

Abstract
A substantial body of evidence has shown that signal transducer and activator of transcription 3 (STAT3) has an important role in the heart in protecting the myocardium from ischemia and oxidative stress. These actions are attributed to STAT3 functioning as a transcription factor in upregulating cardioprotective genes. Loss of STAT3 has been implicated as well in the pathogenesis of heart failure and, in that context and in addition to the loss of a cardioprotective gene program, nuclear STAT3 has been identified as a transcriptional repressor important for the normal functioning of the ubiquitin-proteasome system for protein degradation. The later finding establishes a genomic role for STAT3 in controlling cellular homeostasis in cardiac myocytes independent of stress. Surprisingly, although a well-studied area, very few downstream gene targets of STAT3 in the heart have been definitively identified. In addition, STAT3 is now known to induce gene expression by noncanonical means that are not well characterized in the heart. On the other hand, recent evidence has shown that STAT3 has important nongenomic actions in cardiac myocytes that affect microtubule stability, mitochondrial respiration, and autophagy. These extranuclear actions of STAT3 involve protein–protein interactions that are incompletely understood, as is their regulation in both the healthy and injured heart. Moreover, how the diverse genomic and nongenomic actions of STAT3 crosstalk with each other is unchartered territory. Here we present an overview of what is and is not known about both the genomic and nongenomic actions of STAT3 in the heart from a structure-function perspective that focuses on the impact of posttranslational modifications and oxidative stress in regulating the actions and interactions of STAT3. Even though we have learnt a great deal about the role played by STAT3 in the heart, much more awaits to be discovered.
Keywords: JAK, STAT3, autophagy, cardiac hypertrophy, cardiac remodeling, heart failure, microtubule stability, posttranslational modification, redox
Introduction
First recognized in 1994 as the acute-phase response factor (APRF),1 signal transducer and activator of transcription 3 (STAT3) is one of 7 mammalian STAT transcription factors that play a central role in signaling by growth factors and cytokines. Of the STATs, STAT3 has a critical nonredundant role in cell growth, survival, and differentiation. The unique importance of STAT3 is underscored by the observation that of the STAT family members only disruption of the STAT3 gene causes embryonic lethality.2 Studies over the past 20 years have shown that STAT3 has important actions in protecting the heart under stress.3-6 In addition, human failing hearts were reported to exhibit reduced STAT3 levels and activity.7-9 Yet a cohesive understanding of how STAT3 protects the heart has yet to be achieved. The protection afforded by STAT3 has been ascribed to both traditional genomic actions, i.e., the upregulation of protective genes, and more recently some may say fanciful nongenomic actions that target mitochondrial function and autophagy. Neither action is fully understood. Nor is it known how the two are regulated and integrated under either normal or stress conditions. Here we present an overview of what is known and not known about the protective actions of STAT3 in the heart from a structure-function perspective and how posttranslational modifications and oxidative stress may act as determining factors in regulating the genomic and nongenomic actions of STAT3.
Overview
The STAT3 protein is 770 amino acid in length with 6 distinct domains (Fig. 1).4 A terminal NH2-domain that participates in higher order complex formations that are not well understood is followed by a coiled-coil domain important for protein–protein interaction with other transcription factors and co-regulators. Next, the DNA binding domain canonically interacts with an interferon γ (gamma)-activated sequence (GAS) in the promoter region of specific genes.10 A subsequent linker domain is located just before a Src homology-2 (SH2) domain that is essential for interaction with specific tyrosine-phosphorylated sites such as the YXXQ sites of the gp130 receptor of the IL-6 type cytokines, as well as a specific tyrosine phosphorylated residue of activated STAT1 or STAT3 to form parallel dimers important in canonical STAT3 signaling. Although fairly homologous among all STAT family members, the SH2 domain of STAT3 is distinctive enough to allow for development of specific small molecule inhibitors of STAT3, such as Stattic and S3I-201.11 Two sites of phosphorylation that constitute an “ignition” for canonical STAT3 activation are located in a COOH-terminal region known as the transcription activation domain (TAD).4 The first site is Y705, phosphorylation of which leads to STAT3 dimer formation followed by translocation to the nucleus and induction of gene expression. Y705 is phosphorylated by the Janus kinase (JAK) family members, Src kinase, and epidermal growth factor kinase (EGF).12 The other phosphorylation site of importance in regulating STAT3 activity is S727. Many serine/threonine kinases have been shown to phosphorylate S727 such as protein kinase Cε (PKCε), PKCδ, ERK1/2, mTOR, ZIP kinase, and CDK5.12 S727 phosphorylation can take place either in the cytoplasm or nucleus. In canonical STAT3 signaling S727 phosphorylation has been shown to play a role in boosting the transcriptional activity of STAT3 through the recruitment of transcriptional cofactors, such as the histone acetyltransferase p300/CBP.4,12 STAT3 S727 phosphorylation may also favor STAT3 homodimer formation.13
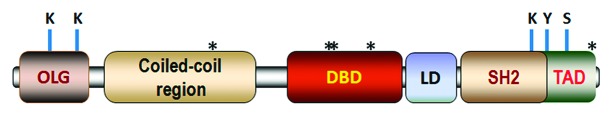
Figure 1. Major domains of STAT3 showing location of critical sites for posttranslational modification. From N-terminus to C-terminus, they are: oligomerization (OLG) domain, coiled-coil domain, DNA binding domain (DBD), linker domain (LD), SH2 domain, and the transcription activation domain (TAD). Phosphorylation of Y705 and S727 within the TAD has a critical role in the canonical genomic actions of STAT3. S727 phosphorylation has also been implicated in the noncanonical genomic and nongenomic actions of STAT3 through either enhanced recruitment of transcriptional co-factors (e.g., p300) and GRIM19, or alterations in STAT3 conformation. Acetylation of lysine residues (K49 and K87) within the NH2-terminal OLG domain are important for transcription by enhancing p300-STAT3 association and stabilizing enhanceosome assembly, as well as for nuclear retention of STAT3. Acetylation of K685 within the SH2 domain helps stabilize STAT3 dimers and enhances transcription. Asterisks (*) indicate redox-sensitive cysteine residues: C259 (within the coiled-coil domain), C418, C426, and C468 (within the DNA binding domain), and C765 within the TAD. Other sites of phosphorylation of STAT3 have been identified, but their importance in controlling the genomic and nongenomic actions of STAT3 is not defined.
STAT3 function is regulated as well by p300-mediated acetylation of lysine residues within the NH2-terminal (K49, K87) and SH2 (K685) domains. Acetylation of the latter, which rather unexpectedly may occur in the cytoplasm, plays a critical role in stabilizing STAT3 dimers and enhancing transcription.12,14,15 Acetylation of K49 and K87 was shown to be important for gene transcription by enhancing or strengthening the interaction of STAT3 with p300 and thus stabilizing enhanceosome assembly.16 Acetylation of these residues is important as well for nuclear retention of STAT3 with deacetylation mediated by histone deacetylases (principally HDAC1 and 4) resulting in STAT3 nuclear exit (and degradation).17,18 By binding p300 and thereby directing acetylation, STAT3 has been shown to positively affect the accessibility of other transcription factors to promoters as well as to enhance transcriptional activity of NFκB.19,20 Finally, K685 acetylation was shown to be important for interaction of STAT3 with DNA methyltransferase 1 and subsequent methylation and silencing of certain promoters;21 Nearly all of these sorts of studies were performed on non-cardiac cells and thus their relevance to cardiac cells is unproven; however, the fundamental aspects of STAT3 signaling and regulation that they define are likely relevant to cardiac myocytes, particularly under conditions of gene induction and genomic plasticity related to stress. For instance, recent evidence was reported that increased p300 acetyltransferase activity in neonatal rat ventricular myocytes due to drug-induced p300 stabilization impacts positively on STAT3 activation and half-life.22
Cardiac Protective Actions of STAT3 Revealed by Genetic Mouse Models
STAT3 has been implicated in the protection of the myocardium produced by different types of preconditioning (ischemic, pharmacological, and remote), as well as postconditioning.4 In the case of preconditioning, STAT3 has been implicated both in the early short-lived phase (not involving gene expression) and the delayed (by upwards of 24 h) longer-lived phase known as the second window of protection (involving gene expression). In many cases, the evidence reported is correlative or based on the use of a JAK2 inhibitor.23-36 More definitive evidence for the importance of STAT3 in the heart has come from mouse genetic models. In 2003, Jacoby et al. explored the effect of postnatal deletion of STAT3 specifically in cardiac myocytes by crossing floxed STAT3 mice with mice expressing Cre recombinase under control of the α-myosin heavy chain (MHC) promoter that is active predominately in mature cardiac myocytes.37 In these mice, myocardial dysfunction and marked cardiac fibrosis were noted with advanced age in the absence of cardiac insult. These hearts were also found to have a greater susceptibility to injury and greater contractile dysfunction was seen with doxorubicin. After lipopolysaccharide (LPS) injection hearts of these mice showed a significant increase in inflammation, fibrosis, and apoptosis, which was attributed to increased production of TNF-α presumably due to increased oxidative stress. The cellular source of TNF-α, specifically whether cardiac myocytes contributed, was not reported. Overall, the findings support an important role for STAT3 in both the aging/aged heart and the heart exposed to oxidative stress, although the exact basis for this was not defined.
Hilfiker-Kleiner et al. also reported progressive fibrosis, along with a reduction in capillary density, in hearts of cardiac myocyte-restricted STAT3 KO mice that became significant at 3–4 mo.38 With advanced age, dilated cardiomyopathy, impaired cardiac function, and premature death was observed. Evidence was reported that STAT3 KO cardiac myocytes produce unidentified paracrine factors that stimulate (mouse embryonic) fibroblast proliferation, but inhibit (mouse lung) endothelial cell proliferation. At 3–4 mo a number of genes associated with fibrosis and anti-angiogenesis were upregulated (COL1A1, COL8A1, OPN, BGN, TNC, PAI-1, CTGF, TSP1, TIMP1, MMP-12, MCP-1, IL2RB, IL15, MCP3, and BCL2A1). Surprisingly, no change was observed in protein levels of VEGF, which previously was linked to the protective actions of STAT3 in heart. In this regard, it is worth noting that the link between the cardioprotective effect of STAT3 and VEGF upregulation, as well as between STAT3 and MnSOD/SOD2 upregulation, was obtained with agonist stimulation (leukemia inhibitory factor/LIF) that induces multiple intracellular signaling pathways, as well as by overexpression of constitutively active STAT3.39-41 The importance of concurrent signaling in shaping the character of STAT3 signaling is described elsewhere.3
Hilfiker-Kleiner et al. also tested the role of STAT3 in ischemia-reperfusion and infarction.38 Infarct size and apoptosis were greater 24 h after reperfusion in KO mice with a significantly impaired fractional shortening 7 d after the insult. After 24 h reperfusion, increased mRNA levels were observed for the pro-apoptotic and pro-autophagy protein BNIP3, while mRNA levels of the prosurvival gene HSP70 were decreased. Again no changes were seen in mRNA or protein for VEGF. With myocardial infarction, a marked increase in mortality was seen: 32% wild-type vs. 100% KO. In a follow up study, this group provided compelling evidence to support the conclusion that the negative effects of cardiac myocyte-targeted STAT3 KO on capillary density and contractile function with advanced age was due to elevated cardiac expression of miR-199a, which in turn compromised the ubiquitin-proteasome system (UPS) of protein degradation by reducing expression of two UPS component proteins (although UPS activity was not directly assessed).42 Transfection of cardiac myocytes with pre-miR-199a resulted in thinning of width and extension of length, as well as reduced levels of α- and β-MHC mRNA and protein levels of total MHC and troponin-T. These effects were recapitulated by knocking down the two UPS components. Expression of miR-199a or pharmacological inhibition of UPS was also associated with increased protein arginine methyltransferase I (PRMT-I) expression and asymmetric dimethylarginine (ADMA) synthesis in cardiac myocytes, which in turn was demonstrated to impair endothelial cell function. In addition, greater miR-199a promoter activity was found in STAT3 knock-down cardiac myocytes. Finally, using the cardiac myocyte-targeted STAT3 deficient mouse model others found that STAT3 is essential for ischemic and pharmacological preconditioning,33 and depending on the protocol is important for ischemic postconditioning as well.43 These findings were mirrored in aged hearts associated with a reduction in STAT3 levels. Bolli et al. developed a tamoxifen-inducible cardiac myocyte-targeted STAT3 knockout (KO) mouse.44 Deletion of STAT3 abrogated the upregulation of cardioprotective (COX-2 and HO-1) and antiapoptotic proteins (e.g., Mcl-1, Bcl-xL, c-FLIPL, and c-FLIPS) that are normally expressed in response to delayed pre-conditioning. However, the impact of STAT3 deletion on the infarct-sparing actions of delayed preconditioning was not reported.
Although cardiac STAT3 is important for limiting infarct damage, unrestricted continuous activation of STAT3 by the gp130-receptor system after myocardial infarction was shown to be detrimental.45 Thus, a precise regulation of STAT3 activity in terms of magnitude, time course, concurrent signals, or other as yet undefined parameters is essential for its beneficial effects. The finding of detrimental consequences after myocardial infarction with unbridled STAT3 activity via gp130 may explain why increased IL-6 serum levels are prognostic markers for adverse outcome in patients with myocardial infarction and heart failure.
We recently assessed whether STAT3 is important in hypertension-induced cardiac remodeling using mice with reduced global STAT3 activity due to a S727A mutation.46 Hearts of SA/SA mice showed signs of developing systolic dysfunction in response to angiotensin II-induced elevated blood pressure after 17 d. With angiotensin II, fibrosis was seen in the left ventricle of both wild-type and SA/SA mice; however, fibrosis in SA/SA mice was largely reparative and was associated with loss of myocytes, while in wild-type hearts reactive fibrosis predominated. Cardiac hypertrophy as indexed by heart to bodyweight ratio and left ventricular anterior wall dimension during diastole was greater in wild-type mice. Altogether, our study and those involving STAT3 KO support the conclusion that the presence of STAT3 in cardiac myocytes is important for normal function and protection from stress.
In 2000, Kunisida et al. published a study highlighting the importance of STAT3 in hypertrophy and cardiac protective signaling.47 Cardiac myocyte-specific STAT3 overexpressing hearts exhibited age-related pathological hypertrophy associated with increased expression of β-myosin heavy chain (MHC) and atrial natriuretic factor (ANF) and protection against doxorubicin induced cardiomyopathy. The later was likely in part the consequence of countering doxorubicin-induced reduction in STAT3 levels, as well as increased cardiotrophin-1 expression (and presumably increased expression of other protective proteins). Subsequently, the same group reported the impact of overexpressing a constitutively active form of STAT3 (caSTAT3) specifically in cardiomyocytes in several studies.39-41 Two studies highlighted the significant role of activated STAT3 in upregulating MnSOD, a mitochondrial superoxide scavenger that protects the heart against increased levels of hypoxia/reoxygenation-induced ROS. Another study emphasized the importance of activated STAT3 in vascular formation by upregulating VEGF synthesis and enhancing VE-cadherin expression, which translated into an increase in capillary density. However, overexpressing STAT3 may not necessarily enhance the normal function of active STAT3 in the heart. At higher unphysiological levels, STAT3 might interact promiscuously with other proteins and artifactually affect cellular events. Even so, Hilfiker-Kleiner et al. presented compelling evidence from cardiac myocyte-restricted STAT3 knockout mice and patients that STAT3 deficiency contributes to the etiology of postpartum cardiomyopathy due to reduced MnSOD expression, increased oxidative stress, and reduced capillary density.9
Arguably, a more reasonable approach to increase STAT3 activity in the heart is to remove the naturally occurring negative feedback inhibitor of STAT3 activation by the JAKs, which is SOCS3. Oba et al. recently reported that progression of left ventricular remodeling over a period of 14 d following an acute myocardial infarction was prevented in cardiac myocyte-targeted SOCS3 KO hearts.48 Enhanced activation of cardioprotective signaling pathways (STAT3, AKT, and ERK1/2) was noted in the KO heart, as well as reduced apoptosis. Compared with the wild-type hearts, following infarction KO hearts exhibited increased expression of antioxidant (MnSOD and HO-1) and anti-apoptotic (Bcl-xL) proteins, as well as decreased levels of pro-apoptotic (Bad and Bax) proteins. Consistent with less fibrosis in the SOCS3 KO hearts, reduced expression of CNTF, TGFβ2, matrix metalloproteinase-9, collagen 1, and collagen 3 and increased expression of TIMP-2 was seen compared with wild-type hearts. Altogether, the findings of this study elegantly demonstrate the key role of STAT3 in cardioprotection.
Genomic Actions of STAT3
STAT3 has been clearly shown to regulate different sets of genes by 3 distinct means: canonical, phosphorylated S727 (pS727)-only, and unphosphorylated STAT3/U-STAT3.49 The latter are 2 aspects of the noncanonical actions of STAT3 in mediating gene expression (Fig. 2). In canonical signaling, Y705 phosphorylation leads to STAT3 dimerization and translocation to the nucleus, where it induces transcription of certain genes by binding to a GAS element (TTCN3GAA).4,50 S727 phosphorylation enhances canonical transcription by recruiting the histone acetylase p300 (Fig. 2).16,51 STAT3 can also bind TTN4–6AA motifs,52,53 broadening its range of action.
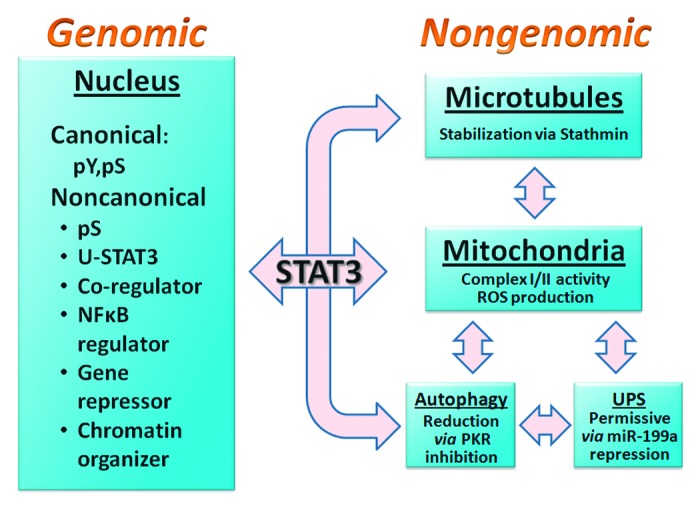
Figure 2. Scheme depicting the genomic and nongenomic actions identified for STAT3 in cardiac myocytes and other cell types. The genomic actions of STAT3 include both canonical and noncanonical events. The former involves STAT3 functioning as a transcription factor in the nucleus by binding TTN4–6AA elements in promoters and enhancing transcription. The noncanonical genomic actions of STAT3, which are diverse and not well understood, include: induction of transcription by pS727 STAT3 (without Y705 phosphorylation) and unphosphorylated STAT3 (U-STAT3); enhancing transcriptional activity of other transcription factors (e.g., nuclear steroid receptors); controlling the processing and nuclear retention of NFκB transcription factors; repressing gene expression; and modulating chromatin structure. Some of these actions may not require DNA binding and some events associated with the regulation of other transcription factors could conceivably occur in the cytoplasm. STAT3 has been shown to exert 3 actions in the cell that are extranuclear and do not involve gene transcription. These nongenomic actions of STAT3 control microtubule stability, mitochondrial function, and autophagy. For most, interaction of STAT3 with a specific protein has been implicated: stathmin (microtubule stability), GRIM19 (mitochondrial function), and PKR (autophagy). For microtubule stability and inhibition of autophagy, the STAT3–protein interaction provides a straightforward mechanistic link. The basis for the mitochondrial role of STAT3 is the least understood and other proteins besides GRIM19 are likely involved. The permissive role of STAT3 in the ubiquitin-proteasome system (UPS) is genomic and results from suppression of miR-199a expression. Understanding of crosstalk between the genomic and mitochondrial actions of STAT3 is limited, as is the likely interplay among the nongenomic actions of STAT3 (for instance, impaired mitochondrial function and enhanced autophagy/mitophagy). Evidence of complex interplay among microtubule stability, mitochondrial function, autophagy, and UPS in various cell types is reported in the literature in general, implying that STAT3 has a central role in cellular homeostasis and stress responsiveness. Differential regulation of posttranslational modifications of STAT3 could form the basis for the integration of the nongenomic and genomic actions of STAT3.
STAT3 is now known to affect transcription noncanonically (Fig. 2),49,54-56 for instance: (1) STAT3 is constitutively present in the nucleus;55,57 (2) likely there are non-consensus binding sites for STAT3 in some promoters in the context of association of STAT3 with other proteins involved in transcription, e.g., NFκB p65;58 (3) STAT3 can bind DNA as a monomer as well,10 although the significance of this to gene transcription is unknown; (4) STAT3 may not necessarily need to bind DNA to enhance transcription;51,59 (5) STAT3 S727 phosphorylation can enhance transcription independent of Y705 phosphorylation;49,60-62 (6) STAT3 contains a nuclear receptor binding motif (LXXLL motif) in the coiled-coil domain and was shown to synergistically enhance transcriptional activity of nuclear receptors;63-66 (7) STAT3 and the shorter STAT3β spliceform lacking the TAD were found to induce a smaller number of genes in common than the numbers of genes induced uniquely by either;67 and (8) U-STAT3, which may increase in nuclei of cardiac myocytes during cardiac hypertrophy, can induce expression of a subset of inflammatory genes.56,68-70 U-STAT3 may function also as a chromatin/genomic organizer.10 Together these observations support the conclusion that STAT3 functions as a transcriptional co-regulator, as well as a transcription factor per se.
How the 3 transcriptional mechanisms of STAT3 are coordinated is unknown, but likely involves regulated STAT3–protein interactions. As mentioned, a number of genes have been implicated in the protective actions of STAT3 in the heart, but to our knowledge definitive evidence for a direct role of STAT3 in their induction based on promoter analysis or ChIP assays is lacking. Three putative STAT3 binding sites were detected in the promoter for miR-199a, but evidence for STAT3 binding to the site was not shown and in this case STAT3 served to repress transcription.42
Nongenomic Actions
STAT3 has been shown to exert 3 actions in the cell affecting microtubule stability, mitochondrial function, and autophagy that are thought to be extranuclear and not involving gene transcription (Fig. 2). Studies on these nongenomic actions of STAT3 are evolving and the findings to date, though perhaps not definitive, are nevertheless intriguing as they could foster development of new therapeutic strategies based on naturally occurring or forced extranuclear STAT3–protein interactions.
STAT3 has been proposed to directly contribute to microtubule (MT) stabilization by interacting with and thereby inhibiting the activity of stathmin (a.k.a. oncoprotein 18), a small ubiquitously expressed and mainly-cytoplasmic protein.71-73 Stathmin binds α/β-tubulin heterodimers causing depolymerization of MTs. This role of STAT3 is reported to be important for cell migration and preventing axonal retraction/degeneration and evidence for this includes: (1) co-immunoprecipitation of STAT3 and stathmin (along with mutational analysis); (2) a sufficient protein ratio of stathmin to STAT3; (3) negative effect of STAT3 deletion on MT stability and cell migration, along with recovery types of experiments with STAT3 mutants that do not bind DNA (which now has shortcomings given the revised understanding of the genomic actions of STAT3); (4) STAT3-mediated reversal of stathmin-inhibition of tubulin polymerization in vitro; and (5) the negative impact of STAT3 inhibitors on MT polymerization.
Excessive MT stabilization in the myocardium has been observed in animal models of pathological cardiac hypertrophy and heart failure,74 but the role of STAT3 in regulating MT stability in cardiac myocytes is not established. However, Ng et al. reported that gp130 family cytokines (established activators of STAT3) induced MT stabilization in neonatal rat ventricular myocytes, which could be inhibited with a JAK2 or STAT3 inhibitor or STAT3 knockdown.74 In contrast, expression of a constitutively active STAT3 (spontaneously dimerizing STAT3) enhanced MT stabilization. A direct role for STAT3 in MT stabilization in this study cannot be ruled in or out. The impact of constitutively active STAT3 would suggest that gene expression was involved, while evidence both for and against a role of Y705 phosphorylation in STAT3-stathmin interaction has been reported.71,73 Given the likely importance in the heart for MT stability in the context of mitochondrial function and autophagy and its possible dysfunction in pathological conditions, the role of STAT3 as a regulator of this process warrants further investigation.
The observations that a component of complex I, GRIM19 can associate with STAT3 prompted investigation into whether STAT3 plays a role in mitochondrial function.75-79 Many (but not all) studies have detected STAT3 in mitochondria from various types of cells and tissues including cardiac myocytes and the heart, and in many (but not all) cases the STAT3 pool is enriched in phosphorylated S727 compared with the cytoplasm.80-85 This enrichment may be dynamically regulated as catecholamine-induced hypertrophy of H9c2 cardiomyoblasts was associated with a reduction in mitochondrial STAT3 phosphorylated S727 levels with no change in total STAT3 levels.86 Recently, the uptake of STAT3 by isolated mitochondria was shown to be mediated by GRIM19 and enhanced by STAT3 S727 phosphorylation,79 which is not unexpected as association of GRIM19 with STAT3 was reported to occur through the TAD of STAT3 and to be positively affected by S727 phosphorylation.87 Interestingly, STAT3 and GRIM19 mutually enhance their translocation to the mitochondria88 and the small heat shock protein HSPB8/HSP22, which is predominately expressed in skeletal/cardiac muscle was reported to be important for the mitochondrial translocation of STAT3.89 STAT3 mitochondrial uptake would also appear to require mitochondrial membrane potential and energy.79
STAT3 has been shown to regulate several aspects of mitochondrial function, including activities of complexes I and II, mitochondrial permeability transition pore (mPTP) opening, and reactive oxygen species (ROS) production. Mitochondria from hearts of mice with postnatal cardiac myocyte-targeted STAT3 KO have reduced ADP-stimulated respiration and complex I and II activities,75,77 and the STAT3 inhibitor Stattic reduced ADP-stimulated respiration of rat mitochondria.77 Calcium-sensitivity of mPTP opening, which can trigger cell death, was enhanced in mitochondria from STAT3-KO mice and Stattic-treated rat mitochondria. Moreover, STAT3 co-immunoprecipitated with pore component cyclophilin D in rat left ventricular mitochondria, suggesting that STAT3 may prevent mPTP opening by binding cyclophilin D similar to the actions of the cardioprotective agent cyclosporine A (CsA).77 In fact, STAT3 was shown to be important for preconditioning-induced infarct size reduction, but not pharmacological conditioning with CsA, suggesting similar modes of action. Intriguingly, in aged mouse hearts mitochondrial STAT3 levels were reduced raising the possibility that loss of STAT3 may contribute to aging-related pathologies of cardiac myocytes.77 Others reported that postconditioning of the hearts of pigs increased mitochondrial levels of STAT3 Y705 phosphorylation (not S727), which was associated with better complex I respiration and calcium retention capacity.84 Finally, mice with cardiac myocyte-specific overexpression of mitochondria-targeted STAT3 bearing a mutation in the DNA-binding domain (MLS-STAT3E) were recently generated.76 MLS-STAT3E expressing mitochondria showed modest decreases in complexes I and II basal activities; however, mitochondria from MLS-STAT3E hearts were protected against ischemic damage to complex I respiratory rates and release of cytochrome c into the cytosol. Compared with wild-type mitochondria, ischemia did not enhance ROS production by MLS-STAT3E mitochondria, which was attributed to partial blockade of electron transport through complex I.
The role of mitochondrial STAT3 in regulating ROS production is intriguing and has been studied primarily in noncardiac cells. Both positive and negative actions of mitochondrial STAT3 on ROS production have been reported and in some cases no parsing of the relative contribution of genomic and nongenomic actions of STAT3 was done: mouse STAT3 null hematopoietic stem/progenitor cells display mitochondrial dysfunction and increased ROS;90 TNF-induced necroptosis of L929 mouse fibrosarcoma cells was attributed to enhanced translocation of STAT3 to the mitochondria by GRIM19 with a subsequent increase in ROS;88 mitochondrial STAT3 was implicated in nerve growth factor (NGF) induced neurite outgrowth and ROS production;80 in astrocytes the absence of STAT3 resulted in decreased mitochondrial membrane potential and ATP production, along with increased ROS production;91 cultured primary osteoblasts from STAT3 KO mice produced elevated levels of ROS;92 and, finally, the key importance of mitochondrial STAT3 to Ras-dependent oncogenic transformation of cancer cells is likely due to increased ROS.93
S727 phosphorylation seems to have special importance in the mitochondrial actions of STAT3, as impaired activity of complexes I and II of mitochondria from STAT3–/– pro-B cells could be restored by expressing a mitochondrial targeted STAT3 bearing a pS727 mimetic while the non-phosphorylatable STAT3 Y705F/S727A was ineffective.75 Moreover, a STAT3 with a phosphorylated Y705 mimetic or a constitutively active STAT3 that spontaneously undergoes dimerization were not effective. Another study reported that a mitochondria-targeted serine-dominant negative mutant of STAT3 attenuated NGF-induced neurite outgrowth, while wild-type and tyrosine-dominant negative mutant STAT3 enhanced NGF induced neurite outgrowth.80 Also, a number of studies report that STAT3 pS727 is enriched in mitochondria compared with the cytoplasm.78-83
While the evidence for a direct role for STAT3 in the function of mitochondria is convincing, the issue of stoichiometry has raised a seemingly insurmountable argument against a direct contribution of (normally expressed levels of) STAT3 to the electron transport chain based on protein–protein interactions. Phillips et al. determined through three different proteomic approaches that the mitochondrial complex I/II ratio to STAT3 in cardiac tissue to be ~105, at least under normal conditions.94 Involvement of other indirect mechanisms, such as regulating in some fashion the posttranslational modifications of mitochondrial proteins, remains a tenable hypothesis. For instance, a mitochondrial pool of GSK-3β localizes to the inner membrane and STAT3 was shown to associate with and negatively affect phosphorylation of GSK3β, thereby enhancing its activity.95 Altogether this would be expected to promote mPTP opening and ROS generation. Evidence was also recently reported that STAT3 tightly associates with inner mitochondrial membrane complexes in rat heart mitochondria, likely complex I.79 Thus, STAT3 functioning in a higher order complex with other proteins that results in posttranslational modification of complex 1 proteins is a possibility. Alternatively, Szczepanek et al. have proposed that STAT3 may function in the redox buffering of the mitochondria given reports that it possesses redox-sensitive cysteines.78
STAT3 may represent a means of communication between the mitochondria and nucleus to regulate cellular metabolism, although evidence to support this conclusion is circumstantial. Recent evidence from cancer research indicates that STAT3 in its capacity as a nuclear transcription factor can act as an “anaerobic switch”, favoring glycolysis and attenuating mitochondrial activity by suppressing genes for mitochondrial proteins.96,97 In fact, STAT3 has been implicated as a causative factor in the well-known Warburg effect observed in cancer cells.97 STAT3 Y705 phosphorylation and canonical signaling has been implicated in these actions of STAT3. A major regulator of mitochondria biogenesis is SIRT1, a NAD-dependent deacetylase that negatively impacts on canonical STAT3 signaling. SIRT1 KO in mouse embryonic fibroblasts was recently reported to increase STAT3 expression and activity, which was accompanied by increased accumulation of S727 phosphorylated STAT3 in mitochondria and mitochondrial respiration.81 Obviously, more research needs to be done for a coherent understanding of the give and take of STAT3 signaling in mitochondria-nucleus crosstalk.
As mentioned, STAT3 was shown to be important for proper protein degradation in the cells by repressing the expression of a suppressor of the expression of UPS components.42 While inhibition of UPS-mediated protein degradation (as would be seen with loss of STAT3) has been shown to enhance autophagy,98 evidence was recently presented that STAT3 tonically inhibits autophagy by physically associating with protein kinase R (PKR).99 PKR is a ubiquitously expressed serine/threonine kinase that phosphorylates eukaryotic translation initiation factor 2-α, thereby inhibiting protein translation and inducing autophagy. PKR is activated (directly or indirectly) by a number of stresses including double stranded RNA, growth factor deprivation, and hydrogen peroxide.100,101 A physical association between the SH2 domain of STAT3 and the catalytic domain of PKR was demonstrated that could be disrupted by long chain saturated fatty acids.99,102 These observations were made with noncardiac cells; however, significant increase in autophagolysosomes and levels of LC3-II were reported in a STAT3-deficient HL-1 mouse atrial cardiac myocyte cell line, indicating enhanced autophagy.103 In summary, normal STAT3 expression would seem to favor UPS over autophagy, while loss of STAT3 would have the converse action.
Proving that STAT3 truly has nongenomic actions in a physiological setting is extremely difficult and whether that has been achieved is debatable. In this regard, two cautionary notes need to be sounded: (1) overexpression studies are not the perfect complement of gene knockout/deletion studies; and (2) what defines the transcriptional activity of STAT3 is a moving target and could conceivably involve just the shuttling of other proteins into the nucleus or the modification of other transcription factors via enhanced posttranslational modification.
The Tail of the Rhino: Distinguishing Feature and Common Link?
The TAD is the least conserved portion of the STAT proteins, although with the exception of STAT2 and 6 all contain a P(M)SP motif that is a target for proline-directed kinases and is involved in recruiting p300 with various potencies.12,104 For STAT3, phosphorylation of the serine residue within this motif (S727) has been implicated in other aspects of STAT3 regulation as well, including: (1) enhancing transcriptional activity by recruiting co-factors;12,104 (2) opposing Y705 phosphorylation by recruiting phosphatases or inducing conformational change by disrupting TAD-coiled-coil domain interaction,105-107 both of which have significance in preventing or terminating canonical STAT3 signaling; (3) determining subcellular distribution of STAT3, specifically uptake by mitochondria;79 (4) terminating STAT3-induced gene expression at a subset of promoters by recruiting histone methyl transferase SET9, which dimethylates STAT3 on a specific residue (K140) thereby causing its dissociation from the promoter;108 and (5) inducing gene expression in its own right.60,61 S727 phosphorylation likely impacts on the conformation of the C-terminus or its accessibility. Approximately 150 associations/interactions with other proteins have been ascribed to STAT3.109,110 For many, the particular region of STAT3 has not been identified, although the coiled-coil domain is likely responsible. Surprisingly, only a few have been reported to involve the TAD of STAT3. Some interactions may be indirect, for instance p300-mediated interaction between STAT3 and Smad1.111Table 1 provides a listing of those that are (seemingly) direct and found from the literature. In one way or another, association of these proteins with STAT3 has been shown to determine the cellular and nuclear actions of STAT3; however, the impact of S727 phosphorylation on the role of STAT3 in the heart is not well studied. We recently showed that hearts of mice with a S727A mutation in both STAT3 alleles adapted poorly to increased blood pressure compared with wild-type mice.46 Whether this was due to differences in the genomic or nongenomic actions of STAT3 will need to be assessed.
Table 1. Proteins shown to interact with the TAD of STAT3.
| Protein | Shown | Involves pS727 | Function | Expressed in heart |
|---|---|---|---|---|
| 14–3-3ζ |
U266 myeloma cell line, T cells |
Yes |
• Protects STAT3 from pS727 dephosphorylation by PP2A112 • Along with STAT3 sequesters pFoxO1 and pFoxO3a in cytoplasm and prolongs T cell activation113 |
Yes |
| GRIM19 |
Various cell types |
Yes |
• Translocation of STAT3 to mitochondria79 • Inhibition of STAT3 transcriptional activity87,114 |
Yes |
| CBP/p300 |
Various cell types |
Yes |
• Enhances STAT3 transcriptional activity51 • STAT3-dependent nuclear retention of NFκB/p6520 • STAT3 acetylation (strengthened p300 binding and dimerization and DNA binding and enhanceosome assembly/transcription factor complexes)14-18 • STAT3-mediated NF-κB p100 processing115 |
Yes |
| Pin1 |
HepG2 cells, MEF, MCF-7 cells |
Yes |
• Promotes STAT3 transcriptional activity and p300 recruitment116 |
NR |
| SET9 |
human colon cancer A4 cells |
Yes |
• Dimethylation and downregulation of STAT3 binding at certain promoters108 |
Likely |
| CDK9 |
HepG2 |
ND |
• localization of CDK9 to proximal promoter so as to phosphorylate RNA pol II switching it from initiation to elongation state117 |
Yes |
| Sp1 |
Rat heart, HUVE |
Yes |
• Upregulation of ICAM-1 transcription after reoxygenation or reperfusion118 • Likely important at numerous genes |
Yes |
| Cyclin D1 |
HepG2 |
ND |
• Inhibition of STAT3 transcriptional activity119 |
Yes (low in normal adult myocardium) |
| SRC-1/ NCoA-1 | HepG2 | No | • Enhanced STAT3 transcriptional activity120 | Yes |
ND, not determined; NR, not reported.
Redox-Sensitivity of STAT3
Perhaps a distinguishing feature of STAT3 compared with the other STATs is redox-sensitivity, although the basis for this selectivity is not known. We recently showed that STAT3 activation in cardiac myocytes is impaired by depleting the major cellular antioxidant molecule and redox-buffer, glutathione (GSH).121 l-Buthionine-sulfoximine (BSO) pretreatment decreased GSH levels, induced ROS formation, and dose-dependently attenuated STAT3 activation by LIF. Glutathione monoethyl ester, which is cleaved to GSH intracellularly, prevented the reduction in STAT3 activation, as did the antioxidant N-acetyl-cysteine; however, LIF-induced STAT1 activation was unaffected by GSH depletion. We also showed that thiophylic compounds inhibit LIF-induced STAT3 activation.122 Pretreatment of human microvascular endothelial cells (HMEC-1), neonatal rat cardiac myocytes, or adult mouse cardiac myocytes with the nitroxyl (HNO) donors Angeli’s salt or nitrosocyclohexyl acetate (NCA) inhibited STAT3 activation. NCA also blocked induction of inflammatory genes (ICAM-1 and CEBPD). The related 1-nitrosocyclohexyl pivalate (NCP), which is not a nitroxyl donor, also inhibited STAT3 activation, indicating that these compounds were acting as thiolate-targeting electrophiles. JAK1 was not a target of acyloxy nitroso compounds, as NCA had no effect on JAK1 catalytic activity. However, pretreatment of recombinant STAT3 with NCA or NCP reduced labeling of free sulfhydryl residues. We also showed that NCP in the presence of diamide enhanced STAT3 glutathionylation in adult cardiac myocytes and altered the SDS-PAGE profile of STAT3 under non-reducing conditions. Finally, we observed that monomeric STAT3 levels are decreased in the Gαq model of heart failure in a redox-sensitive manner,122 supporting the conclusion that the redox-sensitivity of STAT3 has pathophysiological relevance.
Besides us, others have reported that STAT3 has redox-sensitive cysteines that affect its function. Treatment of HepG2 cells with thiol targeting agents was reported to inhibit IL-6-induced STAT3 activation and increase STAT3 glutathionylation.123 These agents also decreased nuclear accumulation of STAT3 and impaired expression of STAT3-target genes. Peroxide was reported to induce STAT3 homodimer formation in HEK293 cells and a cysteine in the N-terminus (C259) was implicated.124 Another study identified 3 redox-sensitive cysteines in the DNA binding domain (C418, C426, and C468) and one within the transcription activation domain (C765).125 In a cell system using IL-6, these residues were found responsible for peroxide-reduced STAT3-mediated reporter gene expression from a consensus GAS element, but not from TTN6AA sites. The latter observation suggests that ROS may differentially affect STAT3 binding to DNA depending upon the promoter.
How might the redox-sensitivity of STAT3 impact on its role in cells? Concerning the genomic actions of STAT3, oxidative stress may alter the gene profile linked to STAT3 because of differential promoter selection or STAT3–protein interactions. For the nongenomic actions of STAT3, protein–protein interactions may be affected. An early observation showed that STAT3 exists in higher order complexes with other proteins in the cytosol.126 Redox-sensitivity might function to determine the binding partners of STAT3, as well as its subcellular distribution. In short, redox-sensitivity may serve as a switch to coordinate the various roles of STAT3 depending upon the stresses placed on the cell.
Reduced STAT3 Activity in the Heart: The Perfect Storm?
Multiple studies have provided evidence that nongenomic STAT3 has a critical role in optimal mitochondrial function and preventing mPTP opening, inhibiting autophagy, proper functioning of the ubiquitin-proteasome system, and microtubule stability. In fact, STAT3 may serve as an integrating factor for these processes. On the other hand, genomic STAT3 has been linked to the upregulation of antioxidant and anti-apoptotic proteins. Thus, reduced protein levels or activity of STAT3, or impediment of its interaction with other proteins may favor the creation of “the perfect storm”, the consequence being unbridled activation of multiple death-inducing processes in the cell simultaneously. That such a scenario may occur in heart failure and contribute to contractile degeneration is plausible, but not yet investigated.
Conclusions and Perspectives
Much has been learned about the beneficial actions of STAT3 in the heart, but often times it is difficult to separate fact from supposition. In any case, what has emerged over the last few years is the central importance of STAT3 in regulating key cellular processes in cardiac myocytes in different cellular compartments by both genomic and nongenomic means. Another preeminent conclusion is that proper regulation of STAT3 in the heart and in cardiac myocytes in either direction, i.e., increased or decreased expression, heightened or depressed activation, is essential for the physiologic (genomic and non-genomic) actions of STAT3. For all these actions, both a housekeeping and a stress-responsive role of STAT3 is likely. STAT3–protein interactions are involved in all these processes and posttranslational modifications surely finesse the actions of STAT3, perhaps akin in naivety at this early stage of discovery to rhinos dancing in stilettos. Nonetheless, understanding how these interactions are regulated so as to coordinate the actions of STAT3 in the healthy and stressed/injured heart could lead to new therapeutic strategies to prevent damage to the heart or improve its performance.
Acknowledgments
This work was supported by a grant to GWB from NHLBI (5R01HL088101-06).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/24352
References
- 1.Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 2.Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94:3801–4. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zgheib C, Zouein F, Kurdi M, Booz G. Differential STAT3 signaling in the heart: Impact of concurrent signals and oxidative stress. JAK-STAT. 2012;1:102–11. doi: 10.4161/jkst.19776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurdi M, Booz GW. Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signaling. J Cardiovasc Pharmacol. 2007;50:126–41. doi: 10.1097/FJC.0b013e318068dd49. [DOI] [PubMed] [Google Scholar]
- 5.Haghikia A, Stapel B, Hoch M, Hilfiker-Kleiner D. STAT3 and cardiac remodeling. Heart Fail Rev. 2011;16:35–47. doi: 10.1007/s10741-010-9170-x. [DOI] [PubMed] [Google Scholar]
- 6.Wagner M, Siddiqui MA. Signaling networks regulating cardiac myocyte survival and death. Curr Opin Investig Drugs. 2009;10:928–37. [PubMed] [Google Scholar]
- 7.Hilfiker-Kleiner D, Hilfiker A, Drexler H. Many good reasons to have STAT3 in the heart. Pharmacol Ther. 2005;107:131–7. doi: 10.1016/j.pharmthera.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Podewski EK, Hilfiker-Kleiner D, Hilfiker A, Morawietz H, Lichtenberg A, Wollert KC, et al. Alterations in Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling in patients with end-stage dilated cardiomyopathy. Circulation. 2003;107:798–802. doi: 10.1161/01.CIR.0000057545.82749.FF. [DOI] [PubMed] [Google Scholar]
- 9.Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, et al. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell. 2007;128:589–600. doi: 10.1016/j.cell.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 10.Timofeeva OA, Chasovskikh S, Lonskaya I, Tarasova NI, Khavrutskii L, Tarasov SG, et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J Biol Chem. 2012;287:14192–200. doi: 10.1074/jbc.M111.323899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debnath B, Xu S, Neamati N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J Med Chem. 2012;55:6645–68. doi: 10.1021/jm300207s. [DOI] [PubMed] [Google Scholar]
- 12.Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, et al. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci. 2009;1171:59–76. doi: 10.1111/j.1749-6632.2009.04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Blenis J, Li HC, Schindler C, Chen-Kiang S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990–4. doi: 10.1126/science.7701321. [DOI] [PubMed] [Google Scholar]
- 14.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–73. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 15.Wang R, Cherukuri P, Luo J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem. 2005;280:11528–34. doi: 10.1074/jbc.M413930200. [DOI] [PubMed] [Google Scholar]
- 16.Hou T, Ray S, Lee C, Brasier AR. The STAT3 NH2-terminal domain stabilizes enhanceosome assembly by interacting with the p300 bromodomain. J Biol Chem. 2008;283:30725–34. doi: 10.1074/jbc.M805941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ray S, Boldogh I, Brasier AR. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology. 2005;129:1616–32. doi: 10.1053/j.gastro.2005.07.055. [DOI] [PubMed] [Google Scholar]
- 18.Ray S, Lee C, Hou T, Boldogh I, Brasier AR. Requirement of histone deacetylase1 (HDAC1) in signal transducer and activator of transcription 3 (STAT3) nucleocytoplasmic distribution. Nucleic Acids Res. 2008;36:4510–20. doi: 10.1093/nar/gkn419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamassia N, Castellucci M, Rossato M, Gasperini S, Bosisio D, Giacomelli M, et al. Uncovering an IL-10-dependent NF-kappaB recruitment to the IL-1ra promoter that is impaired in STAT3 functionally defective patients. FASEB J. 2010;24:1365–75. doi: 10.1096/fj.09-145573. [DOI] [PubMed] [Google Scholar]
- 20.Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–93. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci U S A. 2012;109:7765–9. doi: 10.1073/pnas.1205132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jain S, Wei J, Mitrani LR, Bishopric NH. Auto-acetylation stabilizes p300 in cardiac myocytes during acute oxidative stress, promoting STAT3 accumulation and cell survival. Breast Cancer Res Treat. 2012;135:103–14. doi: 10.1007/s10549-012-2069-6. [DOI] [PubMed] [Google Scholar]
- 23.Cai ZP, Parajuli N, Zheng X, Becker L. Remote ischemic preconditioning confers late protection against myocardial ischemia-reperfusion injury in mice by upregulating interleukin-10. Basic Res Cardiol. 2012;107:277. doi: 10.1007/s00395-012-0277-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian Y, Zhang W, Xia D, Modi P, Liang D, Wei M. Postconditioning inhibits myocardial apoptosis during prolonged reperfusion via a JAK2-STAT3-Bcl-2 pathway. J Biomed Sci. 2011;18:53. doi: 10.1186/1423-0127-18-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higuchi T, Yamauchi-Takihara K, Matsumiya G, Fukushima N, Ichikawa H, Kuratani T, et al. Granulocyte colony-stimulating factor prevents reperfusion injury after heart preservation. Ann Thorac Surg. 2008;85:1367–73. doi: 10.1016/j.athoracsur.2007.12.053. [DOI] [PubMed] [Google Scholar]
- 26.Suleman N, Somers S, Smith R, Opie LH, Lecour SC. Dual activation of STAT-3 and Akt is required during the trigger phase of ischaemic preconditioning. Cardiovasc Res. 2008;79:127–33. doi: 10.1093/cvr/cvn067. [DOI] [PubMed] [Google Scholar]
- 27.Huffman LC, Koch SE, Butler KL. Coronary effluent from a preconditioned heart activates the JAK-STAT pathway and induces cardioprotection in a donor heart. Am J Physiol Heart Circ Physiol. 2008;294:H257–62. doi: 10.1152/ajpheart.00769.2007. [DOI] [PubMed] [Google Scholar]
- 28.Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Bolli R. Endothelial nitric oxide synthase plays an obligatory role in the late phase of ischemic preconditioning by activating the protein kinase C epsilon p44/42 mitogen-activated protein kinase pSer-signal transducers and activators of transcription1/3 pathway. Circulation. 2007;116:535–44. doi: 10.1161/CIRCULATIONAHA.107.689471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishihara M, Miura T, Miki T, Sakamoto J, Tanno M, Kobayashi H, et al. Erythropoietin affords additional cardioprotection to preconditioned hearts by enhanced phosphorylation of glycogen synthase kinase-3 beta. Am J Physiol Heart Circ Physiol. 2006;291:H748–55. doi: 10.1152/ajpheart.00837.2005. [DOI] [PubMed] [Google Scholar]
- 30.Butler KL, Huffman LC, Koch SE, Hahn HS, Gwathmey JK. STAT-3 activation is necessary for ischemic preconditioning in hypertrophied myocardium. Am J Physiol Heart Circ Physiol. 2006;291:H797–803. doi: 10.1152/ajpheart.01334.2005. [DOI] [PubMed] [Google Scholar]
- 31.Lecour S, Suleman N, Deuchar GA, Somers S, Lacerda L, Huisamen B, et al. Pharmacological preconditioning with tumor necrosis factor-α activates signal transducer and activator of transcription-3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal-regulated kinase) Circulation. 2005;112:3911–8. doi: 10.1161/CIRCULATIONAHA.105.581058. [DOI] [PubMed] [Google Scholar]
- 32.Dawn B, Xuan YT, Guo Y, Rezazadeh A, Stein AB, Hunt G, et al. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc Res. 2004;64:61–71. doi: 10.1016/j.cardiores.2004.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith RM, Suleman N, Lacerda L, Opie LH, Akira S, Chien KR, et al. Genetic depletion of cardiac myocyte STAT-3 abolishes classical preconditioning. Cardiovasc Res. 2004;63:611–6. doi: 10.1016/j.cardiores.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 34.Xuan YT, Guo Y, Zhu Y, Han H, Langenbach R, Dawn B, et al. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J Mol Cell Cardiol. 2003;35:525–37. doi: 10.1016/S0022-2828(03)00076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hattori R, Maulik N, Otani H, Zhu L, Cordis G, Engelman RM, et al. Role of STAT3 in ischemic preconditioning. J Mol Cell Cardiol. 2001;33:1929–36. doi: 10.1006/jmcc.2001.1456. [DOI] [PubMed] [Google Scholar]
- 36.Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci U S A. 2001;98:9050–5. doi: 10.1073/pnas.161283798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100:12929–34. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95:187–95. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 39.Funamoto M, Fujio Y, Kunisada K, Negoro S, Tone E, Osugi T, et al. Signal transducer and activator of transcription 3 is required for glycoprotein 130-mediated induction of vascular endothelial growth factor in cardiac myocytes. J Biol Chem. 2000;275:10561–6. doi: 10.1074/jbc.275.14.10561. [DOI] [PubMed] [Google Scholar]
- 40.Osugi T, Oshima Y, Fujio Y, Funamoto M, Yamashita A, Negoro S, et al. Cardiac-specific activation of signal transducer and activator of transcription 3 promotes vascular formation in the heart. J Biol Chem. 2002;277:6676–81. doi: 10.1074/jbc.M108246200. [DOI] [PubMed] [Google Scholar]
- 41.Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, et al. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation. 2001;104:979–81. doi: 10.1161/hc3401.095947. [DOI] [PubMed] [Google Scholar]
- 42.Haghikia A, Missol-Kolka E, Tsikas D, Venturini L, Brundiers S, Castoldi M, et al. Signal transducer and activator of transcription 3-mediated regulation of miR-199a-5p links cardiomyocyte and endothelial cell function in the heart: a key role for ubiquitin-conjugating enzymes. Eur Heart J. 2011;32:1287–97. doi: 10.1093/eurheartj/ehq369. [DOI] [PubMed] [Google Scholar]
- 43.Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, et al. Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res. 2008;102:131–5. doi: 10.1161/CIRCRESAHA.107.164699. [DOI] [PubMed] [Google Scholar]
- 44.Bolli R, Stein AB, Guo Y, Wang OL, Rokosh G, Dawn B, et al. A murine model of inducible, cardiac-specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol. 2011;50:589–97. doi: 10.1016/j.yjmcc.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Hilfiker-Kleiner D, Shukla P, Klein G, Schaefer A, Stapel B, Hoch M, et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010;122:145–55. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- 46.Zouein FA, Zgheib C, Hamza S, Fuseler JW, Hall JE, Soljancic A, et al. Role of STAT3 in angiotensin II-induced hypertension and cardiac remodeling revealed by mice lacking STAT3 serine 727 phosphorylation. Hypertens Res. 2013;36:496–503. doi: 10.1038/hr.2012.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Yamada S, et al. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97:315–9. doi: 10.1073/pnas.97.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oba T, Yasukawa H, Hoshijima M, Sasaki K, Futamata N, Fukui D, et al. Cardiac-specific deletion of SOCS-3 prevents development of left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol. 2012;59:838–52. doi: 10.1016/j.jacc.2011.10.887. [DOI] [PubMed] [Google Scholar]
- 49.Kurdi M, Booz GW. Deciphering STAT3 signaling in the heart: plasticity and vascular inflammation. Congest Heart Fail. 2010;16:234–8. doi: 10.1111/j.1751-7133.2010.00175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Booz GW, Day JN, Baker KM. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol. 2002;34:1443–53. doi: 10.1006/jmcc.2002.2076. [DOI] [PubMed] [Google Scholar]
- 51.Schuringa JJ, Schepers H, Vellenga E, Kruijer W. Ser727-dependent transcriptional activation by association of p300 with STAT3 upon IL-6 stimulation. FEBS Lett. 2001;495:71–6. doi: 10.1016/S0014-5793(01)02354-7. [DOI] [PubMed] [Google Scholar]
- 52.Zhang D, Sun M, Samols D, Kushner I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J Biol Chem. 1996;271:9503–9. doi: 10.1074/jbc.271.16.9503. [DOI] [PubMed] [Google Scholar]
- 53.Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, et al. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem. 2008;283:14665–73. doi: 10.1074/jbc.M707429200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–9. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol. 2008;19:329–40. doi: 10.1016/j.semcdb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18:443–51. doi: 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- 57.Pranada AL, Metz S, Herrmann A, Heinrich PC, Müller-Newen G. Real time analysis of STAT3 nucleocytoplasmic shuttling. J Biol Chem. 2004;279:15114–23. doi: 10.1074/jbc.M312530200. [DOI] [PubMed] [Google Scholar]
- 58.Tiwari P, Tripathi LP, Nishikawa-Matsumura T, Ahmad S, Song SN, Isobe T, et al. Prediction and experimental validation of a putative non-consensus binding site for transcription factor STAT3 in serum amyloid A gene promoter. Biochim Biophys Acta. 2013;1830:3650–5. doi: 10.1016/j.bbagen.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 59.Niehof M, Streetz K, Rakemann T, Bischoff SC, Manns MP, Horn F, et al. Interleukin-6-induced tethering of STAT3 to the LAP/C/EBPbeta promoter suggests a new mechanism of transcriptional regulation by STAT3. J Biol Chem. 2001;276:9016–27. doi: 10.1074/jbc.M009284200. [DOI] [PubMed] [Google Scholar]
- 60.Hazan-Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X, et al. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood. 2010;115:2852–63. doi: 10.1182/blood-2009-10-230060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Courapied S, Sellier H, de Carné Trécesson S, Vigneron A, Bernard AC, Gamelin E, et al. The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition. J Biol Chem. 2010;285:26765–78. doi: 10.1074/jbc.M109.092304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sato N, Kawai T, Sugiyama K, Muromoto R, Imoto S, Sekine Y, et al. Physical and functional interactions between STAT3 and ZIP kinase. Int Immunol. 2005;17:1543–52. doi: 10.1093/intimm/dxh331. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Z, Jones S, Hagood JS, Fuentes NL, Fuller GM. STAT3 acts as a co-activator of glucocorticoid receptor signaling. J Biol Chem. 1997;272:30607–10. doi: 10.1074/jbc.272.49.30607. [DOI] [PubMed] [Google Scholar]
- 64.Matsuda T, Junicho A, Yamamoto T, Kishi H, Korkmaz K, Saatcioglu F, et al. Cross-talk between signal transducer and activator of transcription 3 and androgen receptor signaling in prostate carcinoma cells. Biochem Biophys Res Commun. 2001;283:179–87. doi: 10.1006/bbrc.2001.4758. [DOI] [PubMed] [Google Scholar]
- 65.Aaronson DS, Muller M, Neves SR, Chung WC, Jayaram G, Iyengar R, et al. An androgen-IL-6-Stat3 autocrine loop re-routes EGF signal in prostate cancer cells. Mol Cell Endocrinol. 2007;270:50–6. doi: 10.1016/j.mce.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 66.De Miguel F, Lee SO, Onate SA, Gao AC. Stat3 enhances transactivation of steroid hormone receptors. Nucl Recept. 2003;1:3. doi: 10.1186/1478-1336-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ng IH, Ng DC, Jans DA, Bogoyevitch MA. Selective STAT3-α or -β expression reveals spliceform-specific phosphorylation kinetics, nuclear retention and distinct gene expression outcomes. Biochem J. 2012;447:125–36. doi: 10.1042/BJ20120941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21:1396–408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arany I, Reed DK, Grifoni SC, Chandrashekar K, Booz GW, Juncos LA. A novel U-STAT3-dependent mechanism mediates the deleterious effects of chronic nicotine exposure on renal injury. Am J Physiol Renal Physiol. 2012;302:F722–9. doi: 10.1152/ajprenal.00338.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yue H, Li W, Desnoyer R, Karnik SS. Role of nuclear unphosphorylated STAT3 in angiotensin II type 1 receptor-induced cardiac hypertrophy. Cardiovasc Res. 2010;85:90–9. doi: 10.1093/cvr/cvp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ng DC, Lin BH, Lim CP, Huang G, Zhang T, Poli V, et al. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J Cell Biol. 2006;172:245–57. doi: 10.1083/jcb.200503021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verma NK, Dourlat J, Davies AM, Long A, Liu WQ, Garbay C, et al. STAT3-stathmin interactions control microtubule dynamics in migrating T-cells. J Biol Chem. 2009;284:12349–62. doi: 10.1074/jbc.M807761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Selvaraj BT, Frank N, Bender FL, Asan E, Sendtner M. Local axonal function of STAT3 rescues axon degeneration in the pmn model of motoneuron disease. J Cell Biol. 2012;199:437–51. doi: 10.1083/jcb.201203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ng DC, Ng IH, Yeap YY, Badrian B, Tsoutsman T, McMullen JR, et al. Opposing actions of extracellular signal-regulated kinase (ERK) and signal transducer and activator of transcription 3 (STAT3) in regulating microtubule stabilization during cardiac hypertrophy. J Biol Chem. 2011;286:1576–87. doi: 10.1074/jbc.M110.128157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–7. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, et al. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem. 2011;286:29610–20. doi: 10.1074/jbc.M111.226209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol. 2010;105:771–85. doi: 10.1007/s00395-010-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Szczepanek K, Lesnefsky EJ, Larner AC. Multi-tasking: nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol. 2012;22:429–37. doi: 10.1016/j.tcb.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tammineni P, Anugula C, Mohammed F, Anjaneyulu M, Larner AC, Sepuri NB. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J Biol Chem. 2013;288:4723–32. doi: 10.1074/jbc.M112.378984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou L, Too HP. Mitochondrial localized STAT3 is involved in NGF induced neurite outgrowth. PLoS One. 2011;6:e21680. doi: 10.1371/journal.pone.0021680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bernier M, Paul RK, Martin-Montalvo A, Scheibye-Knudsen M, Song S, He HJ, et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J Biol Chem. 2011;286:19270–9. doi: 10.1074/jbc.M110.200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Park J, Kusminski CM, Chua SC, Scherer PE. Leptin receptor signaling supports cancer cell metabolism through suppression of mitochondrial respiration in vivo. Am J Pathol. 2010;177:3133–44. doi: 10.2353/ajpath.2010.100595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reich NC. STAT3 revs up the powerhouse. Sci Signal. 2009;2:pe61. doi: 10.1126/scisignal.290pe61. [DOI] [PubMed] [Google Scholar]
- 84.Heusch G, Musiolik J, Gedik N, Skyschally A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res. 2011;109:1302–8. doi: 10.1161/CIRCRESAHA.111.255604. [DOI] [PubMed] [Google Scholar]
- 85.Phillips D, Aponte AM, Covian R, Balaban RS. Intrinsic protein kinase activity in mitochondrial oxidative phosphorylation complexes. Biochemistry. 2011;50:2515–29. doi: 10.1021/bi101434x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jeong K, Kwon H, Min C, Pak Y. Modulation of the caveolin-3 localization to caveolae and STAT3 to mitochondria by catecholamine-induced cardiac hypertrophy in H9c2 cardiomyoblasts. Exp Mol Med. 2009;41:226–35. doi: 10.3858/emm.2009.41.4.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang J, Yang J, Roy SK, Tininini S, Hu J, Bromberg JF, et al. The cell death regulator GRIM-19 is an inhibitor of signal transducer and activator of transcription 3. Proc Natl Acad Sci U S A. 2003;100:9342–7. doi: 10.1073/pnas.1633516100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shulga N, Pastorino JG. GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. J Cell Sci. 2012;125:2995–3003. doi: 10.1242/jcs.103093. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 89.Qiu H, Lizano P, Laure L, Sui X, Rashed E, Park JY, et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation. 2011;124:406–15. doi: 10.1161/CIRCULATIONAHA.110.013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mantel C, Messina-Graham S, Moh A, Cooper S, Hangoc G, Fu XY, et al. Mouse hematopoietic cell-targeted STAT3 deletion: stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood. 2012;120:2589–99. doi: 10.1182/blood-2012-01-404004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sarafian TA, Montes C, Imura T, Qi J, Coppola G, Geschwind DH, et al. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS One. 2010;5:e9532. doi: 10.1371/journal.pone.0009532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou H, Newnum AB, Martin JR, Li P, Nelson MT, Moh A, et al. Osteoblast/osteocyte-specific inactivation of Stat3 decreases load-driven bone formation and accumulates reactive oxygen species. Bone. 2011;49:404–11. doi: 10.1016/j.bone.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 93.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–6. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Phillips D, Reilley MJ, Aponte AM, Wang G, Boja E, Gucek M, et al. Stoichiometry of STAT3 and mitochondrial proteins: Implications for the regulation of oxidative phosphorylation by protein-protein interactions. J Biol Chem. 2010;285:23532–6. doi: 10.1074/jbc.C110.152652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee J, Kim JC, Lee SE, Quinley C, Kim H, Herdman S, et al. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in Apcmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J Biol Chem. 2012;287:18182–9. doi: 10.1074/jbc.M111.328831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Darnell JE., Jr. STAT3, HIF-1, glucose addiction and Warburg effect. Aging (Albany NY) 2010;2:890–1. doi: 10.18632/aging.100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Demaria M, Giorgi C, Lebiedzinska M, Esposito G, D’Angeli L, Bartoli A, et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY) 2010;2:823–42. doi: 10.18632/aging.100232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393–8. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 99.Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell. 2012;48:667–80. doi: 10.1016/j.molcel.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 100.Mouton-Liger F, Paquet C, Dumurgier J, Bouras C, Pradier L, Gray F, et al. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochim Biophys Acta. 2012;1822:885–96. doi: 10.1016/j.bbadis.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 101.Bennett RL, Blalock WL, Abtahi DM, Pan Y, Moyer SA, May WS. RAX, the PKR activator, sensitizes cells to inflammatory cytokines, serum withdrawal, chemotherapy, and viral infection. Blood. 2006;108:821–9. doi: 10.1182/blood-2005-11-006817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Niso-Santano M, Shen S, Adjemian S, Malik SA, Mariño G, Lachkar S, et al. Direct interaction between STAT3 and EIF2AK2 controls fatty acid-induced autophagy. Autophagy. 2013;9:415–7. doi: 10.4161/auto.22910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Elschami M, Scherr M, Philippens B, Gerardy-Schahn R. Reduction of STAT3 expression induces mitochondrial dysfunction and autophagy in cardiac HL-1 cells. Eur J Cell Biol. 2013;92:21–9. doi: 10.1016/j.ejcb.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 104.Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–37. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- 105.Wakahara R, Kunimoto H, Tanino K, Kojima H, Inoue A, Shintaku H, et al. Phospho-Ser727 of STAT3 regulates STAT3 activity by enhancing dephosphorylation of phospho-Tyr705 largely through TC45. Genes Cells. 2012;17:132–45. doi: 10.1111/j.1365-2443.2011.01575.x. [DOI] [PubMed] [Google Scholar]
- 106.Zhang T, Seow KT, Ong CT, Cao X. Interdomain interaction of Stat3 regulates its Src homology 2 domain-mediated receptor binding activity. J Biol Chem. 2002;277:17556–63. doi: 10.1074/jbc.M105525200. [DOI] [PubMed] [Google Scholar]
- 107.Booz GW, Day JN, Baker KM. Angiotensin II effects on STAT3 phosphorylation in cardiomyocytes: evidence for Erk-dependent Tyr705 dephosphorylation. Basic Res Cardiol. 2003;98:33–8. doi: 10.1007/s00395-003-0387-x. [DOI] [PubMed] [Google Scholar]
- 108.Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci U S A. 2010;107:21499–504. doi: 10.1073/pnas.1016147107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.BioGRID 3.2 data collection of The Biological General Repository for Interaction Datasets (BioGRID), http://thebiogrid.org/ accessed February 16, 2013.
- 110.I2D (Interologous Interaction Database), http://ophid.utoronto.ca/ophidv2.204/index.jsp accessed February 16, 2013.
- 111.Nakashima K, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Kawabata M, et al. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science. 1999;284:479–82. doi: 10.1126/science.284.5413.479. [DOI] [PubMed] [Google Scholar]
- 112.Zhang J, Chen F, Li W, Xiong Q, Yang M, Zheng P, et al. 14-3-3ζ interacts with stat3 and regulates its constitutive activation in multiple myeloma cells. PLoS One. 2012;7:e29554. doi: 10.1371/journal.pone.0029554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Oh HM, Yu CR, Dambuza I, Marrero B, Egwuagu CE. STAT3 protein interacts with Class O Forkhead transcription factors in the cytoplasm and regulates nuclear/cytoplasmic localization of FoxO1 and FoxO3a proteins in CD4(+) T cells. J Biol Chem. 2012;287:30436–43. doi: 10.1074/jbc.M112.359661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lufei C, Ma J, Huang G, Zhang T, Novotny-Diermayr V, Ong CT, et al. GRIM-19, a death-regulatory gene product, suppresses Stat3 activity via functional interaction. EMBO J. 2003;22:1325–35. doi: 10.1093/emboj/cdg135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nadiminty N, Lou W, Lee SO, Lin X, Trump DL, Gao AC. Stat3 activation of NF-κB p100 processing involves CBP/p300-mediated acetylation. Proc Natl Acad Sci U S A. 2006;103:7264–9. doi: 10.1073/pnas.0509808103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lufei C, Koh TH, Uchida T, Cao X. Pin1 is required for the Ser727 phosphorylation-dependent Stat3 activity. Oncogene. 2007;26:7656–64. doi: 10.1038/sj.onc.1210567. [DOI] [PubMed] [Google Scholar]
- 117.Hou T, Ray S, Brasier AR. The functional role of an interleukin 6-inducible CDK9.STAT3 complex in human γ-fibrinogen gene expression. J Biol Chem. 2007;282:37091–102. doi: 10.1074/jbc.M706458200. [DOI] [PubMed] [Google Scholar]
- 118.Yang XP, Irani K, Mattagajasingh S, Dipaula A, Khanday F, Ozaki M, et al. Signal transducer and activator of transcription 3α and specificity protein 1 interact to upregulate intercellular adhesion molecule-1 in ischemic-reperfused myocardium and vascular endothelium. Arterioscler Thromb Vasc Biol. 2005;25:1395–400. doi: 10.1161/01.ATV.0000168428.96177.24. [DOI] [PubMed] [Google Scholar]
- 119.Bienvenu F, Gascan H, Coqueret O. Cyclin D1 represses STAT3 activation through a Cdk4-independent mechanism. J Biol Chem. 2001;276:16840–7. doi: 10.1074/jbc.M100795200. [DOI] [PubMed] [Google Scholar]
- 120.Giraud S, Bienvenu F, Avril S, Gascan H, Heery DM, Coqueret O. Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J Biol Chem. 2002;277:8004–11. doi: 10.1074/jbc.M111486200. [DOI] [PubMed] [Google Scholar]
- 121.Kurdi M, Sivakumaran V, Duhé RJ, Aon MA, Paolocci N, Booz GW. Depletion of cellular glutathione modulates LIF-induced JAK1-STAT3 signaling in cardiac myocytes. Int J Biochem Cell Biol. 2012;44:2106–15. doi: 10.1016/j.biocel.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zgheib C, Kurdi M, Zouein FA, Gunter BW, Stanley BA, Zgheib J, et al. Acyloxy nitroso compounds inhibit LIF signaling in endothelial cells and cardiac myocytes: evidence that STAT3 signaling is redox-sensitive. PLoS One. 2012;7:e43313. doi: 10.1371/journal.pone.0043313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xie Y, Kole S, Precht P, Pazin MJ, Bernier M. S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology. 2009;150:1122–31. doi: 10.1210/en.2008-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li L, Shaw PEA. A STAT3 dimer formed by inter-chain disulphide bridging during oxidative stress. Biochem Biophys Res Commun. 2004;322:1005–11. doi: 10.1016/j.bbrc.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 125.Li L, Cheung SH, Evans EL, Shaw PE. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res. 2010;70:8222–32. doi: 10.1158/0008-5472.CAN-10-0894. [DOI] [PubMed] [Google Scholar]
- 126.Ndubuisi MI, Guo GG, Fried VA, Etlinger JD, Sehgal PB. Cellular physiology of STAT3: Where’s the cytoplasmic monomer? J Biol Chem. 1999;274:25499–509. doi: 10.1074/jbc.274.36.25499. [DOI] [PubMed] [Google Scholar]