

Abstract
The JAK-STAT pathway is a key regulator of tissue size in Drosophila melanogaster. Here we provide an overview of its roles in processes that regulate the size of Drosophila imaginal discs, epithelia of diploid cells that proliferate and acquire specific fates in the larvae and that become functional in the adult. Drosophila has a single JAK and a single STAT gene, which has facilitated genetic dissection of this pathway. Moreover, the sophisticated genetic tools available in flies for clonal growth assays have made Drosophila an ideal organism in which to dissect the multiple roles of the JAK-STAT pathway in growth control. Studies in flies have revealed JAK-STAT pathway activity as a central node for diverse signals that control proliferation and mass accumulation. In addition, recent work has establish a new role for the pathway in cell competition, a process thought to be akin to the early stages of transformation in which more robust cells kill and take the place of less robust ones.
Keywords: Drosophila, Hippo, JAK-STAT, JNK, Myc, Notch, PRC, Wingless, proliferation
Background
With only one JAK and one STAT, the Janus kinase/signal transducer and activator of transcription (JAK-STAT) pathway has lower complexity in the fruit fly Drosophila melanogaster than in mammals.1 There are 3 interleukin-6 (IL-6)-like cytokines—Unpaired (Upd), also called Outstretched, Upd2, and Upd3. These ligands bind to the receptor Domeless (Dome), which is homologous to gp130, the common chain for the IL-6 receptor family (Fig. 1).2-5 There is a second transmembrane receptor, eye transformer, also called latran, which forms heterodimers with Dome and antagonizes JAK-STAT signaling.6,7 The sole Drosophila JAK, called Hopscotch (Hop), is most similar to JAK2, and the sole STAT, called STAT92E, is most homologous to STATs 3 and 5.8-10 Activated STAT92E dimers modulate expression of target genes, the best characterized of which is Socs36E, which encodes a negative regulator.11 The reduced genetic complexity of the pathway in Drosophila and the observation that numerous human disease genes are conserved in flies,12 make Drosophila an excellent model for studying this pathway.
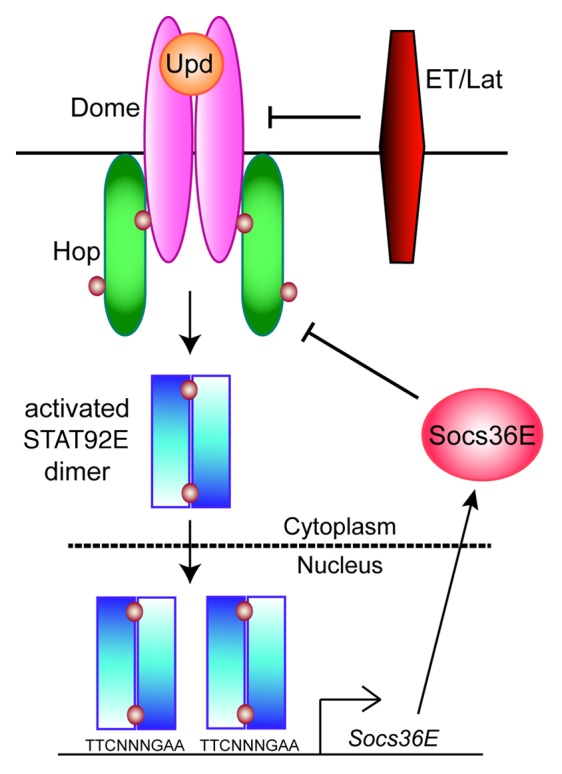
Figure 1. The Drosophila JAK-STAT pathway. Three Unpaired (Upd) ligands here collectively referred to as Upd (orange) activate a dimeric receptor Domeless (Dome) (magenta). This results in activation of the JAK Hopscotch (Hop) (green), leading to tyrosine phosphorylation of Dome. The phosphorylated receptor/JAK complex phosphorylates STAT92E dimer (blue) on Y711, generating an active STAT92E dimer. A consensus TTCNNNGAA site is bound by the activated STAT92E dimer, leading altered gene expression. One of the best-characterized STAT92E target genes is Socs36E, encoding a negative regulator of JAK/receptor activity (pink). A second receptor Eye Transformer (ET) (also called Latran [Lat]), referred to as ET/Lat (red), forms heterodimers with Dome and inhibits JAK-STAT signaling. Brown circles represent phospho-tyrosine residues.
It is well established that dominant-active mutations in JAK2 result in human leukemia and myeloproliferative disorders.13,14 In addition, sustained STAT3 signaling is linked to tumorigenesis in mouse models and a dozen types of human cancer, including all classes of carcinoma.15-17 Cytokine signaling is also important for normal organ size during development as mice deficient for SOCS2, a negative regulator of growth hormone signaling, exhibit gigantism.18 Roles of the JAK-STAT pathway in growth control have been well described in Drosophila. With the advantages of powerful genetic approaches and in vivo clonal growth assays, studies in Drosophila have advanced our knowledge of the importance of this pathway during development, homeostasis and transformation. In this review, we discuss the current understanding of the functions of the JAK-STAT pathway in the growth of imaginal discs.
Growth Control During Development
Imaginal discs are comprised of epithelial cells that give rise to the cuticular structures of the adult, such as compound eyes and wings. Each imaginal disc is formed from a small number of cells (e.g., 50 cells in the case of the wing disc) that are specified in the embryonic ectoderm.19 Once the embryo hatches into the larva, which promptly begins to feed, the discs start to grow rapidly. Larval development lasts ~4 d at 25 °C and consists of three distinct periods called instars, each separated by a molt. Imaginal disc cells proliferate exponentially during larval development to give rise to thousands of cells (e.g., 50 000 cells in the case of the wing disc) at the end of third instar.20,21 Most of the cells in the larva are polyploid, undergoing endoreplication (S phase but not cytokinesis) and increasing their volume substantially. In contrast, imaginal cells are diploid and undergo both S and M phase.
Growth in wild-type eye discs is mediated by an “organizer” which forms during second instar at the dorsal–ventral (D–V) midline through the actions of Iroquois-Complex (Iro-C) genes, which repress the O-glycosyltransferase Fringe (Fng) to the ventral domain (Fig. 2). This juxtaposition of ventral fng+ cells and dorsal fng- cells leads to activation of Notch signaling.22-24 Notch signaling at the D–V midline is required for appropriate disc growth, and the role of Notch in this process is at least partially dependent on JAK-STAT signaling. The upd gene is induced cell-autonomously by Notch, and JAK-STAT signaling has been shown to act downstream of Notch in this context.25,26 For example, misexpression of upd rescued the growth defects observed by decreased Notch signaling and, reciprocally, reduction in upd levels suppressed the eye over-growth caused by increased Notch signaling.25,26
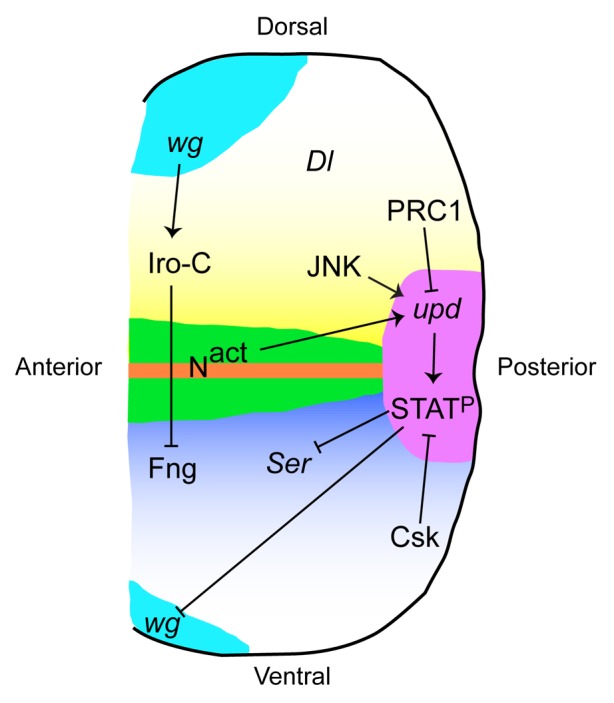
Figure 2. The role of the Drosophila JAK-STAT pathway in eye development. During the second larval instar, Iro-C genes are induced by Wg signaling in the dorsal domain of the eye disc. Iro-C proteins repress Fng expression such that only ventral cells express Fng. This creates a border of Fng-expressing and non-Fng-expressing cells at the midline of the disc, leading to Notch signaling there. Notch induces expression of upd at the posterior midline of the eye disc. JNK signaling can also induce upd during invasive tumor formation. upd is presumed to be repressed by PRC1 in cells outside of the posterior midline. Upd leads to activation of STAT92E (STATP), which represses the Notch ligand Ser to the ventral domain and Wg to the anterior margin. Csk negatively regulates STATP, either through Src or through Dome/Hop. Delta (Dl), another Notch ligand in Drosophila, is expressed only in the dorsal domain.
JAK-STAT pathway is one of the earliest signaling events known to promote eye development; in the eye disc, upd can be detected as early as first instar, approximately 36 h after egg laying.27 As expected by the timing of Upd expression, JAK-STAT pathway activity is detected broadly in young imaginal discs.27-29 Consistent with the expression pattern of Upd, a viable hypomorphic allele in the upd locus outstretched (os) has a small eye phenotype that can be rescued by activating STAT92E.30,31 Indeed, loss of Stat92E is deleterious to eye development. Animals devoid of Stat92E function in the eye disc lack an adult eye.27,31
One important function of JAK-STAT signaling is in formation of the eye field; it promotes proliferation and growth of cells in the eye field while cell-autonomously repressing wingless (wg), which specifies head cuticle fate (see below).27,29,30,32,33 Interestingly, JAK-STAT signaling has been shown to act downstream of Notch signaling. Specifically, activated STAT92E represses expression of Serrate (Ser),34 which encodes a Notch ligand homologous to mammalian Jagged and which is normally restricted to the ventral eye (Fig. 2).22-24 The loss of Stat92E in clones in the dorsal eye results in ectopic expression of Ser there and over-growth of this compartment.34 Finally, the expression of upd prior to the reported formation of the D–V organizer (described above) suggests that either the organizer is actually active in first instar or that upd can be induced independently of Notch signaling. In fact, one study did report an early role of upd in formation of the organizer, implying that JAK-STAT activity can also function upstream of Notch in the eye disc.35
Studies from several labs have since shown that STAT92E is a central regulator of eye size. The functional effects of sustained activation of the JAK-STAT pathway in imaginal discs was forecasted fortuitously by transposon insertion in the Om(1E) gene, a paralog of upd, in the related species D. ananassae, resulting in increased Om(1E) expression and outgrowths in the adult eye.36 Subsequently, the role of sustained JAK-STAT pathway activation in tissue growth was confirmed by targeted mis-expression of upd in the developing eye disc of D. melanogaster; GMR-upd transgenic animals have enlarged eye imaginal discs, resulting in a dramatically enlarged compound adult eye.30 A characterization of GMR-upd and similar transgenic animals revealed that Upd acts as a mitogen for undifferentiated eye cells.30,32 The increased production of Upd ligand in GMR-upd animals expands the number of eye progenitor cells without affecting their patterning, leading to a distinctly larger eye that is otherwise patterned normally.30 The GMR-upd enlarged eye phenotype can be largely suppressed by halving the genetic dose of Stat92E, suggesting that activation of STAT92E downstream of Upd is primarily responsible for the overgrowth.30,32 Shortly after the publication of the GMR-upd animal, another study reported a similar enlarged-eye phenotype resulting from inactivation of C-terminal src kinase (Csk) function in the eye disc. STAT92E is autonomously activated in Csk−/− clones, suggesting that JAK-STAT signaling plays an important role in the Csk−/− over-grown eye.37 Indeed, this phenotype is largely suppressed by reducing Stat92E expression levels.37 The activation of STAT92E in Csk−/− clones is likely due to upregulation of Src kinases in the absence of negative regulation by Csk (as opposed to Csk-dependent activation of Dome or Hop), but this has not been formally shown. Of note, mutations in endosomal sorting complex required for transport (ESCRT) components tsg101 and vps25, which trap the Notch receptor in an activated state, also result in cell-autonomous increases in upd expression and dramatic eye over-growth, a phenotype that depends on STAT92E activation.38-40 In fact, a recent study of Notch-dependent hyperplastic wing imaginal disc tissue has revealed that all three upd ligands are direct targets of Notch signaling and their loci interact directly with the Notch transcriptional effector Suppressor of Hairless.41 As mentioned above, although Notch is activated along the entire midline, upd is induced only at the posterior margin at the midline, i.e., only in a small region of active Notch signaling, indicating that other factors must repress its expression elsewhere in the disc. Interestingly upd genes, particular upd3, are repressed by polycomb-group repressive complex 1 (PRC1) and are ectopically expressed in mutations in any PRC1 component, leading to eye-overgrowth that is suppressed by lowering the dose of Stat92E.42,43 Finally, in the RasV12/scribble (scrib)−/− metastatic tumor model, Upd is upregulated by Jun N-terminal kinase (JNK) signaling, which leads to STAT92E activation in both the tumor and adjacent cells and is required for metastasis of the tumor from the eye disc to the ventral nerve cord.44 Thus, restricting induction of Upd to the posterior midline by balancing Notch activation with PRC1 repression during development and JNK activation during tumorigenesis is important to prevent over-growth (Fig. 2).
JAK-STAT Signaling and Proliferation
Given the significant role of the JAK-STAT pathway in specification of eye size, a key issue is how this pathway controls proliferation. FACS analysis of cells from eye and wing discs with sustained JAK-STAT signaling revealed that these cells appear to progress through G1/S and G2/M cell cycle checkpoints faster than control disc cells.29,30 These results raise the possibility that JAK-STAT signaling controls the expression or activation of factors required for cell cycle progression. In fact, one study reported that cyclin-dependent kinase 4 (Cdk4) functions between Hop and STAT92E in the embryo.45 However, in Drosophila Cdk4 is primarily a regulator of cellular growth (see below) and is dispensable for proliferation.46,47 Cyclin B (CycB), which is required for G2/M progression in the embryo,48 was elevated in a cell-autonomous manner in clones with increased JAK-STAT signaling. It is not known if CycB is a target of JAK-STAT signaling or if increased CycB in JAK-STAT pathway gain-of-function clones simply reflects increased proliferation rates.33 In fact, two independent genetic screens failed to reveal a cell cycle gene that strongly modified the GMR-upd phenotype.30,33 While this could be due to the possibility that cell cycle genes are largely dosage-insensitive in the GMR-upd background, expression profiling of GMR-upd eye discs also did not reveal any potential candidates.34 In addition, three independent whole-genome RNAi screens did not identify a connection between JAK-STAT signaling and genes known to regulate proliferation.49-51
An anti-proliferative role has also been reported for STAT92E. One study reported that Stat92E−/− clones induced late in larval wing development grew to larger sizes than their sibling (+/+) clones.33 The observation that hop−/− clones did not display the same overgrowth phenotype led to the model that in late larval wing discs, STAT92E acts non-canonically (i.e., independently of Upd or Hop) to constrain proliferation.33 This study postulates that Drosophila STAT92E contains both the pro-proliferative function of STAT3 and the anti-proliferative function of STAT1 and that evolutional forces subsequently assigned these roles to distinct mammalian STAT proteins. This paper raises important questions that need to be addressed. First, are there distinct regions of STAT92E that mediate these opposite effects on proliferation? Second, what factors are the pro-proliferative and anti-proliferative targets of STAT92E? Finally, the proposed switch from pro-proliferative to anti-proliferative occurs within a 24-h window. What changes occurs in STAT92E or the chromatin of late larval wing imaginal disc cells to facilitate this switch? Our current understanding of JAK-STAT regulation of proliferation is limited, and more studies will be required at the molecular level to sufficiently answer these questions.
JAK-STAT Signaling and Cell Competition
Local interactions between cells influence their growth and their ability to contribute to the adult. Some of these interactions have been revealed by studying “cell competition”, a process that has been best studied in the Drosophila wing disc52-54 but that also exists in mammals.55,56 In the last 10 years, the field of cell competition has exploded (reviewed in refs. 57 and 58), but a consensus on definitions for each type of competitive interaction has not yet been achieved. In this review, we will use the term “cell competition” to mean the context-specific behavior of cells of a particular genotype: they are killed (out-competed) when surrounded by wild-type cells but viable when placed in the context of slower-growing cells. The first example of cell competition was observed with Minutes (M), dominant mutations in ribosomal protein (Rp) genes that are lethal when homozygous (M/M) but produce viable, slow-developing animals when heterozygous (M/+).52-54 M/+ cells exhibit distinct outcomes depending on the local environment; M/+ clones are viable when residing in a homotypic environment (i.e., when they are surrounded by M/+ cells) but die when grown in the presence of wild-type (+/+) cells. These studies also revealed that death of M/+ cells is associated with proliferation of wild-type cells. The wild-type cells (termed “winners”) subsequently occupy the space of the M/+ cells (termed “losers”), which are eliminated by the winners through cell death to ensure maintenance of normal tissue size. It has been subsequently shown that differences in levels of other growth-regulatory genes such as dMyc, a transcription factor that regulates expression of genes controlling proliferation, cellular growth, and ribosome biogenesis, elicit similar types of competitive interactions; clones with lower levels of dMyc become losers, which are killed by winners that have normal levels of dMyc.59-61
Knowing the dependence of proper cell growth and tissue development on STAT92E activity, clonal growth assays were employed to assess whether modulating the levels of JAK-STAT signaling could induce competitive interactions. Stat92E−/− clones and their wild-type (+/+) sibling clones were induced by FLP/FRT-mediated mitotic recombination62 early in embryonic development and clone size was measured in wing and eye discs after a defined period. Since disc cells are epithelial and remain associated after mitosis, differences in clone size reflect differential growth rates.63 If Stat92E were not required for clonal growth, Stat92E−/− clone areas should comprise ~50% of the total clone area. In one study, control FRT82B wild-type clones and their sibling clones grew to equal sizes and were each ~50% of the total clone area.29 By contrast, Stat92E−/− clones comprised only ~5% of the total clone area in the disc. (In another study, Stat92E−/− clones induced during early larval development were larger [40% of the total clone area].33 The discrepancy in Stat92E−/− clone size is presumably due to the use of weaker Stat92E alleles in the latter study.31,33) By contrast, Stat92E−/− clones in a mosaic background underwent caspase-dependent but JNK-independent cell death and were extruded from the epithelium.29 However, when programmed cell death was blocked in the Stat92E−/− cells, they grew to the same size as sibling clones.29 By contrast, clones lacking dmyc or ribosomal genes like Rpl135 cannot grow even when death is inhibited.64,65 This may represent an important distinction between the function of activated STAT92E and dMyc in losers. The context-specific behavior of cells with reduced JAK-STAT signaling was revealed when Stat92E−/− clones were given a growth advantage. When induced in a Minute background, Stat92E−/− clones grew to large sizes.29 This result reveals that Stat92E−/− cells die in a wild-type background because they have become losers and are out-competed by the more robust wild-type winner cells. Similar results—that loss of Stat92E reduces cellular fitness and renders cells losers—have been observed in a scrib−/− tumor suppressor model in Drosophila.66
Cells with increased dMyc or increased Wingless (Wg) signaling become “supercompetitors”, which we define as a clone of cells overexpressing a particular factor that causes neighboring wild-type cells to experience a growth disadvantage.59,61,67-70 Of note, clones with increased dMyc expression kill losers up to 10 cells away.59 Clonal growth assays, such as the two-clone assay,59,71 which serve as a direct measurement of supercompetitor behavior, revealed that clones with sustained JAK-STAT pathway activation become winners, acquire supercompetitor characteristics, and can kill losers located several cell diameters away through non-autonomous induction of apoptosis.29 This study also demonstrated that, like with dMyc, cells with activated STAT92E require the pro-apoptotic gene head involution defective (hid) to kill surrounding neighbors and achieve supercompetitor status. These results suggest a link between STAT92E and dMyc or between STAT92E and the Wg pathway. Surprisingly, however, no link was found between JAK-STAT signaling and either dmyc mRNA, dMyc protein, or targets of the Hippo pathway,29 which regulate dMyc levels.68,69 In addition, clonal mis-expression of dMyc did not activate STAT, nor did clonal mis-expression of Crumbs,72,73 which is a target of JAK-STAT signaling in the embryo74 and an upstream regulator of Hippo pathway signaling. Finally, hyperactivation of JAK-STAT signaling had no effect on Wg signaling and, reciprocally, Wg did not modulate STAT92E activity.29 These results strongly suggest that at least in the wing imaginal disc JAK-STAT pathway activity functions in parallel to dMyc and Wg in growth and cell competition (Fig. 3).
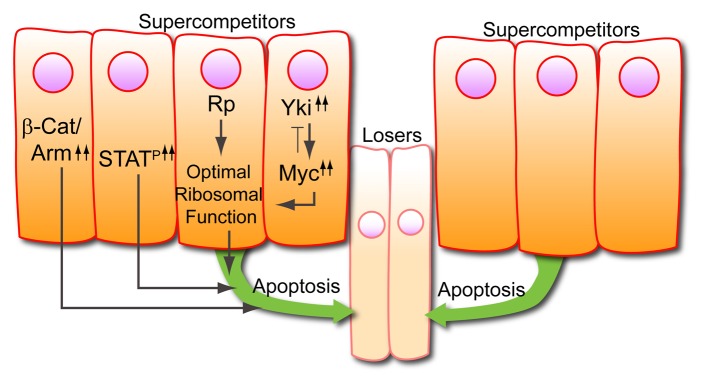
Figure 3. Super-competitors. Super-competitor status can be confirmed by sustained activation of Wg signaling through β-catenin/Armadillo (β-Cat/Arm) or of JAK-STAT signaling (STATP). It can also be achieved through increased ribosome biogenesis, increased Yki expression, which activates the dmyc gene, or through increased dMyc levels. All of these lead to cells (super-competitors) that non-autonomously induce death of neighboring wild-type cells (losers).
JAK-STAT Signaling and Cellular Growth
Tissue growth occurs as a result of both cellular growth (also called mass accumulation) and subsequent proliferation. Studies from Drosophila imaginal discs have shown that cell division and cellular growth are regulated independently.63,75 It follows that in order to get overgrown imaginal tissue, both proliferation and cellular growth must be accelerated concomitantly. Numerous factors affect cell size, including dMyc, CycD/Cdk4, and Hippo.76 Since JAK-STAT signaling in the eye imaginal disc is causal for tissue overgrowth, it is of great interest to unravel how this pathway regulates cellular growth. We define cellular growth as the net production of new proteins, which can occur by a variety of means, including but not limited to increased de novo synthesis of ribosomes (i.e., ribosome biogenesis) or accelerated translation on existing ribosomes. Increased cell size can result from increased cellular growth and can be measured by the forward scatter parameter on a flow cytometer.64 FACS analysis of cells with sustained JAK-STAT signaling revealed no change in cell size.29,30 Consistent with this, there was no change in cell density in clones with activated JAK-STAT signaling.33 The unaltered cell size in cells with sustained JAK-STAT signaling is likely due to the fact that cell division rates are also increased when this pathway is hyper-activated.29,30,32,33 Furthermore, JAK-STAT signaling does not induce genes such as nop5, Nop60B, and Tif-1A (which, incidentally, are targets of dMyc) that regulate de novo ribosome biosynthesis.29 As mentioned above, the JAK-STAT pathway also does not interact with dMyc, which upregulates ribosome biogenesis,65 or regulators of dMyc.68,69 These data suggest that JAK-STAT signaling does not regulate cellular growth by means of increasing ribosome biogenesis. As mentioned above, one study reported that CycD/Cdk4 functions to promote proliferation and acts between Hop and STAT92E in JAK-STAT signaling. However, there are lines of evidence that suggest that this conclusion needs to be re-examined, particularly with respect to JAK-STAT signaling in imaginal discs. First, several groups have reported that CycD/Cdk4 is not required for proliferation but instead promotes growth through mitochondrial biogenesis.46,47,77 Second, we have not found a link between mitochondrial functions and JAK-STAT signaling (Rodrigues and Bach, unpublished data). Taken together, how JAK-STAT signaling controls cellular growth at the molecular level remains a critical area of investigation for the field.
Concluding Remarks
In summary, these studies have revealed that the JAK-STAT pathway is a central regulator of tissue size in Drosophila imaginal discs. In the eye disc, upd is subject to positive and negative regulation, but only Notch-mediated induction of upd has been delineated at the genetic level. Future work should reveal how JNK activates and how PRC1 represses the upd locus. Furthermore, the inhibitory effects of Csk on activated STAT92E also need to be further explored. Despite the central role that JAK-STAT signaling plays in proliferation and cellular growth, the targets of STAT92E required for these processes are yet unknown and need to be determined in future studies. Finally, the recent work showing that cells with sustained JAK-STAT activity become super-competitors raises several outstanding questions, including: (1) Do STAT92E winners secrete a Notum-like molecule, which inhibits neighboring cells from transducing Upd signals? (2) What are non-autonomous signals downstream of STAT92E that cause death in losers? (3) Are these signals regulated by other factors involved in cell competition such as dMyc and Wg signaling? Using Drosophila as a model to study how JAK-STAT signaling regulates proliferation, cellular growth, and cell competition is poised to shed light on mechanisms of tumorigenesis in mammals.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/25408
References
- 1.Arbouzova NI, Zeidler MP. JAK/STAT signalling in Drosophila: insights into conserved regulatory and cellular functions. Development. 2006;133:2605–16. doi: 10.1242/dev.02411. [DOI] [PubMed] [Google Scholar]
- 2.Harrison DA, McCoon PE, Binari R, Gilman M, Perrimon N. Drosophila unpaired encodes a secreted protein that activates the JAK signaling pathway. Genes Dev. 1998;12:3252–63. doi: 10.1101/gad.12.20.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hombría JC, Brown S, Häder S, Zeidler MP. Characterisation of Upd2, a Drosophila JAK/STAT pathway ligand. Dev Biol. 2005;288:420–33. doi: 10.1016/j.ydbio.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 4.Agaisse H, Perrimon N. The roles of JAK/STAT signaling in Drosophila immune responses. Immunol Rev. 2004;198:72–82. doi: 10.1111/j.0105-2896.2004.0133.x. [DOI] [PubMed] [Google Scholar]
- 5.Brown S, Hu N, Hombría JC. Identification of the first invertebrate interleukin JAK/STAT receptor, the Drosophila gene domeless. Curr Biol. 2001;11:1700–5. doi: 10.1016/S0960-9822(01)00524-3. [DOI] [PubMed] [Google Scholar]
- 6.Makki R, Meister M, Pennetier D, Ubeda JM, Braun A, Daburon V, et al. A short receptor downregulates JAK/STAT signalling to control the Drosophila cellular immune response. PLoS Biol. 2010;8:e1000441. doi: 10.1371/journal.pbio.1000441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kallio J, Myllymäki H, Grönholm J, Armstrong M, Vanha-aho LM, Mäkinen L, et al. Eye transformer is a negative regulator of Drosophila JAK/STAT signaling. FASEB J. 2010;24:4467–79. doi: 10.1096/fj.10-162784. [DOI] [PubMed] [Google Scholar]
- 8.Binari R, Perrimon N. Stripe-specific regulation of pair-rule genes by hopscotch, a putative Jak family tyrosine kinase in Drosophila. Genes Dev. 1994;8:300–12. doi: 10.1101/gad.8.3.300. [DOI] [PubMed] [Google Scholar]
- 9.Hou XS, Melnick MB, Perrimon N. Marelle acts downstream of the Drosophila HOP/JAK kinase and encodes a protein similar to the mammalian STATs. Cell. 1996;84:411–9. doi: 10.1016/S0092-8674(00)81286-6. [DOI] [PubMed] [Google Scholar]
- 10.Yan R, Small S, Desplan C, Dearolf CR, Darnell JE., Jr. Identification of a Stat gene that functions in Drosophila development. Cell. 1996;84:421–30. doi: 10.1016/S0092-8674(00)81287-8. [DOI] [PubMed] [Google Scholar]
- 11.Callus BA, Mathey-Prevot B. SOCS36E, a novel Drosophila SOCS protein, suppresses JAK/STAT and EGF-R signalling in the imaginal wing disc. Oncogene. 2002;21:4812–21. doi: 10.1038/sj.onc.1205618. [DOI] [PubMed] [Google Scholar]
- 12.Bier E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat Rev Genet. 2005;6:9–23. doi: 10.1038/nrg1503. [DOI] [PubMed] [Google Scholar]
- 13.Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffé M, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–12. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 14.Levine RL. Janus kinase mutations. Semin Oncol. 2009;36(Suppl 1):S6–11. doi: 10.1053/j.seminoncol.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/S0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 16.Darnell JE. Validating Stat3 in cancer therapy. Nat Med. 2005;11:595–6. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- 17.Migone TS, Lin JX, Cereseto A, Mulloy JC, O’Shea JJ, Franchini G, et al. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science. 1995;269:79–81. doi: 10.1126/science.7604283. [DOI] [PubMed] [Google Scholar]
- 18.Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA, et al. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature. 2000;405:1069–73. doi: 10.1038/35016611. [DOI] [PubMed] [Google Scholar]
- 19.Cohen SM. Imaginal disc development. Cold Spring Harbor, NY: Cold Spring Harbor Press, 1993. [Google Scholar]
- 20.Bryant PJ, Simpson P. Intrinsic and extrinsic control of growth in developing organs. Q Rev Biol. 1984;59:387–415. doi: 10.1086/414040. [DOI] [PubMed] [Google Scholar]
- 21.Madhavan M, Schneiderman HA. Histological analysis of the dynamics of growth of imaginal discs and histoblast nests during the larval development of Drosophila melanogaster. Wilhelm Roux’s Arch. 1977;183:269–305. doi: 10.1007/BF00848459. [DOI] [PubMed] [Google Scholar]
- 22.Papayannopoulos V, Tomlinson A, Panin VM, Rauskolb C, Irvine KD. Dorsal-ventral signaling in the Drosophila eye. Science. 1998;281:2031–4. doi: 10.1126/science.281.5385.2031. [DOI] [PubMed] [Google Scholar]
- 23.Cho KO, Choi KW. Fringe is essential for mirror symmetry and morphogenesis in the Drosophila eye. Nature. 1998;396:272–6. doi: 10.1038/24394. [DOI] [PubMed] [Google Scholar]
- 24.Domínguez M, de Celis JF. A dorsal/ventral boundary established by Notch controls growth and polarity in the Drosophila eye. Nature. 1998;396:276–8. doi: 10.1038/24402. [DOI] [PubMed] [Google Scholar]
- 25.Chao JL, Tsai YC, Chiu SJ, Sun YH. Localized Notch signal acts through eyg and upd to promote global growth in Drosophila eye. Development. 2004;131:3839–47. doi: 10.1242/dev.01258. [DOI] [PubMed] [Google Scholar]
- 26.Reynolds-Kenneally J, Mlodzik M. Notch signaling controls proliferation through cell-autonomous and non-autonomous mechanisms in the Drosophila eye. Dev Biol. 2005;285:38–48. doi: 10.1016/j.ydbio.2005.05.038. [DOI] [PubMed] [Google Scholar]
- 27.Ekas LA, Baeg GH, Flaherty MS, Ayala-Camargo A, Bach EA. JAK/STAT signaling promotes regional specification by negatively regulating wingless expression in Drosophila. Development. 2006;133:4721–9. doi: 10.1242/dev.02675. [DOI] [PubMed] [Google Scholar]
- 28.Bach EA, Ekas LA, Ayala-Camargo A, Flaherty MS, Lee H, Perrimon N, et al. GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr Patterns. 2007;7:323–31. doi: 10.1016/j.modgep.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Rodrigues AB, Zoranovic T, Ayala-Camargo A, Grewal S, Reyes-Robles T, Krasny M, et al. Activated STAT regulates growth and induces competitive interactions independently of Myc, Yorkie, Wingless and ribosome biogenesis. Development. 2012;139:4051–61. doi: 10.1242/dev.076760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bach EA, Vincent S, Zeidler MP, Perrimon N. A sensitized genetic screen to identify novel regulators and components of the Drosophila janus kinase/signal transducer and activator of transcription pathway. Genetics. 2003;165:1149–66. doi: 10.1093/genetics/165.3.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ekas LA, Cardozo TJ, Flaherty MS, McMillan EA, Gonsalves FC, Bach EA. Characterization of a dominant-active STAT that promotes tumorigenesis in Drosophila. Dev Biol. 2010;344:621–36. doi: 10.1016/j.ydbio.2010.05.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai YC, Sun YH. Long-range effect of upd, a ligand for Jak/STAT pathway, on cell cycle in Drosophila eye development. Genesis. 2004;39:141–53. doi: 10.1002/gene.20035. [DOI] [PubMed] [Google Scholar]
- 33.Mukherjee T, Hombría JC, Zeidler MP. Opposing roles for Drosophila JAK/STAT signalling during cellular proliferation. Oncogene. 2005;24:2503–11. doi: 10.1038/sj.onc.1208487. [DOI] [PubMed] [Google Scholar]
- 34.Flaherty MS, Zavadil J, Ekas LA, Bach EA. Genome-wide expression profiling in the Drosophila eye reveals unexpected repression of notch signaling by the JAK/STAT pathway. Dev Dyn. 2009;238:2235–53. doi: 10.1002/dvdy.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutierrez-Aviño FJ, Ferres-Marco D, Dominguez M. The position and function of the Notch-mediated eye growth organizer: the roles of JAK/STAT and four-jointed. EMBO Rep. 2009;10:1051–8. doi: 10.1038/embor.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Juni N, Awasaki T, Yoshida K, Hori SH. The Om (1E) mutation in Drosophila ananassae causes compound eye overgrowth due to tom retrotransposon-driven overexpression of a novel gene. Genetics. 1996;143:1257–70. doi: 10.1093/genetics/143.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Read RD, Bach EA, Cagan RL. Drosophila C-terminal Src kinase negatively regulates organ growth and cell proliferation through inhibition of the Src, Jun N-terminal kinase, and STAT pathways. Mol Cell Biol. 2004;24:6676–89. doi: 10.1128/MCB.24.15.6676-6689.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moberg KH, Schelble S, Burdick SK, Hariharan IK. Mutations in erupted, the Drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non-cell-autonomous overgrowth. Dev Cell. 2005;9:699–710. doi: 10.1016/j.devcel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 39.Vaccari T, Bilder D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev Cell. 2005;9:687–98. doi: 10.1016/j.devcel.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 40.Herz HM, Chen Z, Scherr H, Lackey M, Bolduc C, Bergmann A. vps25 mosaics display non-autonomous cell survival and overgrowth, and autonomous apoptosis. Development. 2006;133:1871–80. doi: 10.1242/dev.02356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Djiane A, Krejci A, Bernard F, Fexova S, Millen K, Bray SJ. Dissecting the mechanisms of Notch induced hyperplasia. EMBO J. 2013;32:60–71. doi: 10.1038/emboj.2012.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Classen AK, Bunker BD, Harvey KF, Vaccari T, Bilder D. A tumor suppressor activity of Drosophila Polycomb genes mediated by JAK-STAT signaling. Nat Genet. 2009;41:1150–5. doi: 10.1038/ng.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.González I, Simón R, Busturia A. The Polyhomeotic protein induces hyperplastic tissue overgrowth through the activation of the JAK/STAT pathway. Cell Cycle. 2009;8:4103–11. doi: 10.4161/cc.8.24.10212. [DOI] [PubMed] [Google Scholar]
- 44.Wu M, Pastor-Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature. 2010;463:545–8. doi: 10.1038/nature08702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X, Oh SW, Zheng Z, Chen HW, Shin HH, Hou SX. Cyclin D-Cdk4 and cyclin E-Cdk2 regulate the Jak/STAT signal transduction pathway in Drosophila. Dev Cell. 2003;4:179–90. doi: 10.1016/S1534-5807(03)00024-8. [DOI] [PubMed] [Google Scholar]
- 46.Meyer CA, Jacobs HW, Datar SA, Du W, Edgar BA, Lehner CF. Drosophila Cdk4 is required for normal growth and is dispensable for cell cycle progression. EMBO J. 2000;19:4533–42. doi: 10.1093/emboj/19.17.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Datar SA, Jacobs HW, de la Cruz AF, Lehner CF, Edgar BA. The Drosophila cyclin D-Cdk4 complex promotes cellular growth. EMBO J. 2000;19:4543–54. doi: 10.1093/emboj/19.17.4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knoblich JA, Lehner CF. Synergistic action of Drosophila cyclins A and B during the G2-M transition. EMBO J. 1993;12:65–74. doi: 10.1002/j.1460-2075.1993.tb05632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Müller P, Kuttenkeuler D, Gesellchen V, Zeidler MP, Boutros M. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature. 2005;436:871–5. doi: 10.1038/nature03869. [DOI] [PubMed] [Google Scholar]
- 50.Baeg GH, Zhou R, Perrimon N. Genome-wide RNAi analysis of JAK/STAT signaling components in Drosophila. Genes Dev. 2005;19:1861–70. doi: 10.1101/gad.1320705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bina S, Wright VM, Fisher KH, Milo M, Zeidler MP. Transcriptional targets of Drosophila JAK/STAT pathway signalling as effectors of haematopoietic tumour formation. EMBO Rep. 2010;11:201–7. doi: 10.1038/embor.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simpson P. Parameters of cell competition in the compartments of the wing disc of Drosophila. Dev Biol. 1979;69:182–93. doi: 10.1016/0012-1606(79)90284-7. [DOI] [PubMed] [Google Scholar]
- 53.Simpson P, Morata G. Differential mitotic rates and patterns of growth in compartments in the Drosophila wing. Dev Biol. 1981;85:299–308. doi: 10.1016/0012-1606(81)90261-X. [DOI] [PubMed] [Google Scholar]
- 54.Morata G, Ripoll P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev Biol. 1975;42:211–21. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- 55.Oertel M, Menthena A, Dabeva MD, Shafritz DA. Cell competition leads to a high level of normal liver reconstitution by transplanted fetal liver stem/progenitor cells. Gastroenterology. 2006;130:507–20, quiz 590. doi: 10.1053/j.gastro.2005.10.049. [DOI] [PubMed] [Google Scholar]
- 56.Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–22. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Beco S, Ziosi M, Johnston LA. New frontiers in cell competition. Dev Dyn. 2012;241:831–41. doi: 10.1002/dvdy.23783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levayer R, Moreno E. Mechanisms of cell competition: themes and variations. J Cell Biol. 2013;200:689–98. doi: 10.1083/jcb.201301051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–16. doi: 10.1016/S0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- 60.Li W, Baker NE. Engulfment is required for cell competition. Cell. 2007;129:1215–25. doi: 10.1016/j.cell.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 61.Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–29. doi: 10.1016/S0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- 62.Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–37. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- 63.Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–93. doi: 10.1016/S0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- 64.Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–90. doi: 10.1016/S0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7:295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- 66.Schroeder MC, Chen CL, Gajewski K, Halder G. A non-cell-autonomous tumor suppressor role for Stat in eliminating oncogenic scribble cells. Oncogene. 2012 doi: 10.1038/onc.2012.476. In press. [DOI] [PubMed] [Google Scholar]
- 67.Vincent JP, Kolahgar G, Gagliardi M, Piddini E. Steep differences in wingless signaling trigger Myc-independent competitive cell interactions. Dev Cell. 2011;21:366–74. doi: 10.1016/j.devcel.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ziosi M, Baena-López LA, Grifoni D, Froldi F, Pession A, Garoia F, et al. dMyc functions downstream of Yorkie to promote the supercompetitive behavior of hippo pathway mutant cells. PLoS Genet. 2010;6:e1001140. doi: 10.1371/journal.pgen.1001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neto-Silva RM, de Beco S, Johnston LA. Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev Cell. 2010;19:507–20. doi: 10.1016/j.devcel.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tyler DM, Li W, Zhuo N, Pellock B, Baker NE. Genes affecting cell competition in Drosophila. Genetics. 2007;175:643–57. doi: 10.1534/genetics.106.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu DC, Johnston LA. Control of wing size and proportions by Drosophila myc. Genetics. 2010;184:199–211. doi: 10.1534/genetics.109.110379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Badouel C, Garg A, McNeill H. Herding Hippos: regulating growth in flies and man. Curr Opin Cell Biol. 2009;21:837–43. doi: 10.1016/j.ceb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 73.Reddy BV, Irvine KD. The Fat and Warts signaling pathways: new insights into their regulation, mechanism and conservation. Development. 2008;135:2827–38. doi: 10.1242/dev.020974. [DOI] [PubMed] [Google Scholar]
- 74.Lovegrove B, Simões S, Rivas ML, Sotillos S, Johnson K, Knust E, et al. Coordinated control of cell adhesion, polarity, and cytoskeleton underlies Hox-induced organogenesis in Drosophila. Curr Biol. 2006;16:2206–16. doi: 10.1016/j.cub.2006.09.029. [DOI] [PubMed] [Google Scholar]
- 75.Weigmann K, Cohen SM, Lehner CF. Cell cycle progression, growth and patterning in imaginal discs despite inhibition of cell division after inactivation of Drosophila Cdc2 kinase. Development. 1997;124:3555–63. doi: 10.1242/dev.124.18.3555. [DOI] [PubMed] [Google Scholar]
- 76.Neto-Silva RM, Wells BS, Johnston LA. Mechanisms of growth and homeostasis in the Drosophila wing. Annu Rev Cell Dev Biol. 2009;25:197–220. doi: 10.1146/annurev.cellbio.24.110707.175242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Icreverzi A, de la Cruz AF, Van Voorhies WA, Edgar BA. Drosophila cyclin D/Cdk4 regulates mitochondrial biogenesis and aging and sensitizes animals to hypoxic stress. Cell Cycle. 2012;11:554–68. doi: 10.4161/cc.11.3.19062. [DOI] [PMC free article] [PubMed] [Google Scholar]