

Background: Proprotein convertases (PCs) activate overexpressed endothelial lipase (EL) inhibitor angiopoietin-like-3 (ANGPTL3) and inactivate EL.
Results: In PC knock-out-mice, analysis of primary hepatocytes and circulating ANGPTL3 and EL fragments revealed that furin is their primary convertase.
Conclusion: However, the lack of hepatocyte furin had no major impact on HDL-cholesterol or EL phospholipase activity.
Significance: Inhibition/silencing of furin in hepatocytes would not affect lipid profiles.
Keywords: Cholesterol, HDL, Lipoprotein, Phospholipase, Protease, Angiopoietin-like 3, Endothelial Lipase, Furin, Knockout Mice, Proprotein Convertases
Abstract
The proprotein convertases (PCs) furin, PC5/6, and PACE4 exhibit unique and/or complementary functions. Their knock-out (KO) in mice resulted in strong and specific phenotypes demonstrating that, in vivo, these PCs are unique and essential during development. However, they also exhibit redundant functions. Liver angiopoietin-like 3 (ANGPTL3) inhibits lipolysis by binding to lipoprotein lipases. It is found in the plasma as full length and truncated forms. The latter is more active and generated by cleavage at a furin-like site. Endothelial lipase (EL) binds heparin sulfate proteoglycans on cell surfaces and catalyzes the hydrolysis of HDL phospholipids. EL activity is regulated by two endogenous inhibitors, ANGPTL3 and ANGPTL4, and by PCs that inactivate EL through cleavage releasing the N-terminal catalytic and C-terminal lipid-binding domains. Herein, because furin and PC5/6 complete KOs are lethal, we used mice lacking furin or PC5/6 specifically in hepatocytes (hKO) or mice completely lacking PACE4. In primary hepatocytes, ANGPTL3 was processed into a shorter form of ANGPTL3 intracellularly by furin only, and extracellularly mainly by PACE4. In vivo, the absence of furin in hepatocytes reduced by ∼50% the circulating levels of cleaved ANGPTL3, while the lack of PACE4 had only a minor effect. Analysis of the EL processing in primary hepatocytes and in vivo revealed that it is mostly cleaved by furin. However, the lack of furin or PC5/6 in hepatocytes and complete PACE4 KO did not appreciably modify plasma HDL levels or EL activity. Thus, inhibition of furin in liver would not be expected to modify the plasma lipid profiles.
Introduction
A large number of secretory proteins are produced as precursors that are cleaved at specific sites to generate mature bioactive products. Most of these specific cleavages occur after basic residues and are achieved by one or more of the seven basic amino acid-specific members of the proprotein convertase (PC)2 family, which share identities with bacterial subtilisins and yeast kexin (genes PCSK1 to PCSK7) (1). Four of them: furin, PC5/6, PACE4, and PC7, are widely or ubiquitously expressed and responsible for most of the processing events occurring in the constitutive secretory pathway: trans Golgi network (TGN), cell surface, and endosomes. This leads to the activation, and less frequently to the inactivation, of receptors, ligands, enzymes, viral glycoproteins, or TGFβ-like growth factors (2, 3). Although these PCs exhibit a functional redundancy ex vivo, their inactivation in mice leads to specific phenotypes revealing that, in vivo, each PC fulfills unique processing events. Furin knock-out (KO) in mice resulted in numerous embryonic malformations, including the absence of axial rotation and heart looping leading to death around embryonic day (E)11 (4). PC5/6 KO leads to death at birth with an altered antero-posterior pattern, including extra vertebrae and lack of tail, kidney agenesis, hemorrhages, collapsed alveoli, and retarded ossification (5, 6). PACE4 KO resulted in an altered left-right patterning, including cyclopism, craniofacial, and cardiac malformations in some embryos (7). Finally, PC7 KO mice exhibit anxiolytic and novelty seeking phenotypes (8).
Endothelial lipase (EL) (Fig. 1B) is the third and last member of the vascular lipoprotein lipase family that also comprises lipoprotein lipase (LPL) and hepatic lipase (HL) (9). EL is produced by many organs including mammary gland, ovary, testis, and intestine (10). EL is the only lipoprotein lipase produced by endothelial cells (11). It plays a crucial role in HDL metabolism as mice either lacking EL (12, 13) or receiving EL antibodies (14) have higher HDL-cholesterol (HDLc) levels. In contrast, EL overexpression reduces HDLc (15, 16). Unlike LPL and HL, EL has a strong phospholipase A1 activity, and a poor triglyceride lipase activity (12, 15, 17).
FIGURE 1.

Processing of ANGPTL3 in COS-1 cells. A, schematic of V5-tagged mouse ANGPTL3 is shown, as well as its canonical PC cleavage site. B and C, COS-1 cells were transiently transfected with an empty vector (V) or vectors encoding indicated proteins (V5-tagged mouse ANGPTL3, PCs and/or the PC inhibitors pro-furin (ppFur) and α1-PDX). D, COS-1 cells were transiently transfected in equal proportions with an empty vector (V) or vectors encoding PC5/6 and/or mouse ANGPTL3 cleavage site mutants. Conditioned media were subjected to Western blotting 24 h post-transfection using a V5 tag mAb.
Several studies indicated that PCs may regulate HDL metabolism by controlling endothelial lipase (EL) activity (18). EL is secreted and binds to heparan sulfate proteoglycans (HSPGs) on the endothelial cell surface, thus favoring HDL retention and delipidation via its phospholipase activity (19, 20). It has been proposed that PCs could enhance HDLc levels, and thus be cardioprotective, through the cleavage-inactivation of EL (21, 22), and cleavage-activation of angiopoietin-like protein 3 (ANGPTL3) (23), an endogenous inhibitor of EL (24). ANGPTL3 belongs to a family of eight secreted factors, encoded by the genes ANGPTL1 to ANGPTL8, exhibiting a signal peptide, an N-terminal coiled-coil domain and a C-terminal fibrinogen-like domain (Fig. 1A). Angiopoietin-like proteins share structural identity with the four known angiopoietins (25, 26) but, different from the latter, they do not bind to Tie1 or Tie2 receptors (27, 28). Unlike other members of the family that are widely expressed, ANGPTL3 is detected only in the liver, where it regulates lipid metabolism (29). In mouse, the loss of ANGPTL3 expression results in lower levels of triglycerides and HDLc (24, 30, 31), whereas ANGPTL3 injection or overexpression increases circulating lipid levels (23, 29). Accordingly, it was shown that ANGPTL3 inhibits lipolysis by binding through its N-terminal region (23, 32) to EL and LPL (24, 30, 33). ANGPTL3 is found in the plasma as a full-length protein and a truncated form generated by cleavage at a furin-like site (RAPR224↓) located in the linker region between the coiled-coil and fibrinogen-like domains (23) (Fig. 1A). This truncated form exhibits enhanced inhibitory activity for EL (18), but not LPL (23), suggesting that PC-mediated cleavage of ANGPTL3 is more important for inhibiting EL versus LPL activity.
To elucidate the in vivo role of hepatic furin, PC5/6 and PACE4 in the processing of ANGPTL3 and EL, we used mice lacking furin or PC5/6 specifically in hepatocytes (hKO) (34), and mice completely lacking PACE4 (7). Our data showed that, in primary hepatocytes, ANGPTL3 was processed intracellularly by furin only, and extracellularly mainly by PACE4. In vivo, the absence of furin in hepatocytes reduced by ∼50% the circulating levels of cleaved ANGPTL3, while the absence of PC5/6 or PACE4 had no or little impact. On the other hand, EL is exclusively processed by furin from hepatocytes both intracellularly and at the cell surface. This conclusion was further supported by the absence of cleaved EL in the plasma of mice lacking furin in hepatocytes. However, circulating HDLc levels and EL activity were not significantly affected in the latter mice, suggesting that in vivo EL cleavage by furin from hepatocytes has no major impact on HDLc.
EXPERIMENTAL PROCEDURES
Mice and Genotyping
Furinflox/flox (35) or Pcsk5flox/flox (5) mice not carrying or carrying a copy of Tg(Alb-cre) (34) were described previously. Mice were housed in a 12 h-light/dark cycle and fed a standard diet (2018 Teklad global 18% protein rodent diet; Harlan Laboratories). All procedures were approved by the IRCM bioethics committee for animal care.
Mice Bleeding and Circulating Cholesterol Measurement
Mice were fasted for 4 h prior to final bleeding by heart punction; the plasma was obtained by centrifugation at 3000 × g for 15 min. Plasma total cholesterol (TC) was determined using the Infinity reagent (Thermo Fisher Scientific, Mississauga, ON). For lipoprotein profiles, a plasma pool from 6 to 9 mice (0.3 ml) was analyzed by FPLC (Pharmacia) on a Superose 6 column with a flow rate of 0.3 ml/min.
Preparation of Primary Hepatocytes, Culture, and Transfection
Hepatocytes were prepared from 8–12-week-old male livers using the two-step collagenase perfusion method (34). After anesthesia of mice by 2% isofluran inhalation, the peritoneal cavity was opened, and the liver was perfused in situ via the inferior vena cava for 6 min at 37 °C with calcium-free HEPES buffer I (142 mm NaCl, 6.7 mm KCl, 10 mm Hepes, pH 7.6), and for 8 min with calcium-supplemented HEPES buffer II (4.7 mm CaCl2, 66.7 mm NaCl, 6.7 mm KCl, 100 mm Hepes, pH 7.4) containing 0.5 mg/ml collagenase Type V (Sigma Aldrich). The perfusion rates were set to 8 and 6 ml/min, respectively. In 3.5 cm Petri dishes coated with fibronectin (0.5 mg/ml, Sigma Aldrich), 5.105 cells were seeded in Williams' medium E supplemented with 10% fetal bovine serum (GIBCO BRL). After 2 h, the medium was replaced with hepatozyme medium (GIBCO BRL) for 12 h prior to treatment. All transfections were performed using 6 μg of DNA and Effectene transfection reagent, as recommended by the manufacturer (Qiagen).
Western Blotting and Antibodies
ANGPTL3 was immunoprecipitated from plasma (50 μl) or hepatocytes culture media (1 ml) using a mouse antibody (R&D system, AF136, 1:200). The proteins were then separated on 8% gels by tricine SDS-PAGE and transferred to a PVDF membrane (Perkin Elmer). ANGPTL3 was revealed using the same ANGPTL3 antibody (1:5000) and anti-goat IgG (1:2000; TrueBlot). For Western blot, plasma or media from primary hepatocytes or transfected cells were analyzed as above, using a mEL polyclonal antibody (1:5,000; graciously provided by D.J. Rader, University of Pennsylvania) or a V5-monoclonal antibody (V5-mAb; 1:5000; Invitrogen). The antigen-antibody complexes were visualized using appropriate HRP conjugated secondary antibodies and enhanced chemiluminescence kit (ECL; Amersham Biosciences or Pierce). Signal quantitation was performed using ImageJ software, and the values given correspond to the ratio CT/(precursor +CT). The presented data show a representative experiment among three or more independent experiments. Signal quantification was then performed on the selected experiment.
Inhibitor Treatment
At 24 h post-transfection, hepatocytes were treated for 6 h with two pan-PC inhibitors: the cell permeable decanoyl-RVKR-choromethylketone (cmk; 50 μm) or the cell surface PC inhibitor hexa-d-arginine (D6R; 20 μm) that also mimicks a PC clevage site (36). Culture media were then replaced with fresh ones containing the inhibitors for an additional 24 h.
Measurement of Phospholipase Activity in Conditioned Media and hKO Plasma
Pre and post-heparin plasma were obtained before and 10 min after intravenous injection of heparin (300 units/kg). The phospholipase activity was measured using EnzChek Phospholipase A1 Assay kit as recommended by the manufacturer (Invitrogen).
RESULTS
Processing of Mouse ANGPTL3 versus Human ANGPTL3 in COS-1 Cells
It has been previously shown that human ANGPTL3 undergoes cleavage at Arg224↓ (23) present in a typical basic amino acid PC-site RXXR224↓ (1). In agreement, R221A substitution prevented processing of ANGPTL3 in HEK293 cells (33) (Fig. 1A). Herein, we tested the ability of various PCs to process both mouse and human sequences.
The V5-tagged mouse ANGPTL3 was co-expressed in COS-1 cells with either furin, PC5/6, PC7, or PACE4. Western blot analysis of the media with a V5-mAb revealed two forms corresponding to the full-length protein (ANGPTL3) and its C-terminal fragment (CT). This substrate, whose partial cleavage by endogenous enzymes is occasionally observed (Fig. 1, B and D, lane 2), was further processed by all tested PCs except PC7 (Fig. 1B). Because very similar data were obtained with human ANGPTL3 (not shown), thereafter we only concentrated on mouse ANGPTL3. Cleavage of ANGPTL3 by PC5/6 as a representative PC was blocked by two general PC-inhibitors, the furin prosegment (ppFur) (37) and α1-PDX (38) (Fig. 1C). Furthermore, analysis the single R224A and R221A or double R224A+R221A mutants revealed that they were not cleaved, reinforcing the hypothesis that cleavage occurs at the typical PC-processing motif RAPR224↓, in which Arg at the P1 and P4 positions are both critical. Although P6 basic residues are sometimes important for PC cleavage, the K219A mutation did not affect ANGPTL3 processing (Fig. 1D).
When overexpressed in mouse, human ANGPTL3 was reported to be also cleaved at ISLSSKPR221↓APR224, a non-canonical site lacking P4, P6, or P8 basic residues (23). This human sequence differs from the mouse one only by the presence of Pro220 instead of Ser220. However, replacement of the Ser by Pro in mouse ANGPTL3-R224A did not lead to cleavage after Arg221 by any of the above PCs or by PC7 (data not shown). Thus, while the cognate processing enzyme of human ANGPTL3 that cleaves at Pro-Arg221 could be thrombin (23) it has yet to be defined.
Subcellular Localization of Mouse ANGPTL3 and Its Processing in Primary Hepatocytes
To elucidate the compartment(s) in which ANGPTL3 is activated by PCs, we performed immunohistochemical analysis in HuH7 cells, as well as cleavage studies in primary hepatocytes lacking specific PCs and/or treated with PC inhibitors.
The subcellular co-localization of the transiently expressed mouse ANGPTL3-V5 using V5-mAb, as well as antibodies against the Golgi marker GM130, the early endosome marker EEA1, and the cell surface marker EGFR revealed that cellular ANGPTL3 was present in the Golgi (likely the trans Golgi network, TGN), early endosomes, as well as at the cell surface (data not shown), as are all the convertases studied (1).
Complete PACE4 KO mice are viable (7), while mice lacking furin (4) or PC5/6 (5, 6) die at E11 or at birth, respectively. However, conditional alleles encoding furin or PC5/6 can be efficiently inactivated in hepatocytes (hKO) in transgenic mice expressing Cre under the control of the albumin promoter (Alb-cre) (34). Thus, we analyzed the processing of endogenous (Fig. 2A) or transiently transfected mouse ANGPTL3 (Fig. 2, B–D) in either wild type (WT) primary hepatocytes, or in those lacking specifically PC5/6 (PC5/6-hKO), PACE4 (PACE4-KO), or furin (Fur-hKO). The data show that the percentage of endogenous secreted CT forms was not affected in the PC5/6-hKO medium, but was reduced by ∼50% in the media of Fur-hKO and [Fur-PC5/6]-hKO primary hepatocytes (13% versus 25% in Fig. 2A). Note that the light band observed below the ANGPTL3 signal in Fig. 2, B–D is likely a migration artifact due to the high concentrations of albumin secreted by hepatocytes. It is not seen in Fig. 2A in which immunoprecipitated products are analyzed.
FIGURE 2.

Cleavage of mouse ANGPTL3 in primary hepatocytes. A, endogenous mANGPTL3 was immunoprecipitated from culture media of primary hepatocytes either WT or lacking PC5/6, furin or both enzymes using a mouse antibody raised against the C-terminal domain of mANGPTL3. B–D, hepatocytes from indicated genotype were transiently transfected with a plasmid expressing V5-tagged mouse ANGPTL3. After 16 h, cells were treated for 6 h with no inhibitor or the PC inhibitors dec-RVKR-cmk (RVKR; 50 μm; cell permeable) and hexa-d-arginine (D6R, 20 μm; cell impermeable). Media were then replaced with fresh ones containing or not the inhibitors for an additional 24 h, and finally analyzed by Western blotting using a mAb V5.
From mouse ANGPTL3 overexpression experiments we deduce the following: 1) Cleavage of ANGPTL3 was completely abrogated by the cell-permeable pan-PC inhibitor RVKR (decanoyl-RVKR-cmk) (36) in all genotypes (Fig. 2, B–D), confirming that it is achieved by PCs. 2) In WT hepatocytes, the cell-surface specific inhibitor D6R prevents ∼50% of the cleavage, revealing that processing occurs both intracellularly and at the cell surface (Fig. 2B). 3) The absence of PC5/6 (PC5/6-hKO) does not affect ANGPTL3 cleavage (Fig. 2B), suggesting that PC5/6 does not participate in the processing of ANGPTL3 in hepatocytes. 4) In contrast, the absence of PACE4 (PACE4-KO) results in ∼50% lower overall processing and the remaining cleavage occurs both intracellularly and at the cell surface in the same proportion since D6R only inhibits ∼50% of the remaining cleavage (Fig. 2C). 5) In the absence of furin (Fur-hKO), or in [Fur-PC5/6]-hKO, processing occurs primarily at the cell surface, as the remaining cleavage is completely abrogated by D6R (Fig. 2D). This suggests that when furin is absent, PACE4 can efficiently substitute for it, resulting in a predominantly cell-surface cleavage. However, in absence of PACE4, furin only partially processes ANGPTL3 at the cell surface and/or in the medium (Fig. 2C). Furthermore, these data show that furin is responsible for most of the intracellular processing of ANGPTL3 (Fig. 2D).
Altogether, the data indicate that, upon overexpression of ANGPTL3 in primary hepatocytes, the CT is mostly generated intracellularly by membrane-bound furin and equally performed by furin and PACE4 extracellularly.
In Vivo Processing of ANGPTL3
Since ANGPTL3 is produced by the liver and secreted into the plasma (39), we used the plasma of the above mouse models to analyze the endogenous circulating forms of ANGPTL3 by immunoprecipitation and Western blotting. Compared with that of WT mice, the percentage of circulating CT forms was unaffected in PC5/6-hKO mice, and decreased by ∼50 and ∼25% in Fur-hKO and PACE4 KO mice, respectively (Fig. 3), Thus, the data reveal the major role played by furin in the processing of ANGPTL3 in vivo, as well as the lesser contribution of PACE4.
FIGURE 3.
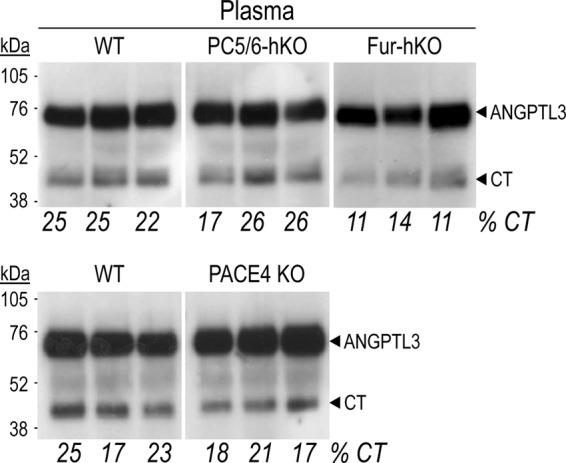
Circulating forms of ANGPTL3 in PC5/6-hKO, Fur-hKO and PACE4 KO mice. Endogenous circulating ANGPTL3 was analyzed in the plasma of 3 mice for each genotype by immunoprecipitation and Western blotting using an antibody raised against ANGPTL3 C-terminal domain. The migration of full-length ANGPTL3 and its C-terminal fragment is shown.
To determine the possible participation of endothelial furin or PC5/6 in the processing of circulating ANGPTL3, analyses were performed in the plasma of newborn mice specifically lacking furin in endothelial cells (Fur-eKO) (40) or in adult PC5/6-eKO mice (41) that carry a copy of the Tie2-cre transgene. Only newborn mice lacking furin in endothelial cells could be assessed as they rapidly die after birth (40). No change in the levels of circulating ANGPTL3 and CT forms (data not shown) were observed, suggesting that endothelial furin and PC5/6 do not affect their circulating levels.
Endothelial Lipase Inactivation in hKO Mice
The plasma levels of HDL are inversely proportional to EL activity (12, 14). Following its secretion, EL binds to the endothelial cell surface HSPGs via basic residues within its heparin-binding motif KMRNKRNSKMYLK337, and hydrolyzes the phospholipids of circulating HDL particles. EL can be inactivated ex vivo within the above motif at KMRNKR330↓NS by all the constitutive PCs (furin, PC5/6, PACE4, and PC7) (Fig. 4A), leading to its release as inactive N- and C-terminal fragments (13, 39).
FIGURE 4.
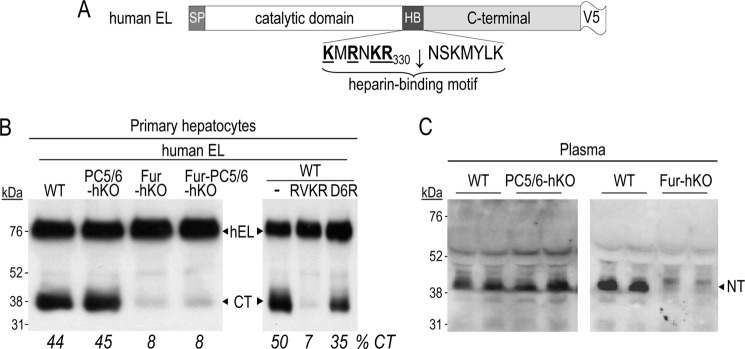
EL is mainly cleaved by furin from hepatocytes. A, schematic of V5-tagged human EL and its canonical PC cleavage site is shown. B, primary hepatocytes, either WT or lacking PC5/6, furin or both, were transiently transfected with a vector expressing V5-tagged human EL and, after 16h, their media were analyzed by Western blot using a mAb-V5. WT primary hepatocytes were also treated for 6h with no inhibitor (−) or the PC inhibitors RVKR (50 μm) or D6R (20 μm). Media were then replaced with fresh ones not containing or containing the inhibitors for an additional 24 h, and finally analyzed by Western blotting using a mAb-V5. C, circulating EL was analyzed in pre-heparin plasma from PC5/6-hKO and Fur-hKO mice and their WT controls by Western blotting using an antibody raised against the N-terminal domain of mEL. The migration of the N-terminal (NT) fragment is shown.
EL was reported to be expressed in many organs (29), including endothelial cells (30), but no report was found on its expression in hepatocytes. Simultaneous qPCR analysis of EL mRNA levels in primary endothelial cells and hepatocytes revealed that EL was equally expressed in both cell types (data not shown). Immunocytochemistry in HuH7 cells expressing V5-tagged human EL was also performed and revealed that EL was present in the Golgi (GM130 marker) and early endosomes (EEA1 marker), but mostly localized at the cell surface (EGFR marker) (data not shown).
To evaluate the role of PCs in EL inactivation in hepatocytes, the 48 h-conditioned media of primary hepatocytes prepared from WT, PC5/6-hKO, or Fur-hKO mice that expressed transiently V5-tagged human EL were analyzed by Western blotting (Fig. 4B). The percentage of C-terminal fragments (CT) generated was unaffected in the medium of PC5/6-hKO hepatocytes, but reduced by more than 80% in that of Fur-hKO or [Fur-PC5/6]-hKO hepatocytes. This indicates that furin in this cell type plays a major role in EL inactivation by cleavage. Treatment with RVKR or D6R revealed that EL is mostly (∼70%) cleaved intracellularly, but also extracellularly as D6R partially (∼30%) inhibited EL cleavage.
In agreement, analysis of mouse EL in plasma by Western blotting using an N-terminal (NT) antibody confirmed the key role of furin, since almost no NT fragment is detected in its absence (Fig. 4C).
Circulating Cholesterol and Cholesterol Profiles in hKO Mice
The absence of furin, PACE4, or PC5/6 did not affect the total cholesterol content (Fig. 5A). To directly assess the physiological role of the PCs from hepatocytes on HDLc metabolism, the plasma lipid profiles of PC5/6, Fur-hKO, and PACE4 KO mice, and their WT counterpart, were analyzed by FPLC (Fig. 5B). Because the lack of furin in hepatocytes resulted in decreased EL and ANGPTL3 cleavages, lower HDLc levels were expected in Fur-hKO plasma. However, FPLC lipid profiles revealed that HDLc levels were reduced by only ∼8% in Fur-hKO (Fig. 5B), suggesting that the overall EL activity in the body was not significantly affected by the absence of furin in hepatocytes.
FIGURE 5.
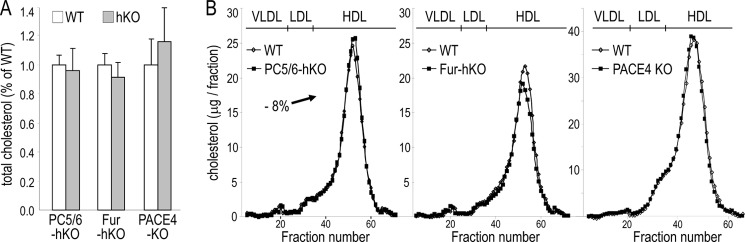
Plasma cholesterol profiles of PC5/6-hKO, Fur-hKO, and PACE4 KO mice and total cholesterol levels. 6–9 mice per genotype were fed a chow diet and fasted 6 h prior to bleeding. A, total cholesterol is expressed as a percentage of WT control values. B, pooled plasma (0.3 ml) was subjected to FPLC and cholesterol levels were measured in each fraction. The elution positions of VLDL, LDL, and HDL are indicated.
Endothelial Lipase Activity Is Unchanged in Fur-hKO Mice
Like phospholipase A1, EL hydrolyzes phospholipids at the sn-1 position and produces phospholipids and free fatty acids. To directly assess the EL-specific activity in Fur-hKO plasma versus that in WT plasma, we used a commercially available fluorogenic substrate and a neutralizing mouse EL antibody to measure specifically the EL activity as recently reported (42). The phospholipase activity was determined in pre- and post-heparin plasma from WT and Fur-hKO mice of 2 months of age, in the presence or absence of a mouse EL antibody (Fig. 6). All lipase activities were normalized to that found in untreated WT plasma. Heparin treatment of WT and Fur-hKO plasma resulted in a ∼45 and ∼55% increase of the lipase activity, respectively. As previously shown (42), the heparin-released activity was essentially due to EL, as mouse EL antibodies reduced it by 91 and 80% in WT and Fur-hKO plasma, respectively. In agreement with the above data on similar cholesterol profiles, the estimated WT and Fur-hKO EL-specific activities did not statistically differ. Altogether, the data indicate that furin in hepatocytes has no major impact on post-heparin plasma EL activity.
FIGURE 6.
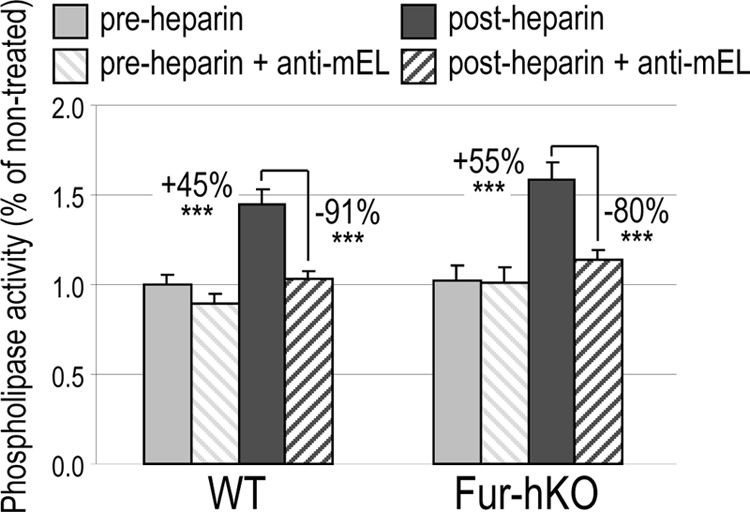
Measurement of EL phospholipase activity in WT and Fur-hKO plasma. Mice (n = 5) fed a chow diet were fasted for 6h. Pre- and post-heparin plasma were obtained before and 10 min after intravenous injection of heparin (0.3 units/g). Phospholipase activity was measured in 0.5 μl of plasma in the absence or presence of 0.2 μl of a mEL antibody. Mean ± S.D.; ***, p < 0.0005 (bilateral Student's t test).
DISCUSSION
The identification of the PCs responsible for the cleavage of ANGPTL3 and EL in vivo has not yet been reported. In this study, we analyzed the role of hepatic PCs in the cleavage-activation of ANGPTL3 (23), and the cleavage-inactivation of EL (21) in COS-1 cells and/or primary hepatocytes and plasma from KO mice.
For ANGPTL3 we showed that: (i) in COS-1 cells, furin, PC5/6 and PACE4 were the 3 candidate convertases for the processing of ANGPTL3 (Fig. 1B). (ii) P1 and P4 Arg are both critical for cleavage (Fig. 1D), in agreement with previously reported P1 Arg221 (33) or P4 Arg224 (18) mutations. (iii) In primary hepatocytes, ANGPTL3 is activated intracellularly by furin only and extracellularly, essentially by PACE4 and furin (Figs. 2, 7). This agrees with the cycling of membrane-bound furin between the TGN and the cell surface (43), and the localization of active PACE4 to the cell surface through its binding to HSPGs (44). The absence of PC5/6 did not affect ANGPTL3 processing. (iv) In plasma, the greatest reduction of ANGPTL3 processing was obtained in Fur-hKO mice that lack furin specifically in hepatocytes (Fig. 3).
FIGURE 7.
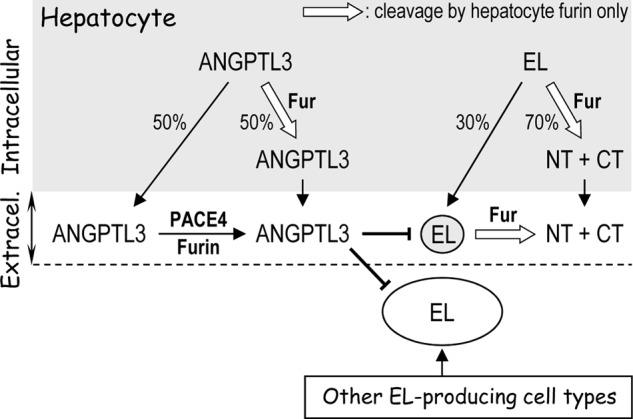
Model proposed for the role of hepatic proprotein convertases in ANGPTL3 and EL processing. ANGPTL3 is mainly secreted by hepatocytes, whereas EL is produced by hepatocytes and other cell types. In the hepatocyte, both molecules are cleaved intracellularly exclusively by furin (white-filled arrow), ANGPTL3 is mainly cleaved extracellularly by PACE4, known to be retained in the extracellular matrix (ECM) and to bind cell-surface HSPGs, whereas extracellular cleavage of EL is exclusively achieved by cell-surface furin from hepatocytes. The circulating fragments of EL would thus originate from hepatocyte EL only upon cleavage by local furin.
The absence of furin in hepatocytes has the strongest effect on ANGPTL3 cleavage, reducing the circulating C-terminal fragment by ∼50%. To determine whether endothelial furin could be responsible for the remaining cleavage of ANGPTL3, we analyzed the plasma of E18.5 embryos of WT and those lacking specifically furin in endothelial cells (40), before their rapid death after birth (data not shown). Although no difference in ANGPTL3 processing could be observed, this analysis led us to reveal an amazingly high level of endogenous processing of ANGPTL3 in E18.5 WT embryos, with 75% of generated CT fragments versus 25% in adults, and a concomitant reduction in the apparent molecular sizes of ANGPTL3 and CT, possibly due to lack of post-translational modification(s). A possible example is O-glycosylation, known to occur at Thr225-Thr226 at the P1′-P2′ positions of the cleavage site of ANGPTL3, which would inhibit PC-processing (45). It is a matter of speculation whether the furin-generated processing of ANGPTL3 is enhanced in E18.5 embryos versus adult liver due to lower O-glycosylation.
In this report, we show for the first time that hepatocytes produce EL. In HuH7 cells, the latter is mostly localized to the cell surface (data not shown). Human EL was previously reported to be inactivated via cleavage at RNKR330↓ by furin, PACE4, and PC5/6, but not PC7, with the P1 and P4 Arg being critical for this processing (13). Herein, we demonstrated that: (i) In primary hepatocytes, cleavage of human EL is exclusively achieved by furin, both intracellularly (∼70%) and extracellularly (∼30%). (ii) In addition, the N-terminal fragment of EL, which is well detected in WT plasma, was not detected in Fur-hKO plasma (Fig. 4).
This key role of furin may reflect its high levels of expression in the liver and hepatocytes. In primary hepatocytes, PC5/6 and PACE4 mRNA levels were, respectively, ∼270- and 7-fold lower than those of furin (data not shown). Furthermore, the inactivation of the PC5/6 gene in hepatocytes (PC5/6-hKO) revealed that this cell type is the only one that expresses PC5/6 in the liver (34). This is not the case for furin whose expression in the liver was only reduced by 2-fold in Fur-hKO livers (34), indicating that other cells in this tissue strongly express furin. However, furin from non-hepatocyte cells does not seem to compensate for the absence of furin in hepatocytes as EL cleavage, which is also achieved at the cell surface, was not detected in the absence of furin in hepatocytes (Fig. 4C).
The limited effect of the absence of furin and PACE4 on ANGPTL3 processing reveals that these PCs, and possibly PC5/6, can compensate for each other. In the absence of furin, the intracellular cleavage ANGPTL3 is expected to be very poor, but to be likely achieved extracellularly in higher proportion. In contrast, there is no redundancy in hepatocytes between furin and other PCs for EL cleavage, both intra- and extracellularly.
Even though hepatocyte-derived furin clearly cleaves both ANGPTL3 and EL in vivo, the plasma HDLc levels (−8%) and total cholesterol were minimally affected by the lack of furin, or not at all affected by the absence of PC5/6 in hepatocytes or by the complete PACE4 deficiency (Fig. 5). Moreover, the level of plasma EL phospholipase activity was unchanged in the Fur-hKO mice, which exhibited reduced plasma ANGPTL3 processing (∼50%) and no detectable EL cleavage (Fig. 6).
These findings raise the question of the physiological importance of EL cleavage into N- and C-terminal fragments by furin. In primary hepatocytes, EL is mainly cleaved intracellularly (∼70%). The absence of compensation by extracellular, shed furin from other cell types suggests that the extracellular cleavage (∼30%) may be rapidly achieved at the hepatocyte cell surface by furin from hepatocytes only. If this is the case, the circulating N- and C-terminal EL fragments would only be generated by furin cleavage of EL in hepatocytes.
This raises the question of the origin of the released post-heparin phospholipase activity measured in the plasma of our mice. Since such EL-related activity is not significantly changed in the presence or absence of furin in hepatocytes (Fig. 6), could it be mostly derived from tissues other than liver hepatocytes (Fig. 7)?
Until very recently, it was thought that ANGPTL3 and ANGPTL4 are the only angiopoietins that can be cleaved and activated by basic-amino acid specific PCs (33, 46). However, a novel paralog of ANGPTL3 has now been identified, called ANGPTL8, which is highly regulated by fasting and re-feeding in mice and humans (26). From its sequence, we predict that ANGPTL8 could be cleaved by a furin-like PC at RQRR175↓EM (human) or RQQR175↓EM (mouse). The function ascribed to ANGPTL8 is to activate ANGPTL3 (26), and both are processed, and likely activated, by a furin-like convertase. Thus, furin-like PCs activate ANGPTL3 either directly by cleavage at RAPR224↓TT, or indirectly by processing its activator ANGPTL8 at a similar RQQR175↓EM site. The putative processing site of ANGPTL8 suggests that furin may also be the major processing enzyme of this factor, especially in view of the presence of Glu at P1′ (1). As this protein is mostly expressed in liver (26), such prediction will now have to be tested in our hKO mice.
Furin levels are often up-regulated in cancers and metastases, and play a role in viral glycoprotein activation, suggesting that inhibition of furin could be potentially beneficial in the treatment of cancers and/or viral infections (reviewed in Ref. 1). Injection of an adenovirus recombinant of the furin prosegment, which inhibits furin, PC5/6 and PACE4 (37), resulted in a dramatic reduction of plasma HDLc in mice (18). In contrast, our results showed that elimination of furin only from hepatocytes results in a minor decrease in HDL (∼8%), suggesting that a specific furin inhibitor or silencer targeting liver may still be contemplated as a therapeutic avenue in some diseases (1), without a major negative effect on HDLc.
In conclusion, this work established that hepatocyte-derived furin is the major processing enzyme of ANGPTL3 and EL in vivo. It would be highly informative in the future to test whether similar conclusions could also be reached for LPL, ANGPTL4, and ANGPTL8.
Acknowledgments
We thank all the members of the Seidah laboratory for helpful discussions and Brigitte Mary for efficacious editorial assistance.
This research was supported by a CIHR Grant 44363, a Strauss Foundation grant, and a Canada Chair Number 216684.
- PCs
- proprotein convertases
- ANGPTL3
- angiopoietin-like 3
- LDL
- low density lipoprotein
- HDL
- high density lipoprotein
- PVDF
- polyvinylidene difluoride
- TGN
- trans Golgi network
- KO
- knockout mice
- hKO
- hepatocytes specific knockout
- EL
- endothelial lipase
- HL
- hepatic lipase
- LPL
- lipoprotein lipase
- E
- embryonic day
- PCSK
- proprotein convertase subtilisins kexin gene
- FBN
- fibrinogen
- HSPGs
- heparin sulfate proteoglycans
- qPCR
- quantitative RT-PCR
- V5-mAb
- V5-monoclonal antibody
- ECL
- enhanced chemiluminescence
- CT
- C-terminal fragment
- ppFur
- the furin prosegment
- RVKR
- decanoyl-RVKR-cmk
- D6R
- hexa-d-arginine
- FPLC
- fast protein liquid chromatography analysis
- P0
- newborn mouse
- NT
- N-terminal fragment.
REFERENCES
- 1. Seidah N. G., Prat A. (2012) The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 11, 367–383 [DOI] [PubMed] [Google Scholar]
- 2. Mayer G., Hamelin J., Asselin M. C., Pasquato A., Marcinkiewicz E., Tang M., Tabibzadeh S., Seidah N. G. (2008) The regulated cell surface zymogen activation of the proprotein convertase PC5A directs the processing of its secretory substrates. J. Biol. Chem. 283, 2373–2384 [DOI] [PubMed] [Google Scholar]
- 3. Seidah N. G., Sadr M. S., Chrétien M., Mbikay M. (2013) The multifaceted Proprotein Convertases: their unique, redundant, complementary and opposite functions. J. Biol. Chem. 288, 21473–21481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roebroek A. J., Umans L., Pauli I. G., Robertson E. J., van Leuven F., Van de Ven W. J., Constam D. B. (1998) Failure of ventral closure and axial rotation in embryos lacking the proprotein convertase Furin. Development 125, 4863–4876 [DOI] [PubMed] [Google Scholar]
- 5. Essalmani R., Zaid A., Marcinkiewicz J., Chamberland A., Pasquato A., Seidah N. G., Prat A. (2008) In vivo functions of the proprotein convertase PC5/6 during mouse development: Gdf11 is a likely substrate. Proc. Natl. Acad. Sci. U.S.A. 105, 5750–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Szumska D., Pieles G., Essalmani R., Bilski M., Mesnard D., Kaur K., Franklyn A., El Omari K., Jefferis J., Bentham J., Taylor J. M., Schneider J. E., Arnold S. J., Johnson P., Tymowska-Lalanne Z., Stammers D., Clarke K., Neubauer S., Morris A., Brown S. D., Shaw-Smith C., Cama A., Capra V., Ragoussis J., Constam D., Seidah N. G., Prat A., Bhattacharya S. (2008) VACTERL/caudal regression/Currarino syndrome-like malformations in mice with mutation in the proprotein convertase Pcsk5. Genes Dev. 22, 1465–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Constam D. B., Robertson E. J. (2000) SPC4/PACE4 regulates a TGFβ signaling network during axis formation. Genes Dev. 14, 1146–1155 [PMC free article] [PubMed] [Google Scholar]
- 8. Besnard J., Ruda G. F., Setola V., Abecassis K., Rodriguiz R. M., Huang X. P., Norval S., Sassano M. F., Shin A. I., Webster L. A., Simeons F. R., Stojanovski L., Prat A., Seidah N. G., Constam D. B., Bickerton G. R., Read K. D., Wetsel W. C., Gilbert I. H., Roth B. L., Hopkins A. L. (2012) Automated design of ligands to polypharmacological profiles. Nature 492, 215–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirata K., Dichek H. L., Cioffi J. A., Choi S. Y., Leeper N. J., Quintana L., Kronmal G. S., Cooper A. D., Quertermous T. (1999) Cloning of a unique lipase from endothelial cells extends the lipase gene family. J. Biol. Chem. 274, 14170–14175 [DOI] [PubMed] [Google Scholar]
- 10. Broedl U. C., Jin W., Rader D. J. (2004) Endothelial lipase: a modulator of lipoprotein metabolism upregulated by inflammation. Trends Cardiovasc. Med. 14, 202–206 [DOI] [PubMed] [Google Scholar]
- 11. Jaye M., Lynch K. J., Krawiec J., Marchadier D., Maugeais C., Doan K., South V., Amin D., Perrone M., Rader D. J. (1999) A novel endothelial-derived lipase that modulates HDL metabolism. Nat. Genet. 21, 424–428 [DOI] [PubMed] [Google Scholar]
- 12. Ma K., Cilingiroglu M., Otvos J. D., Ballantyne C. M., Marian A. J., Chan L. (2003) Endothelial lipase is a major genetic determinant for high-density lipoprotein concentration, structure, and metabolism. Proc. Natl. Acad. Sci. U.S.A. 100, 2748–2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hara T., Ishida T., Kojima Y., Tanaka H., Yasuda T., Shinohara M., Toh R., Hirata K. (2011) Targeted deletion of endothelial lipase increases HDL particles with anti-inflammatory properties both in vitro and in vivo. J. Lipid Res. 52, 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jin W., Millar J. S., Broedl U., Glick J. M., Rader D. J. (2003) Inhibition of endothelial lipase causes increased HDL cholesterol levels in vivo. J. Clin. Invest. 111, 357–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishida T., Choi S., Kundu R. K., Hirata K., Rubin E. M., Cooper A. D., Quertermous T. (2003) Endothelial lipase is a major determinant of HDL level. J. Clin. Invest. 111, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nijstad N., Wiersma H., Gautier T., van der Giet M., Maugeais C., Tietge U. J. (2009) Scavenger receptor BI-mediated selective uptake is required for the remodeling of high density lipoprotein by endothelial lipase. J. Biol. Chem. 284, 6093–6100 [DOI] [PubMed] [Google Scholar]
- 17. McCoy M. G., Sun G. S., Marchadier D., Maugeais C., Glick J. M., Rader D. J. (2002) Characterization of the lipolytic activity of endothelial lipase. J. Lipid Res. 43, 921–929 [PubMed] [Google Scholar]
- 18. Jin W., Wang X., Millar J. S., Quertermous T., Rothblat G. H., Glick J. M., Rader D. J. (2007) Hepatic proprotein convertases modulate HDL metabolism. Cell Metabolism 6, 129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fuki I. V., Blanchard N., Jin W., Marchadier D. H., Millar J. S., Glick J. M., Rader D. J. (2003) Endogenously produced endothelial lipase enhances binding and cellular processing of plasma lipoproteins via heparan sulfate proteoglycan-mediated pathway. J. Biol. Chem. 278, 34331–34338 [DOI] [PubMed] [Google Scholar]
- 20. Qiu G., Hill J. S. (2007) Endothelial lipase enhances low density lipoprotein binding and cell association in THP-1 macrophages. Cardiovasc. Res. 76, 528–538 [DOI] [PubMed] [Google Scholar]
- 21. Gauster M., Hrzenjak A., Schick K., Frank S. (2005) Endothelial lipase is inactivated upon cleavage by the members of the proprotein convertase family. J. Lipid Res. 46, 977–987 [DOI] [PubMed] [Google Scholar]
- 22. Jin W., Fuki I. V., Seidah N. G., Benjannet S., Glick J. M., Rader D. J. (2005) Proprotein convertases are responsible for proteolysis and inactivation of endothelial lipase. J. Biol. Chem. 280, 36551–36559 [DOI] [PubMed] [Google Scholar]
- 23. Ono M., Shimizugawa T., Shimamura M., Yoshida K., Noji-Sakikawa C., Ando Y., Koishi R., Furukawa H. (2003) Protein region important for regulation of lipid metabolism in angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. J. Biol. Chem. 278, 41804–41809 [DOI] [PubMed] [Google Scholar]
- 24. Shimamura M., Matsuda M., Yasumo H., Okazaki M., Fujimoto K., Kono K., Shimizugawa T., Ando Y., Koishi R., Kohama T., Sakai N., Kotani K., Komuro R., Ishida T., Hirata K., Yamashita S., Furukawa H., Shimomura I. (2007) Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler. Thromb. Vasc. Biol. 27, 366–372 [DOI] [PubMed] [Google Scholar]
- 25. Hato T., Tabata M., Oike Y. (2008) The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc. Med. 18, 6–14 [DOI] [PubMed] [Google Scholar]
- 26. Quagliarini F., Wang Y., Kozlitina J., Grishin N. V., Hyde R., Boerwinkle E., Valenzuela D. M., Murphy A. J., Cohen J. C., Hobbs H. H. (2012) Atypical angiopoietin-like protein that regulates ANGPTL3. Proc. Natl. Acad. Sci. U.S.A. 109, 19751–19756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Camenisch G., Pisabarro M. T., Sherman D., Kowalski J., Nagel M., Hass P., Xie M. H., Gurney A., Bodary S., Liang X. H., Clark K., Beresini M., Ferrara N., Gerber H. P. (2002) ANGPTL3 stimulates endothelial cell adhesion and migration via integrin αvβ3 and induces blood vessel formation in vivo. J. Biol. Chem. 277, 17281–17290 [DOI] [PubMed] [Google Scholar]
- 28. Kim I., Moon S. O., Koh K. N., Kim H., Uhm C. S., Kwak H. J., Kim N. G., Koh G. Y. (1999) Molecular cloning, expression, and characterization of angiopoietin-related protein. angiopoietin-related protein induces endothelial cell sprouting. J. Biol. Chem. 274, 26523–26528 [DOI] [PubMed] [Google Scholar]
- 29. Koishi R., Ando Y., Ono M., Shimamura M., Yasumo H., Fujiwara T., Horikoshi H., Furukawa H. (2002) Angptl3 regulates lipid metabolism in mice. Nat. Genet. 30, 151–157 [DOI] [PubMed] [Google Scholar]
- 30. Köster A., Chao Y. B., Mosior M., Ford A., Gonzalez-DeWhitt P. A., Hale J. E., Li D., Qiu Y., Fraser C. C., Yang D. D., Heuer J. G., Jaskunas S. R., Eacho P. (2005) Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology 146, 4943–4950 [DOI] [PubMed] [Google Scholar]
- 31. Fujimoto K., Koishi R., Shimizugawa T., Ando Y. (2006) Angptl3-null mice show low plasma lipid concentrations by enhanced lipoprotein lipase activity. Exp. Anim. 55, 27–34 [DOI] [PubMed] [Google Scholar]
- 32. Lee E. C., Desai U., Gololobov G., Hong S., Feng X., Yu X. C., Gay J., Wilganowski N., Gao C., Du L. L., Chen J., Hu Y., Zhao S., Kirkpatrick L., Schneider M., Zambrowicz B. P., Landes G., Powell D. R., Sonnenburg W. K. (2009) Identification of a new functional domain in angiopoietin-like 3 (ANGPTL3) and angiopoietin-like 4 (ANGPTL4) involved in binding and inhibition of lipoprotein lipase (LPL). J. Biol. Chem. 284, 13735–13745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu J., Afroza H., Rader D. J., Jin W. (2010) Angiopoietin-like protein 3 inhibits lipoprotein lipase activity through enhancing its cleavage by proprotein convertases. J. Biol. Chem. 285, 27561–27570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Essalmani R., Susan-Resiga D., Chamberland A., Abifadel M., Creemers J. W., Boileau C., Seidah N. G., Prat A. (2011) In vivo evidence that furin from hepatocytes inactivates PCSK9. J. Biol. Chem. 286, 4257–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roebroek A. J., Taylor N. A., Louagie E., Pauli I., Smeijers L., Snellinx A., Lauwers A., Van de Ven W. J., Hartmann D., Creemers J. W. (2004) Limited redundancy of the proprotein convertase furin in mouse liver. J. Biol. Chem. 279, 53442–53450 [DOI] [PubMed] [Google Scholar]
- 36. Susan-Resiga D., Essalmani R., Hamelin J., Asselin M. C., Benjannet S., Chamberland A., Day R., Szumska D., Constam D., Bhattacharya S., Prat A., Seidah N. G. (2011) Furin Is the Major Processing Enzyme of the Cardiac-specific Growth Factor Bone Morphogenetic Protein 10. J. Biol. Chem. 286, 22785–22794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhong M., Munzer J. S., Basak A., Benjannet S., Mowla S. J., Decroly E., Chrétien M., Seidah N. G. (1999) The prosegments of furin and PC7 as potent inhibitors of proprotein convertases. In vitro and ex vivo assessment of their efficacy and selectivity. J. Biol. Chem. 274, 33913–33920 [DOI] [PubMed] [Google Scholar]
- 38. Benjannet S., Savaria D., Laslop A., Munzer J. S., Chrétien M., Marcinkiewicz M., Seidah N. G. (1997) Alpha1-antitrypsin Portland inhibits processing of precursors mediated by proprotein convertases primarily within the constitutive secretory pathway. J. Biol. Chem. 272, 26210–26218 [DOI] [PubMed] [Google Scholar]
- 39. Conklin D., Gilbertson D., Taft D. W., Maurer M. F., Whitmore T. E., Smith D. L., Walker K. M., Chen L. H., Wattler S., Nehls M., Lewis K. B. (1999) Identification of a mammalian angiopoietin-related protein expressed specifically in liver. Genomics 62, 477–482 [DOI] [PubMed] [Google Scholar]
- 40. Kim W., Essalmani R., Szumska D., Creemers J. W., Roebroek A. J., D'Orleans-Juste P., Bhattacharya S., Seidah N. G., Prat A. (2012) Loss of endothelial furin leads to cardiac malformation and early postnatal death. Mol. Cell Biol. 32, 3382–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marchesi C., Essalmani R., Lemarié C. A., Leibovitz E., Ebrahimian T., Paradis P., Seidah N. G., Schiffrin E. L., Prat A. (2011) Inactivation of endothelial proprotein convertase 5/6 decreases collagen deposition in the cardiovascular system: role of fibroblast autophagy. J. Mol. Med. 89, 1103–1111 [DOI] [PubMed] [Google Scholar]
- 42. Basu D., Lei X., Josekutty J., Hussain M. M., Jin W. (2013) Measurement of the phospholipase activity of endothelial lipase in mouse plasma. J. Lipid Res. 54, 282–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thomas G. (2002) Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 3, 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seidah N. G., Mayer G., Zaid A., Rousselet E., Nassoury N., Poirier S., Essalmani R., Prat A. (2008) The activation and physiological functions of the proprotein convertases. Int. J. Biochem. Cell Biol. 40, 1111–1125 [DOI] [PubMed] [Google Scholar]
- 45. Schjoldager K. T., Vester-Christensen M. B., Bennett E. P., Levery S. B., Schwientek T., Yin W., Blixt O., Clausen H. (2010) O-Glycosylation Modulates Proprotein Convertase Activation of Angiopoietin-like Protein 3: possible role of polypeptide GalNAc-transferase-2 in regulation of concentrations of plasma lipids. J. Biol. Chem. 285, 36293–36303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lei X., Shi F., Basu D., Huq A., Routhier S., Day R., Jin W. (2011) Proteolytic processing of angiopoietin-like protein 4 by proprotein convertases modulates its inhibitory effects on lipoprotein lipase activity. J. Biol. Chem. 286, 15747–15756 [DOI] [PMC free article] [PubMed] [Google Scholar]