.
Other proteins have been implicated in the basolateral release and transmission of Hh-Np from hh expressing cells [10], [21], [22]. One of these, HMGCoA reductase (hmgcr), contains a transmembrane sterol-sensing domain like disp. HMGCoA reductase is responsible for the synthesis of mevalonic acid, which is a precursor for isoprenoid and sterol biosynthesis, and is a key regulatory target in both biosynthetic pathways [23]. In hmgcr− mutant embryos, Hh-Np is not properly released from sending cells and instead accumulates in punctate aggregates along the basolateral membranes. Unlike vertebrates, flies are unable to synthesize cholesterol from mevalonic acid [24], and Hh is modified by exogenous cholesterol. However, mevalonic acid is nevertheless required for hh signaling because it is a precursor for the synthesis of the isoprenoid geranylgeranyl-pyrophosphate by geranylgeranyl diphosphate synthetase (qm). Like hmgcr, qm mutations impede the release of Hh-Np from hh expressing cells [22]. Geranylgeranyl-pyrophosphate is ultimately used in protein geranylation. In hh signaling, a key target for geranylation is the G protein γ subunit 1 (Gγ1) [22]. Gγ1 is a subunit of the heterotrimeric GαGβGγ1 complex which helps mediate the intracellular trafficking of membrane vesicles and cargo [25], [26]. To be active, this heterotrimeric complex must be anchored to the membrane by Gγ1 geranylation.
Many of the factors implicated in the basolateral release and subsequent transmission of Hh-Np in polarized epithelium also function in the signaling pathway(s) that directs migration of primordial germ cells (PGCs) to the somatic gonadal precursor cells (SGPs) during mid-embryogenesis [8], [21], [22], [24], [27]. The PGCs form at the posterior pole of the embryo during the syncytial blastoderm stage, while the SGPs arise during mid-embryogenesis from the lateral mesoderm in parasegments 10–13. In order to coalesce with the SGPs and form the embryonic gonad, the PGCs must traverse from the posterior end into the middle of the embryo and then subsequently move to the lateral mesodermal cell layer, which harbors the newly formed SGPs [28].
While it is generally agreed that an attractive signal(s) produced by the SGPs is needed to guide PGC migration, the identity of the attractant(s) and how hh or other hh pathway genes like hmgcr, qm and gγ1 function in migration remains unresolved and controversial [8], [28]–[30]. To further elucidate the role of this signaling pathway in PGC migration, we have examined the shifted (shf) gene. shf encodes an extracellular protein that is the Drosophila ortholog of the vertebrate Wnt Inhibitory Factor-1 (WIF-1) [13], [14], [31]–[33]. In vertebrates Wif1 antagonizes wnt signaling by capturing Wnt and anchoring it to the heparan sulfate proteoglycan [31], [34]. Unlike its vertebrate counterpart, Shf has no role in Wnt signaling. Instead, it functions in the hh pathway. The Shf protein is enriched in the basolateral ECM, and it facilitates the long distance transmission of Hh-Np after it has been released from the basolateral membranes of signaling cells. In shf mutants, the basolateral accumulation and subsequent spreading of Hh-Np in the wing disc is disrupted, while apical accumulation appears to be relatively normal. These defects in Hh-Np transmission result in a reduction in the expression of hh targets in the anterior compartment of the wing.
Guerrero and colleagues [35] [Guerrero pers. comm.] have found that the defects in hh signaling in the wings of shf mutants can be suppressed by overexpression of hmgcr. Since hmgcr plays a pivotal role in generating the PGC attractant [27], [29], [31], an intriguing possibility is that the functional connection to shf seen in the wing could also be of significance in PGC migration. Here we have first tested whether shf functions in PGC migration using both ‘loss’ and ‘gain’ of function strategies. We have then used genetic ‘epistasis’ experiments to link shf and the hh pathway gene gγ1 to the functioning of the hmgcr→qm isoprenoid biosynthetic pathway in PGC migration. Finally we use co-expression experiments to show that hmgcr potentiates the transmission of hh-GFP out of the nervous system and into the mesoderm, where it associates with PGCs.
Results
Increase in Shf levels in the sending cells is sufficient to enhance hh target gene expression
Even though shf is expressed during embryogenesis, shf mutant embryos have no discernable patterning defects. The likely reason for this is that Hh-Np needs to spread only 3–4 cell diameters in the embryonic ectoderm [11], [12], [36] whereas a much larger distance (12–13 cell diameters) is required for patterning the wing imaginal disc [11], [15], [20], [32], [33]. To confirm that shf is nevertheless able to promote hh signaling during embryogenesis we asked if ectopic shf can upregulate the hh→wingless (wg)→engrailed (en) autoregulatory loop. In this loop, hh expressing cells in each parasegment signal to the neighboring anterior cells to activate wingless (wg) expression. Wg in turn signals back to the row of hh expressing cells and upregulates en expression. To direct shf expression in hh sending cells we used an hh-Gal4 driver, while we used a patched-Gal4 (ptc-Gal4) driver to express shf in hh receiving (wg expressing) cells. Figure 1 shows that the level of En protein in the stripes is appreciably increased over that seen in wild type stripes (panel C) when excess Shf is produced using the hh-Gal4 driver (panel B). While driving Shf in wg expressing cells using ptc-Gal4 also appears to upregulate the En stripes, the increase over wild type is less pronounced than with the hh-Gal4 driver. To confirm that shf can potentiate hh signaling in the embryo we examined the accumulation of Wg in hh-Gal4:UAS-shf embryos. Figure S1 shows that Wg accumulation in hh receiving cells is upregulated by expressing shf in hh sending cells.
Figure 1. Overexpression of shf upregulates the hh→wg→en autoregulatory loop.
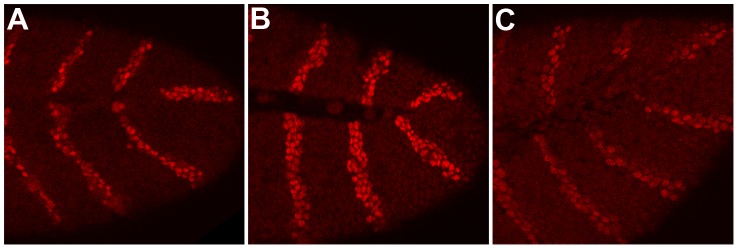
Embryos were generated by mating females homozygous for the UAS-shf transgene with hh-GAL4/TM6 Ubx-LacZ or ptc GAL4/ptc-GAL4 males. The processed embryos were stained with Engrailed (En) or En and β-galactosidase antibodies. (A) ptc-GAL4/UAS-shf, (B) hh-GAL4/UAS-shf (C) control UAS-shf/Ubx-LacZ. It is worth noting that Shf is a secreted protein, and studies on the wing disc indicate that it can rescue the shf mutant phenotype when ectopically expressed in either hh sending or hh receiving cells [30]. In this respect, shf functioning in the embryo would seem to be somewhat different in that it appears to be more effective in potentiating hh signaling when expressed in hh sending than in receiving cells. On the other hand, this difference may simply reflect the fact that shf is expressed in a stripe pattern in the embryonic ectoderm (see Figure 2).
shifted is expressed in the mesoderm
While long distance signaling might not be essential for patterning of the embryonic ectoderm, attractants produced by the SGPs must travel from the posterior mesoderm to the midgut in order to direct migrating PGCs. If Hh-Np functions as a PGC attractant, then shf could have a role in PGC migration, in which case it should be expressed in the mesoderm. Previous studies indicate that shf mRNA is maternally deposited and is uniformly distributed in blastoderm stage embryos [32], [33]. By mid-embryogenesis, shf mRNA is expressed in a segmentally repeating pattern in the ectoderm. This is shown in the stage 10 embryo in Figure 2A. shf expression in the mesoderm (arrow) can also be detected at the posterior. The mesodermal expression is shown more clearly in the stage 12–13 embryo in panel B. Expression in the mesoderm appears to be highest in segments A4–A8, which is the region of the embryonic mesoderm that gives rise to the SGPs, while it is lower in the more anterior segments, which give rise to fat body cells. The upregulation of shf expression in more posterior segments would fit with recent studies of Zhai et al. [37], who showed that shf is regulated by the Bithorax complex gene, Abdominal-B. It should be noted that though shf seems to be preferentially expressed in the posterior mesoderm during the period when PGCs are migrating, its pattern of expression is much broader than hmgcr, which at this stage is only expressed in a subset of mesodermal cells, the SGPs [27].
Figure 2. shf is expressed in the embryonic mesoderm.
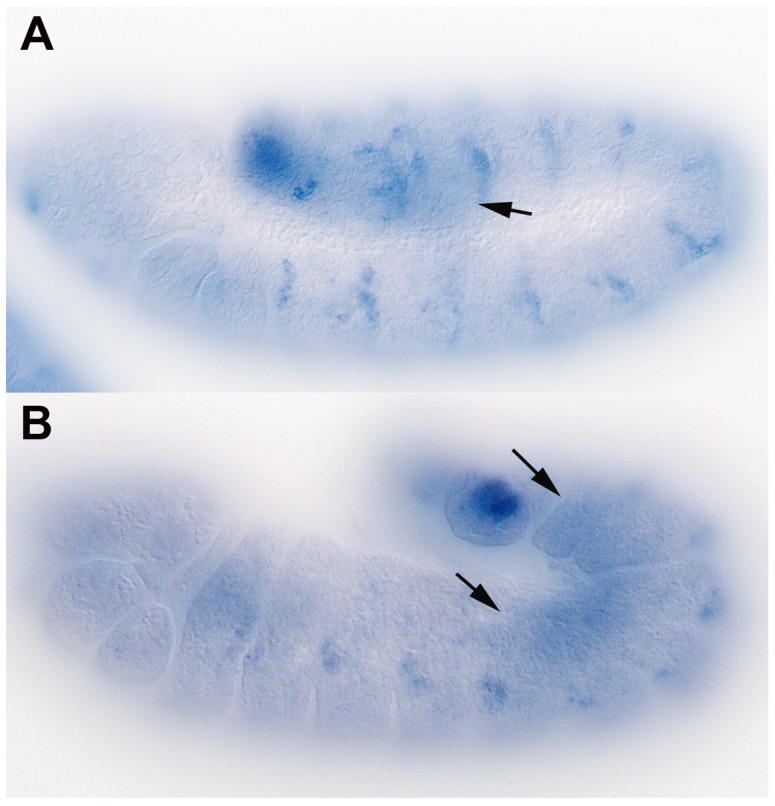
Whole-mount in situ hybridization was performed on staged wild type embryos using anti-sense RNA probes for shf (GH27042) mRNA. A: Stage 11 embryo showing segmental distribution of shf mRNA in the ectoderm. B: Late stage 12 embryo showing that shf mRNA is enriched in the mesoderm in parasegments 10–13. Arrows indicate mesoderm expression.
Embryos compromised for shifted function display germ cell migration defects
We next asked whether shf is needed for proper PGC migration. For this purpose we examined three different shf alleles, shf2, shfx33 and shfEY03173. shf2 has a missense mutation in the third EGF repeat, replacing a conserved cysteine (Cys374) with a serine. shf2 flies are viable and fertile; however, their wings display the typical shf wing phenotype (i.e. a narrowing of the L3–L4 intervein space) [32], [33]. The shfx33 mutant has a 630 bp deletion and is considered a null allele [30]. Male shfx33 flies show slightly stronger shf wing and eye phenotypes than shf2. Finally shfEY03173 is a P-element insertion into the middle of the shf transcription unit. It exhibits the shf wing and eye phenotypes and is semi-lethal.
We found that the early steps in PGC development from their formation through stage 12 are unaltered in shf embryos. However, as shown in Figure 3 for the trans-heterozygous shf2/shfx33 combination, migration phenotypes are evident by the end of stage 13. At this point in wild type embryos, all of the PGCs have migrated from the midgut to the mesoderm and are aligned with the SGPs. By comparison, in shf mutants, a subset of the PGCs lingers behind on the outside surface of the midgut. Interestingly, a similar though more pronounced lingering phenotype is observed in hmgcr embryos. By the time of gonad covalence, we find that over 40% of the shf embryos have 5 or more PGCs scattered in the posterior as compared to two or less for wild type (Figure 3). Similar results were obtained for individual shf mutants (not shown).
Figure 3. shf is required for proper PGC migration.
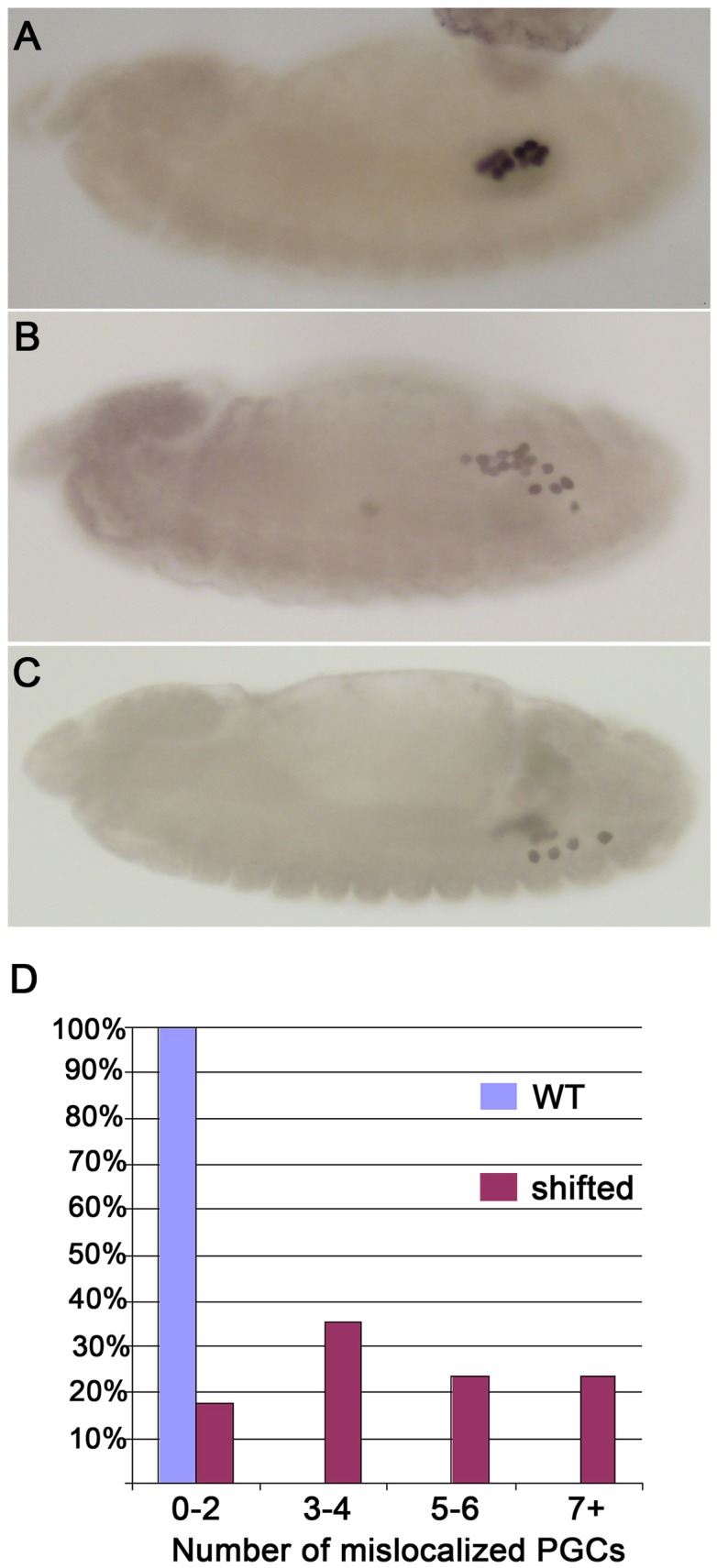
Embryos were generated by mating shfx33/FM7 ftzβ–gal females to shf2 males. PGCs were identified using Vasa antibody, while β-gal antibody was used to identify balancer males and females. The shf embryos shown in B–D are from the cross. (A): Wild type stage 13 embryo with PGCs and SGPs properly aligned. (B) and (C): Stage 13 and stage 14 shf−embryos, respectively. Many of the PGCs are scattered instead of being aligned with the SGPs in the stage 13 embryo and then don't coalesce into the embryonic gonad at stage 14. (D) Plots of the number of scattered or mislocalized PGCs in wild type and shf trans-heterozygous embryos. Wild type: N = 20; shf: N = 17. The embryos were classified as indicated into four different categories based on number of mislocalized PGCs. Migration defects were also observed in shf2 and shfEY03173 embryos (not shown).
Ectopic expression of shifted disrupts germ cell migration
To further substantiate a role for shf in PGC migration, we tested the effects of ectopic expression. Previous studies have shown that misexpression of genes that clearly cannot encode the attractant itself (but instead code for products involved in its production, release or transmission: hmgcr, qm, farnesyl-diphosphate synthase (fpps) and gγ1) can disrupt the migration of PGCs towards the SGPs [22], [24], [27]. Even more remarkable, these genes can be expressed in many unrelated tissues and cell types, yet are still capable of generating an attractant that can compete with the one produced by the SGPs.
To determine if shf also belongs to this special group of genes, we used hh-GAL4 and ptc-GAL4 drivers to ectopically express shf. Since shf ectopic expression using the hh-GAL4 driver was more effective in promoting hh signaling in the embryonic ectoderm than the ptc-GAL4 driver, we anticipated that any effects on PGC migration would likely be stronger with hh-GAL4. Figure 4 shows that this is the case. While about 15% of the ptc-GAL4:UAS-shf embryos had three to four mislocalized PGC, the vast majority resembled wild type (Figure 4A & B). By contrast, nearly 40% of the hh-GAL4:UAS-shf embryos had seven or more mislocalized PGCs (Figure 4E & F).
Figure 4. Ectopic shf expression induces PGC migration defects.

Images in A–F show stage 13–14 (A, B) ptc-GAL4/UAS-shf (C, D), twi-GAL4/UAS-shf and (E, F) hh-GAL4/UAS-shf embryos probed with Vasa antibody to identify PGCs. Only very minor PGC migration defects are evident in ptc-GAL4/UAS-shf, while many scattered PGCs are seen with the twi and hh drivers. Panel at the bottom shows quantitation of the PGC migration defects observed with the 3 different drivers as indicated. The frequency of PGC migration defects typically seen in wild type controls (lacking the GAL4 driver and the UAS-shf transgene) are shown in Figure 3. Blue ptc-GAL4/UAS-shf N = 47. Red twi-GAL4/UAS-shf N = 68. Yellow hh-GAL4/UAS-shf N = 31. D) Plots of the number of scattered or mislocalized PGCs when shf was ectopically expressed with the driver as indicated in the graph.
Germ cell migration is sensitive to ubiquitous shifted expression in the mesoderm
We sought to further confirm the effects of ectopic shf on PGC migration and to compare shf with other genes implicated in this process. For ectopic expression we used the pan-mesodermal driver twist-GAL4. For the purposes of comparison, we selected hmgcr, qm, gγ1, ttv, and hh. As described above, the hmgcr→qm→gγ1 pathway functions in the release of the Hh-Np ligand from sending cells, while ttv provides the proteoglycan matrix that sustains and promotes its transmission.
Previous studies have shown that amongst the genes known to function in germ cell migration hmgcr is a pivotal player in that it encodes a limiting factor for generating an ectopic attractant that can misdirect PGCs. This is illustrated by the dramatic effects of hmgcr misexpression in the mesoderm shown in Figure 5. Over 90% of the embryos have 5 or more mismigrated PGCs, while more than 80% had 7 or more mismigrated PGCs. Similar results have been reported for misexpression in the nervous system ([27]: see below).
Figure 5. Migration defects induced by ectopic expression of hh signaling pathway genes in the mesoderm.

Homozygous twi-GAL4 females were mated with males homozygous for the following UAS transgenes: hmgcr, qm, gγ1, hh (on 2nd and 3rd chromosomes), hh-GFP, ttv, and shf. Purple: 7+ scattered PGCs; Green: 5–6 scattered PGCs; Red: 3–4 scattered PGCs; Blue: 0–2 scattered PGCs. hmgcr: N = 63; qm: N = 86; gγ1: N = 78; hh 2nd: N = 131; hh 3rd: N = 152; hh-GFP: N = 64; UAS-hh-N (3rd chromosome): N = 47); ttv: N = 132; shf: N = 68. Number of scattered PGCs in wild type embryos is shown in Figure 3. Bars show percentage of embryos in each category as indicated.
At least part of the explanation for the unusual potency of hmgcr is that its expression is restricted to the SGPs at the time the PGCs commence their migration. By contrast, all of the other known genes, with the exception of the mesoderm specific MDR49 [38], are more widely expressed in the embryo. Thus, there may only be an incremental increase in the production or transmission of an ectopic PGC attractant when these genes are overexpressed in tissues in which they are already present. With the important caveat that the levels of gene product generated by different UAS transgenes may not be equivalent, Figure 5 shows that while ectopic expression of qm, gγ1, and ttv perturbs PGC migration, their effects are appreciably less severe than hmgcr. For example, whereas over 90% of the twi-GAL4/UAS-hmgcr embryos had 5 or more scattered PGCs, only about 30% of the twi-GAL4/UAS-ttv embryos had 5 or more scattered PGCs. Though ectopic shf causes greater disruptions in PGC migration than some of these other genes, its effects are still less than hmgcr.
Consistent with the results of previous studies [8], ectopic expression of the putative PGC attractant, hh, is also less effective than hmgcr. With two different transgenes encoding wild type Hh-Np, we found that about 40% of the embryos had 5 or more scattered PGCs, while about 25% had 7 or more scattered PGCs. (The effects of hh misexpression on PGC migration are considered further in Text S1.) We also tested transgenes expressing an Hh-GFP fusion protein and the C-terminal truncation Hh-N. The Hh-GFP fusion protein is thought to be processed and transmitted much like the wild type Hh-Np; however, this particular transgene doesn't fully rescue a hh temperature mutant [39]. As shown in Figure 5, the hh-GFP transgene is less effective than either of the hh-Np transgenes. The same is true for a transgene expressing the truncated Hh-N protein, which lacks the C-terminal cholesterol modification.
shifted and gγ1 mutations suppress PGC migration defects induced by ectopic hmgcr
The PGC attractant generated by expression of hmgcr or other genes at ectopic sites is thought to be the same as the attractant produced by these same genes in the SGPs [28], [30]. This means that the synthesis, release and long distance transmission of the ectopically produced attractant should in most instances depend upon precisely the same set of genes and pathways as those involved in generating the bona fide SGP attractant. In principle, it should be possible to exploit this co-dependence to explore the relationship between the different genes involved in generating the attractant. For these experiments we focused on hmgcr, as it plays such a central role in signaling the migrating PGCs [27]. We used an elav driver to express hmgcr in the nervous system.
In order to misdirect migrating PGCs, the attractant generated in the nervous system by ectopic hmgcr must compete with the attractant produced by the SGPs. Thus, reducing the gene dose of a factor that is more critical for the activity of the SGP derived attractant than it is for the nervous system derived attractant should exacerbate the effects of ectopic hmgcr. One example of such a factor would be hmgcr itself. Since expression of the endogenous gene is restricted to the SGPs during PGC migration, reducing the hmgcr gene dose in the nervous system should have little or no effect on the competitive activity of the ectopic attractant. In contrast, production of the attractant by the SGPs should be diminished when there is only a single wild type copy of hmgcr, making it easier for the ectopic nervous system signal to misdirect the migrating PGCs. Figure 6 shows that this prediction is correct—reducing hmgcr gene dose leads to an increase in the number of scattered PGCs. Similar effects would be predicted for other genes that are expressed or function only in SGPs (or mesoderm) but are not expressed and/or not required in the nervous system.
Figure 6. Mutations in gγ1 and shf dominantly suppress the PGC migration defects induced by ectopic expression of hmgcr in the nervous system.

Panel A: Hemizygous stage 15 elav-GAL4, UAS-hmgcr/+ embryo. Panel B: Stage 15 trans-heterozygous elav-GAL4, UAS- hmgcr/gγ1k0817 embryo. C. Quantitation of the PGC migration (visualized with Vasa antibody) defects in embryos hemizygous for the elav-GAL4, UAS-hmgcr recombinant chromosome and heterozygous for mutations in hmgcr, gγ1, and shf. OreR: Homozygous elav-GAL4, UAS- hmgcr males were crossed to wild type females (OreR). hmgcr/+: Homzygous elav-GAL4, UAS- hmgcr males were crossed to hmgcr 1/TM3 Sb Ubx-lacZ females. Embryos heterozygous for hmgcr 1 were identified as β-galactosidase negative. gγ1/+: Homozygous elav-GAL4, UAS- hmgcr males were crossed to the indicated gγ1/CyO Ubx-lacZ mutant. Embryos heterzygous for gγ1 were identifed as β-galactosidase negative. shf/+: Homzygous shf 112 females were mated with homozygous elav-GAL4 UAS hmgcr males. Female embryos heterozygous for the shf mutation were identified as Sxl positive and scored. Male embryos were not scored.
For signaling genes whose expression is not restricted to the SGPs or the mesoderm, reducing their dose could, like hmgcr, have a greater effect on the attractant from the SGPs than on the ectopic attractant. However, it also possible that the factors encoded by these genes might be more limiting for the production or transmission of the ectopic attractant in the nervous system than they are in the SGPs. In this case, reducing their dose should suppress rather than enhance the effects of ectopic hmgcr on PGC migration. For these experiments we selected shf and a second gene gγ1 whose relationship to hmgcr is well defined. The Gγ1 protein must be geranylated in order to be active, and in hh signaling it is an important target for the hmgcr→fpps→qm isoprenoid biosynthetic pathway. Figure 6 shows that the migration defects induced by ectopic hmgcr are mitigated when the embryos are heterozygous for two different gγ1 alleles. Whereas about 60% of the elav-GAL4,UAS-hmgcr embryos have 10 or more scattered PGCs, this number drops to less than 25% for both gγ1 alleles. This finding would argue that gγ1 must also function downstream of hmgcr in the production or transmission of the ectopic attractant. This conclusion would fit with previous studies which showed that ectopic expression of a dominant negative Gγ1 protein that can't be geranylated and poisons the heterotrimeric GαGβGγ1 complex disrupts PGC migration. We next tested shf. Like gγ1 the number of embryos with 10 or more scattered PGCs is reduced, going from 60% to 20%. This finding would argue that production or transmission of the attractant induced at ectopic sites by hmgcr expression depends upon a second hh signaling gene shf.
hmgcr induced migration defects are enhanced by expressing hh pathway genes
To further explore the functional connections between hmgcr and hh pathway genes in the production of an ectopic PGC attractant in the nervous system, we examined the effects of co-expression. For this purpose females carrying a recombinant chromosome containing both elav-GAL4 and UAS-hmgcr were mated to males carrying UAS transgenes for shf, ttv and gγ1. As a positive control we used UAS-hmgcr, as we already had evidence that its effects on germ cell migration were dose dependent. Figure 7A provides a further demonstration that the limiting factor in generating an ectopic PGC attractant is hmgcr. Adding a second copy of UAS-hmgcr essentially eliminates the intermediate migration phenotypes, and almost all embryos have greater than 10 scattered PGCs. Figure 7E shows that co-expressing ttv, gγ1 and shf in the nervous system also exacerbates the effects of ectopic hmgcr; however, none is equivalent to an extra copy of hmgcr.
Figure 7. Enhancement of the elav-GAL4 UAS-hmgcr PGC migration defects by co-expression of hh pathway genes.

Panels A–D. Homozygous elav-GAL4 UAS-hmgcr females were crossed with males homozygous for different transgenes as follows: (A) OreR (wild-type/no transgene), (B) UAS-hmgcr, (C) UAS-hh, and (D) UAS-shf. The embryos shown in (A–D) are at stage 13. Panel E) The bar graph at the bottom of the figure shows the frequency of embryos having different numbers of scattered PGCs in embryos hemizygous for both the recombinant elav-GAL4, UAS-hmgcr chromosome and one of the UAS transgenes (except OreR) as indicated. Ore-R (N = 58); UAS-hmgcr (N = 92); UAS-hh (N = 96); UAS-shifted (N = 62); UAS-ttv (N = 32); UAS-gγ1 (N = 35); UAS-hh-N (3rd chromosome) (N = 39); UAS-hh-N (2nd chromosome) (N = 54); UAS-hh-GFP (N = 60).
hh but not hh-N UAS-transgenes enhance the hmgcr induced migration defects
We next co-expressed hmgcr with wild type Hh-Np or the C-terminal truncation, Hh-N, which lacks the cholesterol modification. In polarized epithelial cells ectopic hmgcr would be expected to promote the basolateral release of Hh-Np and enhance its long distance transmission [13], [21], [22]. By contrast, ectopic hmgcr should have less of an effect on the release or transmission of the partially modified Hh protein Hh-N, which is thought to signal most effectively apically. While it is not clear how the subcellular organization of the secretion machinery in neurons might be related to that in polarized epithelial cells, the hmgcr-dependent PGC attractant must nevertheless be released from neuronal cells in a manner that enables it to move readily into and then through the underlying layer of mesodermal cells so that it can contact the migrating PGCs. If a key function of hmgcr in generating the ectopic attractant in neurons is to enhance the release and transmission of Hh into the underlying mesodermal layer then the same distinction should hold in PGC migration. In this case, co-expression of wild type Hh-Np would be expected to exacerbate the hmgcr induced migration defects, while co-expression of Hh-N should have more modest effects. On the other hand, if the germ cell attractant generated by ectopic hmgcr is completely independent of hh, then co-expression of either Hh ligand should contribute nothing to the hmgcr induced PGC migration defects.
To test these predictions we first compared the effects of driving hh or hh-N expression in the nervous system on PGC migration. Figure S2 shows that expression of a UAS-hh transgene in the nervous system using an elav-GAL4 driver has a greater effect on PGC migration than a UAS-hh-N (3nd chromosome) transgene encoding the truncated and partially modified Hh-N protein. We next examined the effects of co-expressing Hmgcr with either Hh-Np or Hh-N. Figure 7 shows that co-expression of Hh-N (3rd) and Hmgcr leads to only a small increase in the number of mismigrated PGCs. To confirm this finding, we tested a second UAS-hh-N transgene (2nd chromosome). It gave comparable results. In contrast to the UAS-hh-N transgenes, a transgene expressing wild type Hh-Np collaborates with hmgcr, substantially augmenting the effects of ectopic hmgcr on PGC migration. In fact, the effects of the Hh-Np transgene are just about equivalent to that seen with two copies of hmgcr.
hmgcr potentiates the release/transmission of hh-GFP
A strong prediction of the findings described above is that ectopic expression of Hmgcr in neuronal cells will enhance the release and/or transmission of the wild type Hh ligand from these cells into and through the underlying mesodermal cell layer. While we have previously visualized the effects of ectopic Hmgcr on Hh-Np release/transmission in the ectoderm using Hh antibodies [21], it was impossible to obtain reliable images of its transmission through the underlying mesoderm using these antibodies. For this reason, we turned to Hh-GFP to test this prediction. As described above, we found that Hh-GFP is less effective than either Hh-Np or Hh-N in disrupting PGC migration when expressed using the twi-GAL4 driver. A similar result is obtained when the elav-GAL4 driver was used to direct Hh-GFP expression in the embryonic nervous system (Figure S2). On the other hand, Figure 7 shows that Hh-GFP can potentiate the effects of Hmgcr when the two proteins are co-expressed in the nervous system. Thus, though Hh-GFP clearly doesn't fully recapitulate the activity of Hh-Np in this assay, it does resemble Hh-Np in being able to collaborate with Hmgcr.
To determine whether Hmgcr promotes the transmission of Hh into the underlying tissue, we compared the Hh-GFP distribution in embryos expressing this fusion protein in the nervous system with or without Hmgcr. We first detect Hh-GFP in the developing PNS in stage 9–10 embryos, while it subsequently comes on in the developing CNS in stage 11–12 embryos. At these early stages the level of Hh-GFP is relatively low, and the differences in Hh-GFP distribution between hh-GFP and hh-GFP/hmgcr embryos are not very pronounced. This is likely due in part to residual Hmgcr activity from earlier zygotic expression and from maternal deposition and in part because only small amounts of ectopic Hmgcr and Hh-GFP have been synthesized. However, as development proceeds (and Hh-GFP levels increase) the differences between hh-GFP and hh-GFP/hmgcr embryos become more pronounced. This is illustrated in Figure 8. Panels A and B show cross sections of stage 13 hh-GFP and hh-GFP/hmgcr embryos; panels C and D show views of the surface of stage 14 embryos; while panels E and F show cross sections of stage 14 embryos. The same embryos, together with a series of plots of the average pixel density across several ∼3 cell wide sections (as indicated), are shown in Figures S3–S5.
Figure 8. Co-expression of hmgcr with hh potentiates the transmission of the Hh ligand into and through the mesoderm.

Homozygous UAS-Hh-GFP males were mated to females carrying an elav-GAL4 transgene with or without UAS-hmgcr and the progeny probed with GFP antibody. Shown in panels A,C and E are elav-GAL4/UAS-hh-GFP embryos. In this experiment elav-GAL4/CyO,actin-GFP females were mated to homozygous UAS-Hh-GFP males. Embryos of the correct genotype were identified by the unique patterns of GFP expression generated by actin-GFP or the elav/Hh-GFP combination. Shown in panels B, D and F are hh-GFP/hmgcr embryos. Females homozygous for the recombinant elav-GAL4, UAS-hmgcr chromosome were mated to males homozygous for UAS-Hh-GFP. Panels A and B: early stage 13 embryos. Panels C and D: stage 14 embryos. Panels E and F: late stage 14 embryos.
Two differences between hh-GFP and hh-GFP/hmgcr embryos are apparent. First, the overall level of Hh-GFP is elevated in the hh-GFP/hmgcr embryos. In scans of several similarly staged and oriented embryos there was a ∼1.8–2.0 fold difference in the Hh-GFP signal between hh-GFP and hh-GFP/hmgcr. Since different elav-GAL4 transgenes (one on the 2nd and the other recombined with UAS-hmgcr on the 3rd) were used to drive UAS-hh-GFP expression, one explanation for this difference is that the recombined elav-GAL4 transgene drives a higher level of expression. To test this we used the two elav-GAL4 transgenes to express GFP carrying a nuclear localization signal. As shown in Figure S6, the two drivers produce similar amounts of GFP. Thus, a more likely possibility is that hmgcr stabilizes or otherwise promotes Hh accumulation. Consistent with this explanation, we found that the level of endogenous Hh-Np in the ectoderm increased to a similar if not greater extent when hmgcr was ectopically expressed using a hh-GAL4 driver [19]. Second, the transmission of Hh-GFP from expressing cells in the nervous system into the surrounding ectoderm and through the underlying mesoderm is substantially enhanced by co-expression of Hmgcr. This is most obvious in the two sets of stage 14 embryos shown in panels C–F (see also Figures S4 and S5). In both cases, Hh-GFP is largely restricted to the CNS in hh-GFP embryos. In contrast, Hh-GFP is found not only in the nervous system but also in the adjacent ectoderm (Figure 8C and D) and, most importantly, in the underlying mesodermal cell layer in the hh-GFP/hmgcr embryos (Figure 8E and F). Although less pronounced, differences in the level and distribution of Hh-GFP are also evident in stage 13 embryos. This can be seen by comparing panels A and E versus B and F in Figure 8 and the plots of pixel density in Figure S3 and S5.
In addition to being transmitted from the developing nervous system into the mesoderm, we found that Hh-GFP localizes preferentially to PGCs or to tissues containing PGCs. Figure 9 shows PGCs in stage 12–13 embryos expressing Actin-GFP, Hh-GFP or Hh-GFP/Hmgcr. During this period of development PGCs in wild type embryos associate and then align with the gonadal mesodermal SGPs. In both the actin-GFP and hh-GFP embryos, the SGPs are aligned with the gonadal mesoderm, while in the hh-GFP/hmgcr embryos they are scattered in the mesoderm. The middle set of panels shows that PGCs in both the hh-GFP and hh-GFP/hmgcr are distinctly outlined with Hh-GFP, while a GFP signal is not associated with PGCs in the control actin-GFP/UAS-hh-GFP embryos. The PGC associated GFP signal appears earlier in hh-GFP/hmgcr embryos and is (together with the signal in the surrounding mesodermal tissue: see Figure 9) greater than in hh-GFP embryos. In addition, we often observed PGCs in the hh-GFP/hmgcr embryos that have bulges or protrusions. As indicated by the arrows in the hh-GFP/hmgcr panels and in the adjacent enlargements of these PGCs, there are higher levels of Hh-GFP associated with these bulges. However, not all concentrations of Hh-GFP are found associated with obvious bulges or protrusions.
Figure 9. Ectopically expressed Hh-GFP localizes near PGCs.
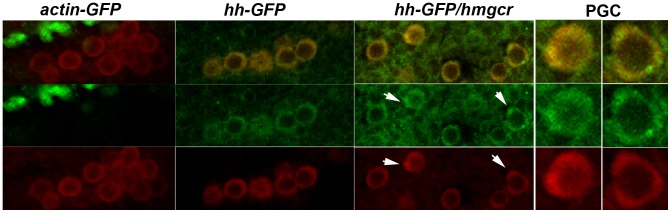
elav-GAL4/CyO,actin-GFP females were mated to homozygous UAS-Hh-GFP males. Embryos carrying the CyO, actin-GFP balancer and UAS-Hh-GFP but no GAL4 driver were identified by their distinctive GFP expression pattern, and the PGCs from one of these embryos are shown in the first set of panels as indicated. The second set of panels show PGCs from the sibling elav-GAL4/UAS-Hh-GFP embryos. In the third set of panels females homozygous for the recombinant elav-Gal4, UAS-hmgcr chromosome were mated to males homozygous for UAS-Hh-GFP. Embryos of the indicated genotypes (actin-GFP: CyO,Actin-GFP/UAS-Hh-GFP; hh-GFP: elav-GAL4/UAS-hh-GFP; hh-GFP/hmgcr: elav-Gal4, UAS-hmgcr/UAS-hh-GFP) were stained with GFP (imaged in green) and Vasa (imaged in red) antibodies. Top panels show merged images, middle panels Hh-GFP, and bottom panels Vasa. The PGCs shown in all three sets are from stage 13 embryos. Arrows in the hh-GFP/hmgcr panels indicate PGCs with bulges/protrusions enriched for Hh-GFP. Panels on far right show close-ups of the two PGCs in the hh-GFP/hmgcr panels that are marked with arrows.
Discussion
The synthesis of mevalonic acid by the enzyme Hmgcr is the rate-controlling step in the biosynthesis of isoprenoids and steroids. In mammals, one end-product of the mevalonate pathway, cholesterol, is used to modify the C-terminus of the processed Hh ligand, and this modification plays an important role in controlling the activity of this signaling molecule. Flies lack the enzymes needed for de novo cholesterol biosynthesis and depend instead upon exogenous cholesterol for this modification of the Hh ligand [24]. Nevertheless, the mevalonate biosynthetic pathway is still used to potentiate the release/transmission of the Hh ligand, in this case through (at least in part) the geranylation of the G protein Gγ1 [21], [22], [35]. Hmgcr as well as the downstream components in the isoprenoid biosynthetic pathway also play a pivotal role in generating the attractant that guides PGC migration both from its native source, the SGPs, and from a variety of different embryonic tissues when ectopically expressed [27]–[30]. However, how hmgcr or the other isoprenoid pathway enzymes function in generating the PGC attractant either in the SGPs or at ectopic sites has remained unresolved and contentious. To address this problem we have focused on the connection between the mevalonate→isoprenoid biosynthetic pathway and two proteins that have been implicated in the long distance basolateral transmission of the Hh-Np ligand, the G protein Gγ1 and the extracellular hh signaling factor Shf.
Previous studies have established that a rate limiting step in generating the PGC attractant either by the SGPs or by other tissues and cell types is the biosynthesis of geranylgeranyl-pyrophosphate by geranylgeranyl diphosphate synthetase (qm) [24], [27], [28]. The control point in the geranylgeranyl-pyrophosphate biosynthetic pathway is the production of mevalonic acid by the enzyme Hmgcr. While hmgcr seems to play a rather similar role in the release/transmission of Hh-Np from hh sending cells, in this case through the geranylation of Gγ1, an important and controversial question is whether the functioning of the hmgcr→qm biosynthetic pathway in hh signaling has any connection to the generation of the PGC attractant. We have addressed this question by determining if the PGC migration defects induced by hmgcr expression in the nervous systems depend upon Gγ1 and Shf. We find that mutations in both gγ1 and shf dominantly suppress the migration defects induced by ectopic hmgcr. In contrast, reducing the dose of the hmgcr gene dominantly enhances the migration defects induced by hmgcr expression in the nervous system. This later finding is expected since reducing hmgcr activity in the SGPs should further compromise the ability of the attractant generated by the SGPs to compete with the attractant generated in the nervous system. The former findings show that the production/activity of the attractant generated in the nervous system by ectopic hmgcr depends on both gγ1 and shf. By themselves, these results do not exclude the possibility that gγ1 and shf only collaborate with hmgcr when it is ectopically expressed in the nervous system while they are not actually needed for the hmgcr-dependent production of the attractant by the SGPs. However, this scenario seems unlikely. For one, there are PGC migration defects in gγ1 and shf mutant embryos. For another, the Gγ1 protein must be geranylated to function in PGC migration [22]. Finally, like hmgcr, ectopic expression of gγ1 and shf in the mesoderm and ectoderm perturbs PGC migration.
Even though Gγ1 and Shf are known to function in the release and transmission of the Hh ligand, it could be argued that these two proteins could also mediate the release/transmission of others molecules, including the “actual” PGC attractant. Indeed, Gγ1 is likely involved in secretion of other molecules, while the fact that Shf homologs in mammals function in Wnt but not Hh signaling raises the possibility that Shf could promote signaling by an as yet unknown ligand (though not Wg: [34], [40]). However, there is evidence that like Gγ1 and Shf, Hh itself depends upon hmgcr and the isoprenoid biosynthetic pathway not only in hh signaling but also in generating an ectopic PGC attractant in the nervous system. This comes from the differences in the effects of ectopically expressed Hh-Np and Hh-N that would be predicted based on the mechanisms proposed for their transmission [15], [20]. First, the apically transmitted Hh-N ligand would be expected to have a smaller effect on PGC migration when ectopically expressed in the nervous system than Hh-Np. With the caveat that expression of different UAS transgene inserts will not be precisely the same, this prediction holds. Second, the geranylation of Gγ1 in response to ectopic Hmgcr would be expected to promote the basolateral release and subsequent spreading of Hh-Np into the mesoderm. By contrast, ectopic Hmgcr should have less influence on Hh-N, which isn't readily internalized by hh sending cells and spreads mostly along the apical surface [20]. With the same caveat, this predicted distinction is also observed. When co-expressed, Hh-Np and Hmgcr collaborate to strongly potentiate PGC migration defects, while there is a more modest collaboration between Hh-N and Hmgcr.
Though an imperfect mimic of Hh-Np, we have taken advantage of a chimeric Hh-GFP fusion protein to analyze the effects of Hmgcr on the transmission of Hh from cells in the embryonic nervous system. We found that Hh-GFP is less effective than Hh-Np (and even Hh-N) in competing with the PGC attractant produced by the SGPs when it is ectopically expressed using the twi or elav GAL4 drivers. Since Hh-GFP appears to have near but not quite wild type activity in morphogenesis [39], it is surprising that it is relatively ineffective in altering PGC migration. However, a plausible reason for this discrepancy is that the demands imposed by the assays used to test Hh-GFP activity in each experimental context are quite different. The morphogenesis assay requires that Hh-GFP substitute for Hh-Np. Since animals can readily tolerate heterozygosity for hh, small deficits in the functioning of the chimeric protein might only have minimal effects on morphogenesis. In contrast, in the PGC migration assay the ectopically expressed Hh-GFP must be able to compete with the attractant(s) produced by the SGPs. If Hh-Np is the relevant endogenous PGC attractant, then even subtle deficiencies in the activity of the chimeric Hh-GFP ligand would be expected to compromise its ability to compete with the wild type protein. It would also follow that it should be possible to “rescue” ectopic Hh-GFP by enhancing its activity. This is the case. While hh-GFP is not very active on its own, it is able to collaborate with hmgcr when co-expressed in the nervous system.
In previous studies we have shown that expressing hmgcr in hh producing cells in the ectoderm increases the overall level of Hh protein and enhances its transmission to adjacent cells [21]. Precisely the same sorts of effects on Hh-GFP are evident when it is “rescued” by co-expression with hmgcr in the nervous system—Hh-GFP levels are elevated, while its transmission into and through the underlying mesodermal cell layer is appreciably enhanced. These hmgcr dependent effects, particularly on the movement of Hh-GFP from the neuroectoderm into the underlying mesoderm, would also provide a plausible explanation for why this biosynthetic enzyme plays such a pivotal role in PGC migration even though it is not directly responsible for the synthesis of the PGC attractant. In the period when PGCs are migrating through the mesoderm, the SPGs are the only cells in the embryo simultaneously expressing both hmgcr and hh [27], [28], [31]. Consequently the accumulation, release and transmission of Hh-Np will be specifically potentiated in SGPs, but not in other hh expressing cells elsewhere in the mesoderm or in the ectoderm. This would provide a mechanism for ensuring that SGP derived Hh-Np out-competes Hh-Np produced elsewhere. Taken together, these findings support the idea that Hh-Np expressed in the SGPs functions as a PGC attractant. With the caveat that the activities of Hh-GFP are not identical to Hh-Np, the fact that Hh-GFP accumulates on the surface and around the PGCs further bolsters this suggestion. Moreover, in a subset of the PGCs Hh-GFP is closely associated with bulges or protrusions that could potentially be of relevance to the process of migration.
On the other hand, a number of critical questions remain. For one, it is not clear how reception of the hh signal could actually translate into directed movement. The endpoint of the signaling cascade in the canonical pathway is the transcriptional activation of target genes, including the hh receptor ptc. However, transcription is likely not involved in this instance, as ptc reporters are not activated in PGCs [29] (unpublished data). Moreover, in mammals hh dependent axonal guidance and fibroblast migration are independent of transcription and involve instead the coupling of Smo activation to pathways that mediate the reorganization of the cytoskeleton [6], [7], [41], [42]. Further studies will clearly be required to establish a connection between hh signaling to the PGCs, changes in the cytoskeleton and directed movement. Another unresolved question is whether SGPs produce any other PGC attractants. Although no other plausible candidates have been identified, our experiments do not exclude the possibility that there are other PGC attractants, even including an attractant(s) whose activity, like Hh-Np, is potentiated by the hmgcr isoprenoid biosynthetic pathway.
Materials and Methods
Immunohistochemistry
The embryo stainings were performed essentially as described in 21. Vasa (from Paul Lasko) and Hh (from Tom Kornberg) antibodies are rabbit polyclonal antibodies. Both were used at a 1∶500 dilution. Engrailed and Wingless antibodies are mouse monoclonal antibodies and were used at 1∶10 dilution. ß-Galactosidase antibody was either a rabbit polyclonal purchased from Cappel (used at 1∶1000 dilution) or a mouse monoclonal antibody from Developmental Studies Hybridoma Bank (used at 1∶10 dilution). GFP antibody is a rabbit polyclonal purchased from Torrey Pines Biolabs (used at 1∶1000 dilution). For con-focal analysis a magnification of 40× was used in almost all the instances, and images were collected using identical settings for the control and experimental samples. Multiple pairs of wild type (sibs) and mutant embryos were imaged in each case, and representative examples are presented.
Mutant and misexpression analysis
Shf stocks including mutants and UAS transgenes were obtained from Seth Blair [32]. UAS-Hh-GFP fusion stocks were a kind gift of Isabel Guerrero [20]. UAS-Hh-Np (two different stocks on the second and third chromosome respectively; from Phil Beachy), UAS-Hh-N: two different stocks on the second (from Tom Kornberg) and third chromosome (from Phil Beachy) respectively, gγ1 mutant stocks, gγ1N159 and gγ1 k0817, were obtained from Fumio Matsuzaki, while UAS- gγ1 stock (gγ1) was kindly provided by the Olson lab [43]. The other UAS and Gal4 stocks used for the misexpression studies were from the Bloomington stock center. hairy-Gal4, elav-Gal4, nanos-Gal4, patched-Gal4, UAS-ß-galactosidase, hh-Gal4/TM6 Ubx-LacZ. In most experiments, males carrying two of the copies UAS transgene were mated with virgin females carrying two copies of the Gal4 transgene. The resulting progeny embryos were fixed and stained for subsequent analysis. elav-Gal4, UAS hmgcr recombinant [38] and UAS-qm stock [24] were obtained from Ruth Lehmann. In many experiments the genotypes of the embryos were unambiguously determined by using balancer chromosomes marked with either GFP or ß-galactosidase.
Supporting Information
Overexpression of shf elevates level of Wingless (Wg) accumulation. Females homozygous for the UAS-shf transgene were mated with hh-GAL4/TM6 Ubx-LacZ males. Embryos generated from the cross were collected and coimmunostained with Wg (imaged in red) and β-galactosidase (imaged in green, not shown) antibodies. (A) control UAS-shf/Ubx-LacZ. (B) hh-GAL4/UAS-shf.
(TIF)
Effects of ectopic expression of different Hh proteins in the nervous system. An elav-GAL4 driver inserted on the 2nd chromosome (as described in the text and the legend to Figure 7) was used to drive expression of UAS-hh, UAS-hh-N (the UAS-hh-N transgenic line on the 3rd chromosome in Figure 7), and UAS-hh-GFP. The graph shows the % of embryos having different numbers of mismigrated PGCs as indicated in the Figure.
(TIF)
Distribution of Hh-GFP in stage 13 hh-GFP and hh-GFP/hmgcr embryos. Embryos in Figure S3 are from panels A and B of Figure 8. This supplemental figure shows a plot of the pixel density (using ImageJ) in an approximately 3 cell wide vertical stripe. The center of each of these stripes is indicated by the arrows below each embryo. There are four plots for each embryo.
(TIF)
Distribution of Hh-GFP in stage14 hh-GFP and hh-GFP/hmgcr embryos. Embryos in Figure S4 are from panels C and D of Figure 8. This supplemental figure shows a plot of the pixel density (using ImageJ) in an approximately 3 cell wide vertical stripe. The center of each of these stripes is indicated by the arrows below each embryo. There are four plots for each embryo.
(TIF)
Distribution of Hh-GFP in stage 14 hh-GFP and hh-GFP/hmgcr embryos. Embryos in Figure S5 are from panels E and F of Figure 8. This supplemental figure has a plot of the pixel density (using ImageJ) in an approximately 3 cell wide vertical stripe. The center of each of these stripes is indicated by the arrows below each embryo. There are four plots for each embryo.
(TIF)
The 2nd and recombined 3rd chromosome elav-GAL4 transgenes drive equivalent levels of expression of a UAS-GFP(NLS) reporter. Females homozygous for the recombined UAS-hmgcr, elav-GAL4 3rd chromosome or the 2nd chromosome elav-GAL4 driver (elav-GAL4/CyO Actin GFP) were mated to UAS-GFP(NLS) males. The resulting embryos were fixed and stained with anti-GFP antibody. (A) elav-GAL4 (2nd)/UAS-GFP(NLS). (B) UAS-hmgcr,elav-GAL4 (3rd)/UAS-GFP(NLS). The signal intensity in each nuclei was estimated by doing pixel counts over a fixed area using ImageJ software. The average pixel intensity per nucleus for elav-Gal4/UAS-GFP(NLS) was 69.8 (S.D.+/−9.7; n = 15) whereas for UAS-hmgcr,elavGal4 embryos was 66 (S.D.+/−13.5; n = 15).
(TIF)
Does ectopic Hh perturb germ cell migration? The data in Figures 5, 7 and S2 showing that hh misexpression perturbs PGC migration are contradicted by experiments reported in Figure 2 of a paper “Hedgehog does not guide migrating Drosophila germ cells” that was published in 2009 by Renault et al. [29] in Developmental Biology. The material presented in Text S1 addresses this controversy.
(PDF)
Acknowledgments
For antibodies and stocks, we thank the Developmental Studies Hybridoma Bank, Drs. P. Lasko, T. Kornberg, S. Blair, I. Guerrero, R. Lehmann, F. Matsuzaki, E. Olson, and the Bloomington Stock Center. We would also like to thank Drs. Blair, Guerrero and also Dr I. Lohmann for very helpful discussions and comments. Gordon Grey provided fly media. Finally, we would like to thank the referees for their insightful comments and suggestions.
Funding Statement
Partial support provided by NIH R01GM043432. PS would also like to thank the Ministry of Education and Science of the Russian Federation for supporting this research: grant #14.B25.31.0022. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Huangfu D, Anderson KV (2006) Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development 33: 3–14. [DOI] [PubMed] [Google Scholar]
- 2. Matise M (2007) Order in the classroom: graded responses to instructive Hh signaling in the CNS. Cell Cycle 10: 1194–9. [DOI] [PubMed] [Google Scholar]
- 3. Wilson CW, Chuang PT (2010) Mechanism and evolution of cytosolic Hedgehog signal transduction. Development 137: 2079–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matise MP, Wang H (2011) Sonic hedgehog signaling in the developing CNS where it has been and where it is going. Curr Top Dev Biol 97: 75–117. [DOI] [PubMed] [Google Scholar]
- 5. Testaz S, Jarov A, Williams KP, Ling LE, Koteliansky VE, et al. (2001) Sonic hedgehog restricts adhesion and migration of neural crest cells independently of the Patched-Smoothen-Gil signaling pathway. Proc Natl Acad Sci U S A 98: 12521–12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hochman E, Castiel A, Jacob-Hirsch J, Amariglio N, Izraeli S (2006) Molecular pathways regulating pro-migratory effects of Hedgehog signaling. J Biol Chem 281 33860–70. [DOI] [PubMed] [Google Scholar]
- 7. Charron F, Stein E, Jeong J, McMahon AP, Tessier-Lavigne M (2003) The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell 113: 11–23. [DOI] [PubMed] [Google Scholar]
- 8. Deshpande G, Swanhart L, Chiang P, Schedl P (2001) Hedgehog signaling in germ cell migration. Cell 106: 759–69. [DOI] [PubMed] [Google Scholar]
- 9. Kato K, Chihara T, Hayashi S (2004) Hedgehog and decapentabplegic instruct polarized growth of cell extensions in the Drosophila trachea. Development (Camb.) 131: 5253–5261. [DOI] [PubMed] [Google Scholar]
- 10. Mann RK, Beach PA (2004) Novel lipid modifications of secreted protein signals. Annu Rev Biochem 2004 73: 891–923. [DOI] [PubMed] [Google Scholar]
- 11. Gallet A (2011) Hedgehog morphogen: from secretion to reception. Trends Cell Biol 21: 238–246. [DOI] [PubMed] [Google Scholar]
- 12. Gallet A, Rodriguez R, Ruel L, Therond PP (2003) Cholesterol modification of hedgehog is required for trafficking and movement, revealing an asymmetric cellular response to hedgehog. Dev Cell 4: 191–204. [DOI] [PubMed] [Google Scholar]
- 13. Avanesov A, Blair SS (2013) The Drosophila WIF1 homolog Shifted maintainsglypican-independent Hedgehog signaling and interacts with the Hedgehog co-receptors Ihog and Boi. Development 140: 107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bilioni A, Sanchez-Hernandez D, Callejo A, Gradilla AC, Ibanez C, et al. (2013) Balancing Hedgehog, a retention and release equilibrium given by Dally, Ihog, Boi and shifted/DmWif. Dev Biol 376: 198–221. [DOI] [PubMed] [Google Scholar]
- 15. Callejo A, Torroja C, Quijada L, Guerrero I (2006) Hedgehog lipid modifications are required for Hedgehog stabilization in the extracellular matrix. Development 133: 471–83. [DOI] [PubMed] [Google Scholar]
- 16. Gallet A, Ruel L, Staccini-Lavenant L, Thérond PP (2006) Cholesterol modification is necessary for controlled planar long-range activity of Hedgehog in Drosophila epithelia. Development1 33: 407–18. [DOI] [PubMed] [Google Scholar]
- 17. The I, Bellaiche Y, Perrimon N (1999) Hedgehog movement is regulated through tout velu-dependent synthesis of a heparan sulfate proteoglycan. Mol Cell 4: 633–9. [DOI] [PubMed] [Google Scholar]
- 18. Häcker U, Nybakken K, Perrimon N (2006) Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol 6: 530–41. [DOI] [PubMed] [Google Scholar]
- 19. Burke R, Nellen D, Bellotto M, Hafen E, Senti KA, et al. (1999) Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell 99: 803–15. [DOI] [PubMed] [Google Scholar]
- 20. Callejo A, Bilioni A, Mollica E, Gorfinkiel N, Andrés G, et al. (2011) Dispatched mediates Hedgehog basolateral release to form the long-range morphogenetic gradient in the Drosophila wing disk epithelium. Proc Natl Acad Sci U S A 108: 12591–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deshpande G, Schedl P (2005) HMGCoA reductase potentiates hedgehog signaling in Drosophila melanogaster . Dev Cell 9: 629–38. [DOI] [PubMed] [Google Scholar]
- 22. Deshpande G, Godishala A, Schedl P (2009) Gγ1, a downstream target for the hmgcr-isoprenoid biosynthetic pathway, is required for releasing the Hedgehog ligand and directing germ cell migration. Plos Genetics 5 (1) e1000333 doi:10.1371/journal.pgen.1000333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DeBose-Boyd RA (2008) Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res 18: 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santos AC, Lehmann R (2004) Isoprenoids control germ cell migration downstream of HMGCoA reductase. Dev Cell 6: 283–93. [DOI] [PubMed] [Google Scholar]
- 25. Anel AMD, Malhotra V (2005) PKCn is required for b1g2/b3g2 and PKD-mediated transport to the cell surface and the organization of the Golgi apparatus. Jour Cell Biol 169: 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bard F, Malhotra V (2006) The formation of TGN to plasma membrane transport carriers. Annu Rev Cell Dev Biol 232: 439–55. [DOI] [PubMed] [Google Scholar]
- 27. Van Doren M, Broihier HT, Moore LA, Lehmann R (1998) HMG-CoA reductase guides migrating primordial germ cells. Nature 396: 466–9. [DOI] [PubMed] [Google Scholar]
- 28. Kunwar PS, Siekhaus DE, Lehmann R (2006) In vivo migration: A germ cell perspective. Annual Review of Cell. And Dev Biol 22: 237–65. [DOI] [PubMed] [Google Scholar]
- 29. Renault AD, Ricardo S, Kunwar PS, Santos A, Starz-Gaiano M, et al. (2009) Hedgehog does not guide migrating Drosophila germ cells. Dev Biol 328: 355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Richardson BE, Lehmann R (2010) Mechanisms guiding primordial germ cell migration: strategies from different organisms. Nat Rev Mol Cell Biol 11: 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsieh JC, Kodjabachian L, Rebbert ML, Rattner A, Smallwood PM, et al. (1999) A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature 398: 431–6. [DOI] [PubMed] [Google Scholar]
- 32. Glise B, Miller CA, Crozatier M, Halbisen MA, Wise S, et al. (2005) Shifted, the Drosophila ortholog of Wnt inhibitory factor-1, controls the distribution and movement of Hedgehog. Dev Cell 8: 255–66. [DOI] [PubMed] [Google Scholar]
- 33. Gorfinkiel N, Sierra J, Callejo A, Ibañez C, Guerrero I (2005) The Drosophila ortholog of the human Wnt inhibitor factor Shifted controls the diffusion of lipid-modified Hedgehog. Dev Cell 8: 241–53. [DOI] [PubMed] [Google Scholar]
- 34. Avanesov A, Honeyager SM, Malicki J, Blair SS (2012) The role of glypicans in Wnt inhibitory factor-1 activity and the structural basis of Wif1's effects on Wnt and Hedgehog signaling. PLoS Genet 8 (2) e1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Callejo A, Culi J, Guerrero I (2007). Lipids and Lipoproteins as carriers for Hedgehog spreading and reception. Fly Meeting Abstracts. Drosophilia Research Conference. March 7–11, 2007. Philadelphia, Pennsylvania, United States of America.
- 36. Delon I, Payre F (2004) Evolution of larval morphology in flies: get in shape with shavenbaby. Trends Genet 20: 305–13. [DOI] [PubMed] [Google Scholar]
- 37. Zhai Z, Fuchs AL, Lohmann I (2010) Cellular analysis of newly identified Hox downstream genes in Drosophila. Eur J Cell Biol 89: 273–8. [DOI] [PubMed] [Google Scholar]
- 38. Ricardo S, Lehmann R (2009) An ABC transporter controls export of a Drosophila germ cell attractant. Science 323: 943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Torroja C, Gorfinkiel N, Guerrero I (2004) Patched controls the Hedgehog gradient by endocytosis in a dynamin-dependent manner, but this internalization does not play a major role in signal transduction. Development 131: 2395–2408. [DOI] [PubMed] [Google Scholar]
- 40. Sánchez-Hernández D, Sierra J, Ortigão-Farias JR, Guerrero I (2012) The WIF domain of the human and Drosophila Wif-1 secreted factors confers specificity for Wnt or Hedgehog. Development 2012 Sep 5. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 41. Bijlsma MF, Borensztajn KS, Roelink H, Peppelenbosch MP, Spek CA (2007) Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell Signal 19 2596–604. [DOI] [PubMed] [Google Scholar]
- 42. Polizio AH, Chinchilla P, Chen X, Kim S, Manning DR, et al. (2011) Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J Biol Chem 286: 19589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yi P, Han Z, Li X, Olson EN (2006) The mevalonate pathway controls heart formation in Drosophila by isoprenylation of Ggamma1. Science 313: 301–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Overexpression of shf elevates level of Wingless (Wg) accumulation. Females homozygous for the UAS-shf transgene were mated with hh-GAL4/TM6 Ubx-LacZ males. Embryos generated from the cross were collected and coimmunostained with Wg (imaged in red) and β-galactosidase (imaged in green, not shown) antibodies. (A) control UAS-shf/Ubx-LacZ. (B) hh-GAL4/UAS-shf.
(TIF)
Effects of ectopic expression of different Hh proteins in the nervous system. An elav-GAL4 driver inserted on the 2nd chromosome (as described in the text and the legend to Figure 7) was used to drive expression of UAS-hh, UAS-hh-N (the UAS-hh-N transgenic line on the 3rd chromosome in Figure 7), and UAS-hh-GFP. The graph shows the % of embryos having different numbers of mismigrated PGCs as indicated in the Figure.
(TIF)
Distribution of Hh-GFP in stage 13 hh-GFP and hh-GFP/hmgcr embryos. Embryos in Figure S3 are from panels A and B of Figure 8. This supplemental figure shows a plot of the pixel density (using ImageJ) in an approximately 3 cell wide vertical stripe. The center of each of these stripes is indicated by the arrows below each embryo. There are four plots for each embryo.
(TIF)
Distribution of Hh-GFP in stage14 hh-GFP and hh-GFP/hmgcr embryos. Embryos in Figure S4 are from panels C and D of Figure 8. This supplemental figure shows a plot of the pixel density (using ImageJ) in an approximately 3 cell wide vertical stripe. The center of each of these stripes is indicated by the arrows below each embryo. There are four plots for each embryo.
(TIF)
Distribution of Hh-GFP in stage 14 hh-GFP and hh-GFP/hmgcr embryos. Embryos in Figure S5 are from panels E and F of Figure 8. This supplemental figure has a plot of the pixel density (using ImageJ) in an approximately 3 cell wide vertical stripe. The center of each of these stripes is indicated by the arrows below each embryo. There are four plots for each embryo.
(TIF)
The 2nd and recombined 3rd chromosome elav-GAL4 transgenes drive equivalent levels of expression of a UAS-GFP(NLS) reporter. Females homozygous for the recombined UAS-hmgcr, elav-GAL4 3rd chromosome or the 2nd chromosome elav-GAL4 driver (elav-GAL4/CyO Actin GFP) were mated to UAS-GFP(NLS) males. The resulting embryos were fixed and stained with anti-GFP antibody. (A) elav-GAL4 (2nd)/UAS-GFP(NLS). (B) UAS-hmgcr,elav-GAL4 (3rd)/UAS-GFP(NLS). The signal intensity in each nuclei was estimated by doing pixel counts over a fixed area using ImageJ software. The average pixel intensity per nucleus for elav-Gal4/UAS-GFP(NLS) was 69.8 (S.D.+/−9.7; n = 15) whereas for UAS-hmgcr,elavGal4 embryos was 66 (S.D.+/−13.5; n = 15).
(TIF)
Does ectopic Hh perturb germ cell migration? The data in Figures 5, 7 and S2 showing that hh misexpression perturbs PGC migration are contradicted by experiments reported in Figure 2 of a paper “Hedgehog does not guide migrating Drosophila germ cells” that was published in 2009 by Renault et al. [29] in Developmental Biology. The material presented in Text S1 addresses this controversy.
(PDF)