

Abstract
Inflammatory bowel disease is associated with an increased risk of colorectal cancer. Colitis-associated cancer is a classical inflammation-driven cancer in which constitutive NFκB and STAT3 activation drive tumorigenesis. Recent findings published by Liang et al. in Cancer Cell demonstrate that sphingosine kinase 1 (SphK1)-mediated upregulation of sphingosine-1-phosphate (S1P) drives a persistent NFκB/IL-6/STAT3/sphingosine-1-phosphate receptor 1 (S1PR1) amplification loop that is critical to the development of chronic colitis and colitis-associated cancer. FTY720, an antagonist of S1PR1, abolished persistent NFκB/IL-6/STAT3 signaling and reduced the development and progression of colitis-associated cancer. Targeting SphK1/S1P/S1PR1 may provide a therapeutic option to prevent the progression of colitis to cancer.
Keywords: colitis-associated cancer, inflammatory bowel disease, STAT3, IL-6, sphingosine-1-phosphate, FTY720, dextran sodium sulfate
Colitis-associated cancer (CAC) is colorectal cancer which is preceded by inflammatory bowel disease (IBD), of which ulcerative colitis and Crohn disease are the 2 most common forms. Although IBD-related cancer accounts for only 2% of all colorectal cancer cases in the general population, IBD localized in the colon is a high-risk factor for colorectal cancer development.1 An association between IBD and colorectal cancer has been well established with a cumulative risk of developing colorectal cancer of 2% at 10 years, 8% at 20 years, and 18% at 30 years of active ulcerative colitis and 8% at 30 years of active Crohn disease.1,2 Increased risk of CAC in IBD patients depends on disease severity, disease duration, efficacy of IBD management, and extent of disease at diagnosis.3 Recent population-based studies suggest that risk of CAC in IBD has decreased over time, which could be partly due to the increased use of maintenance therapy for IBD and surveillance colonoscopy after 8 years of disease duration.4 Several features make CAC distinct from sporadic colon cancer. CAC is often anaplastic, broadly infiltrating, rapidly growing, and develops in flat dysplastic tissue. While genetic alterations are similar to those in sporadic colorectal cancer, the frequency and sequence of these events differ in CAC. Unlike sporadic colorectal cancer, mutations in the adenomatous polyposis coli (APC) tumor suppressor gene happen late during the progression from dysplasia to cancer following earlier mutations in p53 and K-Ras.5 It is thought that DNA damage and mutations caused by inflammatory signaling during IBD may bypass the initial APC mutation step required to initiate sporadic colorectal cancer tumorigenesis.3 Therefore, CAC is a classical inflammation-driven cancer.
The transcription factor STAT3 plays a central role in colitis and CAC due to its regulation of intestinal epithelial cell survival and proliferation. In addition to IL-11, IL-22, HGF, and EGF family members, the proinflammatory cytokine IL-6 is a potent activator of STAT3. Aberrent IL-6/STAT3 signaling is associated with IBD and CAC.6 STAT3 regulates anti-apoptotic genes such as Bcl-xL and Bcl-2, cell cycle regulators such as Cyclin D1 and c-Myc, and angiogenic factors such as VEGF.7 Intestinal epithelial cell-specific deletion of STAT3 diminishes tumor growth and multiplicity during CAC despite increased susceptibility to experimental colitis, mimicking the phenotype of genetic deletion or pharmacological inhibition of IL-6.8-10 STAT3 signaling in the intestinal epithelium is a double-edged sword in that it mediates protection and wound healing during injury and colitis, but can also stimulate tumor cell proliferation and survival.9,10 Constitutive activation of STAT3 has been reported in many cancers including colorectal cancer and CAC.11 The transcription factor NFκB is upregulated in both immune and epithelial cells during active colitis and CAC. NFκB-activating cytokines including TNFα and IL-1β are elevated in IBD and CAC and current treatments for IBD include inhibition of TNFα.12 NFκB is thought to be a major activator of IL-6 production from immune cells, which is a major driver of STAT3 activation in epithelial cells. In tumor cells NFκB and STAT3 can activate one another in a feed-forward fashion, driving persistently active STAT3 and NFκB signaling.13
A recent publication by Liang et al. provide further insight describing sphingosine-1-phosphate (S1P) as an upstream mediator of NFκB and STAT3 persistent activation in colitis and CAC. In addition to their function as critical structural components of cell membranes, sphingolipids serve as bioactive signaling molecules. S1P influences cell fate through promotion of cell proliferation and survival and inhibition of apoptosis.14 The enzymes that regulate levels of S1P are the sphingosine kinases (SphK1 and SphK2) and previous studies have indicated that S1P levels controlled by SphK1 contribute to carcinogenesis.15 Proceeding this study by Liang et al., it was shown that expression of SphK1 is increased in colon of UC-afflicted patients and during colorectal cancer,16,17 mice with targeted deletion of SphK1 exhibited reduced aberrant crypt foci and tumor formation in an experimental model of CAC,16 and S1P activates NFκB signaling.18 Furthermore, it was previously shown that STAT3 induces expression of the S1P receptor S1PR1 in tumors and associated immune cells, which reciprocally activates STAT3 thereby driving persistent IL-6 formation and NFκB signaling and subsequent tumor growth and metastases.13,19 Liang et al. further these findings by demonstrating that the NFκB/IL-6/STAT3/S1PR1 amplification loop is driven by SphK1-mediated upregulation of S1P and that this signaling cascade is critical to the development of chronic colitis and CAC.
Using mice with targeted deletion of SphK2, Liang et al. show that loss of SphK2 enhances SphK1 and S1P expression and increases the severity of dextran sodium sulfate (DSS)-induced colitis and tumorigenesis in the azoxymethane (AOM)-DSS model of CAC compared with wild-type (WT) mice. The associated increase in SphK1 and S1P in SphK2−/− mice was attributed to loss of SphK2-mediated HDA1/2C inhibition, thereby increasing the induction of c-Jun and its target gene SphK1. The severity of colitis in SphK2−/− mice correlated with NFκB activation, IL-6 and TNFα formation, STAT3 activation, and S1PR1 expression. The number of activated Th cells or Tregs was the same between WT and SphK2−/− mice at baseline and during DSS colitis. Mucosal barrier function was not altered by deletion of SphK2 as evidenced by similar rates of FITC-dextran translocation in WT and SphK2−/− mice at baseline and during DSS colitis. A requirement for S1P formation vs. constitutive activation was confirmed using the SphK1 inhibitor, SK1-I, or a competitive S1PR1 antagonist, W146, which reduced the severity of DSS-induced colitis, NFκB and STAT3 activation, IL-6 expression, and S1P expression in SphK2−/− mice.
Using reciprocal bone-marrow chimeric mice, generated by adoptive transfer of bone marrow into lethally irradiated mice, Liang et al. show that induction of colitis and activation of NFκB and STAT3 in SphK2−/− mice is dependent on hematopoietic cells vs. non-hematopoietic cells, such as intestinal epithelial cells. Furthermore, the authors demonstrate that the cellular source of IL-6 in SphK2−/− mice during DSS-induced colitis is macrophages. During the late phase of CAC, macrophages, dendritic cells, and to a lesser extent T cells infiltrate the adenoma and produce IL-6 in SphK2−/− mice.
FTY720 is a S1P mimetic that acts as a functional antagonist of S1PR1 and induces its proteosomal degradation.20 FTY720 alters migration and homing of lymphocytes via S1P receptors and induces activation of CD4+CD25+ Tregs. Previous reports indicate that FTY720 protects against DSS-, trinitrobenzene sulfonic acid (TNBS)-, and oxazolone-induced colitis as well as CD4+CD62L+ T cell transfer colitis.21-23 Liang et al. demonstrate that WT and SphK2−/− mice treated daily with FTY720 exhibit less severe colitis with concurrent lymphopenia. FTY720 treatment abrogated DSS-induced SphK1 and S1P expression in WT and SphK2−/− mice. Furthermore, FTY720 reduced NFκB and STAT3 activation, decreased the elevated levels of IL-6 and S1PR1, and reduced the number of recruited macrophages during DSS colitis in SphK2−/− mice. Since these results suggest that FTY720 ameloriates colitis by impeding the NFκB/IL-6/STAT3/S1PR1 amplification loop initiated by SphK1 and S1P signaling, the authors next assessed the effect of FTY720 on development and progression of CAC. FTY720 administered throughout the CAC protocol reduced tumor number, tumor size, and tumor load in WT and SphK2−/− mice. FTY720 administered only during late-stage CAC proved it can impact tumor progression in WT mice but was not as effective in SphK2−/− mice. Late-stage FTY720 administration reduced proliferation rates of WT and SphK2−/− tumors, suggesting that FTY720 can affect tumor growth and development. This was associated with abrogated STAT3 activation and reduced IL-6 expression in the tumors and infiltrating immune cells as well as NFκB activation in tumors from WT and SphK2−/− mice. Furthermore, late-stage FTY720 administration reduced the elevated SphK1 and S1PR1 in CAC adenomas. These results suggest that FTY720 is effective in abolishing the SphK1/S1P/S1PR1 amplification loop driving persistent STAT3 activation and can even suppress established CAC.
This study by Liang et al. demonstrates a significant advance in our understanding of the molecular pathways driving the transition from chronic inflammation to tumorigenesis. Upregulation of SphK1 during colitis drives the NFκB/IL-6/STAT3/S1PR1 amplification loop linked to tumorigenesis during CAC (Fig. 1). The current study suggests that SphK1 drives tumor infiltrating macrophages and dendritic cells to produce elevated IL-6 levels during CAC, thereby promoting a pro-inflammatory tumor microenvironment. Given the results from this study by Liang et al. demonstrating that SphK2 is an inhibitor of SphK1 expression, it would be worthwhile to determine whether expression of SphK2 is decreased in IBD and/or CAC, subsequently contributing to the increase in SphK1. Targeting upstream mediators of STAT3 activation with FTY720 or other SphK1/S1P/S1PR1 inhibitors may provide a therapeutic option to prevent the progression of colitis to cancer. Alternatively, agonists of SphK2 may provide an additional therapeutic approach to inhibit aberrant SphK1 and downstream signaling. The publication by Liang et al. provides strong evidence that alleviation of a pro-inflammatory tumor microenvironment by ablating constitutive STAT3 activation by targeting SphK1/S1P/S1PR1 signaling holds therapeutic promise in combating CAC.
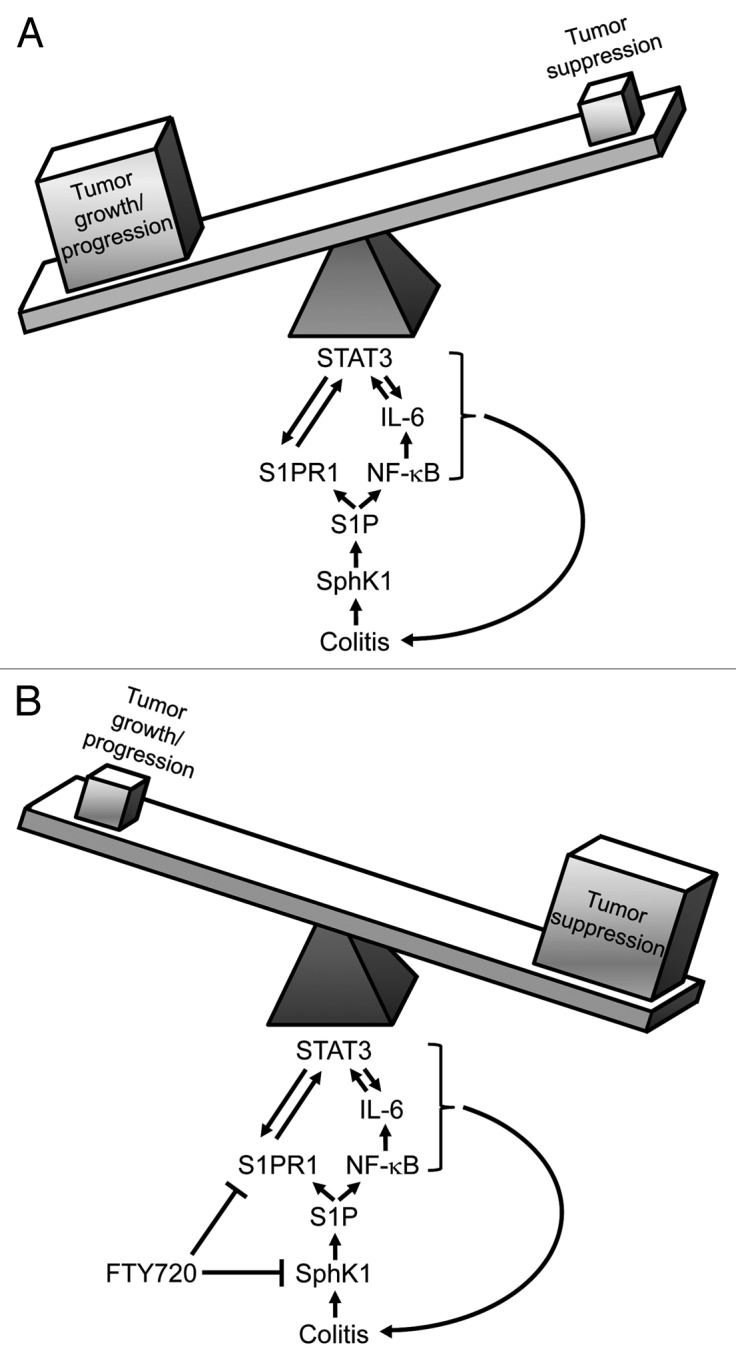
Figure 1. S1P activated by SphK1 is the driver of persistent NFκB and STAT3 activation linked to tumorigenesis during CAC. (A) Increased levels of SphK1 and S1P during colitis lead to a feed-forward amplification loop driving persistent NFκB and STAT3 activation in tumors, tipping the balance toward growth/progression. (B) Inhibition of S1P signaling by the mimetic FTY720, abolishes persistent NFκB and STAT3 activation, suppressing development and progression of tumors during CAC.
Glossary
Abbreviations:
- CAC
colitis-associated cancer
- DSS
dextran sodium sulfate
- IBD
inflammatory bowel disease
- S1P
sphingosine-1-phosphate, SphK, sphingosine kinases
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/24150
References
- 1.Mattar MC, Lough D, Pishvaian MJ, Charabaty A. Current management of inflammatory bowel disease and colorectal cancer. Gastrointest Cancer Res. 2011;4:53–61. [PMC free article] [PubMed] [Google Scholar]
- 2.Canavan C, Abrams KR, Mayberry J. Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment Pharmacol Ther. 2006;23:1097–104. doi: 10.1111/j.1365-2036.2006.02854.x. [DOI] [PubMed] [Google Scholar]
- 3.Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. 2013;35:229–44. doi: 10.1007/s00281-012-0352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol. 2008;14:3937–47. doi: 10.3748/wjg.14.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brentnall TA, Crispin DA, Rabinovitch PS, Haggitt RC, Rubin CE, Stevens AC, et al. Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology. 1994;107:369–78. doi: 10.1016/0016-5085(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 6.Atreya R, Neurath MF. Signaling molecules: the pathogenic role of the IL-6/STAT-3 trans signaling pathway in intestinal inflammation and in colonic cancer. Curr Drug Targets. 2008;9:369–74. doi: 10.2174/138945008784221116. [DOI] [PubMed] [Google Scholar]
- 7.Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10:1314–9. doi: 10.1038/embor.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–72. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7:545–55. doi: 10.1593/neo.04571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holtmann MH, Neurath MF. Anti-TNF strategies in stenosing and fistulizing Crohn’s disease. Int J Colorectal Dis. 2005;20:1–8. doi: 10.1007/s00384-004-0634-0. [DOI] [PubMed] [Google Scholar]
- 13.Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–93. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 15.Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 16.Kawamori T, Kaneshiro T, Okumura M, Maalouf S, Uflacker A, Bielawski J, et al. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009;23:405–14. doi: 10.1096/fj.08-117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, et al. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 2009;23:143–52. doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–8. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421–8. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tonelli F, Lim KG, Loveridge C, Long J, Pitson SM, Tigyi G, et al. FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cell Signal. 2010;22:1536–42. doi: 10.1016/j.cellsig.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. J Immunol. 2007;178:2458–68. doi: 10.4049/jimmunol.178.4.2458. [DOI] [PubMed] [Google Scholar]
- 22.Daniel C, Sartory NA, Zahn N, Schmidt R, Geisslinger G, Radeke HH, et al. FTY720 ameliorates oxazolone colitis in mice by directly affecting T helper type 2 functions. Mol Immunol. 2007;44:3305–16. doi: 10.1016/j.molimm.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 23.Deguchi Y, Andoh A, Yagi Y, Bamba S, Inatomi O, Tsujikawa T, et al. The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncol Rep. 2006;16:699–703. [PubMed] [Google Scholar]