

Abstract
Despite a conceptually simple mechanism of signaling, the JAK-STAT pathway exhibits considerable behavioral complexity. Computational pathway models are tools to investigate in detail signaling process. They integrate well with experimental studies, helping to explain molecular dynamics and to state new hypotheses, most often about the structure of interactions.
A relatively small amount of experimental data is available for a JAK1/2-STAT1 variant of the pathway, hence, only several computational models were developed. Here we review a dominant approach of kinetic modeling of the JAK1/2-STAT1 pathway, based on ordinary differential equations. We also give a brief overview of attempts to computationally infer topology of this pathway.
Keywords: IFN signaling, JAK1/2-STAT1 pathway, computational model, mathematical modeling, kinetics, network inference
Introduction
Computational modeling: mathematical description integrated with experimental data
Computational modeling of signal transduction integrates available knowledge about pathway regulation, and the general chemical and physical principles with experimental data from different biotechnology platforms. Such approach constitutes a powerful solution for formalizing and extending traditional molecular and cellular biology. However, success of the entire project depends crucially on the said integration. Figure 1 illustrates the model’s life cycle paradigm: the recurring processes of molecular experiments, measurements, bioinformatics analyses, and mathematical modeling.

Figure 1. Illustration of the paradigm of an interaction between modeling and experiments. Note that conclusions obtained from a model affect an experimental setup. Similarly, measurements that provide data to bioinformatics analysis allow for model enhancements. In the desired scenario several iterations of such cycle are performed.
In this review we sketch existing attempts to model JAK1/2-STAT1 signaling. There are relatively few such attempts for this particular pathway. The limiting factor is the availability of experimental data. As we will see a new set of molecular data usually allows researchers to formulate more adequate model.
Formal modeling of signaling pathways most often starts with a definition of a topology of the pathway, represented by a biochemical reactions network. Construction of a structure of a model requires systematization of a diverse, incomplete, and usually inconsistent biological knowledge about the underlying phenomena. Consequently, the structure of a model itself is a valuable contribution, provided that it is validated against available experimental data. However, computational modeling of signaling pathways offers much more. It can help to understand roles of designed parts of an investigated pathway. Standard examples include comprehension of a function of cascade layers1 and negative feedback2 in the MAPK pathway or, in case of the JAK-STAT pathway, role of a nuclear export of STATs3 and of a posttranslational negative feedback.4
JAK-STAT signaling
JAK-STAT pathways are highly conserved intracellular signaling pathways of all vertebrates and even found in some more primitive metazoas.5 These pathways have co-evolved with multiple fundamental cellular processes, such as innate and adaptive immune response,6,7 cell growth, differentiation and apoptosis regulation,8,9 maintenance of homeostasis,7 organogenesis,10 or embryonic development.11
JAK-STAT pathways provide a simple and direct route from the membrane receptors to the nucleus for mediating cellular responses to numerous cytokines. Until now, four receptor-associated JAK tyrosine kinases (JAK1/2/3 and TYK2) and seven different STAT family members (STAT1/2/3/4/5A/5B/6) have been identified in mammalian cells.1,12 In short, there are three main stages involved in the transduction of cytokine receptor derived signal through any JAK-STAT pathway: activation of the receptor, direct translocation of the signal into the nucleus and expression of target genes. Binding of cytokine triggers changes to the cognate receptor that permit cross-activation of receptor-associated JAKs. Activated JAKs phosphorylate intracellular tail of the receptor, thereby providing docking sites for latent STAT transcription factors. Recruited STAT monomeric proteins become subsequently phosphorylated by the proximate JAKs, dissociate from the receptor and form homo- or heterodimers directed to enter to the nucleus. Acting as dimers or as more complex oligomers STATs bind to consensus DNA sequences in the regulatory regions of target genes and initiate or enhance the appropriate transcriptional response.5,13
Each stage of JAK-STAT signaling takes different time to execute—it ranges from seconds up to hours, depending on an organism and a tissue. The magnitude and duration of the transduced signal is tightly controlled and can be attenuated at all levels. The key players here are: the protein tyrosine phosphatases (PTPs; e.g., SHP-1 or SHP-2), which dephosphorylate receptors at the membrane and activated STATs in the nucleus;5 protein inhibitors of activated STATs (PIAS), which in principle, block STAT dimers DNA binding or transactivation capacity in the nucleus, both directly (PIAS-1/3) or by recruitment of other co–repressor molecules (PIAS-x/y);14 and expressed as a negative feedback, some members of the family of suppressors of cytokine signaling (SOCS1/2/3), which inhibit receptor signaling by directly inhibiting both JAKs and cytokine receptors.5,15 In principle, these SOCS family members block STAT docking sites on the receptor,16 but they also probably mediate STAT degradation by ubiquitin-proteasome pathway.15 In addition, the transcriptional potency of STATs can be influenced by other proteins, either through direct interaction on a promoter (e.g., on NFκB promoter) or through posttranslational modification, including phosphorylation by growth factor receptors tyrosine kinases or mitogen-activated protein kinases (MAPKs).13
In general, JAK-STAT signaling is involved in a broad spectrum of fundamental processes. However, each cytokine receptor is activated by a small set of cytokines, specific only to this receptor. Consequently, cytokine receptor activates a characteristic combination of individual JAKs and STATs that is determined by the structure of the intracellular domains of the receptor chains.12
Interferons
Interferons, potent regulators of innate immunity, play a key role in the inflammatory response after viral infection and in host defense against microorganisms. Currently, there are three distinct classes recognized: type I (IFN-α/β/ε/κ/ω), type II (IFN-γ), and type III (IFN-λ), distinguishable by their differing target receptors. Types I and II IFN receptors are present in most human cell types while type III IFN-λ receptors are highly expressed in hepatocytes but not in microvascular endothelium, adipocytes, fibroblasts, or CNS cells.17 Types I and III IFN, despite signaling through distinct receptor complexes, stimulate very similar signaling pathways, namely they activate JAK1 and TYK2, which phosphorylate STAT1 and STAT2, respectively. In this pathway variant, the downstream heterotrimer of phosphorylated STAT1, STAT2, and IRF9 is generated to form interferon-stimulated gene factor 3 (ISGF3). ISGF3 acts as a transcription factor within interferon-stimulated response element (ISRE) sites on the promoter or enhancer regions of type I and III IFN-responsive genes.18,19 The focus of this review is modeling of cellular response to type II IFN (IFN-γ), which signals through the IFN-γ receptor (IFNGR), pre-associated with JAK1 and JAK2. Stimulation of the receptor leads to phosphorylation and dimerization of STAT1. Both IFN signaling schemes, including pathway negative regulators are depicted in Figure 2.

Figure 2. Key steps of JAK-STAT signaling in response to IFNs. IFNs induce activation of JAKs, receptors and subsequently, of cytoplasmic STATs; all by phosphorylation. Active STATs essentially form dimers, which then are translocated to nucleus where they act as transcription factors in the specific gene promoter or enhancer regions. IFN signaling negative regulators include multiple cytoplasmic PTPs, nuclear PTP and PIAS proteins, as well as specific to IFN signaling SOCS1 which in principle inhibits STAT1 phosphorylation process. See text for more details.
JAK1/2-STAT1 variant
Active form of IFN-γ receptor is more complex than that for types I and III IFN. IFN-γ signal transduction engages oligomerization of two subunits IFNGR1 and IFNGR2, which triggers activation of receptor-associated kinases JAK1 and JAK2 and phosphorylation of STAT1 on critical tyrosine residue. Phosphorylated STAT1 translocate as transcriptionally active homodimers into the nucleus, bind to the promoter regions via DNA sequences termed GAS (γ-activated site) and, thereby, affect transcription of IFN-γ-induced genes.18 The mechanism that regulates active nuclear import of tyrosine phosphorylated STAT1 dimers was shown to involve a specific carrier, importin α5, and metabolic energy.20 It has been suggested that besides the carrier-dependent transport of phospho-dimers, also unphosphorylated STAT1 molecules shuttle continuously between the nucleus and cytoplasm, in a process mediated by direct interaction with nucleoporins located in the nuclear pore complexes.21 This constitutive nucleocytoplasmic translocation of unphosphorylated STAT1 occurs both in resting and IFN-γ-stimulated cells. The existence of distinct transport pathways allows the controlled nuclear transport of phosphorylated STAT1 dimers during cytokine induction. It was demonstrated that nuclear import and nuclear retention are two separate steps leading up to nuclear accumulation of STAT1. DNA binding protects STAT1 from dephosphorylation, so only once released from DNA, the phosphorylated STAT1 dimers become dephosphorylated by nuclear phosphatases.22 Dimer dephosphorylation is a critical step, which enables the return of STAT1 to the cytosol to participate in additional rounds of the activation and deactivation cycle.23
Recent data have proved that regulation of STAT1 signaling is more complex. There is evidence that unphosphorylated STAT1 also exist as dimers with DNA binding activity and, although less active than phosphorylated forms, may regulate gene expression.24 Besides, STAT1 is known to form larger assemblies than dimers when bound to DNA, including tetramers and extended polymers, which may significantly influence in vivo binding preferences.25
Similarly to other variants of JAK-STAT signal transduction, JAK1/2-STAT1 pathway is negatively regulated by a number of regulatory mechanisms, including activity of various PTPs and PIAS family members, as well as feedback inhibition by SOCSs. However, some of these mechanisms have been recognized as specific or particularly important for attenuation of IFN-γ-induced response.
Among PIAS family members, primarily PIAS1 acts as a partial physiological inhibitor of STAT1.26 It was originally shown to block STAT1 DNA-binding activity,27 but due to its E3 SUMO ligase activity it has been suggested to promote SUMO-conjugation to STAT1.28 Simulation of STAT1, which obstructs tyrosine phosphorylation and affects STAT1 solubility,29 may be a unique mechanism that has evolved to negatively regulate IFN-γ-induced signaling; however the role of PIAS1 in this process has been controversial30 and the identity of the E3 SUMO ligase responsible remains unclear.
The most studied inhibitors of JAK-STAT signaling are the SOCS proteins.31 IFN signaling-specific SOCS1 is known to interact with IFNAR1 receptor and respective TYK2 in type I IFN signaling and with IFNGR1 receptor and JAK2 in type II IFN signaling.15 SOCS1 is itself upregulated via type II IFN signaling, but not known to be upregulated via type I or III IFN signaling. Moreover, as revealed by gene-targeting experiments, loss of SOCS1 results in excessive response to IFN-γ and the majority of inflammatory defects exhibited by SOCS1 knockout mice are related to unbridled IFN-γ signaling.32 Therefore, SOCS1 is recognized as a crucial regulator of IFN-γ induced response.
Whereas type I IFNs are primarily induced in response to viral infection, IFN-γ is produced predominantly by T lymphocytes, NK, and NKT cells, following activation with immune and inflammatory stimuli.33 IFN-γ acts on its receptor to augment innate cellular immunity by activating NK cells and macrophages, upregulating MHC expression, promoting leucocyte migration and inducing the expression of proinflammatory cytokines (e.g., IL-12 p40 subunit, TNF-α, and IL-1) and enzymes involved in elimination of ingested bacterial or protozoan pathogens, such as nitric oxide synthase.34 Some data also implicates role of IFN-γ as a critical immune system component, protecting the host from development of variety of tumors, e.g., in fibrosarcoma, melanoma, and mammary carcinoma models.33 Except from effects on both the innate and adaptive antitumor immune responses as well as inhibition of angiogenesis, one of the major targets of IFN-γ’s antitumor actions is the tumor cell itself. The molecular basis of this effect is the JAK1/2-STAT1-dependent activation of genes encoding cell cycle inhibitors or implicated in promoting tumor cell apoptosis.33
Owing to the central role of IFNγ in immune response to microbial infections and in promoting antitumor protection, JAK1/2-STAT1 pathway exploited by the cytokine is an excellent target for detailed studies. Because of its relative simplicity and high evolutionary conservation it has attracted attention of systems biologists, who have contributed to mathematical models of the signaling cascade. Since dysregulation of JAK-STAT signaling is associated with various immune disorders and cancers, identification of key components and steps in signal transduction pathway provides useful strategy for drug discovery. Computational models can greatly facilitate such predictions.
Computational Models
Most of the experimental data for the JAK-STAT pathways available in the recent literature in vast majority concerns variants activated by ligands other than type II IFN. This data was used in development of many formal models of JAK-STAT signaling; most notably, for the Epo-stimulated pathway variant, a simple model which originated from work of Swameye et al.3 This model was re-used multiple times in mainly methodological case studies, with adjustments such as linear chain approximation of a nuclear transport delay.35-38 For other examples of computational models of JAK-STAT pathway variants activated by Epo, as well as by IL or by type I IFN, see respectively references 39–42. For a broader review see reference 43. In this review we focus on models of the JAK1/2-STAT1 signaling pathway. There are relatively few distinct computational models for this variant of the JAK-STAT pathway.
Please be aware of the fact that in principle mathematical modeling is not suitable for the very detailed description of reality. It is important to maintain an appropriate balance between the number of mechanistic details responsible for the complexity of the model, and their significance in the context of the data available for model validation process (for examples, see refs. 3 and 44). The identifiability of model parameters is one of key issues here: non-identifiable model parameters may require more appropriate experimental design, or model reduction, adapting the complexity of the model to the information content of experimental measurements (compare refs. 35 and 45). Finally, focusing on specific hypotheses while designing the model should also enhance its predictive power (see, e.g., ref. 46).
Kinetic modeling
Mathematical models resulting from a biochemical reactions network are complex, even for the simplest signaling pathways, such as JAK-STAT. Therefore, an in silico analysis is usually based on computationally efficient numerical solutions for a deterministic framework which, despite of a coarse simplification of a spatial distribution and diffusion effects under spatial homogeneity assumption, proves to be very elucidative. In this framework state of a modeled system is represented by the time-dependent vector 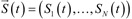 , denoting concentrations of N reacting species S1 (t), …, SN (t). Dynamics of the system is governed by a set of a set of first-order ODE, known as the reaction rate equations (RRE), i.e.:
, denoting concentrations of N reacting species S1 (t), …, SN (t). Dynamics of the system is governed by a set of a set of first-order ODE, known as the reaction rate equations (RRE), i.e.:
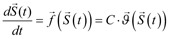 , ,
|
where the right-hand side of the differential equations 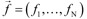 is equal to M state-dependent reactions rates
is equal to M state-dependent reactions rates 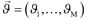 linearly combined according to a fixed stoichiometric matrix of the biochemical reactions network C, i.e.:
linearly combined according to a fixed stoichiometric matrix of the biochemical reactions network C, i.e.:
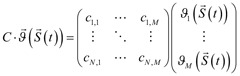 .
.
Stoichiometric matrix directly reflects a structure of the modeled network. Reaction rates, typically, follow the mass action law47 or its enzyme kinetics approximations such as the simplest Michaelis–Menten kinetics.48 Together with the initial state the dynamics of the system is formulated as an initial value problem. Below we briefly review kinetic models for the JAK1/2-STAT1 signaling. All of them describe dynamics using RRE. Development of stochastic counterparts of these models still merits further exploration.
Model by Yamada et al.
Arguably, the first model of the JAK1/2-STAT1 pathway was introduced by Yamada et al.4 This model describes a control mechanism and factors influencing kinetics of the JAK-STAT pathway in IFN-γ-stimulated hepatocytes, recapitulating liver injury associated with increased IFN-γ activity. The Yamada et al.4 model is relatively complex as it captures all essential elements in the JAK1/2-STAT1 signaling, together with many short-lived, intermediate species. These species most likely could be omitted or even systematically eliminated (compare refs. 39 and 49). Scheme of the model is depicted in Figure 3A. The model can be informally divided into three modules: receptor module, transcription factor module (the STAT life-cycle) and posttranslational feedback module. Figure 3B–D shows the numerical solutions of RRE for output species of each module. Respective modules emit output signal for ca. 25 min, 50 min and 1.5 h, with its peak activity in ca. 25th minute, 55th minute, and 2nd and a half hour.

Figure 3. JAK1/2-STAT1 pathway modules denoted by colors on the scheme of a model by Yamada et al.4 (A), and numerical simulations of modules output species representing concentration of, respectively, phosphorylated receptor dimer (B), phosphorylated nuclear STAT1 dimer (C), and SOCS1 (D) with respect to time. Additional decorations of the simulations graphs represent signaling properties, such as: peak activity time τ, duration ϑ, and amplitude α50 at the basal, steady-state level π, corresponding to a constant IFN input of 10 nM. In such in silico experimental setup, unbound active receptor reaches peak in ca. 25 min (B) and from that moment on it is gradually used up in activation of STAT1 proteins. In turn, phosphorylated STAT1 dimers accumulate in nuclei, with maximum concentration reached in ca. 1 h (C). Their slow concentration descent is followed up by delayed expression of SOCS1 proteins (D), which gradually overtake active receptors (B). The remaining small excess of the latter induces a second, much weaker phase of signaling, starting in ca. 5th hour (B–D). After that the signal is completely attenuated. Duration of activity of signaling molecules ν elongates downstream of the pathway, and extent of this effect depends on the strength of signaling α (compare the first and the second phase of signaling; [B–D]).
The model by Yamada et al.4 is currently curated in DOQCS51 and BioModels databases,52,53 for instance, in the machine-readable SBML format.54 It means that this model is basically ready to be simulated and it has been assured that it replicates published behavior. This is a priceless advantage of this model because of its size. Namely, it consists of over 30 variables representing species and of over 60 parameters of reaction rates. In principle, this model was calibrated and validated against the experimental data.4 Kinetic constants and protein concentrations were set based on experimental results mainly from Brysha et al.55 There are noticeable quantitative differences in the kinetics of response to IFN-γ depending on cell type, i.e., mouse liver cells55 and T cells data,56 but the overall qualitative behavior of the JAK1/2-STAT1 pathway is consistent with published biological data.
Yamada et al.4 used their model to underline importance of nuclear phosphatase PTPs and the negative feedback created by SOCS-1 protein, i.e., importance of the JAK1/2-STAT1 pathway negative regulators. Through in silico knockout experiments nuclear PTP was identified as the most important pathway regulator with respect to attenuation of a signal which can arise from a non-significant random appearance of IFN, i.e., in case of an unintended exposure of cells to a small amount of the cytokine. Dephosphorylation of STAT1 dimer by nuclear phosphatase is a prerequisite for the transcription factor export to cytoplasm and thus one of the crucial factors in controlling nuclear accumulation of active STAT1 at the site of regulation of gene expression.23 On the other hand, mechanism of a posttranslational production of SOCS was justified by a requirement of a delay in the activation of the negative feedback. To that end, Yamada et al.4 analyzed hypothetical model, where SOCS was stimulated directly from a downstream of a signal, before the end of transcription and translation of target genes. In such case inhibition turned out not to be effective, and available experimental data clearly show that SOCS-1 expression is induced by IFN-γ and overexpression of SOCS-1 inhibits IFN-γ signaling.57,58
The only obvious component of the JAK-STAT pathway in general, which is missing in the model, is the PIAS inhibitor. Moreover, this model does not take into consideration some currently accepted regulatory mechanisms affecting STAT1:DNA binding activity, such as STAT1 simulation and transcriptional activity of unphosphorylated STAT1. One of the reasons is obviously the fact that these processes had not been clearly defined at the time, when the model was created.
Sensitivity analysis
The same model was analyzed in several following papers,46,59,60 which, to some extent, employed a concept of biological robustness, based on a mathematical technique termed as the sensitivity analysis. Robustness is a property of a system to maintain one or more of its functions under external and internal perturbations.61 On the other hand sensitivity analysis in principle investigates the relation between uncertain parameters of a model and the observable output. For a review of sensitivity analysis methods applicable to chemical reaction networks and signaling pathways in particular see references 62 and 63. Global sensitivity analysis (GSA) is the most suitable for highly nonlinear models, such as RRE models.62 Based on the mathematical model developed by Yamada et al.,4 by means of GSA of the IFN-γ-induced JAK-STAT signaling pathway, Zi et al.59 confirmed that SOCS1 and nuclear phosphatase are critical components for the perturbation of the system output and additionally pointed out an importance of cytoplasmic STAT1 for a general dynamic behavior of the pathway. The result that the nuclear phosphatase and SOCS1 are more critical than SHP-2 and the cytoplasmic phosphatase underscores the importance of downstream (hence, more direct upon STAT1) negative regulators compared with upstream regulators. The SOCS proteins are expressed at low levels in unstimulated cells and become rapidly induced by cytokines, thereby blocking continued signaling and forming a classic negative-feedback loop.31 The finding by Zi et al.59 that the nuclear phosphatase (which promotes the nuclear export) is an important signaling component matches well with the importance of nucleocytoplasmic shuttling process of STAT1, described in biological systems. Along that line of research, Swameye et al.,3 who developed a much simpler model of the JAK2-STAT5 pathway variant, showed that STATs nucleocytoplasmic shuttling parameters are the most sensitive to perturbations with respect to the total amount of nuclear activated STAT.
The Yamada et al.4 model and three hypothetical models, which differ by a form in which STAT protein enters the nucleus and acts as a transcription factor, were compared in work of Shudo et al.60 The original model was selected as an evolutionary preferable one, by presenting the most reasonable strength and time of the response, as well as lowest local sensitivity of kinetic parameters with respect to the input stimuli. This computational reasoning was used to justify that the STAT1 dimerization is an indispensable signaling step in the generation of adequate and robust cellular response to IFN-γ. This is in line with published data implying that active nuclear import induced by cytokine stimulation as well as subsequent STAT1:DNA binding requires STAT1 dimer formation.5,20 However, the model version by Shudo et al.60 differs from the model by Yamada et al.4 by lack of a reaction of dimerization of receptor molecules. This reduction was based on the assumption that cytokine receptors are pre-assembled on the membrane.64-66 We studied consequences of this reduction in reference 46. The receptor activation mechanism is not easily accessible for experimental measurements, thus we performed a comparative computational analysis of four variants of activation of the JAK1/2-STAT1 pathway. Using GSA, complemented with the profile-likelihood based identifiability analysis,35 we showed that the on-membrane pre-assembly of the dimers in the absence of ligand increases the overall environmental robustness of the pathway model. In a more general sense, we evaluated the usefulness of different model selection methods in a frequently encountered, but not much discussed case of a model of a considerable size, which has several variants differing at peripheries.45 With lack of the sufficient experimental data to test against, the generalizability/parsimony principle turned out not to be a sufficient criteria for model selection. The sensitivity analysis based on the robustness concept enabled more conscious, expert-mediated choice of the preferred model.
Hierarchical subsystems identification
On the other hand, Soebiyanto et al.67 applied the multilevel hierarchical systems framework68 to study the coordination principle in the RRE model of JAK1/2-STAT1 signaling by Yamada et al.4 Based on the knowledge of biological functionality they divided the model into five subsystems—4 modules in the cytoplasm and one representing the STAT1 activity in the nucleus. Compartmentalization of the pathway model coupled with in silico inhibition, knockdown/deletion, and perturbation experiments identified SOCS1 as a coordinator. A coordinator’s role is to regulate the subsystems at the lower level to achieve the overall objective of the system. In fact, SOCS1 was shown to regulate functionally independent subsystems related to SHP-2 and cytoplasmic STAT1. This discovery of SOCS1 as a coordinator is in line with the recent publication56 that shows biological data supporting SOCS1 as a crucial signaling component regulator. The model by Soebiyanto also showed that a SOCS1 knockdown leads to high-level activation behavior of the pathway, while SHP2 knockdown results in its constitutive, though pathological, activation. These in silico experimental data agree with the results of STAT1 phosphorylation with SOCS1 knockdown shown by Yamada et al.4 and are consistent with Brysha et al.55 Moreover, it suggests that with abundance of IFN-γ, as it occurs during inflammation, STAT1 can be persistently activated if a SOCS1 mutation/knockdown exists. Without the high availability of cytokines, it is SHP2 that is regulating the constitutive activation of STAT1.
Identifiability of the model
Finally, Quaiser et al.49 also analyzed the model by Yamada et al.,27 focusing, however, on the mathematical problem of model identifiability. Authors proposed an iterative method for model simplification, and demonstrated its use by simplifying the JAK1/2-STAT1 model. Their procedure numerically estimates kinetic parameters of the model, create an identifiability ranking for them, and finally simplify the model based on the identifiability analysis results. A parameter is called identifiable if it is uniquely defined by the model structure and data, simulated or experimental. The simplification steps are applied until the model is identifiable (i.e., parameter variances are small). Fully identifiable version of the original model was obtained after six in silico iterations. The resulting model has only 9 state variables and 10 parameters. Simplifications include: constitutive binding of JAKs and receptors (compare ref. 46); lumping formation of the receptor complex with STAT1 phosphorylation and, conversely, modeling step dissociation of the whole receptor complex in a single step; as well as omitting cytoplasmic STAT1 dephosphorylation (compare ref. 4).
Modeling based on PSC molecular data
A qualitatively different mathematical model of IFN-γ-mediated signaling in pancreatic stellate cells (PSC) was presented by Rateitschak et al.44 It was based on authors own quantitative experimental data. Their relatively small RRE (11 kinetic parameters, 9 species variables) model describes kinetics of chemical reactions. Delayed processes (such as transcription and translation) have been described as a distributed time delay with gamma kernel. Reduced reaction network includes main JAK1/2-STAT1 signaling features, i.e.: binding of the cytokine (IFN) to the receptor (R) combined with its immediate activation, phosphorylation of STAT1 combined with its immediate homodimerization, nuclear cycling of STAT1, transcription of IRF1 and SOCS1 as target genes and negative feedback formed by the latter protein. Most notably this model lacks negative regulation corresponding to cytoplasmic phosphatases, but on the other hand, includes a posttranslational positive feedback loop, representing expression of STAT1 in response to activation of IFN-γ receptors (via unknown underlying mechanism). The scheme of biochemical reactions is shown in Figure 4.
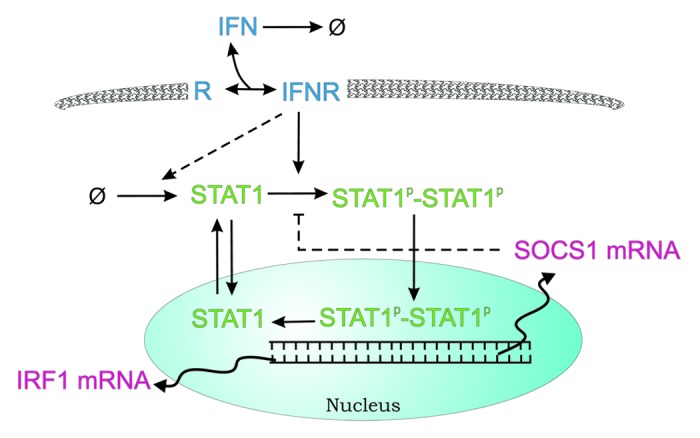
Figure 4. Scheme of the JAK1/2-STAT1 pathway model from the work of Rateitschak et al.44 Waved (transcription and mRNA relocation) and dashed arrows represent delayed processes. Names of variables have been adjusted for a consistency of this review. See text for details.
The results indicate that the contribution of SOCS1 to the termination of STAT1 activation in PSC is limited. The authors suggest exhaustion of IFNγ and dephosphorylation of STAT1 by tyrosine phosphatases as critical steps of STAT1 inactivation in this experimental system.
The proposed JAK1/2-STAT1 model44 successfully reproduced key laboratory findings and more importantly allowed to predict the results of new independent experiments not previously used for model calibration. One of them compared the efficiency of different stimulation modes with respect to the expression/activation of different components of the STAT1 pathway. We considered this experimental design interesting for the following reason: Interferons are cytokines with a variety of clinical applications. Since interferon action also involves induction of negative feedback loops, the application mode (e.g., time between two applications; dose split vs. a single high dose) is likely to influence biological efficiency. Thus, studies at the molecular level may provide deeper insights into the cellular basis of interferon responsiveness or resistance. The model developed specifically for PSC is of potential clinical interests because of the role played by STAT1 in the mediation of antifibrotic effects of IFN.
Network topology reconstruction
In all computational models described up to now one assumed a priori knowledge of the key biochemical reactions involved in the pathway. Consequently it was feasible to model the network dynamics using a set of differential equations. Taking one step back, a different modeling approach that can be found in the literature aims in inference of the network topology from experimental measurements, a methodology that was previously successfully applied in the field of gene regulatory network reconstruction (see, e.g., ref. 69).
In the context of JAK-STAT signaling Kaderali et al.70 reconstructed the network topology from gene knockout data obtained with the use of RNAi screening technology. The pathway is modeled as Bayesian network with probabilistic Boolean threshold functions. Moreover the efficient Monte Carlo procedure to sample from the posterior over model parameters was proposed.
Knockout RNAi data set
The knockdown experiments was conducted for 10 genes involved in two variants of JAK-STAT pathway (JAK1/2-STAT1 and JAK1/TYK2-STAT1/2) in humane hepatoma cell line under three different conditions: no stimulation, IFN-α stimulation, and IFN-γ stimulation. The main obstacle in the network reconstruction task is the fact that the number of possible network topologies increases exponentially with the number of nodes in the network. To reduce the complexity 10 genes of the pathway were manually grouped into six complexes: IFN-α receptor complex, IFN-γ receptor complex, JAK1 and TYK2 kinase complex, JAK1 and JAK2 complex, STAT1, STAT2 and IRF9 complex, and STAT1 homodimer. The proposed method correctly reconstructed the core topology of JAK-STAT pathway. All edges were identified except of STAT1 phosphorylation by JAK1/TYK2.
Constraint-based approach
To reconstruct the JAK-STAT signaling network in a human B cell, Papin and Palsson71 applied the method called extreme pathways analysis. The methodology, proposed originally in the context of metabolic pathways, enables to study various properties of signaling systems such as: input/output relationships, crosstalk, correlated reaction sets, and network redundancy.72 Extreme signaling pathways represent the edges of the steady-state flux cone derived from convex analysis of systems of reaction equations (given by stoichiometric matrix) with (in)equality constraints such as mass balance and reaction irreversibility. The method was used to reconstruct whole JAK-STAT signaling network based on the stoichiometric matrix consisting of 15 different receptors and 15 corresponding ligands identified in human B cells. There were 297 reactions in total that gave rise to 147 extreme pathways characterizing fundamental functional states of the network. The structure of crosstalk in the JAK-STAT network, formally defined as a nonnegative linear combination of extreme pathways, was also analyzed. Such constraint-based approach may provide the tool for the description of biologically and medically significant properties of the pathway.
Discussion
Combination of theoretical and experimental analysis can provide valuable insight into mechanisms of signaling pathways. High quality data are the limiting factor in computational modeling of JAK1/2-STAT1 pathway. We briefly reviewed here analysis performed for existing kinetic models: a set of models derived from the model by Yamada et al.4 and a more recent model of JAK1/2-STAT1 pathway in pancreatic stellate cells build from the scratch on the independent data set.44 Rigorous mathematical approach allows one to study the network dynamics, while techniques of sensitivity analysis and model reduction contribute to identification of key pathway components (potential drug targets).
We would like to emphasize that all reviewed kinetic models of JAK1/2-STAT1 pathway neglect the aspect of intrinsic noise caused by stochastic fluctuations. Our preliminary comparison of mean and standard deviation of the stochastic process with the numerical solution of RRE, both underlying the model by Yamada et al.,4 suggests that the deterministic model is a good approximation.73 However, there is an evident lack of an exhaustive, systematic analysis of influence of both intrinsic and simultaneous intrinsic and extrinsic noise on the behavior of JAK1/2-STAT1 signaling.
In principle, kinetic models building is a tricky and difficult task for many reasons. Except for incomplete biological knowledge, there is no clear notion of model optimality. However, there is a well-established principle of tailoring model complexity to the information content of available experimental data or the research question at hand.3,35,44,49 This parsimonious approach of defining a minimal sufficient model stands as an alternative to including all currently available knowledge of the pathway components and their interactions.4 Although the latter case provides an invaluable systematization of the research subject, the amount of qualitative knowledge about the structure of the biochemical network is usually far in excess of the feasibility of validating its dynamical behavior. In other words, in the case of large models it is practically impossible to fairly assess model relevance to experimental kinetic data at hand, at least with the quantities of data usually available. The obvious drawback of parsimonious models is their limited predictive power. Therefore, we would like to stress that it is absolutely crucial for a modeler to maintain an appropriate balance between the number of mechanistic details and their significance in the context of the data available for validation of the model as well as tools available for the model assessment.
With respect to more mundane aspects of model building, standardization of published models is essential, yet still not widely practiced. This can be achieved, for instance, through common file formats, such as SBML,54 databases of model files, such as BioModels,52,53 and annotation of model components with common vocabularies, such as Systems Biology Ontology.74 In context of computational models of JAK1/2-STAT1 signaling pathway this is perfectly exemplified by variants of the model by Yamada et al.4 In fact, Zi et al.59 and Soebiyanto et al.,67 most likely being unable to exactly reproduce the original model, used a slightly different, smaller variant of the model (with just over 50 instead of 60 rate parameters). Similarly, as already mentioned, Shudo et al.60 applied their own adjustments to the model. Although in a model of such size none of these changes made significant qualitative differences, small quantitative differences are noticeable and confusing.
On the final note, we sketched recent attempts to an even more challenging task of inference of the network topology from experimental measurements. Such approach is invaluable in cases when the network of biochemical reactions is unknown. In the context of JAK1/2-STAT1 modeling the extreme pathway analysis approach71 seems more adequate to understand biochemical reactions network design principles than the Bayesian network inference.70
Concluding, to appreciate the usefulness of a formal model, we need to be conscious that the main role of computational models of signaling pathways is compacting a large pool of detailed knowledge and determining, within the overall glance at the pathway and its dynamics, which of its elements, when, and why are important. It happens that discoveries provided by modeling approach were already known, but their importance lies in the fact that these hypotheses had been confirmed independently by the iterative process of model development and calibration. This is for example the case of nucleocytoplasmic shuttling importance from the model by Swameye et al.3 Likewise, computational model helped to demonstrate the role of dimerization of STAT1 in avoiding undesirable activation caused by the noise in the IFN activity.60 Finally, in a similar manner, it was elucidated that the reported preassembly of cytokine receptors that precedes the appearance of an IFN stimuli strengthens stability of the pathway signal transduction process in slightly varying intracellular conditions.46 Each of these examples of modest computational finding gives a more in-depth understanding of the design principles of JAK-STAT signaling, and signaling pathways in general.
Acknowledgments
The work in this paper was partially supported by the Polish National Center for Science grant no. 2011/01/B/NZ2/00864 and by the Swiss National Science Foundation grant no. 141264. The study was supported by research fellowship within “Information technologies: research and their interdisciplinary applications” agreement number POKL.04.01.01-00-051/10-00.
Glossary
Abbreviations:
- Epo
erythropoietin
- GAS
gamma activation site
- GSA
global sensitivity analysis
- IFN
interferon
- IFNGR
interferon gamma receptor
- IL
interleukin
- IRF
interferon regulatory factor
- ISGF
interferon-stimulated gene factor
- ISRE
interferon stimulated response element
- JAK
Janus kinase
- MAPK
mitogen-activated protein kinase
- MHC
major histocompatibility complex
- NFκB
nuclear factor kappa-B
- NK
natural killer
- ODE
ordinary differential equations
- PIAS
protein inhibitor of activated STAT
- PSC
pancreatic stellate cells
- PTP
protein tyrosine phosphatase
- RRE
reaction rate equations
- SHP-1/2
Src homology region 1/2 domain-containing phosphatase
- SOCS
suppressor of cytokine signaling
- STAT
signal transducer and activator of transcription
- TNF
tumor necrosis factor
- TYK
tyrosine kinase
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/24672
References
- 1.Huang CY, Ferrell JE., Jr. Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1996;93:10078–83. doi: 10.1073/pnas.93.19.10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kholodenko BN. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur J Biochem. 2000;267:1583–8. doi: 10.1046/j.1432-1327.2000.01197.x. [DOI] [PubMed] [Google Scholar]
- 3.Swameye I, Muller TG, Timmer J, Sandra O, Klingmuller U. Identification of nucleocytoplasmic cycling as a remote sensor in cellular signaling by databased modeling. Proc Natl Acad Sci U S A. 2003;100:1028–33. doi: 10.1073/pnas.0237333100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamada S, Shiono S, Joo A, Yoshimura A. Control mechanism of JAK/STAT signal transduction pathway. FEBS Lett. 2003;534:190–6. doi: 10.1016/S0014-5793(02)03842-5. [DOI] [PubMed] [Google Scholar]
- 5.Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/S0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- 6.Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–5. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 7.Pfitzner E, Kliem S, Baus D, Litterst CM. The role of STATs in inflammation and inflammatory diseases. Curr Pharm Des. 2004;10:2839–50. doi: 10.2174/1381612043383638. [DOI] [PubMed] [Google Scholar]
- 8.Bromberg J, Darnell JE., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–73. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 9.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 10.Wang YH, Huang ML. Organogenesis and tumorigenesis: insight from the JAK/STAT pathway in the Drosophila eye. Dev Dyn. 2010;239:2522–33. doi: 10.1002/dvdy.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raz R, Lee CK, Cannizzaro LA, d’Eustachio P, Levy DE. Essential role of STAT3 for embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 1999;96:2846–51. doi: 10.1073/pnas.96.6.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–63. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 13.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–9. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 14.Shuai K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006;16:196–202. doi: 10.1038/sj.cr.7310027. [DOI] [PubMed] [Google Scholar]
- 15.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–65. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 16.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–22. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly C, Klenerman P, Barnes E. Interferon lambdas: the next cytokine storm. Gut. 2011;60:1284–93. doi: 10.1136/gut.2010.222976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 19.Decker T, Stockinger S, Karaghiosoff M, Müller M, Kovarik P. IFNs and STATs in innate immunity to microorganisms. J Clin Invest. 2002;109:1271–7. doi: 10.1172/JCI15770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J. 1997;16:7067–77. doi: 10.1093/emboj/16.23.7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marg A, Shan Y, Meyer T, Meissner T, Brandenburg M, Vinkemeier U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J Cell Biol. 2004;165:823–33. doi: 10.1083/jcb.200403057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer T, Marg A, Lemke P, Wiesner B, Vinkemeier U. DNA binding controls inactivation and nuclear accumulation of the transcription factor Stat1. Genes Dev. 2003;17:1992–2005. doi: 10.1101/gad.268003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haspel RL, Darnell JE., Jr. A nuclear protein tyrosine phosphatase is required for the inactivation of Stat1. Proc Natl Acad Sci U S A. 1999;96:10188–93. doi: 10.1073/pnas.96.18.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterjee-Kishore M, Wright KL, Ting JP, Stark GR. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000;19:4111–22. doi: 10.1093/emboj/19.15.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonham AJ, Wenta N, Osslund LM, Prussin AJ, 2nd, Vinkemeier U, Reich NO. STAT1:DNA sequence-dependent binding modulation by phosphorylation, protein:protein interactions and small-molecule inhibition. Nucleic Acids Res. 2013;41:754–63. doi: 10.1093/nar/gks1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Mink S, Wong KA, Stein N, Getman C, Dempsey PW, et al. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat Immunol. 2004;5:891–8. doi: 10.1038/ni1104. [DOI] [PubMed] [Google Scholar]
- 27.Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–5. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- 28.Ungureanu D, Vanhatupa S, Grönholm J, Palvimo JJ, Silvennoinen O. SUMO-1 conjugation selectively modulates STAT1-mediated gene responses. Blood. 2005;106:224–6. doi: 10.1182/blood-2004-11-4514. [DOI] [PubMed] [Google Scholar]
- 29.Begitt A, Droescher M, Knobeloch KP, Vinkemeier U. SUMO conjugation of STAT1 protects cells from hyperresponsiveness to IFNγ. Blood. 2011;118:1002–7. doi: 10.1182/blood-2011-04-347930. [DOI] [PubMed] [Google Scholar]
- 30.Song L, Bhattacharya S, Yunus AA, Lima CD, Schindler C. Stat1 and SUMO modification. Blood. 2006;108:3237–44. doi: 10.1182/blood-2006-04-020271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–22. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/S0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 33.Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13:95–109. doi: 10.1016/S1359-6101(01)00038-7. [DOI] [PubMed] [Google Scholar]
- 34.de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–20. [PubMed] [Google Scholar]
- 35.Raue A, Kreutz C, Maiwald T, Bachmann J, Schilling M, Klingmüller U, et al. Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics. 2009;25:1923–9. doi: 10.1093/bioinformatics/btp358. [DOI] [PubMed] [Google Scholar]
- 36.Toni T, Stumpf MPH. Simulation-based model selection for dynamical systems in systems and population biology. Bioinformatics. 2010;26:104–10. doi: 10.1093/bioinformatics/btp619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanlier J, Tiemann CA, Hilbers PAJ, van Riel NAW. A Bayesian approach to targeted experiment design. Bioinformatics. 2012;28:1136–42. doi: 10.1093/bioinformatics/bts092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanlier J, Tiemann CA, Hilbers P A J, van Riel N A W. An integrated strategy for prediction uncertainty analysis. Bioinformatics. 2012;28:1130–5. doi: 10.1093/bioinformatics/bts088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bachmann J, Raue A, Schilling M, Böhm ME, Kreutz C, Kaschek D, et al. Division of labor by dual feedback regulators controls JAK2/STAT5 signaling over broad ligand range. Mol Syst Biol. 2011;7:516. doi: 10.1038/msb.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahdavi A, Davey RE, Bhola P, Yin T, Zandstra PW. Sensitivity analysis of intracellular signaling pathway kinetics predicts targets for stem cell fate control. PLoS Comput Biol. 2007;3:e130. doi: 10.1371/journal.pcbi.0030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smieja J, Jamaluddin M, Brasier AR, Kimmel M. Model-based analysis of interferon-beta induced signaling pathway. Bioinformatics. 2008;24:2363–9. doi: 10.1093/bioinformatics/btn400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maiwald T, Schneider A, Busch H, Sahle S, Gretz N, Weiss TS, et al. Combining theoretical analysis and experimental data generation reveals IRF9 as a crucial factor for accelerating interferon α-induced early antiviral signalling. FEBS J. 2010;277:4741–54. doi: 10.1111/j.1742-4658.2010.07880.x. [DOI] [PubMed] [Google Scholar]
- 43.Vera J, Rateitschak K, Lange F, Kossow C, Wolkenhauer O, Jaster R. Systems biology of JAK-STAT signalling in human malignancies. Prog Biophys Mol Biol. 2011;106:426–34. doi: 10.1016/j.pbiomolbio.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 44.Rateitschak K, Karger A, Fitzner B, Lange F, Wolkenhauer O, Jaster R. Mathematical modelling of interferon-gamma signalling in pancreatic stellate cells reflects and predicts the dynamics of STAT1 pathway activity. Cell Signal. 2010;22:97–105. doi: 10.1016/j.cellsig.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 45.Quaiser T, Mönnigmann M. Systematic identifiability testing for unambiguous mechanistic modeling--application to JAK-STAT, MAP kinase, and NF-kappaB signaling pathway models. BMC Syst Biol. 2009;3:50. doi: 10.1186/1752-0509-3-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rybiński M, Gambin A. Model-based selection of the robust JAK-STAT activation mechanism. J Theor Biol. 2012;309:34–46. doi: 10.1016/j.jtbi.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 47.Lund EW. Guldberg and Waage and the law of mass action. J Chem Educ. 1965;42:548. doi: 10.1021/ed042p548. [DOI] [Google Scholar]
- 48.Michaelis L, Menten M. Die Kinetik der Invertinwirkung. Biochem Z. 1913;49:333–69. [Google Scholar]
- 49.Quaiser T, Dittrich A, Schaper F, Mönnigmann M. A simple work flow for biologically inspired model reduction--application to early JAK-STAT signaling. BMC Syst Biol. 2011;5:30. doi: 10.1186/1752-0509-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinrich R, Neel BG, Rapoport TA. Mathematical models of protein kinase signal transduction. Mol Cell. 2002;9:957–70. doi: 10.1016/S1097-2765(02)00528-2. [DOI] [PubMed] [Google Scholar]
- 51.Sivakumaran S, Hariharaputran S, Mishra J, Bhalla US. The Database of Quantitative Cellular Signaling: management and analysis of chemical kinetic models of signaling networks. Bioinformatics. 2003;19:408–15. doi: 10.1093/bioinformatics/btf860. [DOI] [PubMed] [Google Scholar]
- 52.Le Novère N, Bornstein B, Broicher A, Courtot M, Donizelli M, Dharuri H, et al. BioModels Database: a free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 2006;34(Database issue):D689–91. doi: 10.1093/nar/gkj092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C, Donizelli M, Rodriguez N, Dharuri H, Endler L, Chelliah V, et al. BioModels Database: An enhanced, curated and annotated resource for published quantitative kinetic models. BMC Syst Biol. 2010;4:92. doi: 10.1186/1752-0509-4-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, Kitano H, et al. SBML Forum The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19:524–31. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 55.Brysha M, Zhang JG, Bertolino P, Corbin JE, Alexander WS, Nicola NA, et al. Suppressor of cytokine signaling-1 attenuates the duration of interferon gamma signal transduction in vitro and in vivo. J Biol Chem. 2001;276:22086–9. doi: 10.1074/jbc.M102737200. [DOI] [PubMed] [Google Scholar]
- 56.Hanada T, Yoshida T, Kinjyo I, Minoguchi S, Yasukawa H, Kato S, et al. A mutant form of JAB/SOCS1 augments the cytokine-induced JAK/STAT pathway by accelerating degradation of wild-type JAB/CIS family proteins through the SOCS-box. J Biol Chem. 2001;276:40746–54. doi: 10.1074/jbc.M106139200. [DOI] [PubMed] [Google Scholar]
- 57.Sakamoto H, Yasukawa H, Masuhara M, Tanimura S, Sasaki A, Yuge K, et al. A Janus kinase inhibitor, JAB, is an interferon-gamma-inducible gene and confers resistance to interferons. Blood. 1998;92:1668–76. [PubMed] [Google Scholar]
- 58.Song MM, Shuai K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J Biol Chem. 1998;273:35056–62. doi: 10.1074/jbc.273.52.35056. [DOI] [PubMed] [Google Scholar]
- 59.Zi Z, Cho K-H, Sung M-H, Xia X, Zheng J, Sun Z. In silico identification of the key components and steps in IFN-gamma induced JAK-STAT signaling pathway. FEBS Lett. 2005;579:1101–8. doi: 10.1016/j.febslet.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 60.Shudo E, Yang J, Yoshimura A, Iwasa Y. Robustness of the signal transduction system of the mammalian JAK/STAT pathway and dimerization steps. J Theor Biol. 2007;246:1–9. doi: 10.1016/j.jtbi.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 61.Kitano H. Towards a theory of biological robustness. Mol Syst Biol. 2007;3:137. doi: 10.1038/msb4100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saltelli A, Ratto M, Tarantola S, Campolongo F. Sensitivity analysis for chemical models. Chem Rev. 2005;105:2811–28. doi: 10.1021/cr040659d. [DOI] [PubMed] [Google Scholar]
- 63.Charzyńska A, Nałecz A, Rybiński M, Gambin A. Sensitivity analysis of mathematical models of signaling pathways. BioTechnol. 2012;93:291–308. [Google Scholar]
- 64.Krause CD, Mei E, Xie J, Jia Y, Bopp MA, Hochstrasser RM, et al. Seeing the light: preassembly and ligand-induced changes of the interferon gamma receptor complex in cells. Mol Cell Proteomics. 2002;1:805–15. doi: 10.1074/mcp.M200065-MCP200. [DOI] [PubMed] [Google Scholar]
- 65.Schuster B, Meinert W, Rose-John S, Kallen K-J. The human interleukin-6 (IL-6) receptor exists as a preformed dimer in the plasma membrane. FEBS Lett. 2003;538:113–6. doi: 10.1016/S0014-5793(03)00154-6. [DOI] [PubMed] [Google Scholar]
- 66.Krause CD, Lavnikova N, Xie J, Mei E, Mirochnitchenko OV, Jia Y, et al. Preassembly and ligand-induced restructuring of the chains of the IFN-gamma receptor complex: the roles of Jak kinases, Stat1 and the receptor chains. Cell Res. 2006;16:55–69. doi: 10.1038/sj.cr.7310008. [DOI] [PubMed] [Google Scholar]
- 67.Soebiyanto RP, Sreenath SN, Qu CK, Loparo KA, Bunting KD. Complex systems biology approach to understanding coordination of JAK-STAT signaling. Biosystems. 2007;90:830–42. doi: 10.1016/j.biosystems.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mesarovic M, Macko D, Takahara Y. Theory of Hierarchical Multilevel System, Academic Press, 1970. [Google Scholar]
- 69.Dojer N, Gambin A, Mizera A, Wilczyński B, Tiuryn J. Applying dynamic Bayesian networks to perturbed gene expression data. BMC Bioinformatics. 2006;7:249. doi: 10.1186/1471-2105-7-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaderali L, Dazert E, Zeuge U, Frese M, Bartenschlager R. Reconstructing signaling pathways from RNAi data using probabilistic Boolean threshold networks. Bioinformatics. 2009;25:2229–35. doi: 10.1093/bioinformatics/btp375. [DOI] [PubMed] [Google Scholar]
- 71.Papin JA, Palsson BO. The JAK-STAT signaling network in the human B-cell: an extreme signaling pathway analysis. Biophys J. 2004;87:37–46. doi: 10.1529/biophysj.103.029884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schilling CH, Letscher D, Palsson BO. Theory for the systemic definition of metabolic pathways and their use in interpreting metabolic function from a pathway-oriented perspective. J Theor Biol. 2000;203:229–48. doi: 10.1006/jtbi.2000.1073. [DOI] [PubMed] [Google Scholar]
- 73.Rybiński M. Analysis of mathematical models of signalling pathways. M.Sc. thesis, Faculty of Mathematics, Informatics and Mechanics, University of Warsaw, 2008. [Google Scholar]
- 74.Courtot M, Juty N, Knüpfer C, Waltemath D, Zhukova A, Dräger A, et al. Controlled vocabularies and semantics in systems biology. Mol Syst Biol. 2011;7:543. doi: 10.1038/msb.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]