

Abstract
The canonical JAK-STAT signaling pathway transmits signals from the cell membrane to the nucleus, to regulate transcription of particular genes involved in development and many other physiological processes. It has been shown in Drosophila that JAK and STAT also function in a non-canonical mode, to regulate heterochromatin. This review discusses the non-canonical functioning of JAK and STAT, and its effects on biological processes. Decreased levels of activated JAK and increased levels of unphosphorylated STAT generate higher levels of heterochromatin. These higher heterochromatin levels result in suppression of hematopoietic tumor-like masses, increased resistance to DNA damage, and longer lifespan.
Keywords: JAK, STAT, non-canonical JAK-STAT signaling, Drosophila, U-STAT, aging, cancer, tumor suppression, mitochondria
Introduction
The Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway was first characterized in the early 1990s.1 Four JAK and seven STAT genes have been found in humans, one JAK and one STAT in Drosophila2,3 and four STATs but no JAKs in Dictyostelium.4-8 The JAK-STAT pathway is similar in Drosophila and mammals. Binding of extracellular ligands to transmembrane receptors induces the receptors to dimerize and to activate JAKs associated with these receptors. The activated JAKs phosphorylate tyrosine residues on the cytoplasmic tails of the receptors, which function as docking sites for cytoplasmic STAT proteins. The STATs are then phosphorylated on a critical tyrosine residue by the activated JAKS; once phosphorylated, the STATs dimerize and move to the nucleus, where they activate transcription of particular genes.9 This is the canonical JAK-STAT signaling pathway. The simplicity of the Drosophila system, with only one JAK and one STAT gene, has made it an excellent model system for studying this pathway.
Canonical JAK-STAT signaling is involved in many aspects of development and physiology by virtue of its direct transcriptional regulation of target genes.1,3 It turns out, however, that JAK and STAT function in non-canonical modes as well, in both flies and mammals.9,10
Non-Canonical Functions of JAK and STAT
STAT1, STAT3, STAT5, and STAT6 have been reported to function non-canonically in mammals. In 1997 Kumar et al. showed that STAT1 is required for TNFα plus actinomycin induced apoptosis of cultured mammalian cells.11 Cells deficient in STAT1 were resistant to apoptosis, but sensitivity was restored by wild-type STAT1 or a STAT1 variant unable to dimerize. The authors inferred from the results with the STAT1 variant that STAT1 might be functioning non-canonically under these circumstances.
Several non-canonical functions have been attributed to STAT3. In 2000, Wang et al. published data showing that a Src inhibitor prevents the activation of STAT3 by PDGF and PDGFR in Balb/c-3T3 cells, suggesting that STAT3 is activated by Src kinase in this case.12 In 2004, Silver et al. reported the results of immunostaining experiments indicating that phosphorylated STAT3 localizes to focal adhesions, in addition to its nuclear localization; furthermore, STAT3 was co-immunoprecipitated with focal adhesion molecules such as paxillin and focal adhesion kinase.13 In 2006, Ng et al. described a physical interaction between the microtubule binding protein stathmin and STAT3.14 In cells without STAT3, microtubules were disrupted; in cells with STAT3, microtubules were not disrupted, and a physical interaction between STAT3 and stathmin was demonstrated by immunoprecipitation. Transfection of cultured cells with STAT3 attenuated stathmin inhibition of microtubule polymerizaton, as did addition of STAT3 to an in vitro microtubule assembly assay containing stathmin. Thus, by binding to stathmin, STAT3 apparently prevents stathmin binding to microtubules, and promotes microtubule polymerization. Similar findings were described by Verma et al. in 2009.15 Also in 2009, two groups reported that STAT3 is found in mammalian mitochondria.16,17 Wegrzyn et al. localized STAT3 to a subcellular mitochondrial fraction via western blots, and found by means of immunoprecipitation that STAT3 is associated with electron transport chain complex I. Respiration rates were substantially reduced in mitochondria from cells lacking STAT3, but were restored in mitochondria from STAT−/STAT− cells had been reconstituted with wild-type STAT3 or with a STAT3 construct targeted only to mitochondria. Moreover, using mutant STAT3 constructs targeted to mitochondria, Wegrzyn et al. found that neither STAT3 phosphorylation at Tyr705 nor DNA binding nor dimerization was required for restoration of normal respiration rates, though phosphorylation of Ser727 was required. Gough et al. found that mitochondrial, but not nuclear, STAT3 is required for RasV12 transformation of murine and human cells. With immunoblotting, Gough et al. substantiated that STAT3 is found in the mitochondrial fraction; in addition, they showed mitochondrial STAT to be insensitive to proteinase K treatment in the absence of detergent, corroborating that STAT3 was inside of mitochondria. Using procedures similar to those of Wegrzyn et al., they found that neither STAT3 phosphorylation at Tyr705 nor DNA binding nor dimerization was required for its role in RasV12 transformation, though Ser727 phosphorylation was required. Gough et al. also reported that loss of STAT3 resulted in defects in electron transport chain function, though there were some differences between the two reports regarding the particular complexes affected.
In 2012, Lee et al. reported the results of immunostaining experiments indicating that STAT5A and STAT5B are localized to the Golgi apparatus in digitonin-permeabilized human pulmonary arterial endothelial cells (dp-HPAEC).18 GFP-tagged STAT5A appeared to localize to the Golgi apparatus. In 2013, Lee et al. reported the localization of STAT5A to endoplasmic reticulum (ER) sheets in dp-HPEAC; immunoblots with cell extracts showed an association of STAT5a with two ER proteins.19 Deleterious effects on the endoplasmic reticulum were observed when STAT5 was knocked down by means of anti-STAT5 siRNA. These effects were seen in enucleated as well as nucleated cells, suggesting that STAT5 might be functioning non-canonically in the ER. In 2013, Sehgal described an association of STAT6-GFP, but not STAT3, with mitochondria in dp-HPAEC and digitonin-permeabilized human pulmonary smooth muscle cells.20
Non-canonical functions have been demonstrated for JAK2, as well. In 2006, Nilsson et al. reported that prolactin increases stability of the NF1-C2 transcription factor in a mouse mammary epithelial cell line.21 Using a JAK inhibitor, they determined that this stabilizing effect requires JAK2; this result was confirmed when JAK2 levels were decreased with a JAK2-sp.ecific siRNA. Western blot analysis indicated that phosphorylated JAK2 was present in the nucleus of these cells as well as the nuclei of three human breast cancer cell lines, and that NF1-C2 is associated with JAK in the nucleus. Using an in vitro proteasome assay and a JAK inhibitor, it was determined that the association of JAK2 with NF1-C2 prevents its degradation.
In 2009, Dawson et al. demonstrated with immunostaining, as well as subcellular fractionation and western blotting, that JAK2 can be found in the nuclei of human hematopoietic cells.22 Using an in vitro kinase assay followed by western blotting, they showed that JAK2 phosphorylates histone H3 at Tyr 41. Consistent with these results, when a hematopoietic cell line was incubated with JAK2 inhibitors there was a loss of H3Y41 phosphorylation. An unmodified H3 peptide encompassing amino acids 31–56 was found to bind heterochromatin protein 1α (HP1α) in vitro, and this binding was significantly decreased when the Y41 residue was phosphorylated. The biological significance of these observations was investigated by immunoprecipitation of chromatin from a hematopoietic cell line. Treatment with JAK2 inhibitors resulted in a downregulation of the hematopoietic developmental gene lmo2. This downregulation correlated with decreased levels of H3Y41ph and an increase in HP1α binding at sites surrounding the lmo2 transcriptional start site.
In Drosophila, a connection between JAK-STAT signaling and chromatin remodeling has suggested by the observation that the transcriptional repressor Ken recruits a nucleosome remodeling factor, NURF, to STAT/Ken mutual binding sequences to repress STAT-mediated transcription.10,23 Furthermore, the Drosophila protein inhibitor of activated STAT homolog (dPIAS) is known to be a suppressor of variegation, a heterochromatin-mediated phenomenon.24,25 In 2006, Shi et al. published work implicating Drosophila JAK in the regulation of heterochromatin.26 The remainder of this review will focus on what has been learned in Drosophila about the regulation of heterochromatin by JAK and STAT. We will also discuss how changes in heterochromatin levels affect the formation of hematopoietic tumor-like masses, genome stability, and lifespan.
A New Role for JAK
A constitutively activated JAK mutant, hopTum-l, causes hematopoietic tumor-like masses in Drosophila. Interestingly, constitutively activated human JAK2 is also associated with hematopoietic overgrowth and malignancies.27 The discovery of roles for JAK and STAT in heterochromatin regulation, began with a genetic screen in Drosophila to identify genes which affect the formation of the hematopoietic tumor-like masses induced by hopTum-l, that is, genes which modify the hopTum-l phenotype.26 A number of the modifiers identified were genes involved in chromatin modification, including heterochromatin protein 1 (HP1), the histone H3-K9 methyltransferase Su(var)3–9, and the histone deacetylase (HDAC) Rpd3. Flies carrying one copy of the activated JAK mutant hopTum-l and one copy of one of the following loss of function or hypomorphic mutants—Su(var)205, Su(var)3–9, or Rpd3—showed significantly higher numbers of tumors, though animals carrying a single copy of the Su(var)205, Su(var)3–9, or Rpd3 mutant without hopTum-l did not have these masses. HP1 and Su(var)3–9 are essential heterochromatin components that are necessary for heterochromatin-mediated gene silencing.
These results raised the question of how a decrease in heterochromatin components promotes an increase in hematopoietic tumors. Does JAK overactivation actively disrupt HP1/Su(var)3–9 gene silencing, which has been shown to act as a tumor-suppressive mechanism,28 or is there a competition between overactivated JAK and the HP1/Su(var)3–9 silencing system for access to STAT target genes that promote tumorigenesis? To determine whether JAK overactivation disrupts heterochromatic gene silencing, Shi et al. investigated the phenomenon of position effect variegation (PEV), in which active genes normally found in euchromatic regions of the genome can become silenced when placed in conjunction to heterochromatic regions. The silencing occurs in some cells but not in others, in what appears to be a stochastic manner. In Drosophila, PEV can be evaluated easily by inspecting eye color in appropriate fly strains. In wild-type flies, expression of the euchromatic white gene is required for normal red eye color. However, in certain fly strains, the white gene has been juxtaposed, by genetic inversion, to a heterochromatic region of the genome, resulting in suppression of white expression in some eye cells but not others; thus, the eyes are red and white, with a variegated appearance. The more heterochromatic gene silencing there is in such fly strains, the more white the eyes appear, while a de-repression of silencing results in more red color.
Loss-of-function mutations in even a single copy of HP1 or Su(var)3–9 are known to suppress PEV,29-31 that is, give less heterochromatic gene silencing. Shi et al. discovered that the constitutively active hopTum-l mutant suppressed PEV as well, while loss-of-function JAK mutants enhanced PEV, suggesting that overactivated JAK indeed disrupts heterochromatic gene silencing. Shi et al. then assessed more directly the effects of different levels of JAK on heterochromatin. Salivary glands were immunostained with anti-HP1 and anti-H3mK9 antibodies, and centromeric heterochromatin was examined. In support of the idea that overactivated JAK disrupts heterochromatin, Shi et al. found that larvae carrying a hopTum-l (JAK gain-of-function) mutation, showed much lower levels of HP1 and H3mK9 at centromeric heterochromatin in comparison with wild-type larvae; on the other hand, larvae with one JAK loss-of-function allele, exhibited increased areas of HP1 and H3mK9 concentration in their nuclei.
Is the tumorigenesis caused by increased JAK activity related to the decrease in heterochromatin? To address this question, Shi et al. increased the levels of HP1 and assessed the effects on tumor formation. Even a moderate increase in HP1 levels by means of transgene expression, completely suppressed hopTum-l-induced hematopoietic tumor formation. These data strongly suggest that decreased HP1 localization to heterochromatin is required for hopTum-l-induced tumorigenesis. Thus, a previously unknown function for JAK was discovered: the regulation of heterochromatin.
STAT and Heterochromatic Gene Silencing
Shi et al. then asked whether the heterochromatin disruption caused by JAK overactivation was mediated by STAT.32 To answer this question, they reduced the levels of STAT92E, and once again looked at PEV to assess the effect on heterochromatic gene silencing. Since reducing levels of JAK enhanced heterochromatic gene silencing, they expected that lowering STAT92E levels would also enhance silencing. Surprisingly, however, they found that reducing the levels of STAT92E caused a significant de-repression of silencing. In support of these surprising results, when STAT92E levels were increased by means of a STAT92E transgene or a chromosomal duplication, reporter gene expression was completely silenced, that is to say, heterochromatic gene silencing was enhanced. Thus, a paradox had arisen. In the canonical JAK-STAT pathway, JAK activates STAT; higher levels of JAK activity lead to more activated STAT and higher levels of transcription from STAT-activated genes; but heterochromatic gene silencing is enhanced by higher levels of STAT and lower levels of activated JAK.
Shi et al. began to resolve this paradox by characterizing the interaction between STAT92E and HP1; they manipulated the expression levels of both proteins and assessed the effects on PEV. They found that the enhanced heterochromatic gene silencing obtained with increased levels of STAT92E, was countered by lowering HP1 levels, while the reduction in silencing seen with lower HP1 levels was countered by increasing STAT92E levels. Thus, these data show that STAT92E and HP1 function interdependently to effect heterochromatic gene silencing.
Shi et al. then assessed more directly how manipulating STAT92E levels affects heterochromatin in larvae and embryos. They generated larvae with clones of cells having different levels of STAT92E, and detected heterochromatin by immunostaining with antibodies directed against H3mK9 or HP1. Clonal overexpression of stat92E resulted in higher levels of H3mK9 and HP1 staining in comparison with neighboring wild-type cells, corroborating the results of the PEV assays. On the other hand, clones of larval cells with a loss-of-function mutant stat92E, showed a clear reduction in HP1 foci; moreover, in Drosophila embryos with lower levels of STAT92E, localization of HP1 to centromeric heterochromatin was disrupted, and there was less H3mK9 protein. Thus, all of these data support the idea that STAT92E plays an important role in HP1 localization in heterochromatin. Indeed, the immunostaining of larval cells showed STAT92E to be localized mainly in the nucleus, much of it co-localized with HP1.
The role of STAT92E in HP1 localization was confirmed by chromatin immunoprecipitation assays with an anti-HP1 antibody, using cultured Drosophila S2 cells. Immunoprecipitations were done before and after treatment of the cells with RNAi directed against stat92E. The 1360 transposable element served as a heterochromatin marker, because this repetitive sequence is found in constitutively heterochromatic regions of all Drosophila chromosomes; and chromatin immunoprecipitation has demonstrated that HP1 binding to these sequences is enriched.33 The association of HP1 with the 1360 element was indeed significantly reduced after RNAi knockdown of STAT92E.
Co-immunoprecipitation studies then demonstrated that STAT92E and HP1 associate physically; and this interaction was abolished when both HP1-binding motifs of STAT92E were mutated. When immunoprecipitations were done in hopTum-l/+ embryos, less HP1 was co-immunoprecipitated with STAT92E than in wild-type embryos. Thus, in a situation where there was less phosphorylated STAT92E relative to unphosphorylated STAT92E, more STAT92E was found associated with HP1. Taken together, these findings raised the possibility that it is the unphosphorylated form of STAT92E that interacts with HP1. If this hypothesis were in fact correct, the paradox that both more JAK activity and less STAT92E leads to heterochromatin disruption could be resolved, since more JAK activity leads to more phosphorylated, but less unphosphorylated STAT92E.
When immunostaining was done with an anti-phospho-STAT92E antibody, phosphorylated STAT92E did not co-localize with HP1, supporting the idea that it is the unphosphorylated form of STAT92E that co-localizes with HP1. To study how phosphorylation of STAT92E might destabilize heterochromatin, an ex vivo assay was performed in which cultured salivary glands, carrying a STAT92E-GFP transgene, were treated with the tyrosine phosphatase inhibitor pervanadate to increase STAT92E phosphorylation,2 and STAT92E and HP1 localization were studied. Before pervanadate treatment, STAT92E was found in both the cytoplasm and the nucleus, and the nuclear portion was mostly co-localized with HP1. After 20 min of pervanadate treatment, an enrichment of nuclear STAT92E-GFP was observed, but it was no longer co-localized with HP1. After 60 min of pervanadate treatment, HP1 was also dispersed from heterochromatin. If, however, the salivary glands expressed an unphosphorylatable STAT92E-GFP mutant transgene rather than wild-type STAT92E-GFP, the mutant STAT92E remained co-localized with HP1 upon pervanadate treatment. Therefore, these data strongly support the idea that phosphorylation of STAT92E causes it to dissociate from heterochromatin, followed by displacement of HP1.
The dissociation of phosphorylated STAT92E from heterochromatin was corroborated by chromatin immunoprecipitation experiments with an anti-STAT92E antibody that showed an enrichment of 1360 sequences in STAT92E–chromatin complexes before, but not after, pervanadate treatment. Inhibition of protein synthesis by cycloheximide treatment did not prevent the HP1 dispersal induced by pervanadate; heterochromatin loss was actually accelerated in the presence of cycloheximide. These findings suggest that new protein synthesis is not required for the disruption of heterochromatin seen upon STAT92E phosphorylation, and hence imply that heterochromatin destabilization is not dependent upon induction of STAT92E transcriptional targets via the canonical JAK-STAT pathway. Thus, Shi et al. have characterized a new, non-canonical mode of JAK-STAT regulation. In this non-canonical mode, JAK and STAT regulate heterochromatin stability.
Work with mammalian cells has yielded results that are concordant with the findings of Shi et al. Nuclear JAK2 has been detected in hematopoietic cells,22 oocytes,34 and perhaps other cell types.35-37 In hematopoietic cells, JAK2 has been shown to sp.ecifically phosphorylate histone H3 at amino acid Y41, decreasing its association with HP1α; therefore, in this case, JAK2 activity disrupts heterochromatin by phosphorylation of histone H3.22 Unphosphorylated STAT1, STAT3, and low levels of STAT5A have also been found in the nuclei of mammalian cells.38-40 Recently, the tumor suppressor function of unphosphorylated STAT has been extended to a mammalian system. Hu et al. have shown that increased levels of unphosphorylated STAT suppress tumor formation in a xenogeneic mouse model of human colon cancer.41 It would be intriguing to learn whether there is nuclear JAK in Drosophila. Drosophila JAK does contain a putative nuclear localization sequence.3
Genome Stability
Heterochromatin has been associated with the preservation of genome stability.42 A loss of genome stability can result in cell death, premature aging, or cancer.43,44 Do JAK and STAT, then, have a role in preserving genome stability by virtue of their regulation of heterochromatin? Yan et al. addressed this question by genetically manipulating levels of STAT92E and HP1 in Drosophila, and determining how resistant the animals were to radiation-induced DNA damage.45
Upon receiving low levels of ionizing radiation, Drosophila larval cells respond by cell cycle arrest, which results in a dramatic reduction in the number of mitotic cells.46 Cells in mitosis can be readily identified and enumerated by immunostaining of larval imaginal discs, groups of tissue-specific progenitor cells, with antibodies directed against the mitotic marker phosphohistone H3.47 In control animals, with wild-type levels of STAT92E, Yan et al. found the number of mitotic cells in wing imaginal discs to be reduced nearly 10-fold after exposure to low dose γ-irradiation, when compared with non-irradiated discs. When levels of STAT92E were decreased, the number of mitotic cells after irradiation decreased even further, by an average of 3-fold when compared with control larvae. Conversely, an increase in the levels of unphosphorylated STAT92E resulted in a 2-fold increase in mitotic cells compared with controls. These findings are consistent with the idea that the increased levels of heterochromatin seen with higher levels of unphosphorylated STAT92E, impart greater genomic stability and resistance to DNA damage. If this idea is correct, then lowering the levels of HP1 should result in a greater sensitivity to radiation. Yan et al. found that larvae with reduced levels of HP1 did indeed exhibit fewer mitotic cells in their wing discs, indicating a greater sensitivity to radiation.
How do changes in heterochromatin levels affect the response to radiation? Does a decrease in heterochromatin levels result in more DNA damage, or is the response to DNA damaging agents impaired? To answer this question, Yan et al. examined DNA damage in a more direct fashion by assessing the levels of Drosophila phosphorylated H2Av, the equivalent of the mammalian phosphorylated histone variant H2AX (γ-H2AX), which serves as a marker for DNA damage.48,49 Initial experiments were performed with larval brains from non-irradiated animals having reduced levels of either STAT or HP1, or carrying the hopTumL mutation. Immunostaining with anti-γ-H2AX antibodies revealed higher levels of H2Av in these animals than in wild type. Thus, loss of heterochromatin is associated with more DNA damage even in non-irradiated animals. As expected, increasing the levels of heterochromatin, by overexpressing HP1 or an unphosphorylatable STAT92E mutant, suppressed the increase in H2Av associated with hopTumL.
Yan et al. then used immunostaining with anti-γ-H2AX antibodies to assess the effect of increased heterochromatin levels on radiation-induced DNA damage. These experiments utilized larvae expressing unphosphorylatable STAT92E in the posterior but not the anterior compartment of their wing imaginal discs. After low dose irradiation of the larvae, the wing discs were immunostained, and the numbers of H2Av foci in the posterior and anterior compartments were compared. The posterior compartment indeed showed significantly fewer H2Av foci, lending further support to the idea that unphosphorylated STAT92E and heterochromatin protect the genome from DNA damage.
To distinguish between effects on DNA damage and on DNA repair, Yan et al. assessed H2Av levels in Drosophila embryos at different time points after high dose γ-irradiation. Embryos that were either wild type or with reduced levels of heterochromatin were collected, and total protein was extracted and subjected to western blotting with anti-γ-H2AX antibodies. In both mutant and wild-type embryos, levels of H2Av were increased at ten minutes after irradiation (though to different degrees), and then gradually decreased to basal levels by 24 h after irradiation, suggesting that all of the animals had a similar ability to repair DNA damage. Thus, decreased levels of heterochromatin result in more γ-radiation induced DNA damage, but do not seem to impair the DNA repair process.
How might reduced levels of heterochromatin lead to greater DNA damage? Since it is known that loss-of-function HP1 mutants show defects in chromosome compaction,50,51 Yan et al. considered the possibility that lower heterochromatin levels were leading to improper DNA compaction, which in turn was leading to chromosomal breakage. They therefore investigated how reductions in STAT92E or HP1 might affect DNA compaction, by examining metaphase chromosomes in the large diploid nuclei of larval brain neuroblasts. They found that larvae with lower levels of STAT92E or HP1 exhibited significantly longer mitotic chromosomes than wild-type larvae, suggesting that the chromosomes were indeed improperly condensed. They also examined mitotic nuclei in early embryonic syncytia, where others have shown that a reduction in maternally supplied HP1 results in chromosomal condensation and segregation defects.50 In embryos lacking maternally supplied STAT92E, and also in hopTumL embryos, Yan et al. found that a substantial number of the metaphase chromosomes were improperly condensed and frequently exhibited chromosome bridges, phenomena seen only very rarely in wild-type embryos.
Do cells with reduced heterochromatin levels also exhibit chromosome breakage? Direct cytological detection of significant numbers of chromosome breaks poses technical difficulties. Therefore, Yan et al. turned to an indirect detection method. They showed that irradiation during Drosophila larval growth resulted in missing or shortened macrochaetes, the large bristles found on the thorax of adult flies. When levels of heterochromatin were reduced, significantly more flies showed bristle loss than did wild-type flies. With increased levels of heterochromatin, fewer flies exhibited bristle loss. Without irradiation, neither wild-type nor mutant flies showed bristle loss. To determine whether bristle loss was indeed correlated with chromosome breakage, Yan et al. did pulsed-field gel electrophoresis of genomic DNA from irradiated larvae. In animals with reduced levels of heterochromatin, the gel revealed more broken DNA, visualized as faster-migrating DNA fragments, than in wild-type animals. Thus, these results support the idea that lower levels of heterochromatin make flies more susceptible to radiation-induced chromosome breaks.
Higher doses of γ-radiation can cause lethality in Drosophila. Do higher levels of heterochromatin afford protection against such lethality? To answer this question, larvae with different levels of heterochromatin were exposed to a high dose of γ-radiation, and the percentage of larvae giving rise to adult flies was determined. Animals with increased HP1 levels showed a 3-fold increase in viability compared with wild type, while animals with reduced heterochromatin levels showed greater lethality than wild type. These results, then, are consistent with the idea that reduced heterochromatin levels lead to improper chromosomal condensation during mitosis and to DNA breakage, thereby increasing tissue sensitivity to radiation. When genome stability is enhanced by higher levels of heterochromatin, however, a dramatic increase in the survival of irradiated animals ensues.
Heterochromatin and Longevity
Do higher levels of heterochromatin only prolong the lifespan of irradiated animals, or does this salutary effect apply more generally? Larson et al. addressed this issue by genetically manipulating levels of HP1, JAK, and STAT in Drosophila, and determining the effects on lifespan.52
A moderate increase in HP1 levels significantly extended lifespan. On the other hand, reduction of HP1 levels by half resulted in a dramatic shortening of lifespan. Lifespan was also shortened when levels of STAT92E were reduced, or when JAK was overexpressed.
Do aged flies, then, have reduced levels of heterochromatin? To answer this question, Larson et al. using immunostaining to examine HP1 foci in enterocyte nuclei, where pericentric heterochromatin is readily discernible at the chromocenter. In young flies, enterocytes showed a prominent chromocenter enriched in HP1. Old flies, however, showed a dramatic reduction in heterochromatin, with many enterocyte nuclei lacking distinct HP1 foci. To confirm that less HP1 is localized to heterochromatin in old flies than young ones, chromatin immunoprecipitation was done with an anti-HP1 antibody; the amount of immunoprecipitated transposable element 1360, a representative heterochromatic sequence, was then determined by polymerase chain reaction. In young wild-type flies, HP1 was indeed associated with 1360, but not in old wild-type flies. Thus, these data support the concept that heterochromatin levels drop with age. In flies with higher HP1 levels, due to HP1 expression from a transgene, however, HP1 and 1360 were associated in old flies as well as young ones.
Does heterochromatin loss in aged flies result in the de-repression of silent genes? When Larson et al. looked at expression of a heterochromatinized transgene, they found it to be silent in young flies but expressed in old flies, suggesting that lower heterochromatin levels do cause de-repression of silenced genes.
If de-repression of silenced genes occurs during aging, which genes might be critical to the aging process? Larson et al. approached this issue by seeking more information, on the organismal level, about how fruit flies age. Might there be similarities to aging in mammals? To monitor changes in fly behavior over time, video recordings were done. The recordings revealed that aged flies exhibit a gradual loss of mobility, eventually becoming immobile. Fly mobility was quantified by measuring the speed with which flies moved from the bottom of a vial in which they were confined, toward the top. When flies with different levels of heterochromatin were compared, the flies with reduced heterochromatin levels lost mobility significantly faster than wild-type flies, while those with increased heterochromatin levels were mobile for a longer time. Thus, these results suggest a possible association between heterochromatin loss and muscle degeneration in old flies.
To assess muscle integrity, Larson et al. looked at the large intestinal wall muscles of Drosophila, which can be readily visualized in adult flies following minimal dissection. Whole-mount staining with fluorescinated phalloidin was used to assess the integrity of these muscles in young and old flies. In wild-type flies, progressive muscle degeneration was seen with aging; muscle fiber breakage was first detected at 20 d after eclosion, and extensive breakage was seen at 40 d. When heterochromatin levels were reduced, extensive muscle breakage was apparent at 20 d, whereas with increased heterochromatin levels, muscle integrity was preserved beyond 40 d. Furthermore, muscle integrity was found to correlate well with fly mobility. Thus, maintenance of heterochromatin levels is indeed important for maintaining muscle structure, fly mobility, and viability.
What critical genes might be affected by heterochromatin loss? In Drosophila, fragmentation of the nucleolus, the site of rRNA synthesis, has been shown to occur when heterochromatin levels are lowered by a decrease in HP1 or H3mK9.53 When Larson et al., looked at larvae with lower heterochromatin levels resulting from JAK overactivation or a loss of STAT92E, they also detected nucleolar fragmentation. On the other hand, HP1 overexpression suppressed the nucleolar fragmentation associated with JAK overexpression. Nucleolar fragmentation has been attributed to genomic instability at the rDNA locus, presumably from the illegitimate recombination of these highly repetitive sequences. Larson et al. examined illegitimate rDNA recombination by measuring levels of extrachromosomal circular DNA (ECC),53 and indeed found increased ECC in animals with decreased heterochromatin levels. Intriguingly, instability at the rDNA locus in yeast also results in increased ECC, and has been demonstrated to accelerate aging in that organism.54,55
Do changes in heterochromatin levels affect transcription from the rDNA locus? Larson et al. found that heterochromatin loss resulted in increased transcription from the rDNA locus, whereas moderate HP1 overexpression, at levels shown to extend lifespan, resulted in a significant decrease in transcription. Flies with reduced heterochromatin also had greater body weight than wild-type flies, while flies moderately overexpressing HP1 had significantly lower body weight. Body weight is an indication of growth rate, a higher growth rate often being associated with a shorter lifespan.56 The inverse correlation of heterochromatin levels with body weight therefore suggests that heterochromatin regulation of the rDNA locus may be important for viability in Drosophila.
Thus, Larson et al. have identified two means by which maintenance of heterochromatin levels may have a salutary effect on lifespan in Drosophila. Heterochromatin maintenance preserves genomic stability at the rDNA locus, and preserves muscle integrity.
Concluding Remarks
The work discussed here demonstrates that JAK and STAT perform many non-canonical functions in the cell. This review has focused on their important roles in heterochromatin regulation in Drosophila. Activated JAK causes disruption of heterochromatin, while unphosphorylated STAT stabilizes heterochromatin and acts as a tumor suppressor. The higher levels of heterochromatin due to increased levels of STAT, or HP1, extend lifespan. Moreover, higher heterochromatin levels confer greater resistance to radiation-induced DNA damage, by maintaining genomic stability. Lower heterochromatin levels, on the other hand, result in a shorter lifespan and greater sensitivity to radiation-induced DNA damage. The tumor suppressive function of unphosphorylated STAT has recently been demonstrated in mammalian cells as well (Fig. 1).
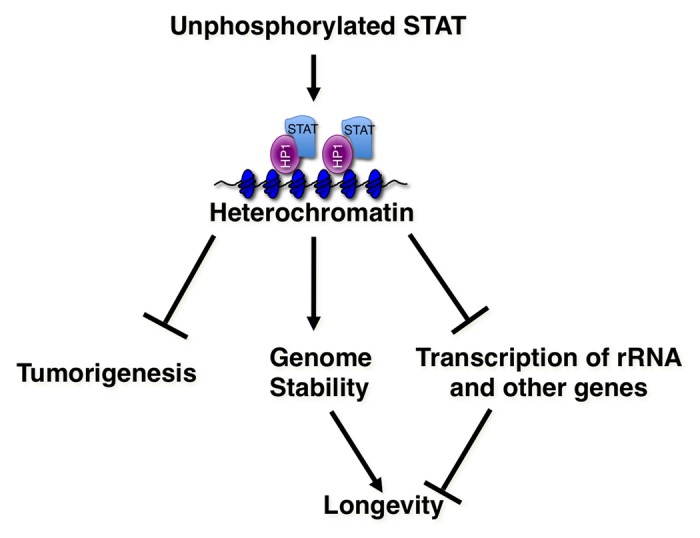
Figure 1. Role of STAT in heterochromatin and in genome stability. In the non-canonical JAK-STAT pathway in Drosophila, unphosphorylated STAT stabilizes heterochromatin, while JAK activation increases STAT phosphorylation and causes heterochromatin disruption. Higher levels of heterochromatin confer greater resistance to radiation-induced DNA damage, and are important for maintaining genomic stability. In addition, heterochromatin formation may lead to the silencing of genes that promote tumorigenesis or aging.
The regulation of heterochromatin by JAK and STAT92E indicates that these molecules have a more global role in the regulation of nuclear processes than was previously appreciated. Interestingly, it has recently been reported that another transcription factor, ATF-2, which is activated by stress-induced kinases, behaves in a similar fashion to STAT.57 Drosophila ATF-2 (dATF-2), apparently in its unphosphorylated form, is involved in heterochromatin assembly; when dATF-2 is phosphorylated, heterochromatin is disrupted. Do other signaling pathways and other transcription factors also regulate heterochromatin in a more global fashion? How is this regulation of heterochromatin coordinated with the other processes occurring in the cell nucleus?
Mammalian STATs, particularly STAT3, appear to perform a number of non-canonical functions in the cytoplasm. Does Drosophila STAT also perform non-canonical functions in the cytoplasm? What is the significance of these non-canonical nuclear and cytoplasmic functions of JAK and STAT? One possibility is that STATs, and to a lesser extent JAKs, have evolved to play many different but unrelated roles in the cell. Another possibility is the existence of a novel level of nuclear and cytoplasmic organization, in which JAK and STAT have important roles. Should the latter possibility be substantiated, it might lead to significant, new insights into cellular function.
Acknowledgments
We thank members of the Li lab for comments and suggestions. This work was supported by the National Institutes of Health (R01CA131326 to WXL).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/26090
References
- 1.Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 2.Yan R, Small S, Desplan C, Dearolf CR, Darnell JE., Jr. Identification of a Stat gene that functions in Drosophila development. Cell. 1996;84:421–30. doi: 10.1016/S0092-8674(00)81287-8. [DOI] [PubMed] [Google Scholar]
- 3.Binari R, Perrimon N. Stripe-specific regulation of pair-rule genes by hopscotch, a putative Jak family tyrosine kinase in Drosophila. Genes Dev. 1994;8:300–12. doi: 10.1101/gad.8.3.300. [DOI] [PubMed] [Google Scholar]
- 4.Kawata T, Shevchenko A, Fukuzawa M, Jermyn KA, Totty NF, Zhukovskaya NV, Sterling AE, Mann M, Williams JG. SH2 signaling in a lower eukaryote: a STAT protein that regulates stalk cell differentiation in dictyostelium. Cell. 1997;89:909–16. doi: 10.1016/S0092-8674(00)80276-7. [DOI] [PubMed] [Google Scholar]
- 5.Fukuzawa M, Araki T, Adrian I, Williams JG. Tyrosine phosphorylation-independent nuclear translocation of a dictyostelium STAT in response to DIF signaling. Mol Cell. 2001;7:779–88. doi: 10.1016/S1097-2765(01)00222-2. [DOI] [PubMed] [Google Scholar]
- 6.Zhukovskaya NV, Fukuzawa M, Tsujioka M, Jermyn KA, Kawata T, Abe T, Zvelebil M, Williams JG. Dd-STATb, a Dictyostelium STAT protein with a highly aberrant SH2 domain, functions as a regulator of gene expression during growth and early development. Development. 2004;131:447–58. doi: 10.1242/dev.00927. [DOI] [PubMed] [Google Scholar]
- 7.Gao Q, Hua J, Kimura R, Headd JJ, Fu XY, Chin YE. Identification of the linker-SH2 domain of STAT as the origin of the SH2 domain using two-dimensional structural alignment. Mol Cell Proteomics. 2004;3:704–14. doi: 10.1074/mcp.M300131-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Goldberg JM, Manning G, Liu A, Fey P, Pilcher KE, Xu Y, Smith JL. The dictyostelium kinome--analysis of the protein kinases from a simple model organism. PLoS Genet. 2006;2:e38. doi: 10.1371/journal.pgen.0020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li WX. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008;18:545–51. doi: 10.1016/j.tcb.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown S, Zeidler MP. Unphosphorylated STATs go nuclear. Curr Opin Genet Dev. 2008;18:455–60. doi: 10.1016/j.gde.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Kumar A, Commane M, Flickinger TW, Horvath CM, Stark GR. Defective TNF-alpha-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases. Science. 1997;278:1630–2. doi: 10.1126/science.278.5343.1630. [DOI] [PubMed] [Google Scholar]
- 12.Wang YZ, Wharton W, Garcia R, Kraker A, Jove R, Pledger WJ. Activation of Stat3 preassembled with platelet-derived growth factor beta receptors requires Src kinase activity. Oncogene. 2000;19:2075–85. doi: 10.1038/sj.onc.1203548. [DOI] [PubMed] [Google Scholar]
- 13.Silver DL, Naora H, Liu J, Cheng W, Montell DJ. Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004;64:3550–8. doi: 10.1158/0008-5472.CAN-03-3959. [DOI] [PubMed] [Google Scholar]
- 14.Ng DC, Lin BH, Lim CP, Huang G, Zhang T, Poli V, Cao X. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J Cell Biol. 2006;172:245–57. doi: 10.1083/jcb.200503021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verma NK, Dourlat J, Davies AM, Long A, Liu WQ, Garbay C, Kelleher D, Volkov Y. STAT3-stathmin interactions control microtubule dynamics in migrating T-cells. J Biol Chem. 2009;284:12349–62. doi: 10.1074/jbc.M807761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–6. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–7. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JE, Yang YM, Liang FX, Gough DJ, Levy DE, Sehgal PB. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am J Physiol Cell Physiol. 2012;302:C804–20. doi: 10.1152/ajpcell.00379.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sehgal PB. Non-genomic STAT5-dependent effects at the endoplasmic reticulum and Golgi apparatus and STAT6-GFP in mitochondria. JAK-STAT. 2013;2:e24860. doi: 10.4161/jkst.24860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan R, Lee JE, Yang YM, Liang FX, Sehgal PB. Live-cell imaging of the association of STAT6-GFP with mitochondria. PLoS One. 2013;8:e55426. doi: 10.1371/journal.pone.0055426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilsson J, Bjursell G, Kannius-Janson M. Nuclear Jak2 and transcription factor NF1-C2: a novel mechanism of prolactin signaling in mammary epithelial cells. Mol Cell Biol. 2006;26:5663–74. doi: 10.1128/MCB.02095-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–22. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arbouzova NI, Bach EA, Zeidler MP. Ken & barbie selectively regulates the expression of a subset of JAK-STAT pathway target genes. Curr Biol. 2006;16:80–8. doi: 10.1016/j.cub.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 24.Betz A, Lampen N, Martinek S, Young MW, Darnell JE., Jr. A Drosophila PIAS homologue negatively regulates stat92E. Proc Natl Acad Sci U S A. 2001;98:9563–8. doi: 10.1073/pnas.171302098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hari KL, Cook KR, Karpen GH. The Drosophila Su(var)2-10 locus regulates chromosome structure and function and encodes a member of the PIAS protein family. Genes Dev. 2001;15:1334–48. doi: 10.1101/gad.877901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi S, Calhoun HC, Xia F, Li J, Le L, Li WX. JAK signaling globally counteracts heterochromatic gene silencing. Nat Genet. 2006;38:1071–6. doi: 10.1038/ng1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 28.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dörken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 29.Sinclair D, Mottus R, Grigliatti T. Genes which suppress position-effect variegation in Drosophila melanogaster are clustered. Mol Gen Genet. 1983;191:326–33. doi: 10.1007/BF00334834. [DOI] [Google Scholar]
- 30.Eissenberg JC, James TC, Foster-Hartnett DM, Hartnett T, Ngan V, Elgin SC. Mutation in a heterochromatin-specific chromosomal protein is associated with suppression of position-effect variegation in Drosophila melanogaster. Proc Natl Acad Sci U S A. 1990;87:9923–7. doi: 10.1073/pnas.87.24.9923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reuter G, Dorn R, Wustmann G, Friede B, Rauh G. Third chromosome suppressor of position-effect variegation loci in Drosophila melanogaster. Mol Gen Genet. 1986;202:481–7. doi: 10.1007/BF00333281. [DOI] [Google Scholar]
- 32.Shi S, Larson K, Guo D, Lim SJ, Dutta P, Yan SJ, Li WX. Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat Cell Biol. 2008;10:489–96. doi: 10.1038/ncb1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Lucia F, Ni JQ, Vaillant C, Sun FL. HP1 modulates the transcription of cell-cycle regulators in Drosophila melanogaster. Nucleic Acids Res. 2005;33:2852–8. doi: 10.1093/nar/gki584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito M, Nakasato M, Suzuki T, Sakai S, Nagata M, Aoki F. Localization of janus kinase 2 to the nuclei of mature oocytes and early cleavage stage mouse embryos. Biol Reprod. 2004;71:89–96. doi: 10.1095/biolreprod.103.023226. [DOI] [PubMed] [Google Scholar]
- 35.Sorenson RL, Stout LE. Prolactin receptors and JAK2 in islets of Langerhans: an immunohistochemical analysis. Endocrinology. 1995;136:4092–8. doi: 10.1210/en.136.9.4092. [DOI] [PubMed] [Google Scholar]
- 36.Lobie PE, Ronsin B, Silvennoinen O, Haldosén LA, Norstedt G, Morel G. Constitutive nuclear localization of Janus kinases 1 and 2. Endocrinology. 1996;137:4037–45. doi: 10.1210/en.137.9.4037. [DOI] [PubMed] [Google Scholar]
- 37.Ram PA, Waxman DJ. Interaction of growth hormone-activated STATs with SH2-containing phosphotyrosine phosphatase SHP-1 and nuclear JAK2 tyrosine kinase. J Biol Chem. 1997;272:17694–702. doi: 10.1074/jbc.272.28.17694. [DOI] [PubMed] [Google Scholar]
- 38.Meyer T, Gavenis K, Vinkemeier U. Cell type-specific and tyrosine phosphorylation-independent nuclear presence of STAT1 and STAT3. Exp Cell Res. 2002;272:45–55. doi: 10.1006/excr.2001.5405. [DOI] [PubMed] [Google Scholar]
- 39.Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci U S A. 2005;102:8150–5. doi: 10.1073/pnas.0501643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyer J, Reich NC. Constitutive nuclear import of latent and activated STAT5a by its coiled coil domain. FASEB J. 2008;22:391–400. doi: 10.1096/fj.07-8965com. [DOI] [PubMed] [Google Scholar]
- 41.Hu X, Dutta P, Tsurumi A, Li J, Wang J, Land H, Li WX. Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc Natl Acad Sci U S A. 2013;110:10213–8. doi: 10.1073/pnas.1221243110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- 43.Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 44.Jefford CE, Irminger-Finger I. Mechanisms of chromosome instability in cancers. Crit Rev Oncol Hematol. 2006;59:1–14. doi: 10.1016/j.critrevonc.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 45.Yan SJ, Lim SJ, Shi S, Dutta P, Li WX. Unphosphorylated STAT and heterochromatin protect genome stability. FASEB J. 2011;25:232–41. doi: 10.1096/fj.10-169367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hari KL, Santerre A, Sekelsky JJ, McKim KS, Boyd JB, Hawley RS. The mei-41 gene of D. melanogaster is a structural and functional homolog of the human ataxia telangiectasia gene. Cell. 1995;82:815–21. doi: 10.1016/0092-8674(95)90478-6. [DOI] [PubMed] [Google Scholar]
- 47.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–60. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 48.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 49.Madigan JP, Chotkowski HL, Glaser RL. DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic Acids Res. 2002;30:3698–705. doi: 10.1093/nar/gkf496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kellum R, Alberts BM. Heterochromatin protein 1 is required for correct chromosome segregation in Drosophila embryos. J Cell Sci. 1995;108:1419–31. doi: 10.1242/jcs.108.4.1419. [DOI] [PubMed] [Google Scholar]
- 51.Perrini B, Piacentini L, Fanti L, Altieri F, Chichiarelli S, Berloco M, Turano C, Ferraro A, Pimpinelli S. HP1 controls telomere capping, telomere elongation, and telomere silencing by two different mechanisms in Drosophila. Mol Cell. 2004;15:467–76. doi: 10.1016/j.molcel.2004.06.036. [DOI] [PubMed] [Google Scholar]
- 52.Larson K, Yan SJ, Tsurumi A, Liu J, Zhou J, Gaur K, Guo D, Eickbush TH, Li WX. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet. 2012;8:e1002473. doi: 10.1371/journal.pgen.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng JC, Karpen GH. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat Cell Biol. 2007;9:25–35. doi: 10.1038/ncb1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91:1033–42. doi: 10.1016/S0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 55.Sinclair DA, Mills K, Guarente L. Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science. 1997;277:1313–6. doi: 10.1126/science.277.5330.1313. [DOI] [PubMed] [Google Scholar]
- 56.Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–25. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 57.Seong KH, Li D, Shimizu H, Nakamura R, Ishii S. Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell. 2011;145:1049–61. doi: 10.1016/j.cell.2011.05.029. [DOI] [PubMed] [Google Scholar]