

Abstract
Maintenance of the cellular redox balance is crucial for cell survival. An increase in reactive oxygen, nitrogen, or chlorine species can lead to oxidative stress conditions, potentially damaging DNA, lipids, and proteins. Proteins are very sensitive to oxidative modifications, particularly methionine and cysteine residues. The reversibility of some of these oxidative protein modifications makes them ideally suited to take on regulatory roles in protein function. This is especially true for disulfide bond formation, which has the potential to mediate extensive yet fully reversible structural and functional changes, rapidly adjusting the protein's activity to the prevailing oxidant levels.
Keywords: Antioxidants, Oxidative Stress, Reactive Oxygen Species (ROS), Redox Signaling, Stress Response
Cysteine: A Highly Versatile Amino Acid
Cysteine residues can take on a diverse set of roles that affect the structure, function, and regulation of proteins. They serve as metal-coordinating residues in countless zinc or iron-sulfur cluster proteins, work as nucleophilic centers in the active site of metabolic enzymes and oxidoreductases, and can form covalent bonds with other cysteines (1–6). This high versatility is in large part due to the reactivity of the cysteine thiol group, which is affected and can be manipulated by the local protein environment. The high reactivity of cysteine thiols also contributes to the fact that cysteines are one of the least abundant amino acids in proteins and, when present, are often very highly conserved and functionally and/or structurally important (7). Although the percentage of cysteines in proteomes increases with the complexity of the organism (from ∼0.4% in certain archaea to ∼2.25% in mammals), it is still significantly lower than the expected 3.28%, which is the probability of cysteine occurrence simply based on its codon usage (8, 9). In addition, bioinformatic analysis revealed that cysteine residues follow a distinct pattern of conservation (10). Whereas surface-exposed cysteines typically show very poor conservation scores, cysteines that occur in clusters have very high conservation scores. This result agrees with experimental data showing that cysteines that confer redox activity or coordinate metals are often found in very close proximity (10, 11), a feature that has been widely used in prediction programs seeking to identify redox-sensitive proteins in silico (12, 13).
What makes cysteine residues so special? Cysteines contain a polarizable sulfur atom, and their oxidation state can range from the fully reduced thiol/thiolate anion to the fully oxidized sulfonic acid (Fig. 1). Most reactions of thiols in biochemical systems involve the nucleophilic attack of the deprotonated thiolate anion (RS−) on an electrophilic center, hence making the overall reactivity of a thiol group strongly dependent on the pKa of the cysteine side chain. In free cysteine, the pKa of the thiol is 8.3–8.5, indicating that it is almost fully protonated at physiological pH (14). In proteins, pKa values of cysteine thiols are dramatically influenced by their local environment. Although the pKa values of cysteines buried in the core are on average ∼9.5 (10), values as low as 3.4 have been reported for some active site cysteines (15). Low pKa values appear to be primarily due to stabilizing hydrogen bonds between the cysteine sulfur and the polypeptide backbone or nearby side chains: the higher the number of stabilizing hydrogen bonds, the lower the pKa of the cysteine (16–18). Positively charged amino acids in close vicinity to the cysteine, which have long been thought to play a major role in stabilizing the deprotonated thiolate anion (15, 19), seem to take on a smaller role in reducing the cysteine pKa unless hydrogen bonds are involved as well (20). Helix dipoles also appear to contribute to a decrease in the pKa of cysteine residues, making cysteines near the N-terminal end of helices more likely to have lower pKa values than those in other parts of the protein (21). A third parameter that substantially decreases the pKa of cysteines is the coordination of metal ions such as Zn2+ and Fe2+/3+, which stabilize the negatively charged thiolate anions (22, 23). pKa prediction programs such as PROPKA (24) are helpful in identifying proteins with cysteine thiols with particularly low pKa. These proteins are potentially crucial players in redox-regulated processes because the thiol reactivity might enable them to sense small alterations in the levels of endogenous reactive oxygen species (ROS)2 and respond with changes in gene expression, metabolism, or signaling (17, 25). It should be noted, however, that although the fraction of reactive thiolates increases with decreasing pKa values, the reactivity of thiolates either appears to decrease with decreasing pKa or is unaffected by pKa depending on the substrate tested (18).
FIGURE 1.
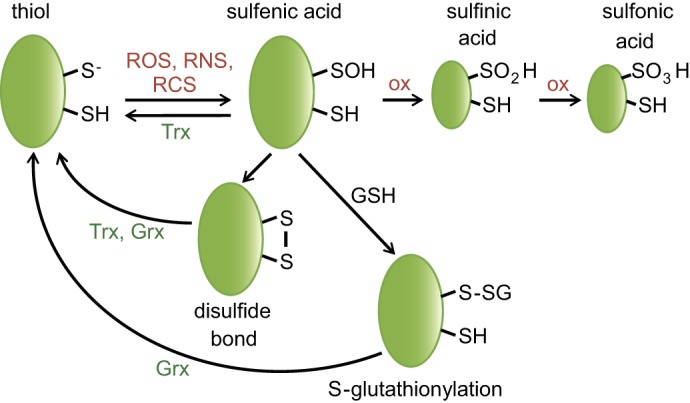
Reversible and irreversible cysteine modifications. Oxidation of cysteine thiol (RSH/RS−) by ROS, RNS, or RCS leads to the formation of highly reactive sulfenic acid (RSOH), which can react either with another thiol to form a disulfide bond (RSSR) or with GSH to become S-glutathionylated (RSSG). These oxidative modifications are reversible, and reduction is catalyzed by the Trx and/or Grx system. Further oxidation of sulfenic acid to sulfinic acid (RSO2H) and sulfonic acid (RSO3H) is thought to be generally irreversible in vivo.
Selenocysteine, the 21st amino acid, is structurally very similar to cysteine apart from the selenium atom, which is in place of sulfur. This single atom change increases the reactivity of selenocysteine compared with cysteines (26). Selenocysteines are almost exclusively found in the catalytic sites of enzymes, either performing thiol-selenodisulfide exchange reactions or scavenging oxidants (27).
Cysteine Thiol: A Popular Player in Redox Biology
ROS such as superoxide (O2⨪) and peroxide (H2O2) are constantly produced by members of the electron transport chain (28), in NADPH oxidases (29), and in peroxisomes (30). Moreover, UVA radiation and certain xenobiotics induce endogenous ROS generation via photochemical reactions with chromophores or cytochrome P450, respectively (31, 32). In addition, organisms are exposed to high levels of exogenously produced ROS. For instance, bacteria encounter bactericidal concentrations of hypochlorous acid (HOCl) once engulfed by neutrophils (33, 34).
Whether endogenously produced or taken up by diffusion, ROS have the potential to oxidize and unfold proteins, contributing to the damaging effects of oxidative stress. In some proteins, ROS transiently modulate protein activity, most commonly via the formation of sulfenic acids that lead to disulfide bonds (see below). Reversible ROS-induced thiol modifications were originally identified as sensing mechanisms in proteins involved in oxidative stress defense. It is now becoming clear, however, that ROS are not simply toxic species that inevitably cause oxidative stress. Instead, ROS such as H2O2 are often transiently and locally produced as part of signaling processes, reversibly modulating redox-sensitive proteins in signaling pathways. ROS-mediated signaling has recently been shown to be involved in growth, development, and differentiation processes (35), making redox-regulated disulfide bond formation an important alternative post-translational control mechanism on par with phosphorylation/dephosphorylation reactions.
Mechanism of Disulfide Bond Formation
When reactive cysteine thiols meet reactive oxygen, nitrogen, or chlorine species, sulfenic acid (RSOH) formation occurs (Fig. 1). Although stabilized in protein environments that lack nearby nucleophiles, such as in NADPH peroxidases and human serum albumin (36, 37), sulfenic acids are generally considered to be highly unstable oxidation intermediates. They rapidly interact with nearby cysteines to form inter- or intramolecular disulfide bonds (RSSR), making this the primary route for oxidant-mediated disulfide bond formation (38). Alternatively, sulfenic acids react with the small tripeptide GSH, leading to S-glutathionylation. The rate with which protein thiols react with oxidants depends on the oxidant as well as on the reactivity of the respective cysteine (18, 36). Although the pKa values and accessibility of the cysteines are clearly contributing factors in thiol reactivity, additional factors such as desolvation of reactive cysteines and steric hindrance seem to be involved as well. Moreover, substrate binding might affect a protein's reactivity by influencing the environment of the nucleophile. For instance, the caspase-3 active site cysteine shows a significantly higher reactivity toward H2O2 in the presence of substrates than in the absence of client proteins (39). That other factors apart from pKa affect thiol reactivity is also illustrated by the fact that the reaction rate of protein thiols with oxidants such as H2O2 spans over 7 orders of magnitude without any detectable correlation to the acidity of the respective active site thiol (18).
One class of enzymes that undergo oxidant-mediated disulfide bond formation within the reducing environment of the cytosol is oxidant-detoxifying enzymes such as 2-Cys peroxiredoxins, which use reversible disulfide bond formation as a mechanism to decompose H2O2 and organic peroxides (40). Most other proteins that undergo reversible oxidant-mediated disulfide bond formation use this mechanism to regulate their activity in response to oxidants. These proteins are commonly referred to as redox-regulated proteins. Like peroxiredoxins, many of these proteins function in the first line of oxidative stress defense, acting as transcriptional regulators (e.g. OxyR, Yap1p), which rapidly induce the expression of antioxidant genes to detoxify ROS and repair the damage (41, 42); chaperones (e.g. Hsp33), which prevent oxidative stress-induced protein aggregation (3, 43); or metabolic enzymes (e.g. GAPDH), which use reversible oxidation to reroute metabolites, thereby increasing the cell's capacity to defend against oxidative stress (44). Other redox-regulated proteins such as Keap1 and PKC are involved in signal transduction cascades (45, 46). These proteins use reversible disulfide bond formation to rapidly adapt cellular signaling processes to the oxidation status of the cell.
Oxidant-mediated disulfide bond formation, which typically occurs within the reducing environment of the cytosol, is distinct from the formation of structural disulfide bonds, which takes place within the oxidizing milieu of the eukaryotic endoplasmic reticulum or the bacterial periplasm. Structural disulfide bonds are typically introduced through direct thiol-disulfide exchange reactions, catalyzed by enzymes with redox catalytic cysteines (47–49). Notable exceptions are the Erv1/Erv2 proteins, which belong to the family of multidomain sulfhydryl oxidases (50). These oxidases introduce disulfide bonds into client proteins in the endoplasmic reticulum and mitochondrial intermembrane space, transferring the electrons directly onto molecular oxygen, generating considerable amounts of H2O2 in this process (51, 52).
Reversibility: An Important Aspect of Redox Regulation
Specialized reductases are responsible for restoring the original redox state of cysteines (2). Members of the highly conserved thioredoxin (Trx) and glutaredoxin (Grx) family fulfill this role in nearly all organisms studied. Like other oxidoreductases, Trx and Grx contain two redox-active cysteines arranged in a Cys-X2-Cys motif, which is embedded within a characteristic Trx fold (53). Thiol-disulfide exchange reactions are initiated by the more N-terminally located cysteine, which has a low pKa and attacks the disulfide bond in client proteins, forming a transient mixed disulfide intermediate in this process. This mixed disulfide bond is subsequently cleaved by a second nucleophilic attack of the more C-terminally located cysteine, resulting in the reduction of the client protein and the oxidation of the oxidoreductase (54). Oxidized Trx is then directly reduced by Trx reductase, which uses NADPH for its own reduction (55). Oxidized Grx initially forms a mixed disulfide with GSH. This mixed disulfide bond is subsequently attacked by a second GSH molecule, forming GSSG and reduced Grx (56). In turn, glutathione reductase reduces oxidized GSSG, also utilizing cellular NADPH as the electron donor. The major functions of Trx and Grx include the reduction of protein sulfenic acids, disulfide bonds, S-nitrosylations, and S-glutathionylations (57, 58). Although the two systems are able to functionally complement each other in vivo (59), some degree of specificity has been observed for the two protein families. Trx appears to be the main system for reducing sulfenic acids. In contrast, Grx seems to be primarily responsible for the reduction of S-glutathionylated proteins (60). In this reaction, the N-terminal active site cysteine of Grx attacks the glutathionylated cysteine in client proteins, leading to the formation of glutathionylated Grx and the release of the reduced substrate (61). Grx is subsequently reduced by a second molecule of glutathione.
Cysteine-coordinating Metal Centers as Redox Switches in Oxidative Stress Defense
The ability of proteins to use reversible disulfide bond formation as a functional switch requires high reactivity of the participating cysteine thiols (i.e. low pKa), close proximity to other cysteines, and sufficient disulfide-mediated conformational rearrangements to affect the function of the protein. An almost ideal combination of these features is found in cysteine-coordinating Zn2+ centers, where the positive charges of Zn2+ serve to stabilize the reactive thiolates, and Zn2+ coordination enables often distant cysteines to come into very close spatial proximity. Moreover, high affinity zinc binding confers considerable stability to protein domains that would otherwise be too unstable to fold. Hence, major structural rearrangements can result from the formation of disulfide bonds and zinc release. It is therefore not surprising that zinc centers, originally thought of as redox-inert and purely structural features in proteins, are now increasingly recognized as common redox units in redox-regulated proteins (see examples in supplemental Table S1) (3, 45).
One well characterized redox-regulated protein that uses a zinc-binding motif as a redox switch is the molecular chaperone Hsp33 (Fig. 2A). Specifically activated by oxidative disulfide bond formation, Hsp33 protects organisms against the lethal consequences of oxidative stress. In the reducing non-stress environment of the bacterial cytosol, Hsp33 is reduced, zinc-coordinated, and chaperone-inactive. Zinc binding is mediated by four tetrahedrally arranged thiolate anions, which coordinate one Zn2+ ion with attomolar affinity (3). Exposure of Hsp33 to fast-acting oxidants such as HOCl leads to the rapid formation of two disulfide bonds, zinc release, and the unfolding of the C-terminal redox switch domain. Unfolding of this domain leads to the activation of Hsp33 as a chaperone (Fig. 2A) (43, 62), making it capable of binding oxidatively unfolding proteins and preventing their irreversible aggregation. Once oxidative stress conditions subside, the cysteines of Hsp33 are re-reduced, and zinc is re-coordinated. Refolding of Hsp33 appears to trigger the release of client proteins for refolding by the ATP-dependent chaperone system DnaK/DnaJ/GrpE, closing the activation cycle of Hsp33 (63).
FIGURE 2.
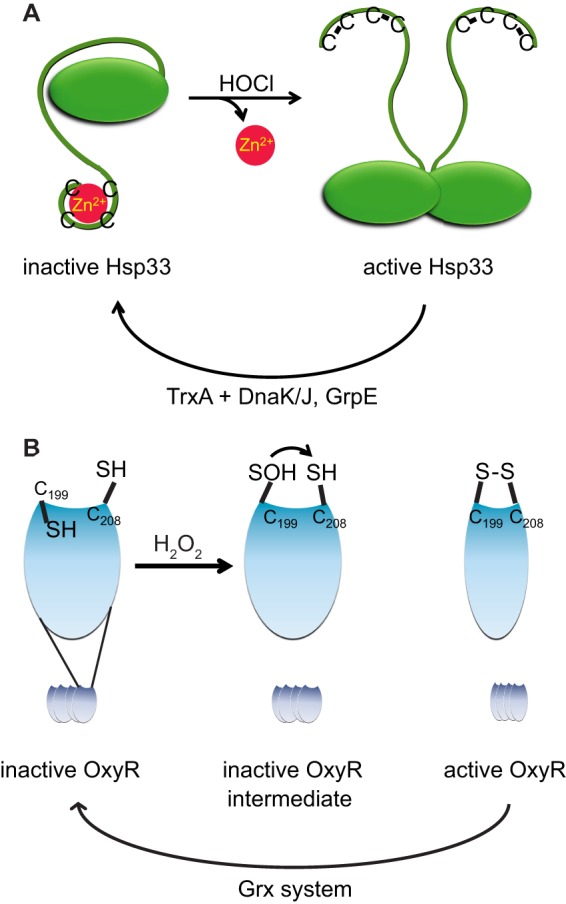
Redox-mediated activation of oxidative stress defense proteins. A, HOCl-mediated activation of Hsp33 chaperone activity involves formation of two intramolecular disulfide bonds, zinc release (red spheres), and unfolding of the C-terminal redox switch domain. Once activated, Hsp33 functions as an ATP-independent chaperone holdase, able to bind numerous unfolding client proteins to prevent their aggregation. When oxidative stress conditions subside, the disulfide bonds of Hsp33 are re-reduced by the Trx system, triggering the transfer of client proteins to the DnaK/DnaJ/GrpE system for refolding. B, H2O2-mediated activation of the transcriptional activator OxyR involves formation of an intramolecular disulfide bond between two cysteines, which are separated by 17 Å in the reduced form. These extensive conformational changes allow binding of RNA polymerase and transcription of target genes. The oxidative modifications are reversed by the Grx system, whose expression is under OxyR control.
It is noteworthy that incubation of Hsp33 with slow-acting oxidants such as H2O2 is not sufficient to activate the chaperone. Although H2O2 appears to be sufficient to induce formation of one disulfide bond in the distal cysteine pair, the second disulfide does not form unless unfolding conditions such as heat shock are present as well (5, 62). Structural studies suggest that limited accessibility of the remaining cysteines prevents H2O2 from forming the second disulfide bond. These results serve to illustrate that the reactivity and accessibility of cysteines can differ even within the same Zn2+ coordination site, conferring additional layers of regulation and oxidant specificity to zinc-mediated redox switches. However, the very same features make predictions about the potential redox sensitivity of cysteines in zinc centers challenging and experimental evaluation on a case-by-case basis necessary.
Many other zinc-binding proteins have been shown to be sensitive to oxidation. Examples of redox-regulated eukaryotic zinc finger proteins include transcriptional regulators such as MTF-1 (metal regulatory transcription factor 1), whose oxidative stress-mediated disulfide bond formation and homodimerization increase DNA binding affinity and thus transcription of target genes (supplemental Table S1) (64). Most other regulators, including SP-1 (specificity protein 1) or the tumor suppressor protein p53, use their redox-responsive zinc centers directly for DNA binding. Therefore, these proteins lose transcriptional activity upon oxidative disulfide bond formation and zinc release (65–67).
PKC, a eukaryotic kinase with two zinc-binding domains, has also been shown to be redox-regulated (6). Exposure of PKC to oxidants such as H2O2 causes oxidation of the two regulatory zinc finger motifs, thereby relieving PKC autoinhibition. This mechanism promotes activation of PKC in the absence of Ca2+ or phospholipids, second messengers that are otherwise necessary for its activation (6, 45, 68). Oxidative activation of PKC appears to lead to either prosurvival signaling (NF-κB, Akt/ASK1 (apoptosis signaling kinase 1)) or cell death (SAPK/JNK, ERK1/2, p38) depending on the isoform of PKC that is activated, its cellular localization, and the cell type affected (6).
Thiol-Disulfide Switches in Redox-regulated Transcription Factors
One very well characterized member of the group of non-metal-binding redox-regulated proteins is OxyR, a prokaryotic transcription factor whose reactive cysteine shows exquisite sensitivity to H2O2 (Fig. 2B). OxyR, which forms a homotetrameric DNA-binding structure (69), negatively regulates its own transcription and positively regulates the expression of OxyS, a small non-translated RNA, as well as ∼30 other genes (including ahpC/F and katG) involved in H2O2 stress defense (70, 71). OxyR contains six cysteines, with Cys199 and Cys208 being highly conserved (4). The first step in OxyR activation occurs when Cys199 is oxidized by peroxide to a sulfenic acid. The rate constant for this reaction is extremely high (1.1 × 105 to ∼107 m−1 s−1), ensuring activation of OxyR at H2O2 concentrations barely exceeding the physiological levels of H2O2 (∼100 nm) (4, 41, 72). However, activation of OxyR will be a competitive process with respect to other peroxide-detoxifying enzymes such as catalases and peroxiredoxins, which react with H2O2 with equally high reaction rates. Sulfenic acid formation appears to induce an extrusion of Cys199 from its hydrophobic pocket while introducing flexibility into the loop containing Cys208, a residue that is 17 Å away from Cys199 in the reduced form (Fig. 2B) (73). A second slower exchange leads to the formation of a disulfide bond between these two cysteines, locking OxyR in a transcriptionally active state (4, 73). Rotation of the OxyR dimeric interface by 30° compared with the monomers in reduced OxyR changes the interdimer orientation of the tetramer, necessary for the cooperative binding of RNA polymerase and subsequent transcription of the target sequences (41, 73). The unusual stability of the OxyR disulfide bond against re-reduction by the Grx system allows gene transcription to proceed for extended periods of time despite a reducing redox environment (41, 72).
Since the discovery of the OxyR redox-regulatory mechanism, numerous other pro- and eukaryotic transcriptional regulators have been identified that use reversible disulfide bond formation to mount effective responses against reactive oxygen and nitrogen species (supplemental Table S1). Their transcriptional activation is achieved by a variety of different means. Transcriptional repressors such as the global regulator SarZ of Staphylococcus aureus or CprK of Desulfitobacterium spp. dissociate from DNA upon disulfide-mediated conformational changes, causing derepression of gene expression (74, 75). Oxidative disulfide bond formation in the transcriptional activator Yap1p leads to its nuclear accumulation, causing increased antioxidant gene expression (42). Similarly, transcriptional activation of FOXO/Daf16 is also controlled by nuclear translocation. Under oxidative stress conditions, an intermolecular disulfide bond between FOXO and the nuclear import factor Tnpo1 forms, which appears to be crucial for the peroxide-mediated translocation of FOXO and the activation of antioxidant gene expression (76). This activation mechanism of FOXO is distinct from the insulin/PI3K/PKD signaling pathway and represents an alternative mechanism of FOXO activation. Another strategy is employed by redox-regulated prokaryotic anti-sigma factors and eukaryotic inhibitors. Their disulfide-mediated degradation causes the release and activation of the respective transcriptional regulators, resulting in the activation of gene expression (77, 78).
Redox-sensitive Active Site Cysteines as Functional Switches in Metabolic and Signaling Enzymes
Active site cysteines are, by definition, highly reactive and hence prone to undergo oxidative modifications. One excellent example is the active site cysteine of GAPDH, which plays a crucial role in glycolysis. Upon exposure of GAPDH to a variety of different ROS, sulfenic acid formation occurs at the active site cysteine, followed by the formation of a disulfide bond with a nearby cysteine (5, 79). This oxidation blocks glycolysis and appears to be responsible for the decrease in ATP levels observed in a variety of different species upon oxidative stress treatment (80, 81). Moreover, it contributes to the rerouting of glucose to the pentose phosphate pathway, generating NADPH instead of NADH (82). NADPH is a key factor in the reduction of the Trx and Grx systems, and accordingly, increasing the levels of NADPH raises the cell's antioxidant potential. Note that oxidative modification of the active site cysteines of GAPDH not only modulates its glycolytic activity but also leads to its nuclear translocation. In the nucleus, GAPDH interacts with heterodimeric RNA and DNA-binding proteins, suggesting that GAPDH may function as a transcriptional regulator during oxidative stress conditions (44).
The active site cysteine of protein-tyrosine phosphatase (PTP) is another well known target of oxidants, making tyrosine dephosphorylation a redox-regulated signaling event in eukaryotes (83). Although protein-tyrosine kinases are often activated under oxidative stress conditions (84), oxidation of PTPs generally leads to their inactivation. Under non-stress conditions, the active site thiolate of PTP (85) attacks the phosphorylated protein substrate, generating a cysteinyl phosphate intermediate, which is subsequently hydrolyzed (86). Exposure of PTP to oxidants such as H2O2 leads initially to the formation of sulfenic acid, followed by the formation of reversible oxidation products, which differ among different PTPs. Disulfide formation with a nearby resolving cysteine appears to be the predominant oxidation product in the case of SHP1/2, PTEN, and Cdc25 (supplemental Table S2) (87–89). Oxidation inactivates the phosphatase activity, thus enhancing the signaling intensity achieved by substrate phosphorylation and therefore leading to the activation of signaling pathways such as like NF-κB-inducing kinase/IκB kinase and MAPKs (ERK, p38, JNK). Their activation ultimately results in the phosphorylation and activation of NF-κB, causing expression of genes involved in antioxidant defense (84). Disulfide bond formation and other known oxidative PTP modifications such as S-glutathionylation and sulfenamide formation lead to conformational changes in the PTP active site, which appear necessary for the subsequent re-reduction and reactivation of PTPs by members of the Trx or Grx system. This reduction terminates the signaling event.
Peroxidases: Proteins of Many Traits
Peroxiredoxins are a family of sulfhydryl-dependent peroxidases and are remarkable proteins with a variety of functions associated with oxidative stress defense. The primary function of peroxiredoxins appears to be the decomposition of H2O2. Driven by extremely high sensitivity and reactivity toward H2O2 (∼104 to ∼2 × 107 m−1 s−1) (90, 91), peroxiredoxins rapidly undergo sulfenic acid formation at their active site cysteine. Subsequent disulfide bond formation with a nearby resolving cysteine and reduction of the disulfide bond by members of the Trx system catalyze the conversion of H2O2 to water (Fig. 3) (92).
FIGURE 3.
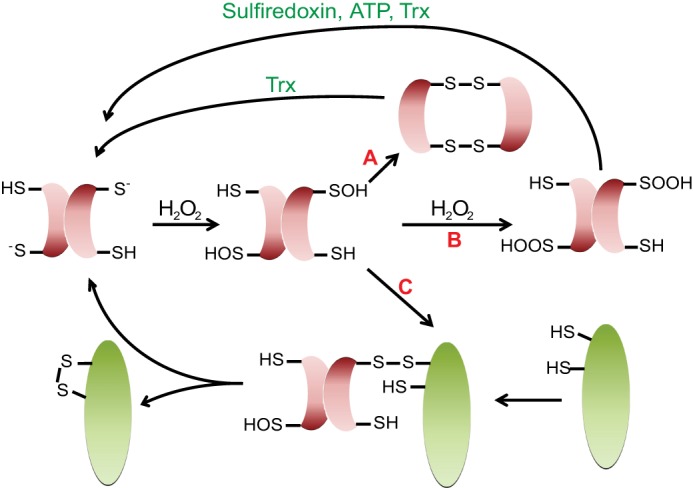
Versatile life of peroxiredoxins. Peroxiredoxins (red) catalyze the decomposition of H2O2, transiently forming a sulfenic acid in this process. Disulfide bond formation and subsequent reduction by Trx regenerate the active site cysteine thiol in peroxiredoxin (arrow A). In selected eukaryotic peroxiredoxins, high levels of H2O2 lead to the overoxidation of sulfenic acid to sulfinic acid, causing inactivation of the peroxidase function and concomitant activation as a chaperone holdase (arrow B). ATP-dependent sulfiredoxins convert the sulfinic acid back to sulfenic acid, which is subsequently reduced by Trx. Peroxiredoxins also undergo thiol-disulfide exchange reactions, leading to the oxidative activation of selected proteins. After oxidation of their active site cysteine to sulfenic acid, an intermolecular disulfide bond forms between peroxiredoxin (e.g. Orp1) and the client protein (e.g. Yap1p) (green). This disulfide bond is subsequently resolved, leading to the formation of an intramolecular disulfide bond in the client protein and the release of reduced peroxiredoxin (arrow C).
Most typical reaction rates of cysteine thiols with H2O2 are extremely slow (∼5–15 m−1 s−1) (14, 17), which raises the intriguing question as to how cysteines in redox-regulated proteins other than peroxidases are so rapidly oxidized by H2O2 in vivo (17, 90). Several studies have now demonstrated that peroxidases have the capacity to perform catalytic thiol-disulfide exchange reactions, thereby influencing DNA transcription and cellular signaling (93–96). One well studied example is the oxidative modification and activation of Yap1p by Gpx3 (glutathione peroxidase 3)/Orp1 in yeast. Gpx3, whose active site cysteine is rapidly oxidized to sulfenic acid, reacts with a cysteine (Cys598) in Yap1p, forming a transient Gpx3-Yap1p intermolecular disulfide bond (97). The intermolecular disulfide bond is subsequently resolved by a thiol-disulfide exchange reaction, which results in the reduction of Gpx3 and an intramolecular disulfide bond in Yap1p, leading to the activation of Yap1p (94, 97). A similar mechanism for oxidation equivalent transfer was reported for the oxidative activation of the ASK1/p38 pathway (93). In this case, oxidized peroxiredoxin-1 forms a disulfide exchange intermediate with ASK1. This leads to the oxidation and activation of ASK1 and the reduction of peroxiredoxin-1, making it ready for another round of peroxide detoxification (93, 96). It now remains to be seen how many proteins are oxidized by peroxidases and what criteria make them client proteins.
Conclusion
Reversible disulfide bond formation is a highly versatile post-translational mechanism employed by an ever-growing list of proteins to rapidly adjust their functional activity the moment ROS start to accumulate. With the development of highly sensitive and quantitative redox proteomic techniques (5, 98, 99), we have now entered an era in which it will be feasible to determine the complete inventory of oxidation-sensitive protein thiols in organisms. This analysis will determine how redox-regulated checkpoints are embedded into a variety of metabolic pathways and how cells rapidly switch between distinct catabolic or anabolic processes, protect particularly vulnerable intermediates, and activate survival pathways in response to oxidative stress. The discovery of conserved redox-regulated pathways that improve oxidative stress tolerance in eukaryotes will be invaluable in understanding how proteins and pathways can be manipulated to decrease or increase their oxidative stress resistance, thereby facilitating the discovery of new treatment possibilities for diseases in which the redox balance is compromised.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM065318 (to U. J.). This is the fourth article in the Thematic Minireview Series on Redox-active Protein Modifications and Signaling.

This article contains supplemental Tables S1 and S2 and additional references.
- ROS
- reactive oxygen species
- Trx
- thioredoxin
- Grx
- glutaredoxin
- PTP
- protein-tyrosine phosphatase.
REFERENCES
- 1. Alam M. S., Garg S. K., Agrawal P. (2009) Studies on structural and functional divergence among seven WhiB proteins of Mycobacterium tuberculosis H37Rv. FEBS J. 276, 76–93 [DOI] [PubMed] [Google Scholar]
- 2. Holmgren A., Johansson C., Berndt C., Lönn M. E., Hudemann C., Lillig C. H. (2005) Thiol redox control via thioredoxin and glutaredoxin systems. Biochem. Soc. Trans. 33, 1375–1377 [DOI] [PubMed] [Google Scholar]
- 3. Jakob U., Eser M., Bardwell J. C. A. (2000) Redox switch of Hsp33 has a novel zinc-binding motif. J. Biol. Chem. 275, 38302–38310 [DOI] [PubMed] [Google Scholar]
- 4. Lee C., Lee S. M., Mukhopadhyay P., Kim S. J., Lee S. C., Ahn W. S., Yu M. H., Storz G., Ryu S. E. (2004) Redox regulation of OxyR requires specific disulfide bond formation involving a rapid kinetic reaction path. Nat. Struct. Mol. Biol. 11, 1179–1185 [DOI] [PubMed] [Google Scholar]
- 5. Leichert L. I., Gehrke F., Gudiseva H. V., Blackwell T., Ilbert M., Walker A. K., Strahler J. R., Andrews P. C., Jakob U. (2008) Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 8197–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giorgi C., Agnoletto C., Baldini C., Bononi A., Bonora M., Marchi S., Missiroli S., Patergnani S., Poletti F., Rimessi A., Zavan B., Pinton P. (2010) Redox control of protein kinase C: cell- and disease-specific aspects. Antioxid. Redox Signal. 13, 1051–1085 [DOI] [PubMed] [Google Scholar]
- 7. Brooks D. J., Fresco J. R., Lesk A. M., Singh M. (2002) Evolution of amino acid frequencies in proteins over deep time: inferred order of introduction of amino acids into the genetic code. Mol. Biol. Evol. 19, 1645–1655 [DOI] [PubMed] [Google Scholar]
- 8. Miseta A., Csutora P. (2000) Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 17, 1232–1239 [DOI] [PubMed] [Google Scholar]
- 9. Jones D. P., Go Y.-M. (2011) Mapping the cysteine proteome: analysis of redox-sensing thiols. Curr. Opin. Chem. Biol. 15, 103–112 [DOI] [PubMed] [Google Scholar]
- 10. Marino S. M., Gladyshev V. N. (2010) Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J. Mol. Biol. 404, 902–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wong J. W., Ho S. Y., Hogg P. J. (2011) Disulfide bond acquisition through eukaryotic protein evolution. Mol. Biol. Evol. 28, 327–334 [DOI] [PubMed] [Google Scholar]
- 12. Sanchez R., Riddle M., Woo J., Momand J. (2008) Prediction of reversibly oxidized protein cysteine thiols using protein structure properties. Protein Sci. 17, 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun M. A., Wang Y., Cheng H., Zhang Q., Ge W., Guo D. (2012) RedoxDB–a curated database for experimentally verified protein oxidative modification. Bioinformatics 28, 2551–2552 [DOI] [PubMed] [Google Scholar]
- 14. Luo D., Smith S. W., Anderson B. D. (2005) Kinetics and mechanism of the reaction of cysteine and hydrogen peroxide in aqueous solution. J. Pharm. Sci. 94, 304–316 [DOI] [PubMed] [Google Scholar]
- 15. Grauschopf U., Winther J. R., Korber P., Zander T., Dallinger P., Bardwell J. C. (1995) Why is DsbA such an oxidizing disulfide catalyst? Cell 83, 947–955 [DOI] [PubMed] [Google Scholar]
- 16. Roos G., Foloppe N., Messens J. (2013) Understanding the pKa of redox cysteines: the key role of hydrogen bonding. Antioxid. Redox Signal. 18, 94–127 [DOI] [PubMed] [Google Scholar]
- 17. Winterbourn C. C., Metodiewa D. (1999) Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 27, 322–328 [DOI] [PubMed] [Google Scholar]
- 18. Ferrer-Sueta G., Manta B., Botti H., Radi R., Trujillo M., Denicola A. (2011) Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem. Res. Toxicol. 24, 434–450 [DOI] [PubMed] [Google Scholar]
- 19. Foloppe N., Nilsson L. (2004) The glutaredoxin -C-P-Y-C- motif: influence of peripheral residues. Structure 12, 289–300 [DOI] [PubMed] [Google Scholar]
- 20. Billiet L., Geerlings P., Messens J., Roos G. (2012) The thermodynamics of thiol sulfenylation. Free Radic. Biol. Med. 52, 1473–1485 [DOI] [PubMed] [Google Scholar]
- 21. Sengupta D., Behera R. N., Smith J. C., Ullmann G. M. (2005) The α helix dipole: screened out? Structure 13, 849–855 [DOI] [PubMed] [Google Scholar]
- 22. Hightower K. E., Fierke C. A. (1999) Zinc-catalyzed sulfur alkylation: insights from protein farnesyltransferase. Curr. Opin. Chem. Biol. 3, 176–181 [DOI] [PubMed] [Google Scholar]
- 23. Rozema D. B., Poulter C. D. (1999) Yeast protein farnesyltransferase. pKas of peptide substrates bound as zinc thiolates. Biochemistry 38, 13138–13146 [DOI] [PubMed] [Google Scholar]
- 24. Li H., Robertson A. D., Jensen J. H. (2005) Very fast empirical prediction and rationalization of protein pKi values. Proteins 61, 704–721 [DOI] [PubMed] [Google Scholar]
- 25. Claiborne A., Miller H., Parsonage D., Ross R. P. (1993) Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB J. 7, 1483–1490 [DOI] [PubMed] [Google Scholar]
- 26. Hondal R. J., Marino S. M., Gladyshev V. N. (2013) Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid. Redox Signal. 18, 1675–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rahmanto A. S., Davies M. J. (2012) Selenium-containing amino acids as direct and indirect antioxidants. IUBMB Life 64, 863–871 [DOI] [PubMed] [Google Scholar]
- 28. Dröse S., Brandt U. (2012) Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 748, 145–169 [DOI] [PubMed] [Google Scholar]
- 29. Lambeth J. D. (2004) NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189 [DOI] [PubMed] [Google Scholar]
- 30. Gabaldón T. (2010) Peroxisome diversity and evolution. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 365, 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aroun A., Zhong J. L., Tyrrell R. M., Pourzand C. (2012) Iron, oxidative stress and the example of solar ultraviolet A radiation. Photochem. Photobiol. Sci. 11, 118–134 [DOI] [PubMed] [Google Scholar]
- 32. Zangar R. C., Davydov D. R., Verma S. (2004) Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol. 199, 316–331 [DOI] [PubMed] [Google Scholar]
- 33. Leto T. L., Geiszt M. (2006) Role of Nox family NADPH oxidases in host defense. Antioxid. Redox Signal. 8, 1549–1561 [DOI] [PubMed] [Google Scholar]
- 34. Winterbourn C. C., Kettle A. J. (2013) Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 18, 642–660 [DOI] [PubMed] [Google Scholar]
- 35. Schippers J. H. M., Nguyen H. M., Lu D., Schmidt R., Mueller-Roeber B. (2012) ROS homeostasis during development: an evolutionary conserved strategy. Cell. Mol. Life Sci. 69, 3245–3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Poole L. B., Karplus P. A., Claiborne A. (2004) Protein sulfenic acids in redox signaling. Annu. Rev. Pharmacol. Toxicol. 44, 325–347 [DOI] [PubMed] [Google Scholar]
- 37. Turell L., Botti H., Carballal S., Ferrer-Sueta G., Souza J. M., Durán R., Freeman B. A., Radi R., Alvarez B. (2008) Reactivity of sulfenic acid in human serum albumin. Biochemistry 47, 358–367 [DOI] [PubMed] [Google Scholar]
- 38. Rehder D. S., Borges C. R. (2010) Cysteine sulfenic ACid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry 49, 7748–7755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hampton M. B., Stamenkovic I., Winterbourn C. C. (2002) Interaction with substrate sensitises caspase-3 to inactivation by hydrogen peroxide. FEBS Lett. 517, 229–232 [DOI] [PubMed] [Google Scholar]
- 40. Hall A., Karplus P. A., Poole L. B. (2009) Typical 2-Cys peroxiredoxins–structures, mechanisms and functions. FEBS J. 276, 2469–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zheng M., Aslund F., Storz G. (1998) Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 279, 1718–1721 [DOI] [PubMed] [Google Scholar]
- 42. Tachibana T., Okazaki S., Murayama A., Naganuma A., Nomoto A., Kuge S. (2009) A major peroxiredoxin-induced activation of Yap1 transcription factor is mediated by reduction-sensitive disulfide bonds and reveals a low level of transcriptional activation. J. Biol. Chem. 284, 4464–4472 [DOI] [PubMed] [Google Scholar]
- 43. Winter J., Ilbert M., Graf P. C., Ozcelik D., Jakob U. (2008) Bleach activates a redox-regulated chaperone by oxidative protein unfolding. Cell 135, 691–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hwang N. R., Yim S. H., Kim Y. M., Jeong J., Song E. J., Lee Y., Lee J. H., Choi S., Lee K. J. (2009) Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem. J. 423, 253–264 [DOI] [PubMed] [Google Scholar]
- 45. Gopalakrishna R., Jaken S. (2000) Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 28, 1349–1361 [DOI] [PubMed] [Google Scholar]
- 46. Dinkova-Kostova A. T., Holtzclaw W. D., Wakabayashi N. (2005) Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry 44, 6889–6899 [DOI] [PubMed] [Google Scholar]
- 47. Bardwell J. C., McGovern K., Beckwith J. (1991) Identification of a protein required for disulfide bond formation in vivo. Cell 67, 581–589 [DOI] [PubMed] [Google Scholar]
- 48. Ziegler D. M., Poulsen L. L. (1977) Protein disulfide bond synthesis–possible intracellular mechanism. Trends Biochem. Sci. 2, 79–81 [Google Scholar]
- 49. Frand A. R., Kaiser C. A. (1999) Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol. Cell 4, 469–477 [DOI] [PubMed] [Google Scholar]
- 50. Lange H., Lisowsky T., Gerber J., Mühlenhoff U., Kispal G., Lill R. (2001) An essential function of the mitochondrial sulfhydryl oxidase Erv1p/ALR in the maturation of cytosolic Fe/S proteins. EMBO Rep. 2, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Daithankar V. N., Wang W., Trujillo J. R., Thorpe C. (2012) Flavin-linked Erv-family sulfhydryl oxidases release superoxide anion during catalytic turnover. Biochemistry 51, 265–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dabir D. V., Leverich E. P., Kim S. K., Tsai F. D., Hirasawa M., Knaff D. B., Koehler C. M. (2007) A role for cytochrome c and cytochrome c peroxidase in electron shuttling from Erv1. EMBO J. 26, 4801–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martin J. L. (1995) Thioredoxin–a fold for all reasons. Structure 3, 245–250 [DOI] [PubMed] [Google Scholar]
- 54. Roos G., Foloppe N., Van Laer K., Wyns L., Nilsson L., Geerlings P., Messens J. (2009) How thioredoxin dissociates its mixed disulfide. PLoS Comput. Biol. 5, e1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Williams C. H., Jr. (1995) Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J. 9, 1267–1276 [DOI] [PubMed] [Google Scholar]
- 56. Holmgren A., Aslund F. (1995) Glutaredoxin. Methods Enzymol. 252, 283–292 [DOI] [PubMed] [Google Scholar]
- 57. Berndt C., Lillig C. H., Holmgren A. (2007) Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 292, H1227–H1236 [DOI] [PubMed] [Google Scholar]
- 58. Holmgren A. (1989) Thioredoxin and glutaredoxin systems. J. Biol. Chem. 264, 13963–13966 [PubMed] [Google Scholar]
- 59. Prinz W. A., Aslund F., Holmgren A., Beckwith J. (1997) The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 272, 15661–15667 [DOI] [PubMed] [Google Scholar]
- 60. Gallogly M. M., Mieyal J. J. (2007) Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 7, 381–391 [DOI] [PubMed] [Google Scholar]
- 61. Peltoniemi M. J., Karala A. R., Jurvansuu J. K., Kinnula V. L., Ruddock L. W. (2006) Insights into deglutathionylation reactions. Different intermediates in the glutaredoxin and protein disulfide isomerase catalyzed reactions are defined by the γ-linkage present in glutathione. J. Biol. Chem. 281, 33107–33114 [DOI] [PubMed] [Google Scholar]
- 62. Ilbert M., Horst J., Ahrens S., Winter J., Graf P. C., Lilie H., Jakob U. (2007) The redox-switch domain of Hsp33 functions as dual stress sensor. Nat. Struct. Mol. Biol. 14, 556–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Reichmann D., Xu Y., Cremers C. M., Ilbert M., Mittelman R., Fitzgerald M. C., Jakob U. (2012) Order out of disorder: working cycle of an intrinsically unfolded chaperone. Cell 148, 947–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Günther V., Davis A. M., Georgiev O., Schaffner W. (2012) A conserved cysteine cluster, essential for transcriptional activity, mediates homodimerization of human metal-responsive transcription factor-1 (MTF-1). Biochim. Biophys. Acta 1823, 476–483 [DOI] [PubMed] [Google Scholar]
- 65. Wu X., Bishopric N. H., Discher D. J., Murphy B. J., Webster K. A. (1996) Physical and functional sensitivity of zinc finger transcription factors to redox change. Mol. Cell. Biol. 16, 1035–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hutchison K. A., Matić G., Meshinchi S., Bresnick E. H., Pratt W. B. (1991) Redox manipulation of DNA-binding activity and BuGR epitope reactivity of the glucocorticoid receptor. J. Biol. Chem. 266, 10505–10509 [PubMed] [Google Scholar]
- 67. Rainwater R., Parks D., Anderson M. E., Tegtmeyer P., Mann K. (1995) Role of cysteine residues in regulation of p53 function. Mol. Cell. Biol. 15, 3892–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lin Y. L., Shivji M. K. K., Chen C., Kolodner R., Wood R. D., Dutta A. (1998) The evolutionarily conserved zinc finger motif in the largest subunit of human replication protein A is required for DNA replication and mismatch repair but not for nucleotide excision repair. J. Biol. Chem. 273, 1453–1461 [DOI] [PubMed] [Google Scholar]
- 69. Kullik I., Stevens J., Toledano M. B., Storz G. (1995) Mutational analysis of the redox-sensitive transcriptional regulator OxyR–regions important for DNA-binding and multimerization. J. Bacteriol. 177, 1285–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Altuvia S., Weinstein-Fischer D., Zhang A., Postow L., Storz G. (1997) A small, stable RNA induced by oxidative stress: role as a pleiotropic regulator and antimutator. Cell 90, 43–53 [DOI] [PubMed] [Google Scholar]
- 71. Zheng M., Wang X., Doan B., Lewis K. A., Schneider T. D., Storz G. (2001) Computation-directed identification of OxyR DNA binding sites in Escherichia coli. J. Bacteriol. 183, 4571–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Aslund F., Zheng M., Beckwith J., Storz G. (1999) Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc. Natl. Acad. Sci. U.S.A. 96, 6161–6165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Choi H., Kim S., Mukhopadhyay P., Cho S., Woo J., Storz G., Ryu S. E. (2001) Structural basis of the redox switch in the OxyR transcription factor. Cell 105, 103–113 [DOI] [PubMed] [Google Scholar]
- 74. Levy C., Pike K., Heyes D. J., Joyce M. G., Gabor K., Smidt H., van der Oost J., Leys D. (2008) Molecular basis of halorespiration control by CprK, a CRP-FNR type transcriptional regulator. Mol. Microbiol. 70, 151–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chen P. R., Nishida S., Poor C. B., Cheng A., Bae T., Kuechenmeister L., Dunman P. M., Missiakas D., He C. (2009) A new oxidative sensing and regulation pathway mediated by the MgrA homologue SarZ in Staphylococcus aureus. Mol. Microbiol. 71, 198–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Putker M., Madl T., Vos H. R., de Ruiter H., Visscher M., van den Berg M. C., Kaplan M., Korswagen H. C., Boelens R., Vermeulen M., Burgering B. M., Dansen T. B. (2013) Redox-dependent control of FOXO/DAF-16 by transportin-1. Mol. Cell 49, 730–742 [DOI] [PubMed] [Google Scholar]
- 77. Song T., Dove S. L., Lee K. H., Husson R. N. (2003) RshA, an anti-sigma factor that regulates the activity of the mycobacterial stress response sigma factor SigH. Mol. Microbiol. 50, 949–959 [DOI] [PubMed] [Google Scholar]
- 78. Thakur K. G., Praveena T., Gopal B. (2010) Structural and biochemical bases for the redox sensitivity of Mycobacterium tuberculosis RslA. J. Mol. Biol. 397, 1199–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cotgreave I. A., Gerdes R., Schuppe-Koistinen I., Lind C. (2002) S-Glutathionylation of glyceraldehyde-3-phosphate dehydrogenase: role of thiol oxidation and catalysis by glutaredoxin. Methods Enzymol. 348, 175–182 [DOI] [PubMed] [Google Scholar]
- 80. Colussi C., Albertini M. C., Coppola S., Rovidati S., Galli F., Ghibelli L. (2000) H2O2-induced block of glycolysis as an active ADP-ribosylation reaction protecting cells from apoptosis. FASEB J. 14, 2266–2276 [DOI] [PubMed] [Google Scholar]
- 81. Spragg R. G., Hinshaw D. B., Hyslop P. A., Schraufstätter I. U., Cochrane C. G. (1985) Alterations in adenosine triphosphate and energy charge in cultured endothelial and P388D1 cells after oxidant injury, J. Clin. Invest. 76, 1471–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shenton D., Grant C. M. (2003) Protein S-thiolation targets glycolysis and protein synthesis in response to oxidative stress in the yeast Saccharomyces cerevisiae. Biochem. J. 374, 513–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ostman A., Frijhoff J., Sandin A., Böhmer F. D. (2011) Regulation of protein tyrosine phosphatases by reversible oxidation, J. Biochem. 150, 345–356 [DOI] [PubMed] [Google Scholar]
- 84. Jung K. J., Lee E. K., Yu B. P., Chung H. Y. (2009) Significance of protein tyrosine kinase/protein tyrosine phosphatase balance in the regulation of NF-κB signaling in the inflammatory process and aging. Free Radic. Biol. Med. 47, 983–991 [DOI] [PubMed] [Google Scholar]
- 85. Peters G. H., Frimurer T. M., Olsen O. H. (1998) Electrostatic evaluation of the signature motif (H/V)CX5R(S/T) in protein-tyrosine phosphatases. Biochemistry 37, 5383–5393 [DOI] [PubMed] [Google Scholar]
- 86. Denu J. M., Dixon J. E. (1998) Protein tyrosine phosphatases: mechanisms of catalysis and regulation. Curr. Opin. Chem. Biol. 2, 633–641 [DOI] [PubMed] [Google Scholar]
- 87. Caselli A., Marzocchini R., Camici G., Manao G., Moneti G., Pieraccini G., Ramponi G. (1998) The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J. Biol. Chem. 273, 32554–32560 [DOI] [PubMed] [Google Scholar]
- 88. Lee S. R., Yang K. S., Kwon J., Lee C., Jeong W., Rhee S. G. (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277, 20336–20342 [DOI] [PubMed] [Google Scholar]
- 89. Sohn J., Rudolph J. (2003) Catalytic and chemical competence of regulation of Cdc25 phosphatase by oxidation/reduction. Biochemistry 42, 10060–10070 [DOI] [PubMed] [Google Scholar]
- 90. Budde H., Flohé L., Hecht H. J., Hofmann B., Stehr M., Wissing J., Lünsdorf H. (2003) Kinetics and redox-sensitive oligomerisation reveal negative subunit cooperativity in tryparedoxin peroxidase of Trypanosoma brucei brucei. Biol. Chem. 384, 619–633 [DOI] [PubMed] [Google Scholar]
- 91. Parsonage D., Youngblood D. S., Sarma G. N., Wood Z. A., Karplus P. A., Poole L. B. (2005) Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry 44, 10583–10592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Trujillo M., Ferrer-Sueta G., Thomson L., Flohé L., Radi R. (2007) Kinetics of peroxiredoxins and their role in the decomposition of peroxynitrite. Subcell. Biochem. 44, 83–113 [DOI] [PubMed] [Google Scholar]
- 93. Jarvis R. M., Hughes S. M., Ledgerwood E. C. (2012) Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 53, 1522–1530 [DOI] [PubMed] [Google Scholar]
- 94. Delaunay A., Pflieger D., Barrault M. B., Vinh J., Toledano M. B. (2002) A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 111, 471–481 [DOI] [PubMed] [Google Scholar]
- 95. Fomenko D. E., Koc A., Agisheva N., Jacobsen M., Kaya A., Malinouski M., Rutherford J. C., Siu K. L., Jin D. Y., Winge D. R., Gladyshev V. N. (2011) Thiol peroxidases mediate specific genome-wide regulation of gene expression in response to hydrogen peroxide. Proc. Natl. Acad. Sci. U.S.A. 108, 2729–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Veal E. A., Findlay V. J., Day A. M., Bozonet S. M., Evans J. M., Quinn J., Morgan B. A. (2004) A 2-Cys peroxiredoxin regulates peroxide-induced oxidation and activation of a stress-activated MAP kinase. Mol. Cell 15, 129–139 [DOI] [PubMed] [Google Scholar]
- 97. Ma L. H., Takanishi C. L., Wood M. J. (2007) Molecular mechanism of oxidative stress perception by the Orp1 protein. J. Biol. Chem. 282, 31429–31436 [DOI] [PubMed] [Google Scholar]
- 98. Leonard S. E., Reddie K. G., Carroll K. S. (2009) Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem. Biol. 4, 783–799 [DOI] [PubMed] [Google Scholar]
- 99. Held J. M., Danielson S. R., Behring J. B., Atsriku C., Britton D. J., Puckett R. L., Schilling B., Campisi J., Benz C. C., Gibson B. W. (2010) Targeted quantitation of site-specific cysteine oxidation in endogenous proteins using a differential alkylation and multiple reaction monitoring mass spectrometry approach. Mol. Cell. Proteomics 9, 1400–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]