

Abstract
Signal transducers and activators of transcription 3 (STAT3) proteins are cytoplasmic transcription factors that translocate into the nucleus to induce transcription following growth factor or cytokine stimulation. Besides their normal functions, these proteins play an important role in cancer cells through the abnormal activation of cell cycle progression and the deregulation of survival and senescence pathways. New data obtained from the laboratory of Guido Kroemer identifies STAT3 as a new autophagy regulator. In the cytoplasm, in the absence of conventional phosphorylation on the tyrosine 705 residue, STAT3 interacts with the PKR kinase to inhibit eIF2A phosphorylation and so reduce autophagic pathways. This new and nonconventional function of STAT3 has an important role in normal cells but we suggest that it might also affect cancer cells and the response to chemotherapy treatment.
Keywords: STAT3, autophagy, cell death, oncogene, senescence
Whereas ubiquitination is well known to induce the degradation of soluble proteins, macroautophagy (also called autophagy) has recently emerged as an evolutionarily conserved process that degrades misfolded protein aggregates, damaged organelles, and abnormal mitochondria. This process relies on the activation of ATG genes and the upregulation of intracellular proteins that specifically recognize aggregates, target them to specific vesicles known as autophagosomes and allow lysosomal fusion.1 Protein degradation can be followed by catabolic recycling and therefore autophagy can be seen as a powerful mechanism that allows cell survival in a stressful environment such as the one cancer cells encounter.
The role of autophagy in cancer is complex. It has been demonstrated that the inactivation of Beclin-1 (the human ortholog of Atg6, which induces the formation of autophagosomes) is correlated with tumor development using mouse genetic models or human tumor samples. These observations described Beclin-1 as a haploinsufficient tumor suppressor gene, suggesting that autophagy functions as a suppressive pathway.2,3 These results were further confirmed by Atg5 and Atg7 deletions in mice, which also led to increased tumor initiation.4 This is probably related to the accumulation of the p62/SQSTM1 sequestosome protein, which is normally degraded during autophagy. The failure of autophagy-deficient cells to eliminate p62 leads to DNA damage and the abnormal activation of the Nrf2 and NFκB proteins.5-7 The upregulation of the NFκB transcription factor probably explains why autophagy default also leads to inflammation, as a consequence of the abnormal production of cytokines and chemokines mediated by NFκB. Interestingly, p62/SQSTM1 plays an important role in ras-mediated transformation and this is also correlated with the production of cytokines that are induced during oncogenic stress and senescence escape.8,9 This implies that some ras-expressing tumors might depend on p62/SQSTM1, whose expression can be viewed as a consequence of the autophagy downregulation and reduced tumor suppression that occur during the initial stage of cell transformation.
Important results have also demonstrated that autophagy is necessary for oncogene-induced senescence (OIS). OIS is a tumor-suppressor mechanism that induces permanent cell cycle arrest in response to oncogenic signals.10,11 It relies on the combined activation of the p53-p21waf1 and p16-Rb pathways to induce cell cycle arrest and the transcriptional repression of proliferative genes through heterochromatin formation. Young et al. have demonstrated that OIS induction is preceded by autophagy and the activation of ATG genes.12,13 In response to the ras oncogene, the inactivation of these genes by RNA interference reduced senescence as a consequence of autophagy inhibition. During the successive steps that lead to protective growth arrest in response to the ras oncogene, the Akt-mTOR pathway is inactivated and this inhibits phosphorylation of the Foxo3a transcription factor and the ATG proteins, ULK1, Atg13, and Beclin-1.14,15 The lack of Akt/mTOR-mediated phosphorylation of Foxo3 allows its nuclear translocation and the transcriptional activation of the ATG genes such as ULK1. In parallel, the lack of Akt/mTOR-mediated phosphorylation of ULK1, Atg13, and Beclin-1 promotes Ambra/Beclin/PI3KC3 association, which is essential for autophagosome formation. These results have been confirmed by a different study showing that ras leads to the upregulation of Beclin-1 and the consequent formation of autophagosomes.16 In these different experimental conditions, growth arrest was also inhibited following Beclin-1, Atg5, and Atg7 inactivation. Altogether, these observations illustrate the role of autophagy in the context of tumor suppression if we agree that senescence is always a suppressive pathway.17
As stated above, the role of autophagy in cancer is complex and important studies have demonstrated that its upregulation is necessary for tumor cell survival. Autophagy is expected to play an important role in the condition of hypoxic growth or nutrient privation. In this context, catabolic recycling may be a powerful mechanism that allows cell growth in a difficult cancer microenvironment. In line with this hypothesis, when performed in growing cell lines expressing the ras oncogene, the inactivation of ATG genes is associated with a significant decrease in cell survival.18 These observations do not necessarily contradict the role of autophagy as a tumor suppressor in the initial stages of cell transformation. Suppressive functions are generally characterized during acute ras signaling, in the early stage of oncogenic signaling. This is a different situation compared with established cell lines, which are growing with established ras mutations. These cells have by definition inactivated the suppressive mechanisms induced by the oncogene19,20 and are addicted to various survival, metabolic, and dedifferentiation pathways as we recently demonstrated in colorectal cancer cells.21 In these two different contexts, we can speculate that autophagy has different functions, suppressive in the early stages of cancer initiation and oncogenic in cells that have bypassed senescence and cell death protections.
In addition to many studies on ras signaling, the role of autophagy has also been characterized in other intracellular signaling pathways. Signal transducers and activators of transcription 3 (STAT3) proteins are latent cytoplasmic transcription factors that translocate into the nucleus to induce gene transcription.22 Binding growth factors to their receptors activates intracellular kinases such as Janus or Src, which then phosphorylate STAT3 on its 705 tyrosine residue (Y705). Although it was initially believed that dimerization and Y705 phosphorylation lead to the only active form of STAT3, it is now recognized that STAT3 can exert numerous nuclear or cytoplasmic functions in the absence of this phosphorylation.23,24 STAT3 plays an important role in tumorigenesis and its constitutive activation has been reported in several primary cancers and many oncogene-transformed cells.22,25-27 Importantly, we and others have also shown that this oncogene plays an important role in the response to chemotherapy treatment.28,29 Many studies have described the effect of STAT3 on cell transformation, apoptosis deregulation, and angiogenesis, using in vitro approaches, growth in two dimensions, in spheroid30 or in vivo mice models.31 As an oncogene, STAT3 is expected to deregulate suppressive pathways32 but the transcription factor’s effect on autophagy largely remains to be characterized. A study by Kroemer and colleagues has recently shown that the cytoplasmic form of STAT3 plays a key role in regulating this catabolic mechanism.33 Using a chemical library, they have observed that many drugs known to block the conventional activation of the transcription factor induce a significant activation of autophagy. Drugs such as Stattic, JSI-124, and WP1066 induce this mechanism and the effect was confirmed using either RNA interference or knockout cells. STAT3 inhibition leads to LC3+ dots, p62/SQSTM1 degradation, LC3 conversion, and autophagosome formation, which are all hallmarks of the autophagic program. In contrast, STAT3 overexpression induces a downregulation of this pathway and, most importantly, this was also observed when the authors used an Y705 mutated form of the transcription factor. Further suggesting a cytoplasmic role, no effect was noticed when the transcription factor was fused to a specific sequence that induces its constitutive nuclear localization. Interestingly, the authors identified several potential autophagy regulators such as HSP90, mTOR, and PKR kinase (EIF2AK2) within the STAT3 interactome. Through co-immunoprecipitation, mutagenesis, and molecular modeling, they show that STAT3 interacts with PKR in the cytoplasm and that this interaction is mediated by specific residues within its SH2 domain. In fact, it appears that this SH2 sequence is similar to a specific domain within the eIF2A translational regulator, one of the main targets of the PKR kinase. Consequently, the authors demonstrate that the PKR-eIF2A pathway is an important inducer of autophagy and that STAT3 inactivates this pathway through its binding to PKR and the inactivation of eIF2A phosphorylation. Interestingly, other inducers of autophagy such as palm oil are also regulated by the cytoplasmic form of the transcription factor and their effect on autophagy induction correlates with the dissociation of the STAT3-PKR complex. Other reports have proposed that STAT3 can regulate this pathway and as stated above we can speculate that this transcription factor regulates the transcription of ATG genes, perhaps in association with NFκB.23 The study by Kroemer and colleagues describes the protein’s nonconventional cytoplasmic role in the regulation of autophagy, further illustrating that STAT3 has functions in the absence of Y705 phosphorylation and does not always act as a classic transcription factor.
This new study concerns the normal functions of STAT3 and does not address the oncogene’s role in autophagy regulation. This is an important issue since this protein allows cell survival through the upregulation of proteins such as Bcl-Xl, mcl1, and survivin. In addition, it has recently been suggested that the cytoplasmic form of STAT3, not phosphorylated on its Y705 residue, plays an important role in cells expressing the ras oncogene.34 Considering the links between autophagy and cancer, it will be very interesting to determine the effect of the oncogenic form of STAT3 on autophagy. If we are convinced that its oncogenic form is phosphorylated on its 705 residue and dimeric, and that its main role is to activate cancer genes, then the nuclear form of STAT3 is not expected to interact with PKR. Consequently, tumor cells expressing this dimeric form should express an active PKR and show enhanced phosphorylation of eIF2-α. This would probably be the same if STAT3 interacts with NFκB to exert its oncogenic activity. In this case, the formation of the complex is also expected to prevent the interaction with PKR since its main function would be to regulate the transcription of genes involved in cancer progression. Interestingly, it has recently been demonstrated that NFκB plays an important role in the process of p27-mediated autophagy through the regulation of the skp2 protein.35,36 Whether this is related to its interaction with STAT3 and the consequent activation of PKR remains to be clarified. By allowing the cytoplasmic activation of the PKR kinase, these two oncogenic forms of STAT3 are expected to induce a normal activation of autophagy and therefore reduce the expression of p62/SQSTM1. Although this remains to be demonstrated, it can be suspected that some ATG genes are also targeted by the STAT3-NFκB complex. If this hypothesis is correct, it will be interesting to determine if autophagy is a protective or tumor-suppressor mechanism when tumor cells express a conventional tyrosine-phosphorylated form of STAT3.
However, if we believe that the oncogenic form of STAT3 is cytoplasmic and not phosphorylated on its Y705 residue, as recently shown in the case of ras-expressing cells, then we can speculate that STAT3 could in this case interact with PKR to block the induction of autophagy. In ras-expressing cells, it is striking to note that the cytoplasmic form of STAT3 has been described as a regulator of mitochondrial functions.34 Given the very close links between autophagy, mitochondrial functions, and apoptosis, these observations could lead to the hypothesis that the STAT3-mediated inhibition of PKR during autophagy might somehow be connected to its mitochondrial activity in ras-transformed cells. In addition, if a monomeric form of STAT3 that interacts with PKR is effectively oncogenic, we expect that the resulting reduced autophagy would lead to an enhanced expression of p62/SQSTM1. As stated above, this protein activates the NFκB pathway and a permanent autocrine loop induced by NFκB maintains STAT3 activation in tumor cells.37 In this context, the STAT3-mediated activation of p62/SQSTM1 would provide an additional means of reactivating NFκB and maintaining a secreted survival loop.
Given the very complex role of autophagy in cancer cells, we believe that it will be important to distinguish two cases in these further studies. During cancer initiation and senescence induction, the role of autophagy is probably not the same as in the functions in transformed cells that inactivate tumor suppressive pathways. Consequently, we also expect STAT3 phosphorylation, localization, and functions to vary during the successive stages of tumor progression. Therefore, further experiments are required to determine if STAT3 regulates autophagy to the same extent in primary cells, during senescence and in established cells that have escaped suppressive pathways (see Fig. 1 for a proposed hypothesis). The characterization of STAT3 in the different cell compartments based on its post-translational modifications and partners might be the key to understanding different STAT3 functions during the successive stages of cancer progression.
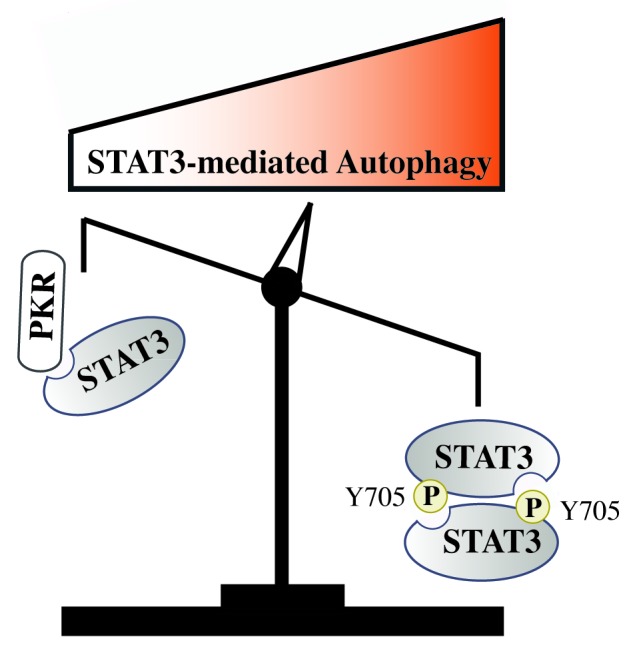
Figure 1. Schematic representation of autophagy regulation by STAT3 and proposed hypothesis. In normal conditions, latent cytoplasmic STAT3 binds to PKR, inhibits its activity, and reduces autophagy levels through eIF2A inhibition. Conventional STAT3 oncogenic activation relies on its 705 phosphorylation, dimerization, and consequent nuclear translocation. Further experiments are therefore needed to determine if the STAT3 dimer releases the PKR kinase, which would then become available to phosphorylate eIF2A and induce macroautophagy.
Finally, this new function of the STAT3 oncogene in autophagy might also play an important role in chemotherapy response. As stated above, we and others have shown that STAT transcription factors play an important role in the response to treatment, either through their expected role on survival or through nonconventional functions on DNA repair or DNA damage signaling.29,38-40 Since autophagy can also be considered an adaptation mechanism allowing chemotherapy escape, it will be interesting to determine if autophagy inhibitors can potentiate the effects of the various drugs developed to inactivate the STAT3 pathway.41 These drugs might also be useful to prevent the survival effect of this oncogene in response to conventional genotoxic treatments. These are important issues to address, given the problem of tumor resistance in the field of oncology.
Acknowledgments
Work in our laboratory is supported by grants from the Ligue Contre le Cancer (comité du Maine et Loire), the Rotary Club, the Canceropole Grand Ouest, the Pays de la Loire region, and the French National Research Agency (ANR).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/24353
References
- 1.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–10. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–4. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–54. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 9.Coppé JP, Patil CK, Rodier F, Krtolica A, Beauséjour CM, Parrinello S, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One. 2010;5:e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–7. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 12.White E, Lowe SW. Eating to exit: autophagy-enabled senescence revealed. Genes Dev. 2009;23:784–7. doi: 10.1101/gad.1795309. [DOI] [PubMed] [Google Scholar]
- 13.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338:956–9. doi: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 17.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Carné Trécesson S, Guillemin Y, Bélanger A, Bernard AC, Preisser L, Ravon E, et al. Escape from p21-mediated oncogene-induced senescence leads to cell dedifferentiation and dependence on anti-apoptotic Bcl-xL and MCL1 proteins. J Biol Chem. 2011;286:12825–38. doi: 10.1074/jbc.M110.186437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–42. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21:1396–408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–47. [PubMed] [Google Scholar]
- 25.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/S0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 26.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 28.Courapied S, Sellier H, de Carné Trécesson S, Vigneron A, Bernard AC, Gamelin E, et al. The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition. J Biol Chem. 2010;285:26765–78. doi: 10.1074/jbc.M109.092304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vigneron A, Gamelin E, Coqueret O. The EGFR-STAT3 oncogenic pathway up-regulates the Eme1 endonuclease to reduce DNA damage after topoisomerase I inhibition. Cancer Res. 2008;68:815–25. doi: 10.1158/0008-5472.CAN-07-5115. [DOI] [PubMed] [Google Scholar]
- 30.Leslie K, Gao SP, Berishaj M, Podsypanina K, Ho H, Ivashkiv L, et al. Differential interleukin-6/Stat3 signaling as a function of cellular context mediates Ras-induced transformation. Breast Cancer Res. 2010;12:R80. doi: 10.1186/bcr2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Barré B, Vigneron A, Perkins N, Roninson IB, Gamelin E, Coqueret O. The STAT3 oncogene as a predictive marker of drug resistance. Trends Mol Med. 2007;13:4–11. doi: 10.1016/j.molmed.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell. 2012;48:667–80. doi: 10.1016/j.molcel.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 34.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–6. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barré B, Coqueret O, Perkins ND. Regulation of activity and function of the p52 NF-κB subunit following DNA damage. Cell Cycle. 2010;9:4795–804. doi: 10.4161/cc.9.24.14245. [DOI] [PubMed] [Google Scholar]
- 36.Barré B, Perkins ND. The Skp2 promoter integrates signaling through the NF-kappaB, p53, and Akt/GSK3beta pathways to regulate autophagy and apoptosis. Mol Cell. 2010;38:524–38. doi: 10.1016/j.molcel.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 37.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Courapied S, Cherier J, Vigneron A, Troadec MB, Giraud S, Valo I, et al. Regulation of the Aurora-A gene following topoisomerase I inhibition: implication of the Myc transcription factor. Mol Cancer. 2010;9:205. doi: 10.1186/1476-4598-9-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barry SP, Townsend PA, Knight RA, Scarabelli TM, Latchman DS, Stephanou A. STAT3 modulates the DNA damage response pathway. Int J Exp Pathol. 2010;91:506–14. doi: 10.1111/j.1365-2613.2010.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Townsend PA, Cragg MS, Davidson SM, McCormick J, Barry S, Lawrence KM, et al. STAT-1 facilitates the ATM activated checkpoint pathway following DNA damage. J Cell Sci. 2005;118:1629–39. doi: 10.1242/jcs.01728. [DOI] [PubMed] [Google Scholar]
- 41.Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol. 2012;30:1005–14. doi: 10.1200/JCO.2010.31.8907. [DOI] [PMC free article] [PubMed] [Google Scholar]