

Abstract
The identification of a constitutively active JAK2 mutant, namely JAK2-V617F, was a milestone in the understanding of Philadelphia chromosome-negative myeloproliferative neoplasms. The JAK2-V617F mutation confers cytokine hypersensitivity, constitutive activation of the JAK-STAT pathway, and cytokine-independent growth. In this study we investigated the mechanism of JAK2-V617F-dependent signaling with a special focus on the activation of the MAPK pathway. We observed JAK2-V617F-dependent deregulated activation of the multi-site docking protein Gab1 as indicated by constitutive, PI3K-dependent membrane localization and tyrosine phosphorylation of Gab1. Furthermore, we demonstrate that PI3K signaling regulates MAPK activation in JAK2-V617F-positve cells. This cross-regulation of the MAPK pathway by PI3K affects JAK2-V617F-specific target gene induction, erythroid colony formation, and regulates proliferation of JAK2-V617F-positive patient cells in a synergistically manner.
Keywords: JAK2-V617F, JAK, Gab1, PI3K, MAPK, STAT, signal transduction, myeloid disorders, polycythemia vera, essential thrombocythemia
Introduction
Most cytokines signal through the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway. Depending on the cytokine or growth factor, the mitogen activated protein kinase (MAPK) cascade, the phosphatidyl-inositol-3-kinase (PI3K) cascade as well as the phospholipase C (PLC) pathway may be additionally activated. Maintaining the balance between STAT-dependent and STAT-independent pathways is very crucial to prevent severe inflammatory and neoplastic disorders.1 Constitutive cytokine signaling is often associated with inflammatory, autoimmune, or neoplastic diseases. The identification of a constitutively active mutant of JAK2, namely JAK2-V617F, in the year 20052-7 was a milestone in the understanding of Philadelphia chromosome-negative myeloproliferative neoplasms. The activating JAK2-V617F mutation is found in approximately 95% of polycythemia vera (PV) patients, in about 55% of patients with essential thrombocythemia (ET) or primary myelofibrosis (PMF) and rarely in other myeloid disorders.8,9 Myeloproliferative neoplasms (MPN) are clonal hematopoietic disorders arising either from a hematopoietic stem cell or in some cases at the level of a pluripotent hematopoietic progenitor cell. MPN are characterized by uni- or multi-lineage hematopoietic hyperplasia resulting in an overproduction of terminally differentiated myeloid cells such as erythrocytes, platelets, or granulocytes.10 The JAK2-V617F mutation confers cytokine hypersensitivity, constitutive activation of the JAK-STAT pathway, and cytokine-independent growth of so-called endogenous erythroid colonies (EEC) in the majority of patients with PV.5,11,12
Currently, only limited knowledge about the underlying molecular mechanisms as well as the resulting dysregulations associated with JAK2-V617F expression exists. Data derived both from patients and mouse models suggest misbalanced signaling of the mutated kinase. In addition, there is strong evidence for deregulated signaling to be dependent upon the expression level of JAK2-V617F.13 During the last decade, tyrosine kinase inhibitors (TKI) specifically targeting oncogenic kinases have emerged as highly effective and well tolerated anti-cancer drugs.14-18 Thus, mutant JAK2 and its downstream signaling components constitute attractive therapeutic targets for MPN patients.
Currently, multiple clinical trials employing JAK2 tyrosine kinase inhibitors for the treatment of MPN are under way. However, several problems with JAK2 TKI treatment are becoming more and more evident: JAK2 signaling is crucial for both normal hematopoiesis and neoplastic cells, leaving no therapeutic window to completely eradicate the malignant clone while saving normal hematopoiesis. In addition, due to the highly conserved tyrosine kinase domains within the JAK family most, if not all currently available JAK2 inhibitors also inhibit other JAK family members, thus leading to adverse effects such as immunosuppression.19-21 To develop more disease-specific treatment strategies, it is of great relevance to understand the mechanisms underlying the misbalanced signaling and pathophysiological action of JAK2-V617F. As a result, a combination of inhibitors against the dysregulated pathways could lead to a substantial improvement in the treatment of patients with MPN.
The regulatory signaling network is orchestrated by the cytokine receptors themselves as well as by adaptor proteins and multi-site docking proteins which coordinate and convey the balance of signaling processes. In this study, we focus on the MAPK activation in JAK2-V617F expressing cells and found deregulation of the multi-site docking protein Gab1. Gab1 is known to regulate the PI3K and the MAPK pathway.22,23 Both pathways play an important role in the proliferation and differentiation of hematopoietic cells.
Gab family proteins harbour binding sites for the regulatory subunit p85 of PI3K, the SH2 containing phosphatase 2 (SHP2), the RasGTPase activating protein (RasGAP), the phospholipase Cγ (PLCγ), the adaptor proteins Crk, CRKL, and Grb2.23-25 The timely cellular translocation of Gab proteins to the plasma membrane by binding PtdIns(3,4,5)P3 is vital for the physiological coordination of the involved signaling pathways. However, recent own studies indicate that the presence of PtdIns(3,4,5)P3 at the plasma membrane is not sufficient to recruit the Gab1 protein. Instead, PH domain-mediated recruitment of full length Gab1 to this membrane strongly depends on phosphorylation at serine 552 (S552) by ERK1/2.26 In the study presented here, we identified deregulated Gab1 activation and downstream STAT-independent signaling in JAK2-V617F-expressing cells. Finally, we demonstrate that PI3K signaling and MAPK activation in JAK2-V617F-expressing cells are connected and regulate JAK2-V617F-specific gene induction. Furthermore both pathways control erythroid colony formation and act synergistically on cell proliferation of JAK2-V617F-positive patient cells.
Results
JAK2 and the MAPK cascade act synergistically on the proliferation of JAK2-V617F positive patient cells
It is known that JAK2-V617F activates the JAK-STAT and the MAPK pathway, both of which are involved in the regulation of cell proliferation.5,27-30 In order to investigate whether inhibitors of the MAPK cascade influence the proliferation of JAK2-V617F-positive cells we incubated in vitro differentiated erythroid cells (amplified from CD34+ cells from JAK2-V617F-positive patients) with the MEK1 inhibitor U0126 and the JAK inhibitor 1 (Fig. 1A). JAK inhibitor 1 (2 µM) strongly suppressed cell proliferation. U0126 suppressed cell proliferation already at 5 μM. Inhibition was even more pronounced at higher concentrations (10 and 20 µM) of U0126. Additionally, we investigated whether signaling through JAK and MEK/ERK exerts any synergistic, additive or antagonistic activity on JAK2-V617F-positive primary cell growth. Thus, we performed growth assays in the presence of both JAK inhibitor 1 and U0126 (ratio of concentrations was kept constant at 4:1). Dose-effect analyses of the drug combinations were performed according to Chou and Talalay using the CompuSyn software.31-34 All combination index (CI) values measured for cells from three different patients varied between 0.257 and 0.618 indicating strong synergistic to moderate synergistic effects (see also Materials and Methods) (Fig. 1B for one of three patients). These results indicate that in JAK2-V617F-expressing primary cells activity of JAK2 and of the MAPK cascade cooperate to drive proliferation.

Figure 1. JAK2-V617F-specific cell proliferation and gene induction. (A) In vitro differentiated erythroid cells (from CD34+ cells of three JAK2-V617F positive patients) were subjected to proliferation assays and were treated with JAK inhibitor 1 (JI1, 2 μM) and different concentrations of U0126 (5, 10, and 20 μM) in 96 well plates. After 72 h 10 μl of WST-1 reagent was added and the relative proliferation of cells was assessed according to the manufacturer’s instructions using an absorbance reader. (B) In vitro differentiated erythroid cells were treated with JAK inhibitor 1 (JI1), U0126 (U0) or a combination thereof (JI1 + U0, ratio of concentrations was kept constant at 4:1). Proliferation was investigated as described in (A) and a dose-effect analysis of the drug combination was performed using the CompuSyn software. Combination index (CI) values lower than 0.9 indicate synergistic effects. (C) HEK JAK2 and HEK JAK2-V617F cells were cultivated for 11 h in the absence (−) or presence (+) of doxycycline (dox) (1 ng/ml) to induce JAK2 and JAK2-V617F gene expression. Cells were lysed and whole cell lysates were separated by SDS-PAGE and analyzed by western blot immunodetection for expression of JAK2 and STAT3 to control loading of the gel. (D) Cells were treated as for (C). Where indicated 10 µM U0126 was added for the last 6.5 h of cultivation (lower panels). Gene induction of MMP-10 and Serpin B2 was detected by qRT-PCR
JAK2-V617F-specific gene induction
To identify MAPK-dependent mechanisms involved in JAK2-V617F-mediated deregulation of cell signaling and proliferation we screened for JAK2-V617F-specific MAPK-dependent gene induction. For this aim we utilized HEK293 cells expressing JAK2 wild-type or JAK2-V617F under control of a doxycycline-inducible promoter.27 Doxycycline-dependent expression of JAK2 and JAK2-V617F is shown in Figure 1C. First, we investigated JAK2-V617F-specific gene induction by gene array analyses in the absence and presence of the MEK inhibitor U0126 (data not shown). These analyses identified the matrix metalloprotease (MMP)-10 and the serine protease inhibitor Serpin B2 as JAK2-V617F-inducible and MAPK-dependent genes. Serpins and MMPs are involved in fibrin and matrix remodeling, as well as in blood clotting. Plasminogen activator inhibitors [PAI-1 and PAI-2 (also known as Serpin B2)] inhibit plasminogen activation, fibrinolysis and ECM degradation. The activity or upregulation of PAIs is correlated with bleeding or thrombosis in patients with PV, ET, or other MPNs who often suffer from hemostasis disorders as a result of disordered fibrinolytic activity.35-37 Increased Serpin B2 levels were also reported in atypical myeloproliferative diseases.38 Importantly, Serpin B2 exerts cytoprotective activities by inhibiting retinoblastoma protein degradation and inhibition of procaspase 3 activation.39,40 Matrix metalloproteases (MMPs) have been described to be involved in bone marrow remodelling in myelofibrosis, PV, and ET41,42 and are also associated with the mobilization of CD34+ cells to peripheral blood in myelofibrosis.43 Although overexpression of MMP-10 (also known as Stromelysin-2) is mainly associated with solid tumors, it has recently been found upregulated in T lymphoma.44 As MMP-10 is induced by JAK-utilizing cytokines such as IL-4, IL-6, and IL-1344 a JAK2-V617F-dependent MMP-10 gene induction as shown here is likely to be relevant.
Induction of MMP-10 and Serpin B2 genes in response to JAK2-V617F expression was confirmed by RT-qPCR (Fig. 1D, upper panels). In clear contrast, expression of JAK2 wild-type did not induce MMP-10 or Serpin B2 gene induction arguing for JAK2-V617F-mediated gene expression. MAPK-dependent induction of MMP-10 and Serpin B2 was confirmed by the observation that both genes were not induced in the presence of U0126 (Fig. 1D, lower panels).
JAK2-V617F-specific activation of signaling components
We speculated that the molecular basis for JAK2-V617F-specific gene induction may be reflected by activation of specific signaling pathways. Thus, we first monitored activation/phosphorylation of ERK, STAT3, STAT5, and Gab1 in HEK cells expressing JAK2 or JAK2-V617F (Fig. 2A). Phosphorylation of these proteins was only detectable in cells expressing JAK2-V617F but not in cells expressing JAK2 or in the absence of doxycycline. Importantly, activation of these signaling components could be blocked by the JAK inhibitor 1, further supporting the idea that activation of STAT proteins and of the Gab1/ERK pathway in JAK2-V617F-expressing cells is due to the enzymatic activity of JAK2-V617F. To further confirm these findings in the model system, we additionally monitored activation of the observed proteins in patient-derived human erythroleukemia (HEL) cells, which express JAK2-V617F endogenously (Fig. 2B). Similar to JAK2-V617F expressing HEK cells, phosphorylation of Gab1, ERK, STAT3, and STAT5 was detected and could be prevented by JAK inhibitor 1 in HEL cells. These observations recapitulate that JAK2-V617F activates STAT transcription factors as well as the MAPK cascade. Furthermore, we demonstrate for the first time that Gab1 is constitutively phosphorylated in JAK2-V617F-expressing cells. Gab proteins contribute to hematopoiesis and coordinate MAPK and PI3K signaling in response to many cytokines and growth factors. Both pathways are frequently deregulated in cancer cells.45 Furthermore cancer-associated mutations in Gab1 have recently been identified.46 Thus, the potential involvement of Gab1 in the pathways activated by JAK2-V617F was further investigated.

Figure 2. JAK2-V617F-specific activation of signaling components. (A) HEK JAK2 and HEK JAK2-V617F cells were cultivated for 12 h in the absence or presence of doxycycline (1 ng/ml) to induce JAK2 and JAK2-V617F gene expression. Where indicated 500 nM JAK inhibitor 1 was added to the cells for 4 h. Subsequently, cells were lysed and whole cell lysates were separated by SDS-PAGE and analyzed by western blot immunodetection for phosphorylated Gab1, ERK, STAT3, and STAT5. After stripping the membranes were stained with antibodies against JAK2 to monitor JAK2 gene induction and with antibodies against Gab1, ERK, STAT3, and STAT5 to control for equal loading of the gel. (B) HEL cells were treated with 500 nM JAK inhibitor 1 for 4 h or left untreated. Subsequently, cells were lysed and whole cell lysates were separated by SDS-PAGE and analyzed by western blot immunodetection for phosphorylated Gab1, ERK, STAT3, and STAT5. After stripping the membranes were detected with antibodies against Gab1, ERK, STAT3, and STAT5 to control for equal loading of the gel.
JAK2-V617F affects the cellular distribution of Gab1
In response to growth factor or cytokine stimulation Gab1 translocates from the cytoplasm to the plasma membrane to coordinate signal transduction. As we detected constitutive phosphorylation of Gab1 in JAK2-V617F-expressing cells, we tested whether JAK2-V617F expression also affects the cellular distribution of Gab1. Gab1-GFP was expressed in HEK JAK2 or HEK JAK2-V617F cells. Figure 3 shows Gab1 residing in the cytoplasm in HEK JAK2 cells in the absence and presence of doxycycline arguing that induction of wild-type JAK2 expression does not influence the cellular distribution of Gab1. However, the expression of JAK2-V617F results in the recruitment of Gab1 to the plasma membrane. These observations indicate that JAK2-V617F leads to deregulated cellular distribution of the multi-site docking protein Gab1.
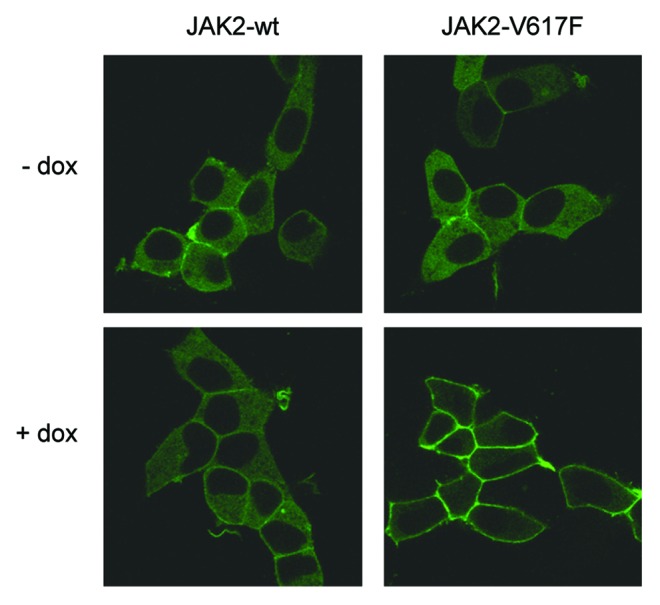
Figure 3. JAK2-V617F induces membrane localization of Gab1. HEK JAK2/Gab1-GFP and HEK JAK2-V617F/Gab1-GFP cells were seeded and cultivated on poly-l-lysine-coated glass bottom dishes. On the following day the medium was replaced by phenol red-free DMEM/10% FCS and cells were left for 24 h in the incubator. Cells were left untreated or treated with doxycycline (2.5 ng/ml) for 12 h to induce JAK2 and JAK2-V617F expression, respectively. Cells were transferred into the incubation chamber of a laser scanning microscope and left there for 30 min before confocal imaging.
JAK2-V617F-dependent membrane recruitment of Gab1 requires sustained MAPK activity
In a previous study we demonstrated that cytokine-induced membrane recruitment of Gab1 requires MAPK-dependent phosphorylation of serine residues 552 and 568 of Gab126 which releases a block of the PH-domain (Wolf et al., unpublished data). Here, we tested whether membrane recruitment of Gab1 initiated by JAK2-V617F expression is accomplished by the same molecular mechanisms. HEK cells expressing JAK2-V617F were treated with the MEK inhibitor U0126 for 2.5 h or with solvent alone. Figure 4A shows membrane binding of Gab1-GFP in JAK2-V617F-expressing HEK cells in the absence of U0126. However, administration of U0126 almost completely induces re-localization of Gab1 to the cytoplasm. In line with these results, Gab1 S552, 568A mutants are not found at the plasma membrane in cells expressing JAK2-V617F, whereas Gab1 S552, 568E mutants, mimicking phosphorylation of both serine residues, are located at the membrane independently of JAK2-V617F expression (Fig. 4B). Therefore, we conclude that JAK2-V617F-dependent persistent membrane binding of Gab1 is regulated by continuous MAPK activity and phosphorylation of serine residues 552 and 568 of Gab1.

Figure 4. JAK2-V617F-dependent membrane binding of Gab1 is MAPK dependent. (A) HEK JAK2-V617F/Gab1-GFP cells were seeded and cultivated on poly-l-lysine-coated glass bottom dishes. On the following day the medium was replaced by phenol red-free DMEM/10% FCS and cells were left for 24 h in the incubator. Cells were treated with doxycycline (2.5 ng/ml) for 9 h to induce JAK2-V617F expression. Cells were transferred into the incubation chamber of a laser scanning microscope and left there for 30 min. Subsequently, U0126 (20 µM) or solvent alone (DMSO) was added for additional 2.5 h in the presence of doxycycline. Imaging was performed before (−) and after (+) treatment with the inhibitor or solvent. (B) HEK JAK2/Gab1-GFP, HEK JAK2/Gab1-S552A-S568A, HEK JAK2/Gab1-S552E-568E, HEK JAK2-V617F/Gab1-GFP, HEK JAK2-V617F/Gab1-S552A-S568A, and HEK JAK2-V617F/Gab1-S552E-S568E cells were seeded and cultivated on poly-l-lysine-coated glass bottom dishes. On the following day the medium was replaced by phenol red-free DMEM/10% FCS and cells were left for 24 h in the incubator. Cells were treated with doxycycline (2.5 ng/ml) for 12 h to induce JAK2 and JAK2-V617F expression, respectively. Cells were transferred into the incubation chamber of a laser scanning microscope and left for 30 min before confocal imaging.
JAK2-V617F-dependent membrane recruitment of Gab1 requires the PH domain within Gab1 and sustained PI3K activity
Cytokine-induced membrane recruitment of Gab1 is facilitated through the N-terminal PH domain of Gab1. However, the isolated PH domain of Gab1 was found at the plasma membrane also in the absence of cytokines26 due to the lack of PH domain-blocking regions found in Gab1 (Wolf et al., unpublished data). Here we tested the cellular distribution of Gab1-ΔPH, lacking the PH domain and Gab1-PH (the isolated Gab1 PH domain) in the presence or absence of JAK2-V617F. Figure 5A demonstrates that JAK2-V617F expression had no effect on the cellular distribution of Gab1-ΔPH or Gab1-PH. This demonstrates that the PH domain is necessary for correct Gab1 localization and that JAK2-V617F signaling does not change Gab1 localization by directly affecting this domain.

Figure 5. JAK2-V617F-dependent membrane binding of Gab1 is PI3K dependent. (A) HEK JAK2-V617F/Gab1-GFP, HEK JAK2-V617F/Gab1-ΔPH-GFP, and HEK JAK2-V617F/Gab1-PH-GFP cells were seeded and cultivated on poly-l-lysine-coated glass bottom dishes. On the following day the medium was replaced by phenol red-free DMEM/10% FCS and cells were left for 24 h in the incubator. Cells were left untreated or treated with doxycycline (2.5 ng/ml) for 12 h, transferred and subjected to confocal imaging as described in Figure 4B. (B) HEK JAK2-V617F/Gab1-GFP cells were cultivated, induced with doxycycline (2.5 ng/ml) and transferred into the incubation chamber as described in (A) and Figure 4B. Subsequently, LY294002 (40 µM, 10 min), Wortmannin (100 nM, 30 min), or solvent (10 min) was added. Imaging was performed before (-) and after (+) treatment with the inhibitors or solvent.
PH domains are known to bind PtdIns(3,4,5)P3 at the plasma membrane. Therefore, we asked whether JAK2-V617F-dependent Gab1 membrane binding could be blocked by inhibitors of the PI3K. Figure 5B demonstrates that in JAK2-V617F expressing cells Gab1 moves from the plasma membrane back to the cytoplasm after addition of Wortmannin or LY294002. These data show that persistent membrane recruitment of Gab1 in response to JAK2-V617F expression requires its PH domain and sustained PI3K activity.
PI3K affects JAK2-V617F-dependent Gab1 phosphorylation and MAPK activation
Since PI3K inhibitors affected the JAK2-V617F-dependent cellular distribution of Gab1, we asked whether they also influence signaling pathways coordinated by Gab1 in JAK2- or JAK2-V617F-expressing cells. We analyzed Gab1, ERK, and also STAT3 phosphorylation in HEK cells expressing JAK2-V617F or JAK2 (Fig. 6A). As already shown in Figure 2A, JAK2-V617F expression induced phosphorylation of Gab1, ERK, and STAT3, whereas expression of wild-type JAK2 did not induce phosphorylation of these proteins. Interestingly, the PI3K inhibitor Wortmannin reduced phosphorylation of Gab1 and ERK in JAK2-V617F-expressing cells while maintaining the activation of the STAT3 pathway (Fig. 6A). Similar observations were made in the HEL cells (expressing JAK2-V617F endogenously) (Fig. 6B): Treatment of the cells with JAK inhibitor 1 demonstrated STAT3, STAT5, Gab1, and ERK activation/phosphorylation depending on JAK2-V617F enzymatic activity. Both the PI3K inhibitor Wortmannin and the MAPK inhibitor U0126 reduced persistent phosphorylation of Gab1 and ERK, whereas STAT activation was not affected in HEL cells. These data identified PI3K and MAPK activity to be crucial for persistent Gab1 tyrosine phosphorylation in JAK2-V617F-expressing cells. Interestingly, inhibition of the PI3K pathway also inhibited persistent MAPK activation in HEK JAK2-V617F cells. These data demonstrate that JAK2-V617F-mediated MAPK activation is regulated by the PI3K pathway.

Figure 6. PI3K-dependent activation of MAPK and Gab1 phosphorylation in JAK2-V617F expressing cells. (A) HEK JAK2 and HEK JAK2-V617F cells were cultivated for 12 h in the absence or presence of doxycycline (1 ng/ml) to induce JAK2 and JAK2-V617F gene expression. Where indicated, Wortmannin (100 nM) was added to the cells for 20 min. Subsequently, cells were lysed, whole cell lysates were separated by SDS-PAGE and transferred to a membrane by western blot immunodetection. The blots were detected for phosphorylated Gab1, ERK, STAT3, and Akt. After stripping the membranes were stained with antibodies against JAK2, Gab1, ERK, STAT3, and Akt to control for equal loading of the gel. (B) HEL cells were treated with 500 nM JAK inhibitor 1 for 4 h, with U0126 (10 µM) for 20 min, with Wortmannin (100 nM) for 20 min or left untreated. Western blots were generated and detected for the phosphorylated and non-phosphorylated forms of Gab1, ERK, STAT3, STAT5 and p70 as described in (A).
PI3K activity is involved in JAK2-V617F-induced, MAPK-dependent gene induction of MMP-10 and Serpin B2
Since PI3K inhibitors affected JAK2-V617F-dependent MAPK activation we asked whether this cross-regulation has an impact on JAK2-V617F-specific gene induction of MMP-10 and Serpin B2 demonstrated in Figure 1D. Thus, we compared JAK2-V617F-dependent gene induction in the absence or presence of JAK inhibitor 1, U0126, or Wortmannin in HEK JAK2-V617F cells (Fig. 7A). Expression of MMP-10 and Serpin B2 was only observed after induction of JAK2-V617F with doxycycline. JAK inhibitor 1 as well as U0126 completely blocked gene expression of both JAK2-V617F target genes. Interestingly and in line with the biochemical analyses above, Wortmannin diminished JAK2-V617F-dependent induction of MMP-10 and Serpin B2 to a similar extent as Wortmannin reduced ERK-phosphorylation in Figure 6. These results underline the importance of PI3K for the induction of MAPK-dependent JAK2-V617F-specific genes.

Figure 7. Regulation of JAK2-V617F-specific gene induction and erythroid colony formation by MAPK and PI3K pathways. (A) HEK JAK2-V617F cells were cultivated for 11 h in the absence or presence of doxycyline (1 ng/ml) to induce JAK2-V617F gene expression. JAK inhibitor 1 (JI1, 500 nM) was added together with doxycycline, U0126 (U0, 10 µM) or Wortmannin (100 nM) were added for the last 6.5 h of cultivation. Gene induction MMP10 and SerpinB2 was detected by quantitative real time PCR. (B) CD34+ cells (isolated from JAK2-V617F positive MPN patients) were subjected to CFC assays (see Materials and Methods) in the presence or absence of the JAK inhibitor 1 (JI1, 2 μM), the MEK1 inhibitor U0126 (U0, 10 μM), or the Akt inhibitor MK2206 (MK, 2 μM). The erythroid colonies were counted using an inverted microscope after 12 to 16 d of culture. The amount of colonies grown from inhibitor-treated cells was calculated as percentage of maximum number of colonies ( = untreated control).
JAK2-V617F-dependent erythroid colony formation depends on PI3K and MAPK activity
To study the relevance of MAPK and PI3K pathway activation for blood cell differentiation in JAK2-V617F-expressing cells, we compared erythroid colony formation in the absence or presence of JAK, PI3K or MEK inhibitors. Hematopoietic progenitor cells (CD34+) derived from two JAK2-V617F-positive MPN patients were cultured in the absence or presence of inhibitors of the JAK2, PI3K/Akt, and MAPK pathway (JAK inhibitor 1, MK2206, and U0126, respectively). MK2206 (an Akt inhibitor) had to substitute for Wortmannin because of its longer half-life in aqueous solution, appropriate for long-term experiments. MK2206 showed similar potency in suppressing Akt phosphorylation when compared with Wortmannin (data not shown). As shown in Figure 7B erythroid colony formation was reduced by all three inhibitors demonstrating that all three pathways act together in driving red blood cell differentiation.
In summary, all these data support the hypothesis that deregulated cell proliferation of JAK2-V617F-expressing cells is strongly influenced by MAPK and PI3K activity.
Inhibition of PI3K/Akt and the MAPK cascade affect proliferation of JAK2-V617F-positive patient cells synergistically
Due to the regulation of MAPK by the PI3K pathway in JAK2-V617F-expressing cells we sought to investigate whether these two pathways act synergistically on the proliferation of in vitro differentiated erythroid cells (amplified from CD34+ cells of JAK2-V617F-positive MPN patients). We incubated the cells with MK2206, U0126, or a combination thereof (the ratio of concentrations was kept constant at 3:1). A dose-effect analysis was performed as for Figure 1A. All CI values measured for cells from three different patients varied between 0.110 and 0.610 indicating strong synergistic to moderate synergistic effects (see also Materials and Methods) (Fig. 8 shows representative results of one of three patients). These observations indicate that proliferation of JAK2-V617F-positive primary cells is induced by synergistic cooperation of the PI3K and MAPK pathways.

Figure 8. The MEK/Erk1/2- and PI3K/Akt-pathway inhibitors U0126 and MK2206 act synergistically to supress growth of cells isolated from JAK2-V617F-positive patients. In vitro differentiated erythroid cells (from CD34+ cells from three JAK2-V617F positive patients) were subjected to proliferation assays and were treated with the MEK inhibitor U0126, the Akt inhibitor MK2206 or a combination thereof (ratio of concentrations was kept constant at 3:1) (The concentration in μM is indicated in brackets). After 72 h 10 μl of WST-1 reagent was added and the relative proliferation of cells was assessed according to the manufacturer’s instructions using an absorbance reader. A dose-effect analysis of the drug combination was performed using the CompuSyn software. Combination index (CI) values lower than 0.9 indicate a synergistic effect of the drug combination.
Discussion
Most therapeutic strategies based on interfering with signaling cascades rely on the classical linear concept of signal transduction pathways. JAK inhibitors have been described to have beneficial effects in clinical trials by alleviating constitutional symptoms observed in MPN patients47,48 while there is no evidence that conventional treatment [by cytoreductive drugs (e.g., hydroxyurea, IFNα), chemotherapeutic agents or radiation] improves the constitutional symptoms.49 Unfortunately, treatment of myelofibrosis patients with ruxolitinib (the first FDA-approved JAK inhibitor) neither leads to the reduction of bone marrow fibrosis or JAK2-V617F allele burden nor does it improve overall survival compared with the best available conventional therapy. Instead, the treatment with JAK inhibitors commonly induces adverse effects as (severe) anemia and thrombocytopenia probably resulting from the inhibition of downstream signaling of cytokine receptors employing JAK1 or JAK2. Therefore, the elucidation of underlying molecular mechanisms including the interplay between the JAK-STAT signaling pathway with other signaling pathways or other players to identify new target molecules remains a major mission of research in the field of MPN. Consequently, a detailed analysis of the signaling events downstream of JAK2-V617F is mandatory.
In the study presented here, we demonstrate that the PI3K pathway regulates JAK2-V617F-dependent MAPK activation (Fig. 6) and MAPK-dependent gene induction in JAK2-V617F-positive cells (Fig. 7). Furthermore, we show deregulated phosphorylation (Fig. 2) and cellular translocation of Gab1 (Fig. 3) which is known to regulate MAPK activation in response to cytokines PI3K-dependently.23,50 Finally, we demonstrate synergistic action of the PI3K and MAPK pathway on the proliferation of JAK2-V617F-positive patient cells (Fig. 8). These new insights could help to develop more specific, combinatorial therapeutic approaches to treat MPN patients.
Development of therapies concomitantly inhibiting JAK2 and ideally synergistically other important cellular regulators might contribute to reach this goal. Combining specific kinase inhibitors with JAK inhibitors has beneficial effects on growth inhibition of JAK2-V617F-expressing cells. Aurora kinase inhibitors,51 PI3K/Akt/mTOR pathway inhibitors,52-54 or MEK inhibitors55 (Fig. 1) act synergistically with JAK inhibitors on the proliferation of JAK2-V617F-positive cells. Additionally, other compounds e.g., histone deacetylase (HDACs) inhibitors,56,57 demethylating agents,58 or HSP90 inhibitors59,60 have been tested for their potential therapeutic activity in MPN. However, here we present data which indicate that JAK2-V617F-dependent MAPK and PI3K activation are connected. MAPK and PI3K signaling pathways are coupled through Gab1 and thus represent novel potential targets for interfering with JAK2-V617F-dependent pathological cell proliferation. Interestingly, not only simultaneous inhibition of JAK and MEK1 (Fig. 1) but also the combination of PI3K/Akt/mTOR pathway inhibitors with a MEK inhibitor synergistically suppresses proliferation of JAK2-V617F-expressing cells from MPN patients (Fig. 8). These data suggest that also a specific inhibition of STAT-independent pathways and their mutual regulation need to be considered to design novel therapeutic strategies to cope with JAK2-V617F-dependent neoplasia.
Our study demands to consider crosstalk mechanisms as potential new targets. This may increase the specificity for intervention approaches compared with the interference in very upstream signaling elements such as receptor kinases or receptor-associated kinases. Furthermore, combined targeting of downstream elements which act synergistically may increase the efficiency for the treatment, compared with single inhibitor approaches. The study presented here elucidates the crosstalk of the MAPK and PI3K cascade both initiated in JAK2-V617F-expressing cells.
Materials and Methods
Materials
All compounds were dissolved in DMSO at a concentration of 10 mM. MK2206 was from Selleck chemicals and JAK inhibitor 1 (JI1), U0126, LY294002, and Wortmannin were from Calbiochem (Merck Biosciences).
Cells
The HEK Flp-In stable transfectants inducibly expressing JAK2 (wild-type) and JAK2-V617F were described previously27 and maintained in Dulbecco’s modified Eagle’s medium (DMEM, Lonza), supplemented with 10% fetal calf serum (FCS), 100 mg/l streptomycin and 60 mg/l penicillin. The HEL (human erythroleukemia) cell line was purchased at DSMZ and maintained in RPMI-1640 medium (Lonza) supplemented with 10% FCS, 100 mg/l streptomycin, 60 mg/l penicillin, and 2 mM l-glutamine.
Peripheral blood mononuclear cells (PBMC) from JAK2-V617F-positive MPN patients were isolated by a Ficoll-Paque PLUS (GE Healthcare) gradient centrifugation according to the manufacturer’s instructions. CD34+ cells were purified using the CD34 MicroBead Kit on LS columns and a QuadroMACS Separator (all from Miltenyi Biotec) according to the manufacturer’s protocol.
Prior to use in proliferation assays CD34+ cells were amplified by cultivation in StemSpan H3000 medium (StemCell Technologies) supplemented with 20% serum substitute (BIT9500, StemCell Technologies) and 100 ng/ml FLT3-L, Tpo, and SCF at a density of 3 × 105 to 1 × 106 cells/ml. To achieve differentiation into the erythroid lineage, the cells were subsequently cultured in the presence of 50 ng/ml SCF, 50 ng/ml IGF-1, and 3U/ml Epo at a density of 5 × 105 to 1 × 106 cells/ml.61-63
Microarray analysis
HEK JAK2 and HEK JAK2-V617F cells were cultivated in the absence or presence of doxycycline (1 ng/ml) for 11 h to induce JAK2 or JAK2-V617F expression. Total RNA was isolated using the RNeasy Kit (Qiagen) according to manufacturer’s instructions. RNA quality was assessed using the RNA 6000 Nano Assay (Agilent 2100 Bioanalyzer, Agilent) and RNA quantity was assessed using the NanoDrop 1000. Total RNA (200 ng, RNA integrity number of 10) was reverse-transcribed and amplified using the Applause WT-Amp ST System as described in the manufacturer’s manual (NuGEN Technologies). Subsequently, sense strand cDNA fragmentation and labeling was performed according to the Encore™ Biotin Module (NuGEN Technologies). The fragmented labeled sample was hybridized to an Affymetrix GeneChip (Affymetrix) Human Gene 1.0 ST Array (36 079 ref. seq. transcripts covered). Gene expression levels were pre-processed with the robust multi-array average algorithm.64 Only probe sets that have a mean detection above background, a P value less than 0.05, and a mean expression value greater than 20 in at least one treatment condition were considered for analysis. Transcripts with a fold induction more than 2 and P value less than 0.01 after doxycyclin treatment were addressed as inducible genes.
RT-qPCR
Total RNA was isolated using the RNeasy Kit (Qiagen) according to manufacturer’s instructions. One µg of RNA was reverse-transcribed into cDNA with Omniscript (Qiagen) using random hexameric primers according to manufacturer’s instructions. Taqman gene expression assays for human MMP-10 (Hs00233987_m1), human Serpin B2 (Hs01010736_m1) and human HPRT (Hs99999909_m1) were obtained from Applied Biosystems and PCR was performed using qPCR Mastermix plus (Eurogentec). The PCR was performed in a final volume of 20 µl containing 2 µl cDNA and 1 µl Taqman gene expression assay solution. Following a 15 min denaturing step at 94 °C amplification was performed in 40 cycles (15 sec at 94 °C, 60 sec at 60 °C, 30 sec at 70 °C) on a Rotorgene (Qiagen). The gene of interest and the reference gene were amplified in duplicates. The quantification of gene expression was calculated using the ∆∆Ct method.65 The figure shows the average of three biological replicates ± SD. Maximal gene induction was set 100.
Confocal live cell microscopy
HEK JAK2 and HEK JAK2-V617F cells stably transfected with the indicated Gab1-eGFP constructs26 were seeded on poly-l-lysine-coated glass bottom dishes (5160-168 Imaging Dish 1.0, MoBi Tec). Twenty-four hours later the culture medium was exchanged to DMEM without phenol red and 10% FCS. On the next day dishes were transferred into the pre-heated chamber of the microscope. Live cell imaging was performed with a confocal laser-scanning microscope (LSM700, Zeiss). The temperature of the chamber and of the microscope’s objective was adjusted to 37 °C; the atmosphere was set to 5% CO2. Cells were treated as indicated in the figure legends. Before detection, the cells were incubated for 30 min in the incubation chamber of the confocal microscope. The eGFP-fusion proteins were excited using laser light with a wavelength of 488 nm. Emission was detected in the range of 493–700 nm.
Cell lysis and western blot analysis
For the isolation of cellular proteins, confluent HEK cell cultures were lysed in 300 μl RIPA-lysis buffer (50 mM TRIS-HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40, 15% glycerol, 1 mM NaF, and 1 mM Na3VO4) supplemented with 10 μg/ml of each aprotinin, pepstatin (Sigma-Aldrich) and leupeptin (MP Biochemicals) as well as 0.8 μM Pefabloc (Carl Roth). HEL cells were lysed directly in 1× Laemmli buffer. Proteins were separated by SDS-PAGE and transferred to a polyvinylidene-difluoride (PVDF) membrane (PALL). After blocking the membrane, antigens were detected by incubation with the specific primary antibodies as indicated (1:1000) and horseradish-peroxidase (HRP)-coupled secondary antibodies (1:7500). The membranes were incubated with an Immobilon Western Chemiluminescent HRP Substrate (Millipore Corporation). The phospho-STAT3 (9145), phospho-ERK1/2 (7383), phospho-Gab1 (3233), phospho-JAK2 (3776), JAK2 (3230), p70 (2708), and phospho-p70 (9234) antibodies were purchased from Cell Signaling Technology. The STAT5 (835), STAT3 (482), ERK1 (94, 93-G), and ERK2 (154-G) antibodies were obtained from Santa Cruz Biotechnology. The phospho-STAT5 (611964) antibody was from BD Biosciences, while the Gab1 antibody (06-579) was from Upstate Biotechnology. The horseradish peroxidase-conjugated secondary antibodies were purchased from Southern Biotech. Luminescent signals were detected using an ECL solution as described before.66
Cell proliferation assay
The WST-1 Cell Proliferation Assay (Roche) was used according to the provider’s protocol. Shortly, 1–3 × 104 of the in vitro differentiated erythroid cells were plated in 100 μl growth medium (StemSpan H3000 medium with 20% serum substitute BIT9500 and 20 ng/ml Epo and Tpo) with the indicated concentrations of inhibitor into wells of microtiter plates. After 72 h, 10 μl of WST-1 reagent was added per well and the cells were incubated for 30 min to 4 h at 37 °C. The relative proliferation of cells was assessed according to the manufacturer’s instructions using an absorbance reader (Tecan). At least three replicates with cells isolated from three patients were performed.
Colony forming cell (CFC) assay
CD34+ cells (500 cells per 35 mm dish) were seeded in methylcellulose medium MethoCult® H4230 (StemCell Technologies) supplemented with 1 U/ml Epo (Calbiochem, Merck4Biosciences), 10 ng/ml IL-3, 10 ng/ml G-CSF and 10 ng/ml GM-CSF (all from PeproTech) according to the manufacturer’s instructions. Cells were incubated with inhibitors at indicated concentrations or left untreated. The erythroid colony numbers were counted using an inverted microscope after 12 to 16 d of culture. The amount of colonies grown from inhibitor-treated cells was calculated as percentage of the maximum number of colonies ( = untreated control). Three replicates with cells isolated from three patients were performed.
Analysis of drug combinations according to the Chou–Talalay method
The inhibitory effect on proliferation by two single drugs and their combination was assessed using the WST-1 proliferation assay (see above). The compounds were applied in various concentrations while keeping the concentration ratio between the two inhibitors constant (for details see figure legends). A dose-effect analysis of the drug combination was performed using the CompuSyn software31-33 according to the Chou–Talalay method.34 The Combination Index (CI) value is the quantitative measure of the dose-effect analysis of a drug combination. CI values < 1 represent synergism, CI > 1 corresponds to antagonism and CI ≈1 represents an additive effect for the drug combination. Within the synergistic range the following classification is used. CI < 01: very strong synergism; 0.1 < CI < 0.3: strong synergism; 0.3 < CI < 0.7: synergism; 0.7 < CI < 0.85: moderate synergism; 0.85 < CI < 0.9: slight synergism. Three replicates with cells isolated from three patients were performed.
Acknowledgments
This project has been funded by the German Federal Ministry for Education and Science (FORSYS-Centre MaCS: 0313922, JAKSys: 031 6167 A) and the German Federal State of Saxony-Anhalt (Research Centre Dynamic Systems [CDS]) providing grants to FS. This work was also supported by the grants “FSC-PUL09-MyeloJak” of the University of Luxembourg. KG was funded by the grant “Aides à la Formation-Recherche” of the Fonds National de la Recherche Luxembourg (FNR, AFR PHD-08-030). We also thank Drs Guy Berchem and Valérie Palissot of the Laboratory of Experimental Hemato-Oncology of the CRP Santé in Luxembourg for providing patient material for this study. The authors of this article are responsible for the content of this publication.
Glossary
Abbreviations:
- CI
combination index
- Crk
adapter molecule crk/v-crk sarcoma virus CT10 oncogene homolog
- CRKL
Crk-like protein
- EEC
endogenous erythroid colonies
- ERK
extracellular signal regulated kinase
- ET
essential thrombocythemia
- FCS
fetal calf serum
- Gab1
Grb2-associated binder 1
- Grb2
growth factor receptor bound protein 2
- JAK
Janus kinase
- HDAC
histone deacetylase
- HEK
human embryonic kidney
- HEL
human erythroleukemia
- PI3K
phosphatidyl-inositol-3-kinase
- MAPK
mitogen activated protein kinase
- MMP
matrix metalloproteinase
- MPN
myeloproliferative neoplasm
- PAI
plasminogen activator inhibitor
- PBMC
peripheral blood mononuclear cells
- PCR
polymerase chain reaction
- PH
pleckstrin homology (domain)
- PLC
phospholipase C
- PMF
primary myelofibrosis
- PV
polycythemia vera
- RasGAP
Ras-GTPase activating protein
- PVDF
polyvinylidene-difluorid
- Serpin
serine protease inhibitor
- SHP2
Src-homology domain containing phosphatase 2
- STAT
signal transducer and activator of transcription
- TKI
tyrosine kinase inhibitor
Ethics Statement
All experiments on patient samples were approved by the Comité National d’Etique de Recherche (CNER) in Luxembourg according to the Declaration of Helsinki. An informed, written consent of every patient included in the study has been obtained.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/24574
References
- 1.Ernst M, Jenkins BJ. Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet. 2004;20:23–32. doi: 10.1016/j.tig.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–8. doi: 10.1182/blood-2005-03-1320. [DOI] [PubMed] [Google Scholar]
- 3.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 4.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Cancer Genome Project Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 5.James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 6.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 7.Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280:22788–92. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steensma DP. JAK2 V617F in myeloid disorders: molecular diagnostic techniques and their clinical utility: a paper from the 2005 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn. 2006;8:397–411, quiz 526. doi: 10.2353/jmoldx.2006.060007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tefferi A. Classification, diagnosis and management of myeloproliferative disorders in the JAK2V617F era. Hematology Am Soc Hematol Educ Program. 2006;2006:240–5. doi: 10.1182/asheducation-2006.1.240. [DOI] [PubMed] [Google Scholar]
- 10.Constantinescu SN, Girardot M, Pecquet C. Mining for JAK-STAT mutations in cancer. Trends Biochem Sci. 2008;33:122–31. doi: 10.1016/j.tibs.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Prchal JF, Axelrad AA. Letter: Bone-marrow responses in polycythemia vera. N Engl J Med. 1974;290:1382. doi: 10.1056/NEJM197406132902419. [DOI] [PubMed] [Google Scholar]
- 12.Ugo V, Marzac C, Teyssandier I, Larbret F, Lécluse Y, Debili N, et al. Multiple signaling pathways are involved in erythropoietin-independent differentiation of erythroid progenitors in polycythemia vera. Exp Hematol. 2004;32:179–87. doi: 10.1016/j.exphem.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111:3931–40. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 14.Hochhaus A, O’Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L, et al. IRIS Investigators Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23:1054–61. doi: 10.1038/leu.2009.38. [DOI] [PubMed] [Google Scholar]
- 15.Kane RC, Farrell AT, Saber H, Tang S, Williams G, Jee JM, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res. 2006;12:7271–8. doi: 10.1158/1078-0432.CCR-06-1249. [DOI] [PubMed] [Google Scholar]
- 16.Keedy VL, Temin S, Somerfield MR, Beasley MB, Johnson DH, McShane LM, et al. American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) Mutation testing for patients with advanced non-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol. 2011;29:2121–7. doi: 10.1200/JCO.2010.31.8923. [DOI] [PubMed] [Google Scholar]
- 17.Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116:5089–102. doi: 10.1182/blood-2010-04-261867. [DOI] [PubMed] [Google Scholar]
- 18.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. SHARP Investigators Study Group Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 19.Haan C, Behrmann I, Haan S. Perspectives for the use of structural information and chemical genetics to develop inhibitors of Janus kinases. J Cell Mol Med. 2010;14:504–27. doi: 10.1111/j.1582-4934.2010.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29:789–96. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–27. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eulenfeld R, Dittrich A, Khouri C, Müller PJ, Mütze B, Wolf A, et al. Interleukin-6 signalling: more than Jaks and STATs. Eur J Cell Biol. 2012;91:486–95. doi: 10.1016/j.ejcb.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 23.Wöhrle FU, Daly RJ, Brummer T. Function, regulation and pathological roles of the Gab/DOS docking proteins. Cell Commun Signal. 2009;7:22. doi: 10.1186/1478-811X-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonald CB, Bhat V, Mikles DC, Deegan BJ, Seldeen KL, Farooq A. Bivalent binding drives the formation of the Grb2-Gab1 signaling complex in a noncooperative manner. FEBS J. 2012;279:2156–73. doi: 10.1111/j.1742-4658.2012.08600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harkiolaki M, Tsirka T, Lewitzky M, Simister PC, Joshi D, Bird LE, et al. Distinct binding modes of two epitopes in Gab2 that interact with the SH3C domain of Grb2. Structure. 2009;17:809–22. doi: 10.1016/j.str.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 26.Eulenfeld R, Schaper F. A new mechanism for the regulation of Gab1 recruitment to the plasma membrane. J Cell Sci. 2009;122:55–64. doi: 10.1242/jcs.037226. [DOI] [PubMed] [Google Scholar]
- 27.Haan S, Wüller S, Kaczor J, Rolvering C, Nöcker T, Behrmann I, et al. SOCS-mediated downregulation of mutant Jak2 (V617F, T875N and K539L) counteracts cytokine-independent signaling. Oncogene. 2009;28:3069–80. doi: 10.1038/onc.2009.155. [DOI] [PubMed] [Google Scholar]
- 28.Lu X, Levine R, Tong W, Wernig G, Pikman Y, Zarnegar S, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci U S A. 2005;102:18962–7. doi: 10.1073/pnas.0509714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mercher T, Wernig G, Moore SA, Levine RL, Gu TL, Fröhling S, et al. JAK2T875N is a novel activating mutation that results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood. 2006;108:2770–9. doi: 10.1182/blood-2006-04-014712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–68. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 32.Chou TC. Preclinical versus clinical drug combination studies. Leuk Lymphoma. 2008;49:2059–80. doi: 10.1080/10428190802353591. [DOI] [PubMed] [Google Scholar]
- 33.Chou TC, Martin N. CompuSyn for Drug Combinations and for General Dose-Effect Analysis, Software and User’s Guide: A Computer Program for Quantitation of Synergism and Antagonism in Drug Combinations, and the Determination of IC50 and ED50 and LD50 Values. Paramus, NJ, USA: ComboSyn Inc, 2005:www.combosyn.com
- 34.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 35.Pósán E, Ujj G, Kiss A, Telek B, Rák K, Udvardy M. Reduced in vitro clot lysis and release of more active platelet PAI-1 in polycythemia vera and essential thrombocythemia. Thromb Res. 1998;90:51–6. doi: 10.1016/S0049-3848(98)00005-X. [DOI] [PubMed] [Google Scholar]
- 36.Cancelas JA, García-Avello A, García-Frade LJ. High plasma levels of plasminogen activator inhibitor 1 (PAI-1) in polycythemia vera and essential thrombocythemia are associated with thrombosis. Thromb Res. 1994;75:513–20. doi: 10.1016/0049-3848(94)90226-7. [DOI] [PubMed] [Google Scholar]
- 37.Senno SL, Pechet L. Clinical implications of elevated PAI-1 revisited: multiple arterial thrombosis in a patient with essential thrombocythemia and elevated plasminogen activator inhibitor-1 (PAI-1) levels: a case report and review of the literature. J Thromb Thrombolysis. 1999;8:105–12. doi: 10.1023/A:1008907001042. [DOI] [PubMed] [Google Scholar]
- 38.Kasyapa CS, Kunapuli P, Hawthorn L, Cowell JK. Induction of the plasminogen activator inhibitor-2 in cells expressing the ZNF198/FGFR1 fusion kinase that is involved in atypical myeloproliferative disease. Blood. 2006;107:3693–9. doi: 10.1182/blood-2005-04-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delhase M, Kim SY, Lee H, Naiki-Ito A, Chen Y, Ahn ER, et al. TANK-binding kinase 1 (TBK1) controls cell survival through PAI-2/serpinB2 and transglutaminase 2. Proc Natl Acad Sci U S A. 2012;109:E177–86. doi: 10.1073/pnas.1119296109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tonnetti L, Netzel-Arnett S, Darnell GA, Hayes T, Buzza MS, Anglin IE, et al. SerpinB2 protection of retinoblastoma protein from calpain enhances tumor cell survival. Cancer Res. 2008;68:5648–57. doi: 10.1158/0008-5472.CAN-07-5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen MK, Riisbro R, Holten-Andersen MN, Brown PdeN, Junker P, Brünner N, et al. Collagen metabolism and enzymes of the urokinase plasminogen activator system in chronic myeloproliferative disorders: correlation between plasma-soluble urokinase plasminogen activator receptor and serum markers for collagen metabolism. Eur J Haematol. 2003;71:276–82. doi: 10.1034/j.1600-0609.2003.00134.x. [DOI] [PubMed] [Google Scholar]
- 42.Jensen MK, Holten-Andersen MN, Riisbro R, de Nully Brown P, Larsen MB, Kjeldsen L, et al. Elevated plasma levels of TIMP-1 correlate with plasma suPAR/uPA in patients with chronic myeloproliferative disorders. Eur J Haematol. 2003;71:377–84. doi: 10.1034/j.1600-0609.2003.00096.x. [DOI] [PubMed] [Google Scholar]
- 43.Xu M, Bruno E, Chao J, Huang S, Finazzi G, Fruchtman SM, et al. MPD Research Consortium Constitutive mobilization of CD34+ cells into the peripheral blood in idiopathic myelofibrosis may be due to the action of a number of proteases. Blood. 2005;105:4508–15. doi: 10.1182/blood-2004-08-3238. [DOI] [PubMed] [Google Scholar]
- 44.Van Themsche C, Alain T, Kossakowska AE, Urbanski S, Potworowski EF, St-Pierre Y. Stromelysin-2 (matrix metalloproteinase 10) is inducible in lymphoma cells and accelerates the growth of lymphoid tumors in vivo. J Immunol. 2004;173:3605–11. doi: 10.4049/jimmunol.173.6.3605. [DOI] [PubMed] [Google Scholar]
- 45.Vaughan TY, Verma S, Bunting KD. Grb2-associated binding (Gab) proteins in hematopoietic and immune cell biology. Am J Blood Res. 2011;1:130–4. [PMC free article] [PubMed] [Google Scholar]
- 46.Ortiz-Padilla C, Gallego-Ortega D, Browne BC, Hochgräfe F, Caldon CE, Lyons RJ, et al. Functional characterization of cancer-associated Gab1 mutations. Oncogene. 2013;32:2696–702. doi: 10.1038/onc.2012.271. [DOI] [PubMed] [Google Scholar]
- 47.Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–98. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 48.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reilly JT, McMullin MF, Beer PA, Butt N, Conneally E, Duncombe A, et al. Writing group: British Committee for Standards in Haematology Guideline for the diagnosis and management of myelofibrosis. Br J Haematol. 2012;158:453–71. doi: 10.1111/j.1365-2141.2012.09179.x. [DOI] [PubMed] [Google Scholar]
- 50.Gu H, Neel BG. The “Gab” in signal transduction. Trends Cell Biol. 2003;13:122–30. doi: 10.1016/S0962-8924(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 51.Gäbler K, Rolvering C, Kaczor J, Eulenfeld R, Méndez SA, Berchem G, et al. Cooperative effects of Janus and Aurora kinase inhibition by CEP701 in cells expressing Jak2V617F. J Cell Mol Med. 2013;17:265–76. doi: 10.1111/jcmm.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vannucchi AM. Management of myelofibrosis. Hematology Am Soc Hematol Educ Program. 2011;2011:222–30. doi: 10.1182/asheducation-2011.1.222. [DOI] [PubMed] [Google Scholar]
- 53.Vannucchi AM, Biamonte F. Epigenetics and mutations in chronic myeloproliferative neoplasms. Haematologica. 2011;96:1398–402. doi: 10.3324/haematol.2011.052068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bogani C, Bartalucci N, Martinelli S, Tozzi L, Guglielmelli P, Bosi A, et al. Associazione Italiana per la Ricerca sul Cancro AGIMM Gruppo Italiano Malattie Mieloproliferative mTOR inhibitors alone and in combination with JAK2 inhibitors effectively inhibit cells of myeloproliferative neoplasms. PLoS One. 2013;8:e54826. doi: 10.1371/journal.pone.0054826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fiskus W, Ganguly S, Kambhampati S, Bhalla KN. Role of additional novel therapies in myeloproliferative neoplasms. Hematol Oncol Clin North Am. 2012;26:959–80. doi: 10.1016/j.hoc.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 56.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 57.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 58.Harrison C, Verstovsek S, McMullin MF, Mesa R. Janus kinase inhibition and its effect upon the therapeutic landscape for myelofibrosis: from palliation to cure? Br J Haematol. 2012;157:426–37. doi: 10.1111/j.1365-2141.2012.09108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fiskus W, Verstovsek S, Manshouri T, Rao R, Balusu R, Venkannagari S, et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin Cancer Res. 2011;17:7347–58. doi: 10.1158/1078-0432.CCR-11-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marubayashi S, Koppikar P, Taldone T, Abdel-Wahab O, West N, Bhagwat N, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010;120:3578–93. doi: 10.1172/JCI42442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bruchova H, Yoon D, Agarwal AM, Mendell J, Prchal JT. Regulated expression of microRNAs in normal and polycythemia vera erythropoiesis. Exp Hematol. 2007;35:1657–67. doi: 10.1016/j.exphem.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bruchova H, Yoon D, Agarwal AM, Swierczek S, Prchal JT. Erythropoiesis in polycythemia vera is hyper-proliferative and has accelerated maturation. Blood Cells Mol Dis. 2009;43:81–7. doi: 10.1016/j.bcmd.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gaikwad A, Nussenzveig R, Liu E, Gottshalk S, Chang K, Prchal JT. In vitro expansion of erythroid progenitors from polycythemia vera patients leads to decrease in JAK2 V617F allele. Exp Hematol. 2007;35:587–95. doi: 10.1016/j.exphem.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 65.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haan C, Behrmann I. A cost effective non-commercial ECL-solution for Western blot detections yielding strong signals and low background. J Immunol Methods. 2007;318:11–9. doi: 10.1016/j.jim.2006.07.027. [DOI] [PubMed] [Google Scholar]