

Background: Bacteria require the enzyme indole-3-glycerol phosphate synthase for the production of Trp.
Results: Glu-51 and Lys-53 are identified as the base and acid acting in the dehydration step of enzyme catalysis.
Conclusion: Ring closure and dehydration steps are catalyzed by distinct active-site surfaces.
Significance: Enzyme inhibitors targeted against these active-site surfaces may serve as novel antibiotics.
Keywords: Bacterial Metabolism, Drug Development, Enzyme Kinetics, Enzyme Mechanisms, Protein Engineering, Tryptophan
Abstract
The tryptophan biosynthetic enzyme indole-3-glycerol phosphate synthase is a proposed target for new antimicrobials and is a favored starting framework in enzyme engineering studies. Forty years ago, Parry proposed that the enzyme mechanism proceeds through two intermediates in a series of condensation, decarboxylation, and dehydration steps. X-ray crystal structures have suggested that Lys-110 (numbering according to the Sulfolobus solfataricus enzyme) behaves as a general acid both in the condensation and dehydration steps, but did not reveal an efficient pathway for the reprotonation of this critical residue. Our mutagenesis and kinetic experiments suggest an alternative mechanism whereby Lys-110 acts as a general acid in the condensation step, but another invariant residue, Lys-53, acts as the general acid in the dehydration step. These studies also indicate that the conserved residue Glu-51 acts as the general base in the dehydration step. The revised mechanism effectively divides the active site into discrete regions where the catalytic surfaces containing Lys-110 and Lys-53/Glu-51 catalyze the ring closure (i.e. condensation and decarboxylation) and dehydration steps, respectively. These results can be leveraged toward the development of novel inhibitors against this validated antimicrobial target and toward the rational engineering of the enzyme to produce indole derivatives that are highly prized by the pharmaceutical and agricultural industries.
Introduction
The indole moiety is one of the most common heterocycles in nature and is considered a “privileged” structure involved in a plethora of compounds within the pharmaceutical, agricultural, and materials science industries (1). As such, the synthesis of indole and its many derivatives remains a very active area of research (2). New insights into indole synthesis can be gleaned from the study of the fifth enzyme in the tryptophan biosynthetic pathway, indole-3-glycerol phosphate synthase (IGPS).2 This enzyme is responsible for catalyzing the ring closure of 1-(o-carboxyphenylamino)-1-deoxyribulose 5-phosphate (CdRP) to form indole-3-glycerol phosphate (IGP) and occurs through chemistry that is a departure from the more common organic synthetic routes. An engineered IGPS enzyme may be able to catalyze the synthesis of new indole derivatives or improve the economical feasibility of indole production through the use of more temperate, environmentally friendly conditions. IGPS has also been suggested as a novel target for antibacterial drugs aimed at combating multidrug-resistant tuberculosis (3–5) and potentially other bacterial pathogens (6). Despite the importance of IGPS in several active fields of research, critical issues regarding the molecular level details of its catalytic mechanism have yet to be satisfactorily explained.
Forty years ago, Parry proposed that IGPS catalysis proceeds through two intermediates (I1 and I2) in a series of condensation, decarboxylation, and dehydration steps (Fig. 1) (7, 8). Crystal structures of IGPS from Sulfolobus solfataricus (ssIGPS) bound with reduced CdRP shows that the substrate docks with the enzyme in an extended unproductive conformation (at least at lower temperatures) in which the C1 and C2′ sites are separated by a distance too great (4.5 Å) to initiate bond formation (9). This finding suggests that a substantial rearrangement of the substrate must occur prior to or during these chemical events. Initiation of the enzyme-catalyzed reaction is proposed to begin with the protonation of the ketone in CdRP via general acid catalysis through Lys-110 (numbering according to ssIGPS), an absolutely conserved and essential amino acid residue (9–11). The resulting secondary carbocation is then a feasible candidate for electrophilic attack from the benzyl ring, creating a sterically strained I1 intermediate. Consistent with this proposed mechanism, tandem mass spectrometry has identified fragment patterns that were attributed to the transient formation of I1 during both acid- and IGPS-catalyzed reactions (12). Following decarboxylation, general acid and base chemistry (predicted to occur through Lys-110 and Glu-159 based on crystallographic evidence) catalyzes the dehydration of the newly formed alcohol and the direct removal of an alkyl hydrogen to create the pyrrole carbon-carbon double bond of the indole moiety.
FIGURE 1.
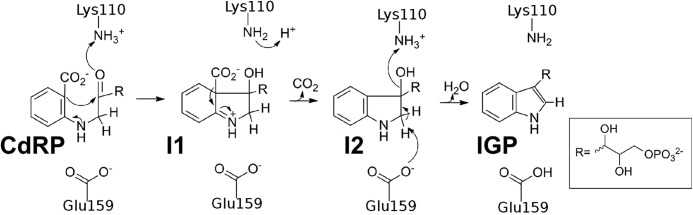
The previously proposed chemical mechanism for the conversion of CdRP to IGP by IGPS has chemical inconsistencies, including the need to reprotonate Lys-110 following the first step.
Although functional and structural studies are mostly consistent with the above mechanism (5, 9–14), there are still some critical issues that are not yet resolved. For example, Lys-110 is predicted to be a general acid in both the ring closure and dehydration steps, yet there is no clear mechanism by which Lys-110 could become reprotonated for the second proton transfer event. An alternative hypothesis is that another active-site residue, such as Lys-53, also acts as a general acid, likely during the dehydration step. Indeed, site-directed mutagenesis of Escherichia coli IGPS demonstrated that Lys-53 (Lys-55 in E. coli IGPS) is highly important to IGPS catalysis (13). Lys-53 may also play roles in substrate binding and structural rearrangements occurring during catalysis. Crystal structures of ssIGPS have indicated that Lys-53 makes an ionic interaction with the carboxylate moiety in the anthranilate ring. Molecular dynamic simulations undertaken by Bruice and co-workers (15, 16) have also led to the proposition that the increased flexibility of the β1α1 loop and Lys-53 at higher temperatures allows the substrate to form a more productive near attack conformation.
In this work, we have undertaken a series of kinetic experiments aimed at elucidating the specific roles of each of the charged conserved active-site residues in ssIGPS. In particular, our kinetic experiments revealed that it is Lys-53 (and not Lys-110) that donates a proton to the C2′ hydroxyl group to catalyze the dehydration of I2 to IGP product. We also identify Glu-51 as the general base in ssIGPS catalysis, rather than the previously proposed assignment of either Glu-159 or Glu-210 as suggested from crystallographic experiments (9) and molecular dynamics simulations (15, 16), respectively. This work provides a strong foundation toward modifying IGPS for new catalytic function and/or developing next-generation inhibitors against IGPS for antimicrobial purposes.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis of ssIGPS
Initial site-directed mutagenesis of the A/T-rich ssIGPS gene cloned into pET21b (17) proved problematic. To help solve this problem, we obtained an E. coli codon usage-optimized version of ssIGPS (GenScript) that had a richer G/C sequence and cloned it into pET101 using the Champion pET Directional TOPO expression kit (Invitrogen). Unfortunately, preliminary protein expression trials indicated a substantial overexpression of the β-lactamase protein. Thus, the ssIGPS gene was subsequently subcloned from pET101 into pET26 (KanR) with the restriction endonucleases XbaI and SacI using standard procedures. Site-directed mutagenesis was performed to obtain the E51Q, K53R, K53Q, E159Q, R182A, and E210Q variants of ssIGPS using the QuikChange Lightning mutagenesis kit (Stratagene) with appropriate PCR primers and the pET26 construct. All sequences for WT and mutant ssIGPS enzymes were confirmed through DNA sequencing (Nucleic Acid Facility, The Pennsylvania State University).
The original WT ssIGPS overexpressed from pET21b (17) was hexahistidine-tagged at the N terminus, but ssIGPS from pET26 is not His-tagged. It was important to these studies that the His-tagged and non-His-tagged versions of WT ssIGPS gave essentially identical kinetic parameters.
Protein Overexpression, Purification, and Characterization
The overexpression and purification of WT and variant ssIGPS enzymes followed protocols described previously (11, 17, 18). Steady-state enzyme assays for ssIGPS followed previously established procedures (11, 19, 20). In short, ssIGPS activity was measured via fluorescence by monitoring the formation of the product IGP. IGP was excited at 278 nm, and emission was measured at 340 nm. Initial rate data were fit to the Michaelis-Menten equation using nonlinear regression with the program KaleidaGraph (Equation 1),
where v is the initial reaction velocity, ET is the total amount of enzyme in the assay, and [S] is the substrate concentration.
The solvent viscosity, solvent deuterium kinetic isotope, and pH effects for WT and variant ssIGPS enzymes were determined by varying the buffer conditions of the standard assay. Solvent viscosity effects (SVEs) were determined by enzyme assays in 50 mm HEPPS and 4 mm EDTA (pH 7.5) with 0–30% (w/v) glycerol. Studies with sucrose gave similar results as those with glycerol (data not shown). The relative viscosities of the buffer solutions were measured using an Ostwald viscometer. The addition of microviscogen did not change Km values, but it had effects on kcat values where noted. PEG 8000 was used as a macroviscogen control, but it did not alter the kinetic parameters of WT or mutant enzymes (i.e. kcat(PEG 8000)/kcat(no viscogen) = 1.0 ± 0.2 for 1.9% (w/v) PEG 8000 at a relative viscosity of 1.7). SVEs were not performed for K53Q ssIGPS because of this mutant's very low catalytic activity. SVEs were obtained from the slope of a plot of relative viscosity versus rate0/rateviscogen.
Solvent deuterium kinetic isotope effects (SDKIEs) were measured for kcat in D2O for WT and variant ssIGPS enzymes using saturating concentrations of CdRP (800 nm for WT, 4000 nm for E210Q, and 8000 nm for E51Q and K53R). pD values were determined by measurement of the pH (pD = pH + 0.4). The SDKIE was defined as kH2O/kD2O.
pH studies for ssIGPS were performed using the following buffers: MES (pH 5.0–6.5), HEPES (pH 6.5–8.0), Bicine (pH 8.0–8.5), and CHES (pH 8.6–10.0). None of the buffers gave any significant nonspecific effects. The pH-rate profiles were fit using Kaleidograph. For WT and K53R ssIGPS enzymes, which display two ionizations, data were fit to Equation 2 by nonlinear regression.
For E51Q ssIGPS, which displays only a single ionization, data were fit to Equation 3 by nonlinear regression,
where v is the estimated first-order (kcat) or second-order (kcat/Km) rate constant, C is the pH-independent rate value, and pKa1 and pKa2 are the pKa values associated with the ascending and descending limbs of the pH-rate profile, respectively. In our studies, pH changes did not significantly affect Km values.
RESULTS
Solvent Viscosity and Isotope Effects Reveal the Rate-determining Step(s) in ssIGPS Catalysis
There are six highly conserved, charged residues in the active site of IGPS: Glu-51, Lys-53, Lys-110, Glu-159, Arg-182, and Glu-210. Previous mutagenesis experiments have indicated that these residues are important for enzyme catalysis (10, 13) but were not able to assign specific roles for these residues because the kinetic steps that were most affected by these mutations were not elucidated. We have previously used SDKIEs and SVEs to illuminate the rate-determining step(s) for WT ssIGPS under different temperature conditions (11); the SVEs and SDKIEs reported herein are on the steady-state turnover rate constant, kcat. In ssIGPS, the SDKIE reports on the proton transfer event in the condensation reaction (see supplemental “Experimental Procedures”), and the SVE reports on diffusion-controlled events, such as substrate binding and product release.
It should be noted that the dehydration step is not expected to produce a substantial SDKIE. Within the dehydration step, it is likely that the removal of the non-exchangeable alkyl hydrogen is rate-limiting over the loss of water, causing the step to be isotope-insensitive. A similar reaction involving the reduction of spiro-(cyclopentane-1,2′-pseudo-indoxyl) is known to produce a dihydro derivative that is highly analogous to I2 (21). Conversion of this dihydro derivative into tetrahydrocarbazole occurs readily in the presence of trace acid. Kinetic studies on the rearrangement of the pyrrole-substituted structure (22) between 11-hydroxytetrahydrocarbazole and spiro-(cyclopentane-1,2′-pseudo-indoxyl) further demonstrated that although the rearrangement process can be either acid- or base-catalyzed, the reaction rate is still notably slower than the solvent exchange processes of secondary alcohols or an indole amine.
A careful analysis of SDKIEs and SVEs revealed the rate-determining step (Fig. 2A). For example, the rate-determining step for WT ssIGPS at 75 °C must be at the ring closure step because there is a substantial SDKIE, but no SVE (i.e. not diffusion-limited) (Table 1). This finding is in contrast to what happens at 25 °C, at which there is a SVE close to the theoretical maximum of 1, but no SDKIE, and is consistent with product release being rate-determining at this lower temperature (11). It should be noted that pre-steady-state kinetic experiments for ssIGPS also indicated that product release is rate-determining at 25 °C, but these experiments were not able to assign micro-rate constants to each of the individual chemical steps (23). As such, SVE and SDKIE experiments are required to determine which step of the mechanism is impacted (mostly) by the amino acid substitutions.
FIGURE 2.
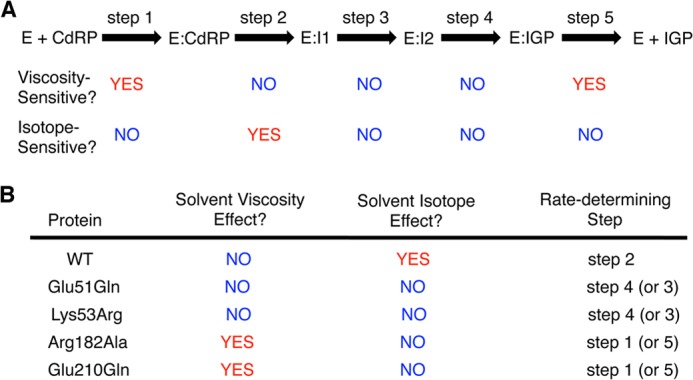
Solvent viscosity and isotope effects reveal the rate-determining step(s) in WT and variant ssIGPS enzymes. A, the kinetic mechanism of IGPS. There is a substantial SVE when the rate-determining step is diffusion-controlled, such as in product release, similar to what occurs at low temperatures (11). A substantial SDKIE is present only when the condensation step is rate-determining (see “Experimental Procedures” and supplemental “Experimental Procedures”). B, summary of the SVEs and SDKIEs for WT and variant ssIGPS enzymes at 75 °C indicates that single amino acid substitutions can change the rate-determining step and hence what kcat reports on. A change in the rate-determining step also means that changes to kcat may underreport the impact of amino acid substitutions on the individual kinetic steps.
TABLE 1.
Steady-state kinetics (at 75 °C) indicate that Lys-53 and Glu-51 are the general acid and base of the dehydration step in IGPS catalysis
| Protein | kcata | Kma | kcat/Km | SDKIE (kH2O/kD2O) | SVE |
|---|---|---|---|---|---|
| s−1 | nm | ×106m−1 s−1 | |||
| WT | 0.67 ± 0.03 | 44 ± 9 | 15 | 3.6 ± 0.3 | −0.2 ± 0.1 |
| E51Q | 0.007 ± 0.001 | 13.0 ± 4.4 | 0.54 | 0.9 ± 0.1 | NDb |
| K53R | 0.06 ± 0.01 | 2440 ± 3 | 0.03 | 1.0 ± 0.1 | 0.2 ± 0.1 |
| K53Qc | 0.001 ± 0.001 | ND | ND | ND | ND |
| R182A | 0.41 ± 0.07 | 1062 ± 499 | 0.38 | 1.1 ± 0.6 | 0.7 ± 0.3 |
| E210Q | 0.97 ± 0.07 | 386 ± 96 | 2.5 | 1.0 ± 0.3 | 0.7 ± 0.2 |
a Errors in kcat and Km are reported as S.E.
b ND, not determined.
c Due to the low activity of K53Q, the reported kcat was estimated using only one concentration of CdRP (12 μm), and we were not able to determine Km, kcat/Km, and SDKIE values.
Mutational Analysis Suggests That Lys-53 Plays Roles in Both Substrate Binding and Chemical Catalysis in ssIGPS
The original mechanism based on the x-ray crystal structures of ssIGPS (9) suggested that Lys-110 acts as a general acid both in the ring closure and dehydration steps (Fig. 1). Because there was no obvious reprotonation mechanism for Lys-110, we predicted that another residue acts as a general acid in one of the steps. Excluding Lys-110, the only remaining cationic residues in the active site of IGPS are Lys-53 and Arg-182, both of which are strongly conserved. We analyzed ssIGPS variants at both positions to determine whether either Lys-53 or Arg-182 could act as a general acid.
Our kinetic results indicated that the R182A substitution had only a minor effect on the overall catalytic rate (kcat) but led to a 20-fold increase in the Michaelis-Menten constant (Table 1). These changes in the kinetic parameters were accompanied by a substantial SVE, suggesting that the rate-determining step for the R182A variant is diffusion-controlled. The 40-fold decrease in kcat/Km for the R182A variant suggests that Arg-182 plays an important role in substrate capture.
To test the importance of the positive charge on Lys-53 and its ability to act as a general acid, we assayed ssIGPS variants with Arg and Gln substituted at position 53; Arg retains the positive charge and some ability to donate a proton, whereas Gln does not retain either property but would still be able to participate in CdRP binding through a hydrogen bond with the carboxylate moiety. The Lys-53 substitutions had substantial effects on both the kcat and Km kinetic parameters (Table 1). In particular, the 10-fold decrease in maximum catalytic turnover suggests a role for Lys-53 in the chemical steps of IGPS catalysis. The K53Q substitution had an even more dramatic effect on IGPS catalysis, resulting in an almost 700-fold decrease in activity even at a CdRP concentration of 12 μm (50 × Km for WT IGPS). These results suggest that the positive charge and/or the ability of Lys-53 to act as a general acid is critical for IGPS catalysis.
To further determine which kinetic steps are most affected by the Lys-53 substitutions, we determined SVE, SDKIE, and pH effects for the K53R variant (Figs. 2 and 3 and Table 1). K53R ssIGPS had a slightly larger SVE (at 75 °C) compared with WT ssIGPS, consistent with substrate binding or product release making a slightly larger contribution to the overall rate. The pH and SDKIE results indicate a direct and specific role for Lys-53 in IGPS catalysis. The bell-shaped pH-rate curves for WT ssIGPS are consistent with acid/base catalysis (at 25 °C, pKa1 = 5.4 ± 0.2 and pKa2 = 8.9 ± 0.2; at 75 °C, pKa1 = 5.7 ± 0.1 and pKa2 = 8.7 ± 0.1) (Fig. 3A). For K53R IGPS, the pKa for the descending limb of the pH-rate profile has been shifted substantially compared with the WT enzyme (at 25 °C, pKa1 = 6.9 ± 0.1 and pKa2 > 9.5; at 75 °C, pKa1 = 7.1 ± 0.1 and pKa2 > 9.5) (Fig. 3B). These results, together with the finding that the Gln substitution is much more detrimental than the Arg substitution (Table 1), suggests that Lys-53 is behaving as a general acid during the IGPS-catalyzed reaction. It should be noted that Arg would still be capable of acting as a general acid, albeit a poorer one than Lys, which would help explain why the catalytic rate was still substantial even with this substitution at Lys-53. The placement of an amino acid incapable of acting as a general acid (i.e. Gln) at this position led to an almost 700-fold reduction in enzyme activity. The lack of a SDKIE for the K53R variant further suggests that Lys-53 acts as the general acid in the dehydration step of the reaction.
FIGURE 3.

pH-rate profiles indicate that Glu-51 and Lys-53 act as the general base and acid, respectively, in the dehydration step of IGP production. Shown are the pH-rate profiles for kcat at 25 °C (left) and 75 °C (right) for WT ssIGPS (A) and variants K53R (B) and E51Q (C). Km values were pH-independent. Note the logarithmic scale on the y axes of these plots. In the K53R variant, the pKa2 is substantially larger, as expected if the residue at position 53 acts as a general acid. The lack of the ascending limb in the pH-rate profile of the E51Q variant is consistent with Glu-51 acting as a general base in ssIGPS catalysis. Both the K53R and E51Q variants show shifts in their other pKa values. These effects are likely caused by changes to the local microenvironment. For example, the loss of the negatively charged carboxylate group of Glu-51 would tend to lower the pKa of the nearby side chain of Lys-53.
Glu-51 Acts as a General Base in the Dehydration Step of the Reaction
Both Glu-159 and Glu-210 were previously proposed to act as the general base involved in the dehydration of I2 to IGP. The assignment of Glu-159 in this role was based on the following observations. Glu-159 was essential to the reaction with the corresponding amino acid substitution in E. coli IGPS (i.e. E163Q), yielding a 540-fold decrease in kcat (10); Lys-110 was believed to be acting as general acid in the dehydration step; and Glu-159 appeared to be positioned to act as the general base. In contrast, molecular dynamics studies predicted that Glu-210, which is directly adjacent to Glu-159, would be a better choice for the general base in the dehydration step (16).
In light of our findings that it is Lys-53, but not the originally proposed Lys-110, that acts as the general acid in the dehydration step, it seemed unlikely that either Glu-159 or Glu-210 could be acting as the general base in the dehydration step given that the large distance between them and Lys-53 would prevent simultaneous contact with the appropriate regions of I2. To investigate this further, we examined amino acid substitutions at Glu-159 and Glu-210 to test their potential roles as general bases (9, 14, 18, 24). Similar to the analogous change in E. coli IGPS, the E159Q substitution in ssIGPS led to a substantial decrease in catalytic activity (kobs < 1.5 × 10−5 s−1) (Table 1). This change is on the same order of magnitude as previously seen for the K110R variant (11), which likely indicates that Glu-159 and Lys-110 are operating in the same chemical step of the reaction. The E210Q variant did not show any decrease in kcat, indicating that Glu-210 is not involved directly in the chemistry of the reaction (Table 1). However, the 6-fold decrease in kcat/Km for E210Q ssIGPS, along with its substantial SVE, suggests that Glu-210 plays a role in substrate capture (Fig. 2B).
Because experimental analysis of Glu-159 could not conclusively confirm the role of Glu-159 as a general base, we also examined the invariant and catalytically important residue Glu-51. This residue is the only other viable candidate for this role; it is located close enough to Lys-53 such that simultaneous contact between Glu-51 and Lys-53 and I2 is feasible, and its functional role in IGPS catalysis was previously ambiguous. Our kinetic analysis of the E51Q variant showed a 100-fold decrease in kcat compared with the WT enzyme (Table 1). The kinetic parameters are on the same order of magnitude as those seen for K53Q, as we expect for residues acting in the same step of the chemical mechanism. The E51Q variant also showed a complete loss of the SDKIE for kcat, as was also observed for the K53R variant. The pH dependence of E51Q displays a loss in the ascending limb that is seen in the WT enzyme (at 25 °C, pKa2 = 5.7 ± 0.2; at 75 °C, pKa2 = 6.5 ± 0.1) (Fig. 3C). It should be noted that the E51Q and K53R substitutions also affected the pKa of the other limb in the pH-rate profile, likely because of the close proximities of these residues and their abilities to alter the local microenvironment of the other ionizable group. Altogether, our results implicate Lys-53 and Glu-51 as the general acid and base, respectively, in the dehydration step. It is also important to point out that the pH results for WT, K53R, and E51Q were similar at 25 °C and 75 °C (Fig. 3), suggesting that the identities of the general acid and base for the dehydration step are the same at both temperatures.
DISCUSSION
The biological production of indole, tryptophan, and their derivatives is a critical process necessary for life. The IGPS enzyme catalyzes the fifth step in the tryptophan biosynthetic pathway, the ring closure of CdRP to form IGP. In this study, we have reported kinetic and mutagenesis data that delineate the roles of the highly conserved, charged amino acid residues in the active site of IGPS (Fig. 4A). Consistent with previous suggestions (9, 11), Lys-110 acts as the general acid in the ring closure reaction and is likely aided by Glu-159. Our studies indicate that the general acid and base in the dehydration reaction are Lys-53 and Glu-51, respectively (Fig. 4B). Arg-182 and Glu-210 primarily participate in substrate capture. In our investigation of the pH dependence of the reaction, we also found similar profiles at 25 °C and 75 °C, suggesting that the nature of the chemical steps is largely invariant with temperature.
FIGURE 4.

The revised chemical mechanism reconciles structural, kinetic, and mutagenesis data for IGPS. A, assigned roles for the charged residues within the active site of ssIGPS. B, proposed revision to the chemical mechanism of ssIGPS, highlighting the roles of Lys-53 and Glu-51 during the dehydration step. Hydrogen extraction may occur from the alkyl hydrogen, as originally proposed (see Fig. 1), or may potentially occur from the amine hydrogen. C, the condensation/decarboxylation and dehydration steps occur in two different catalytic pockets. The first is shown in cyan and involves Lys-110 and Glu-159, and the second is shown in yellow and involves Lys-53 and Glu-51. The crystallographic docked structure of IGP is shown in the left panel. I2 was approximated using this docked structure through manual rotation within the ribose chain (right panel). These rotations show a plausible alignment of both the 2′-alcohol and the secondary amine of I2 with the Lys-53/Glu-51 catalytic surface. This rearrangement of the ribose chain can be obtained without considering thermal flexibility from either the phosphate anchor or the surrounding protein. D, surface rendering of the active site of ssIGPS docked with the reaction product IGP (Protein Data Bank code 1A53). The product is orientated with the indole ring facing inward toward to the core of the protein and the phosphate anchor located near the mouth of the cavity. The second catalytic surface is not located as deep within the pocket as the first. This positioning, along with the loss of the positive charge on Lys-53 following the dehydration, may aid in product release. Molecular graphics and analysis were performed using the UCSF Chimera package (25).
The functional identification of Glu-51 and Lys-53 as the critical residues in the dehydration step is in contrast to previous proposals based on x-ray crystal structures and molecular dynamic simulations (9, 15). Our proposal is seemingly at odds with the crystal structure of IGP docked into ssIGPS because, in this conformation, the side chains of Glu-51 and Lys-53 are not properly positioned for this type of chemistry. The docked conformation of I2 within the active site is not experimentally known, however. Rotation along the C3′-C4′ bond of the ribose moiety in IGP relocates the amine group of the indole ring within contact distance of Glu-51 while also positioning the C2′ site (along with the corresponding 2′-hydroxyl site of I2) near Lys-53, potentially resolving the conflict between our kinetic data and the available crystallographic structures. Correspondingly, we believe that it is reasonable to hypothesize that there is sufficient thermal flexibility of the substrate within the active site to reorient I2 into a reactive geometry with Glu-51 and Lys-53. This hypothesis is also supported by the observations that CdRP must undergo substantial internal bond rotations to close the initial 4.5-Å distance between the C1 and C2′ atoms necessary for ring closure and the weak crystallographic electron density of the ribulose moiety in the bound protein complex. Recent pre-steady-state kinetic experiments in which fluorescent dyes have been attached to the β1α1 loop (i.e. Lys-53–Asp-65) have also indicated that structural rearrangements around this highly flexible loop are critical for catalysis (23).
We suggest then that following ring closure, internal rotations in the ribose moiety reorient I2 into a position where the 2′-hydroxyl is in close proximity to Lys-53 and the amine group of the substrate is in close proximity to Glu-51 (Fig. 4C). During the final step of the reaction, the electron lone pair of Glu-51 attacks the hydrogen bound to the amine group of I2. The loss of the hydrogen from I2 may occur at the N-H site rather than at the adjacent C-H site, considering the differences in their pKa values (i.e. for N-H, pKa ∼ 21; for C-H, pKa ∼ 44–51). This event initiates an electron cascade that creates a carbon double bond between the C1′ and C2′ atoms and concludes with Lys-53 donating a proton to the O2′ hydroxyl to release H2O and produce IGP. The reaction leaves the side chain of Lys-53 neutralized, weakening the electrostatic attraction between the protein and the phosphate anchor.
Mapping this revised mechanism onto the active site reveals three distinct regions of function (Fig. 4D). The first region contains the surfaces formed by the combination of Lys-110 and Glu-159 and is likely responsible for the ring closure/decarboxylation step of the catalytic pathway. The second region is located adjacent to the first, is composed of the surfaces from Glu-51 and Lys-53, and is responsible for completing the dehydration in the second step of the mechanism. The third region is the phosphate-anchoring pocket. After the dehydration reaction, Lys-53 is rendered neutral, likely causing a reduction in the binding affinity by 2–3 orders of magnitude and potentially aiding in the release of IGP from the enzyme.
This revised catalytic mechanism for IGPS can be leveraged toward rational drug design efforts against this previously validated antimicrobial target by targeting one or more of these catalytic surfaces. IGPS has also been used as a favored starting scaffolding in enzyme engineering research. The revised catalytic mechanism presented here should find utility in efforts to engineer new enzymes for the production of indole derivatives that have important medical and agricultural applications.
Acknowledgments
We thank Drs. Stephen Benkovic, Phil Bevilacqua, and Craig Cameron for careful reading of and comments on the manuscript. We also thank Dr. Stephen Benkovic and Michelle Spiering for the kind use of the spectrofluorometer and the Department of Research Computing and Cyberinfrastructure at The Pennsylvania State University for use of the facility.
This work was supported by National Science Foundation Career Grant MCB1053993 (to D. D. B.).

This article contains supplemental “Experimental Procedures,” Equations 1–18, and additional references.
- IGPS
- indole-3-glycerol phosphate synthase
- CdRP
- 1-(o-carboxyphenylamino)-1-deoxyribulose 5-phosphate
- IGP
- indole-3-glycerol phosphate
- I1
- intermediate 1
- I2
- intermediate 2
- ssIGPS
- S. solfataricus IGPS
- SVE
- solvent viscosity effect
- HEPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid
- SDKIE
- solvent deuterium kinetic isotope effect
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid.
REFERENCES
- 1. Barden T. C. (2010) Indoles: industrial, agricultural and over-the-counter uses. Top. Hetercycl. Chem. 26, 31–46 [Google Scholar]
- 2. Humphrey G. R., Kuethe J. T. (2006) Practical methodologies for the synthesis of indoles. Chem. Rev. 106, 2875–2911 [DOI] [PubMed] [Google Scholar]
- 3. Shen H., Wang F., Zhang Y., Huang Q., Xu S., Hu H., Yue J., Wang H. (2009) A novel inhibitor of indole-3-glycerol phosphate synthase with activity against multidrug-resistant Mycobacterium tuberculosis. FEBS J. 276, 144–154 [DOI] [PubMed] [Google Scholar]
- 4. Shen H., Yang E., Wang F., Jin R., Xu S., Huang Q., Wang H. (2010) Altered protein expression patterns of Mycobacterium tuberculosis induced by ATB107. J. Microbiol. 48, 337–346 [DOI] [PubMed] [Google Scholar]
- 5. Czekster C. M., Neto B. A. D., Lapis A. A. M., Dupont J., Santos D. S., Basso L. A. (2009) Steady-state kinetics of indole-3-glycerol phosphate synthase from Mycobacterium tuberculosis. Arch. Biochem. Biophys. 486, 19–26 [DOI] [PubMed] [Google Scholar]
- 6. Sasaki T., Mizuguchi S., Honda K. (2012) Growth inhibitory effects of anthranilic acid and its derivates against Legionella pneumophila. J. Biosci. Bioeng. 113, 726–729 [DOI] [PubMed] [Google Scholar]
- 7. Houlihan W. J. (ed) (1972) Indoles, Wiley-Interscience, New York [Google Scholar]
- 8. Smith M., March J. (2001) March's Advanced Organic Chemistry: Reactions, Mechanisms, And structure, 5th Ed., John Wiley & Sons, Inc., New York [Google Scholar]
- 9. Hennig M., Darimont B. D., Jansonius J. N., Kirschner K. (2002) The catalytic mechanism of indole-3-glycerol phosphate synthase: crystal structures of complexes of the enzyme from Sulfolobus solfataricus with substrate analogue, substrate, and product. J. Mol. Biol. 319, 757–766 [DOI] [PubMed] [Google Scholar]
- 10. Darimont B., Stehlin C., Szadkowski H., Kirschner K. (1998) Mutational analysis of the active site of indoleglycerol phosphate synthase from Escherichia coli. Protein Sci. 7, 1221–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zaccardi M. J., Mannweiler O., Boehr D. D. (2012) Differences in the catalytic mechanisms of mesophilic and thermophilic indole-3-glycerol phosphate synthase enzymes at their adaptive temperatures. Biochem. Biophys. Res. Commun. 418, 324–329 [DOI] [PubMed] [Google Scholar]
- 12. Czekster C. M., Lapis A. A. M., Souza G. H. M. F., Eberlin M. N., Basso L. A., Santos D. S., Dupont J., Neto B. A. D. (2008) The catalytic mechanism of indole-3-glycerol phosphate synthase (IGPS) investigated by electrospray ionization (tandem) mass spectrometry. Tetrahedron Lett. 49, 5914–5917 [Google Scholar]
- 13. Eberhard M., Kirschner K. (1989) Modification of a catalytically important residue of indole-3-glycerol phosphate synthase from Escherichia coli. FEBS Lett. 245, 219–222 [DOI] [PubMed] [Google Scholar]
- 14. Knöchel T. R., Hennig M., Merz A., Darimont B., Kirschner K., Jansonius J. N. (1996) The crystal structure of indole-3-glycerol phosphate synthase from the hyperthermophilic archaeon Sulfolobus solfataricus in three different crystal forms: effects of ionic strength. J. Mol. Biol. 262, 502–515 [DOI] [PubMed] [Google Scholar]
- 15. Mazumder-Shivakumar D., Bruice T. C. (2004) Molecular dynamics studies of ground state and intermediate of the hyperthermophilic indole-3-glycerol phosphate synthase. Proc. Natl. Acad. Sci. U.S.A. 101, 14379–14384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mazumder-Shivakumar D., Kahn K., Bruice T. C. (2004) Computational study of the ground state of thermophilic indole glycerol phosphate synthase: structural alterations at the active site with temperature. J. Am. Chem. Soc. 126, 5936–5937 [DOI] [PubMed] [Google Scholar]
- 17. Schneider B., Knöchel T., Darimont B., Hennig M., Dietrich S., Babinger K., Kirschner K., Sterner R. (2005) Role of the N-terminal extension of the (βα)8-barrel enzyme indole-3-glycerol phosphate synthase for its fold, stability, and catalytic activity. Biochemistry 44, 16405–16412 [DOI] [PubMed] [Google Scholar]
- 18. Hennig M., Darimont B., Sterner R., Kirschner K., Jansonius J. N. (1995) 2.0 Å structure of indole-3-glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus: possible determinants of protein stability. Structure 3, 1295–1306 [DOI] [PubMed] [Google Scholar]
- 19. Eberhard M., Tsai-Pflugfelder M., Bolewska K., Hommel U., Kirschner K. (1995) Indoleglycerol phosphate synthase-phosphoribosyl anthranilate isomerase: comparison of the bifunctional enzyme from Escherichia coli with engineered monofunctional domains. Biochemistry 34, 5419–5428 [DOI] [PubMed] [Google Scholar]
- 20. Merz A., Knöchel T., Jansonius J. N., Kirschner K. (1999) The hyperthermostable indoleglycerol phosphate synthase from Thermotoga maritima is destabilized by mutational disruption of two solvent-exposed salt bridges. J. Mol. Biol. 288, 753–763 [DOI] [PubMed] [Google Scholar]
- 21. Witkop B., Patrick J. B. (1951) Addition reactions and Wagner-Meerwein rearrangements in the indoxyl series. J. Am. Chem. Soc. 73, 713–718 [Google Scholar]
- 22. Witkop B., Patrick J. B. (1951) The course and kinetics of the acid-base-catalyzed rearrangements of 11-hydroxytetrahydrocarbazolenine. J. Am. Chem. Soc. 73, 2188–2195 [Google Scholar]
- 23. Schlee S., Dietrich S., Kurćon T., Delaney P., Goodey N. M., Sterner R. (2013) Kinetic mechanism of indole-3-glycerol phosphate synthase. Biochemistry 52, 132–142 [DOI] [PubMed] [Google Scholar]
- 24. Wilmanns M., Priestle J. P., Niermann T., Jansonius J. N. (1992) Three-dimensional structure of the bifunctional enzyme phosphoribosylanthranilate isomerase:indoleglycerolphosphate synthase from Escherichia coli refined at 2.0 Å resolution. J. Mol. Biol. 223, 477–507 [DOI] [PubMed] [Google Scholar]
- 25. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]