

Background: Methyllycaconitine is an antagonist at subtypes of the nicotinic acetylcholine receptor.
Results: A reactive methyllycaconitine probe was covalently trapped by a cysteine introduced on the complementary face of the α4 subunit and only in the (α4)3(β2)2 nAChR stoichiometry.
Conclusion: The α4-α4 interface on the α4β2 nAChR contains a methyllycaconitine binding site.
Significance: Defining the molecular interactions of nAChR ligands at the α4-α4 interface may lead to superior therapeutics.
Keywords: Cys-loop Receptors, Homology Modeling, Molecular Pharmacology, Nicotinic Acetylcholine Receptors, Receptor Structure-Function, Cysteine Trapping, Receptor Inhibition
Abstract
The α4β2 nicotinic acetylcholine receptors (nAChRs) are widely expressed in the brain and are implicated in a variety of physiological processes. There are two stoichiometries of the α4β2 nAChR, (α4)2(β2)3 and (α4)3(β2)2, with different sensitivities to acetylcholine (ACh), but their pharmacological profiles are not fully understood. Methyllycaconitine (MLA) is known to be an antagonist of nAChRs. Using the two-electrode voltage clamp technique and α4β2 nAChRs in the Xenopus oocyte expression system, we demonstrate that inhibition by MLA occurs via two different mechanisms; that is, a direct competitive antagonism and an apparently insurmountable mechanism that only occurs after preincubation with MLA. We hypothesized an additional MLA binding site in the α4-α4 interface that is unique to this stoichiometry. To prove this, we covalently trapped a cysteine-reactive MLA analog at an α4β2 receptor containing an α4(D204C) mutation predicted by homology modeling to be within reach of the reactive probe. We demonstrate that covalent trapping results in irreversible reduction of ACh-elicited currents in the (α4)3(β2)2 stoichiometry, indicating that MLA binds to the α4-α4 interface of the (α4)3(β2)2 and providing direct evidence of ligand binding to the α4-α4 interface. Consistent with other studies, we propose that the α4-α4 interface is a structural target for potential therapeutics that modulate (α4)3(β2)2 nAChRs.
Introduction
The neuronal nicotinic acetylcholine receptors (nAChRs)4 are a family of ligand-gated ion channels that mediate fast synaptic transmission between cells both in the central and peripheral nervous system (1). These receptors are a pentameric protein assembly that can be formed by up to 12 different subunits in mammals (α2–10 and β2–4) in different combinations, leading to a wide variety of individual subtypes, each with its own pharmacology (2). Of these receptors, the heteromeric α4β2 nAChRs are the most widely expressed in the brain and are implicated in processes as diverse as cognition, consciousness, mood, nicotine addiction, and nociception among others (1). The homomeric α7 nAChR is not limited to expression in the central nervous system and is implicated in processes that include the cholinergic anti-inflammatory pathway and schizophrenia (3, 4).
Each subunit within the pentameric nAChR complex contains a large extracellular domain, four transmembrane segments (M1–4) of which the second (M2) lines the channel pore, two short loops M1-M2 and M2-M3 that mediate signal transduction, and a large intracellular loop M3-M4 that contributes to channel conductance (1, 2). Acetylcholine (ACh) binds at the interface of two subunits in the extracellular domain, which results in a series of conformational changes, culminating in the rotation of the physically distant M2 region that forms the channel pore. Thus, a conserved central hydrophobic leucine residue that is thought to physically occlude the channel pore in the closed state is moved out of the way to allow passive ion flow across the membrane (5–7).
Considerable structural information has been gained from the x-ray crystal structures of related bacterial (8–10) and invertebrate (11) ligand-gated ion channels, soluble ACh-binding proteins (AChBPs) (12, 13), and the electron microscopy structure of the muscle nAChR from Torpedo marmorata (6, 14). Furthermore, the similarities in the structure at the ACh binding site between the α7, α4β2 nAChRs and AChBPs have allowed researchers to use the AChBPs as a structural template to create homology models of receptor subtypes. This has been combined with protein-ligand docking studies and molecular dynamics simulations to understand and explain differences in ligand selectivity (15–18). Recently the co-crystallization of a range of partial agonists and antagonists of α7 and α4β2 nAChRs with the Lymnaea stagnalis and Aplysia californica AChBP has revealed ligand-receptor interactions in detail (19–22).
Although the α7 and α4β2 nAChRs share similar gene sequence, there are functional differences including ion selectivity, gating kinetics, and pharmacological profile (1, 2). Furthermore, the α4β2 nAChR is known to exist in two stoichiometries, (α4)2(β2)3 and (α4)3(β2)2, which respond to ACh with different sensitivities thought to be mediated through binding to the α4-α4 interface that is present in the (α4)3(β2)2 stoichiometry, in addition to the consensus α4-β2 agonist binding site (23–27). The binding site located in the α4-α4 subunit interface has not been thoroughly investigated in terms of its binding to nAChR pharmacological agents and the functional effect these agents have on the receptor. Recently, it has been suggested that the selective binding of ligands to the α4-α4 site could serve to allosterically modulate the receptor, positively or negatively influencing the response to ACh, similar to the benzodiazepine binding site in the GABAA receptors (26, 28). These studies join a body of emerging evidence that pharmacological agents can exert their effects by binding to alternative subunit interfaces, distinct from the α-β interface containing the consensus agonist binding site (28–30).
In this study we sought to determine the binding interactions of the known nAChR antagonist methyllycaconitine (MLA) at the α4β2 nAChR. By measuring the inhibition of ACh responses by MLA on α4β2 nAChRs, we demonstrate that binding of MLA reduces ACh activation in an apparently insurmountable manner. Using an MLA analog (MLA-maleimide) that is reactive to cysteine and by changing the ratio of subunits injected into the oocyte cell expression system, we demonstrate that MLA-maleimide is covalently trapped at an introduced cysteine (α4(D204C)) on the complementary side of the α4-α4 interface. We propose that MLA binds to the α4-α4 site to inhibit ACh-mediated activation of the receptor. This interface and amino acid (α4[D204]) has not been previously identified to interact with MLA.
EXPERIMENTAL PROCEDURES
Mutagenesis and Transcription of Recombinant α7 and α4β2 Receptors
DNA encoding for the rat α4 subunit previously subcloned into the plasmid pSP64, rat β2 subunit subcloned into the plasmid pSP65, and the rat α7 subunit subcloned into the pBS SK(+) vector were generous gifts from Professor Jim Boulter, University of California, Los Angeles, CA. DNA encoding for the hRIC-3 chaperone protein was a generous gift from Professor Millet Treinin, Hebrew University of Jerusalem. Site-directed mutagenesis was performed to create cysteine substitutions using the QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA). Single colonies of Escherichia coli containing plasmid DNA were isolated, grown, and then the DNA-purified and sequenced to identify that the mutations were successfully incorporated.
α7 DNA was linearized using SmaI, α4 was linearized with EcoRI, and β2 was linearized with HindIII before transcription. The α7 and RIC-3 mRNAs were transcribed in vitro using T7 mMessage mMachineTM transcription kit, and α4 and β2 mRNAs were transcribed using the SP6 mMessage mMachineTM transcription kit (Ambion Inc., Austin, TX). α7 mRNA was polyadenylated using the poly-A-tailing kit (Ambion). RNA was treated with DNase before purification, and RNA concentrations were measured by spectrophotometry using the Nanodrop (Thermo Fisher Scientific). RNA was mixed to the ratios stated in the text for α4β2 nAChR expression.
Xenopus laevis Surgery, Oocyte Extraction, and Injection
The experiments were performed with animal ethics approvals from The University of Sydney. Female X. laevis was anesthetized with tricaine (850 mg/500 ml). Several ovarian lobes were surgically removed by a small incision on the abdomen. The X. laevis were allowed to recover from the surgery, and the time interval between surgical procedures on each frog was 12 months. Three recoverable surgeries were performed on each X. laevis, and a final terminal surgery was performed with the frog exposed to a lethal dose of anesthetic. The lobes were cut into small pieces and were rinsed thoroughly with oocyte-releasing buffer 2 (OR2 (Roche Applied Science); 82.5 mm NaCl, 2 mm KCl, 1 mm MgCl2, 5 mm HEPES (hemi-Na+)). The lobes were digested with collagenase A (2 mg/ml in OR2) at room temperature. The oocytes were further washed with OR2 and stored in ND96 wash solution (96 mm NaCl, 2 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2, 5 mm HEPES (hemi-sodium salt) supplemented with 2.5 mm sodium pyruvate and 0.5 mm theophylline) until ready for injection. Stage V-VI oocytes were selected and microinjected with 2 ng of mRNA in 50.6 nl of nuclease-free water. After injection, the oocytes were maintained at 18 °C in the presence of ND96 wash solution augmented with 50 μg/ml kanamycin.
Electrophysiological Recording of Recombinant Receptors
Whole-cell currents were measured using a two-electrode voltage clamp with a Geneclamp 500B amplifier together with a Powerlab/200 (AD Instruments, Sydney, Australia) and Chart Version 3.5 for PC as previously described (31). The recording microelectrodes were filled with 3 m KCl and had resistances between 0.2 and 1 megaohms. 1–5 days post-injection oocytes held at −60 mV (unless otherwise stated) were used for recording. To prevent the activation of Ca2+-activated chloride currents, oocytes were superfused with calcium-free buffer (115 mm NaCl, 2.5 mm KCl, 1.8 mm BaCl2, 10 mm HEPES, pH 7.4) while recording until a stable base current was reached.
Concentration-response curves for agonist were constructed by measuring the peak current responses elicited from a range of agonist concentrations (10 nm–10 mm). Responses were normalized to the maximum current (I/Imax) to compare data from different oocytes. Oocytes were washed for 5 min between agonist applications for the α7(L9′T), 10 min for the α4β2, and 20 min for the α7 and α4(D204C)β2 receptors, to ensure receptors were not desensitized from the previous agonist application. To evaluate the mode of inhibition, ACh concentration-response curves were determined in the presence of MLA at the concentrations given under “Results.” Where the curve was determined with preincubation, MLA was applied for 3 min before the solution was switched to contain the same concentration of MLA with ACh. Inhibitory concentration-response curves to MLA were determined with co-application of a range of MLA concentrations and the EC50 concentration of ACh for the appropriate nAChR. Peak responses were normalized to the EC50 current (I/I50).
Rate of Reaction
MLA-maleimide was synthesized as previously described (32). The rate of covalent trapping of the reactive MLA-maleimide by the cysteine residue was measured by application of an EC50 concentration of ACh to the receptor at regular 20-min intervals. Before the application of 1 μm reactive MLA compound, MLA-maleimide, the peak currents (I50) were required to be within 10% of each other. Once this was achieved, MLA-maleimide was applied for 30 s at 2.5 min post ACh application followed by ACh (EC50) after a 17-min wash. This sequence was repeated until there was no longer a reduction in the peak currents to ACh.
Concentration-Response Curves
The amplitude of each current response to ACh (I) was normalized to the amplitude of the maximum current response to ACh (Imax). Normalized concentration-response curves were constructed and analyzed using GraphPad “Prism” version 5.0 according to Equation 1,
where [A] is the ligand concentration, and nH is the Hill slope. Mean parameters of each curve were derived from at least three oocytes and are stated under “Results.” Unless otherwise stated, the 95% confidence interval derived from the curve fitting is given in brackets.
Rate of MLA-maleimide Reaction to Introduced Cysteine
The change in response to an EC50 concentration of ACh after cumulative time of MLA-maleimide addition (t) was expressed relative to the response to an EC50 concentration of ACh before reagent addition at t = 0. The data expressed in this way were fitted to a single exponential decay to obtain an estimate of the first order rate constant (k) and final current (I∞) according to Equation 2,
These were compared with the wild type using a Student's t test.
α4-α4 nAChR Homology Model
Homology models of the rat extracellular domain α4-α4 subunit interface was prepared with MODELLER Version 9.10 (33) essentially as previously described (26) with the following changes: (i) the human sequence for α4 was substituted by the rat sequence (entry P09483 at the Protein Knowledgebase (34) as aligned by T-COFFEE Version 9.02 (35), (ii) the AChBP crystal structure with MLA in the binding site (22) (PDB code 2BYR) was substituted for the previously used structure with epibatidine, (iii) loop C was modeled only on the AChBP structure with MLA, and (iv) MLA was included in the homology models instead of nicotine. The best of 100 models according to the DOPE score (36) included in MODELLER were selected. The mutagenesis wizard in PyMOL (The PyMOL Molecular Graphics system, Version 1.5.0.1 Schrödinger LLC, New York) was used to change Asp-204 to a cysteine and select the rotamer placing the thiol closest to the maleimide reactive site. Next, a bond was added between the sulfur of the cysteine and the C4 (CH) of MLA-maleimide. The structure with the covalently bound MLA-maleimide was pre-processed with default settings and subjected to a restrained minimization with an root mean square deviation cutoff of 0.5 Å using the Protein Preparation Wizard of the Schrödinger Suite 2012 (Schrödinger Suite 2012 Epik Version 2.2, Impact Version 5.7, and Prime version 2.3, Schrödinger LLC, New York) to relax the strain caused by the added covalent bond.
RESULTS
MLA Inhibition Is Surmountable at α7 nAChRs but Is Apparently Insurmountable at α4β2 nAChRs
MLA is reported to be highly selective for the α7 nAChR compared with other receptor subtypes (37, 38). To demonstrate this with the rat nAChR proteins, we performed a concentration-response curve to ACh for α7 and α4β2 and then performed an inhibition concentration-response curve of MLA to the approximate EC50 concentrations of ACh (100 and 300 μm, respectively; Fig. 1, Table 1). As MLA and ACh would be expected to have different binding kinetics, we preincubated with MLA for 3 min to ensure that the MLA was at equilibrium before co-application with ACh (Fig. 1). After a 17-min washout of the MLA, the current elicited by ACh was unchanged, demonstrating that the MLA inhibition was reversible for both subtypes. The IC50 was ∼1000-fold higher for α4β2 (200 nm) than for α7 (140 pm). The rapid desensitization of the α7 nAChR can lead to erroneous measurement of peak currents without a fast-application system, as receptors may be in a desensitized state before the ACh solution, reaching equilibrium in the bath. To account for this, we introduced a pore mutation α7(L9′T) that abolishes rapid desensitization and increases the sensitivity to ACh. The EC50 of ACh was decreased (α7 = 92 μm, α7(L9′T) = 1.3 μm), but the IC50 of MLA was unchanged (α7 = 140 pm, α7[L9′T] = 150 pm) (Fig. 1, Table 1), demonstrating that this mutation alters the intrinsic gating properties of the receptor but not the binding of MLA.
FIGURE 1.

A, concentration-response curves to ACh of oocytes injected with a 1:1 ratio of α4β2 (■) and α7 (●) nAChR mRNA are shown. B, concentration-response curves for ACh against oocytes injected with α7(L9′T) (■) and α7 (●) nAChR mRNA are shown. C, inhibition of 100 μm ACh with 1 μm MLA incubated for various time periods before co-application of 100 μm ACh and 1 μm MLA are shown. D, MLA inhibition concentration-response curves after a 3-min incubation of oocytes injected with a 1:1 ratio of α4β2 (■) and α7 (●) nAChR mRNA are expressed as a fraction of the current elicited by 300 and 100 μm ACh, respectively. E, MLA inhibition concentration-response curves of oocytes injected with α7(L9′T) (■) and α7(●) nAChR mRNA are expressed as a fraction of the current elicited by 1 and 100 μm ACh, respectively. Data are represented as the means ± S.E., and the curve-fitting procedure is as explained under “Experimental Procedures.”
TABLE 1.
ACh concentration-response and MLA inhibition concentration-response curve parameters derived from the curve-fitting procedures
CI, confidence interval.
| Receptor (ratio) | ACh |
MLA |
||||
|---|---|---|---|---|---|---|
| EC50 (95% CI) | nH (95% CI) | n | IC50 (95% CI) | −nH (95% CI) | n | |
| m | m | |||||
| α7 + RIC-3 | 9.2 × 10−5 (6.9–12.5 × 10−5) | 1.2 (0.8–1.6) | 5 | 1.4 × 10−10 (0.8–2.5 × 10−10) | 1.3 (0.6–2.0) | 8 |
| α7(L9'T) | 1.3 × 10−6 (1.1–1.5 × 10−6) | 2.1 (1.5–2.6) | 5 | 1.5 × 10−10 (0.8–3.0 × 10−10) | 1.2 (0.6–1.9) | 6 |
| α4β2 (1:1) | 1.5 × 10−4 (1.1–1.9 × 10−4) | 0.91 (0.75–1.1) | 16 | 2.0 × 10−7 (1.1–3.5 × 10−7) | 1.2 (0.6–1.9) | 6 |
| α4β2 (1:10) | 1.7 × 10−6 (1.2–2.4 × 10−6) | 0.88 (0.6–1.2) | 4 | 1.3 × 10−7 (0.5–3.7 × 10−7) | 0.8 (0.4–1.2) | 3 |
| α4(D204C)β2 (1:1) | 2.6 × 10−5 (1.4–5 × 10−5) | 0.53 (0.36–0.7) | 9 | 2.7 × 10−8 (1.5–5.1 × 10−8) | 0.5 (0.3–0.6) | 7 |
| α4(D204C)β2 (1:10) | 1.9 × 10−6 (1.5–2.3 × 10−6) | 0.97 (0.8–1.1) | 6 | 1.2 × 10−7 (1.1–1.4 × 10−7) | 1.0 (0.9–1.2) | 5 |
To determine the mode of inhibition, increasing concentrations of ACh were applied in the presence of the IC50 concentration of MLA. In the α7(L9′T) receptor, the maximum current elicited by ACh in the absence of MLA could be reached in the presence of MLA if high enough concentrations of ACh were applied such that the inhibition was surmountable (Fig. 2, Table 2).
FIGURE 2.

A, shown are concentration-response curves to ACh of oocytes injected with α7(L9′T) nAChR mRNA in the absence of MLA (■) and in the presence of 100 pm MLA (□) after a 3 min incubation. B, concentration-response curves to ACh of oocytes injected with a 1:1 shown is a ratio of α4β2 nAChR mRNA in the absence of MLA (■) and in the presence of 10 nm (▵), 30 nm (○), 200 nm (□), and 1 μm (♢) MLA after a 3-min incubation. Data are represented as the means ± S.E., and the curve-fitting procedure is as explained under “Experimental Procedures.”
TABLE 2.
ACh concentration-response curve in the presence of a fixed concentration of MLA; parameters derived from curve fitting
Error is from Student's t test two-tailed, unequal variance, logEC50 of individual experiments used for statistical comparisons.
| Receptor (ratio) | Incubation | [MLA] | I/IACh (95% CI)a | EC50 (95% CI) | nH (95% CI) | n |
|---|---|---|---|---|---|---|
| m | m | |||||
| α7(L9'T) | 3 min | 1 × 10−10 | 0.97 (0.87–1.06) | 6.6 × 10−6 (5–9 × 10−6)b,c | 1.8 (1–2.8) | 4 |
| α4β2 (1:1) | 3 min | 2 × 10−7 | 0.46 (0.35–0.58)b,d | 9.5 × 10−5 (3–30 × 10−5) | 0.8 (0.2–1.4) | 5 |
| α4β2 (1:1) | 0 min | 2 × 10−6 | 1.12 (0.99–1.26) | 6.7 × 10−4 (4.1–11 × 10−4)e,f | 0.98 (0.6–1.4) | 5 |
| α4β2 (1:1) | 3 min | 1 × 10−8 | 1.14 (1.05–1.2) | 1.9 × 10−4 (1.4–2.6 × 10−4) | 0.8 (0.6–1.0) | 4 |
| α4β2 (1:1) | 3 min | 3 × 10−8 | 0.71 (0.61–0.8)b,f | 1.0 × 10−4 (0.6–1.8 × 10−4) | 1.1 (0.5–1.6) | 3 |
| α4β2 (1:1) | 3 min | 1 × 10−6 | 0.22 (0.18–0.25)b,d | 8.2 × 10−5 (4.6–15 × 10−5)b,f | 1.5 (0.3–2.8) | 5 |
a I/IACh normalized to 10 μm ACh for α7(L9'T); 1 mm for all others.
b Compared to value without MLA application.
c p < 0.01.
d p < 0.001.
e Compared to the value without incubation.
f p < 0.05.
When increasing concentrations of ACh were co-applied with an IC50 concentration of MLA to the α4β2 nAChR, the maximum current that was elicited in the presence of MLA was significantly lower than that with ACh alone, but the EC50 was not significantly different (Fig. 2, Table 2). We repeated this experiment with a range of MLA concentrations, demonstrating that even at low MLA concentrations (>30 nm) the maximum ACh response was significantly different with the EC50 unchanged, consistent with insurmountable inhibition (Fig. 2, Table 2). At the highest concentration of MLA applied, 1 μm, the maximum current was significantly reduced by 78% and the EC50 was significantly reduced, albeit by only 1.8-fold (p = 0.041, Student's t test).
To determine the subtype stoichiometry that we are measuring, we injected α4 and β2 subunit mRNA in a 1:1 and 1:10 injection ratio and measured the ACh concentration-response curves. For the 1:10 injection ratio the α4β2 nAChR showed high ACh sensitivity (EC50 1.7 μm, Fig. 3, Table 1), indicating that the 1:10 population consisted of an essentially homogenous (α4)2(β2)3 stoichiometry. The 1:1 ratio showed a low ACh sensitivity (EC50 150 μm, p = 0.21 Student's t test, Fig. 3). We are, therefore, able to conclude that in our system the current elicited by ACh at oocytes injected with a 1:1 ratio predominantly flows across receptors of the (α4)3(β2)2 stoichiometry. This compares favorably to EC50 values reported for human α4β2 nAChRs of 1.6 and 83 μm for the (α4)2(β2)3 and (α4)3(β2)2 stoichiometries, respectively (26).
FIGURE 3.

A, MLA inhibition concentration-response curves of oocytes injected with a 1:1 ratio of α4β2 nAChR mRNA after a 3-min incubation (■) and without incubation (□) is expressed as a fraction of the current elicited by 100 μm ACh. B, shown are concentration-response curves to ACh of oocytes injected with a 1:1 ratio of α4β2 nAChR mRNA in the absence of MLA (■), in the presence of 200 nm MLA after a 3-min incubation (□), and in the presence of 2 μm MLA without incubation (○). C, shown are concentration-response curves of ACh of oocytes injected with α4β2 mRNA in a 1:1 (■) and 1:10 (□) ratio (solid lines) and α4(D204C)β2 mRNA in a 1:1 (●) and 1:10 (○) ratio (dashed lines). Currents are normalized to the maximum current of each individual cell. D, shown are MLA inhibition concentration-response curves of oocytes injected with α4β2 mRNA in a 1:1 (■) and 1:10 (□) ratio (solid lines) and α4(D204C)β2 mRNA in a 1:1 (●) and 1:10 (○) ratio (dashed lines). Currents are normalized to the current elicited by 300 μm ACh (1:1), 1 μm (1:10), 30 μm ACh (D204C 1:1), or 1 μm (D204C 1:10). Data are represented as the means ± S.E., and the curve-fitting procedure is as explained in the data analysis under “Experimental Procedures.”
Mode of Antagonism Is Altered by MLA Preincubation
To further investigate the mode of inhibition of the α4β2 nAChR, we compared the inhibition with and without a 3-min preincubation of MLA. Without preincubation, the IC50 of MLA was significantly increased (IC50 = 1.5 μm no incubation, cf. 200 nm with incubation, p < 0.05, Fig. 3, Table 2). When increasing concentrations of ACh were co-applied with MLA, the maximum current elicited was not significantly changed compared with the maximum current in the absence of MLA, such that the inhibition was surmountable (Fig. 3, Table 2). This result suggests that the insurmountable mode of inhibition depends on the binding of MLA to the receptor having reached equilibrium.
As MLA contains a tertiary amine, it would be expected to be charged at physiological pH and may exhibit voltage-dependent pore-block. To determine whether MLA binds to the channel lumen or to an alternative site leading to insurmountable inhibition, we determined a current-voltage (I-V) curve with preincubation of MLA and demonstrated that there was no change in the relative inhibition of ACh-induced currents by MLA at various holding potentials (Fig. 4). This observation suggests that MLA does not block the pore of the channel in the transmembrane region, in contrast to other simple analogues of MLA that inhibit α4β2 nAChRs (31).
FIGURE 4.

Current-voltage (I-V) relationship of oocytes injected with a 1:1 ratio of α4β2 nAChR mRNA in the absence of MLA or after a 3 min incubation with MLA. A, the currents normalized to the response to 100 μm ACh at −90 mV are shown for different voltages when 100 μm ACh (■), 300 nm MLA (▴), or 300 nm MLA and 100 μm ACh (▾) is applied. B, the fraction of the ACh response that is inhibited by 300 nm MLA at different holding potentials is shown. Slope = 0.0023 ± 0.001, p = 0.19 cf. 0, z-test, n = 4). Data points are given as the means ± S.E.
We instead focused on the subunit interfaces of the nAChR that do not contain the classical α4-β2 ACh binding site. Due to the heteromeric nature of the α4β2 nAChR, the alternative interfacial binding sites depend on subunit stoichiometry (Fig. 5). In addition to the α4-β2 interfaces, the (α4)3(β2)2 nAChR contains α4-α4 and β2-α4 interfaces, whereas the (α4)2(β2)3 nAChR contains β2-α4 and β2-β2 interfaces. Given that irreversible inhibition was observed for the (α4)3(β2)2 nAChR stoichiometry, the unique α4-α4 interface suggested itself as a possible site of MLA binding. The high affinity for MLA binding to the α7-α7 interface of the α7 nAChR also prompted investigation of the structurally related α4-α4 interface present in the (α4)3(β2)2 nAChR. In addition, recent investigations (26, 27) have demonstrated that occupation of the α4-α4 interface modulates the agonist response of the (α4)3(β2)2 nAChR stoichiometry.
FIGURE 5.
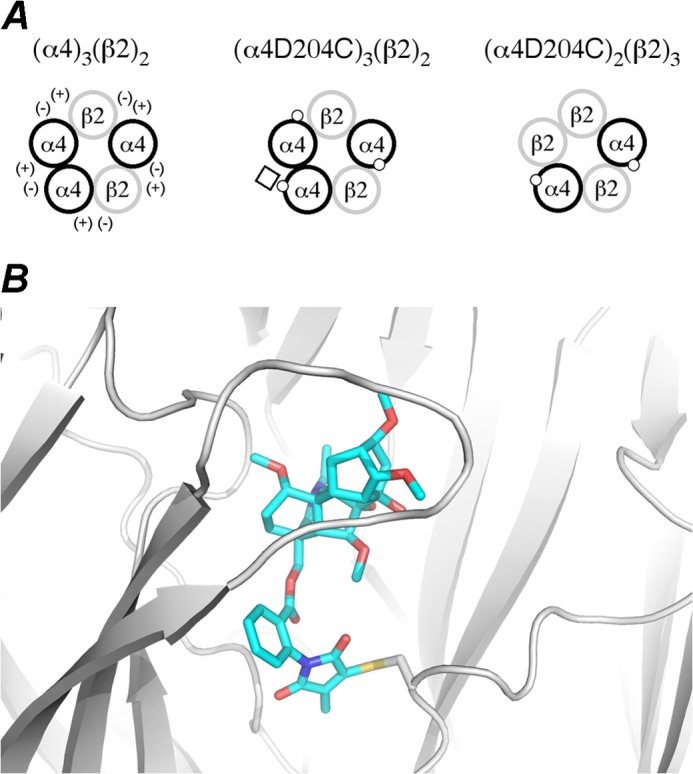
A, schematics of α4β2 nAChR expression with principal (+) and complementary (−) sides of the receptor interfaces are shown. The (α4)3(β2)2, (α4[D204C])3(β2)2 (1:1 mRNA injection ratio), and (α4[D204C])2(β2)3 (1:10 mRNA injection ratio) are represented with α subunits (black circles), with the D204C mutation (white dot) shown, and with β subunits (gray circles). The D204C mutation is located at the α4-α4 and β2-α4 interfaces of the (α4[D204C])3(β2)2 receptor and at the β2-α4 interfaces only on the (α4[D204C])2(β2)3 receptor. The reactive MLA-maleimide (white square) is covalently trapped with the D204C mutant residue only at the α4-α4 interface of the (α4[D204C])3(β2)2 receptor. B, shown is a structural model of the α4-α4 interface with the MLA-maleimide covalently trapped by cysteine (D204C).
To experimentally investigate MLA binding at the α4-α4 interface, we elected to employ covalent trapping, a methodology that uses cysteine mutagenesis in combination with synthetically derived reactive probes and computational models to interrogate the location and nature of ligand binding at functional nAChRs expressed in Xenopus oocytes (31, 32). To this end we manipulated the MLA molecule such that it contained a reactive maleimide group suitable for covalent trapping with cysteine mutants (Fig. 6). Maleimides react rapidly, selectively, and irreversibly with thiolate-bearing residues such as the amino acid cysteine even in the presence of other nucleophiles such as lysine (39). We previously demonstrated that MLA-maleimide reacts covalently to α7 nAChR cysteine mutants in a time-dependent manner (32). In the α7 nAChR, the cysteine mutation that reacted with MLA-maleimide was located in loop F. Because this loop is highly flexible and in the α4 subunit is different in sequence and length compared with the α7 subunit, the corresponding residue in α4 could not be reliably identified from a sequence alignment alone. Thus, to determine the optimal position for the cysteine to react with MLA-maleimide in the α4-α4 interface, we constructed a dimeric α4-α4 homology model (see “Experimental Procedures”) with MLA in the binding site. The residue located closest to the MLA-succinimide moiety was aspartate 204 on the complementary face of the binding site. To test the feasibility of covalent bond formation, we introduced the cysteine in this position computationally, created the bond to the MLA-maleimide moiety, and relaxed the structure with a root mean square deviation cutoff of 0.5 Å on all heavy atoms. The small relaxation required of the complex suggested that the covalent bond could form between the introduced cysteine and MLA-maleimide without marked changes in the inhibitor binding mode (Fig. 5).
FIGURE 6.

MLA-maleimide inhibition of α4β2 nAChRs is identical to MLA. A, the chemical structure of MLA (i) and MLA-maleimide (ii) shows the location of the reactive maleimide group. B, shown are raw traces from a single experiment demonstrating inhibition of ACh-activation of (α4)3(β2)2 (i) and (α4)2(β2)3 (ii) receptors by 100 nm MLA or 100 nm MLA-maleimide after a 3-min incubation. C, shown is the mean ± S.E. of current responses normalized to 100 or 1 μm ACh for (α4)3(β2)2 and (α4)2(β2)3 nAChRs, respectively. Inhibition of ACh (filled bars) by 100 nm MLA or 100 nm MLA-maleimide (open bars) after 3 min of incubation was not significantly different at (α4)3(β2)2 or (α4)2(β2)3 nAChRs (p > 0.05; Student's t test; respectively, n = 4).
The generation of the MLA-maleimide-reactive probe involves the formal removal of two hydrogen atoms to afford the unsaturated maleimide unit. This small structural change was not expected to significantly change the potency of the ligand. To demonstrate this, we performed an experiment on oocytes expressing (α4)3(β2)2 nAChRs, where the ACh-elicited current was measured. After a 10-min washout period, 100 nm MLA-maleimide was preincubated for 3 min then co-applied with 100 μm ACh, then the same procedure was repeated with 100 nm MLA (Fig. 6). There was no significant difference in the inhibition of ACh-elicited currents with either 100 nm MLA or 100 nm MLA-maleimide (I/IACh = 0.57 ± 0.07 100 nm MLA, 0.57 ± 0.08 100 nm MLA-maleimide; p > 0.05, paired Student's t test). This experiment was repeated on oocytes expressing (α4)2(β2)3 nAChRs. There was no significant difference in currents elicited by 1 μm ACh when inhibited by 100 nm MLA compared with 100 nm MLA maleimide (I/IACh = 0.44 ± 0.03, 100 nm MLA; 0.45 ± 0.02, 100 nm MLA-maleimide; p > 0.05, paired Student's t test). This demonstrates that MLA-maleimide inhibits either receptor stoichiometry with the same potency as MLA (Fig. 6).
Effect of Cysteine Mutations to Receptor Function
The target α4(D204C) mutation was introduced using site-directed mutagenesis. To ensure that the α4(D204C) mutation did not cause any gross changes to receptor function, we performed concentration-response curves to ACh and inhibition concentration-response curves to MLA for a 1:1 and 1:10 injection ratio. There was a small shift (∼5-fold reduction in EC50 compared with wild type) in the concentration-response curve to ACh for the α4(D204C)β2 nAChR injected in a 1:1 ratio, with an EC50 of 26 μm. When a 1:10 injection ratio was evaluated, the EC50 of ACh (EC50 = 1.9 μm) was not significantly different from wild type (p > 0.05, Fig. 3, Table 1). When the inhibition curve to MLA was performed for the 1:1 ratio, the IC50 was 27 nm, ∼10-fold more potent than the wild-type α4β2 nAChR. When a 1:10 injection ratio was performed, the IC50 of MLA (IC50 = 120 nm) was not significantly different from wild type (p > 0.05; Fig. 3, Table 1). These results suggest that the introduction of the cysteine mutation did not make gross changes to the structure that might interfere with receptor function and that the potency of MLA to the mutants was similar to the wild type for either stoichiometry.
MLA-maleimide Is Covalently Trapped at the (α4[D204C])3(β2)2 nAChR
Based on the α4-α4 homology model with MLA in the binding site, we hypothesized that MLA-maleimide would react with the introduced cysteine to form a covalent bond, irreversibly reducing the current elicited by ACh (Fig. 5). We tested this hypothesis by applying 10 μm ACh to oocytes injected with α4(D204C) and wild-type β2 subunits in a 1:1 ratio to predominantly express the (α4[D204C])3(β2)2 stoichiometry and then re-applied ACh to ensure the current was reproducible. We then applied 1 μm MLA-maleimide for 30 s and washed with buffer for 17 min to remove any MLA within the perfusion system. We then applied 10 μm ACh again and measured the reduction in the ACh-elicited current before MLA-maleimide addition (Fig. 7). This MLA-maleimide concentration was ∼10-fold greater than the IC50 for MLA. A similar protocol for MLA IC50 concentration has previously been employed to covalently trap MLA-maleimide in the binding site of α7 receptor cysteine mutants (32). After 30 s of incubation with the MLA-maleimide, the ACh-elicited current was reduced by 51 ± 0.08% (mean ± S.E., n = 5) (Fig. 7). As MLA-maleimide was not present during the ACh application, we conclude that the inhibition in current was due to an irreversible reaction of the maleimide group with the introduced cysteine at D204C.
FIGURE 7.

MLA-maleimide irreversibly inhibits α4(D204C)β2 nAChRs injected in a 1:1 but not 1:10 ratio. A, shown is the initial trapping experiment of MLA-maleimide to the introduced cysteine. i, 10 μm ACh was applied to α4(D204C)β2 nAChRs injected in a 1:1 ratio (open bar). 1 μm MLA-maleimide was applied for 30 s followed by a 17-min wash with buffer to remove excess MLA-maleimide. 10 μm ACh was applied again, demonstrating an irreversible reduction in the ACh-elicited response. ii, shown is an identical experiment, except 10 μm MLA were incubated for 3 min then co-applied with 1 μm MLA-maleimide followed by a 17-min wash and application of 10 μm ACh. iii, 1 μm ACh was applied to α4(D204C)β2 nAChRs injected in a 1:10 ratio (open bar). 1 μm MLA-maleimide was applied for 30 s followed by a 17-min wash with buffer to remove excess MLA-maleimide. 1 μm ACh was applied again, blocking the irreversible reduction in the ACh-elicited response. B, shown is the mean ± S.E. of ACh-elicited responses after 30 s of treatment of 1 μm MLA-maleimide and a 17-min wash, normalized to the initial ACh-elicited response. 1 μm MLA-maleimide significantly reduced the ACh-elicited response in α4(D204C)β2 nAChRs injected in a 1:1 ratio (filled bar, middle) compared with a 1:10 (open bar) ratio (p < 0.01, n = 3–4; Student's t test). Preincubation for 3 min and co-application of 10 μm MLA with 1 μm MLA-maleimide prevented the MLA-maleimide reaction (filled bars, left), significantly blocking the reduction of the ACh-elicited response at the α4(D204C)β2 nAChR injected in a 1:1 ratio (p < 0.01, n = 4–6; Student's t test). C, rate of reaction experiments with individual oocytes expressing α4(D204C)β2 nAChRs injected in a 1:1 ratio (i and ii), α4β2 nAChRs injected in a 1:1 ratio (iii), and α4(D204C)β2 nAChRs injected in a 1:10 ratio (iv). An EC50 concentration of ACh (10, 10, 100, and 1 μm for (i–iv), open bars) was applied before application of 1 μm MLA (i) or) MLA-maleimide (filled bars) (ii–iv. Each successive 30-s application of MLA or MLA-maleimide was followed by a 17-min wash with buffer to remove excess MLA and then an ACh application to measure the reduction in current resulting from trapping of the MLA-maleimide with the cysteine. D, shown is the mean ± S.E. of ACh normalized to the initial response to ACh after the cumulative time of successive 1 μm MLA or MLA-maleimide applications. Rates of reaction of MLA-maleimide to (α4[D204C])3(β2)2 (●), (α4[D204C])2(β2)3 (○), and α4β2 wild type (■) and MLA to (α4[D204C])3(β2)2 (□) are plotted with a single exponential fit to the average responses shown as described under “Experimental Procedures” (n = 3–6 for each rate of reaction). E, shown is the mean ± S.E. of the maximum reduction of ACh-elicited current as a fraction of the initial ACh-elicited current. The maximum reduction in current was determined either by fitting each individual experiment to a single exponential decay (α4(D204C)β2 1:1 and 1:10 ratio and MLA-maleimide; open bars) or the reduction in ACh-elicited response after 120 s of cumulative MLA or MLA-maleimide addition (α4β2 and MLA or MLA-maleimide and α4(D204C)β2 1:1 and MLA). The reduction in ACh-elicited response is significantly greater for α4D204Cβ2 injected in the 1:1 compared with the 1:10 ratio (p < 0.001, n = 3- 5), indicating that MLA-maleimide is being trapped at the D204C residue in the α4-α4 interface (white bars). The ACh-response recovers up to 90% of the initial response, significantly different to the application of MLA-maleimide applied to the (α4[D204C])3(β2)2 nAChR, injected in a 1:1 ratio (p < 0.01, n ≥ 3; Student's t test).
To illustrate that the maleimide group was required for the irreversible inhibition, we evaluated whether MLA bound to the receptor could inhibit the reaction between the cysteine mutant and MLA-maleimide. 10 μm ACh was applied to oocytes injected with a 1:1 ratio of α4(D204C) and β2 mRNA at least twice to ensure a consistent response. 10 μm MLA was applied for 3 min to equilibrate MLA binding followed by co-application of 10 μm MLA and 1 μm MLA-maleimide for 30 s. After a 17-min washout to remove MLA and MLA-maleimide, 10 μm ACh was re-applied, and the response was compared with 10 μm ACh before MLA and MLA-maleimide application. After a 3-min preincubation of MLA (10 μm) followed by a co-application of MLA (10 μm) and MLA-maleimide (1 μm), the inhibition by MLA-maleimide was significantly attenuated, and the response to ACh (10 μm) after a 17-min washout was unchanged relative to the initial response to ACh (I/IACh = 1.08 ± 0.08, p < 0.01; paired Student's t test). This competition experiment demonstrates that MLA binds to the (α4[D204C])3(β2)2 nAChR and blocks the reaction of MLA-maleimide at the cysteine residue, presumably by preventing MLA-maleimide binding to the nAChR.
To measure the full extent of the reaction of MLA-maleimide to the (α4[D204C])3(β2)2 receptor, we performed a rate of reaction experiment. We applied 10 μm ACh to oocytes injected with α4(D204C) and wild-type β2 subunits in a 1:1 ratio to predominantly express the (α4[D204C])3(β2)2 stoichiometry and then re-applied ACh to ensure the current was reproducible. We then applied 1 μm MLA-maleimide for 30 s and washed with buffer for 17 min to remove any MLA within the perfusion system. We then applied 10 μm ACh again and measured the reduction in the ACh-elicited current before MLA-maleimide addition (Fig. 7). This process was repeated until the current reached a plateau. The decrease in the maximum current was plotted against the cumulative time and fitted with an exponential decay. The maximum current inhibition was 82% (I∞ = 0.18 ± 0.04, n = 5), and the first-order decay constant was k = 0.03 s−1 (Fig. 7, Table 3). This was significantly different from the wild-type α4β2 nAChR, where MLA-maleimide application and wash inhibited the ACh-elicited current by 38% (I∞ = 0.62 ± 0.12, n = 4, p < 0.01) after 120 s incubation, and experiments from individual oocytes did not fit a single exponential decay curve (Fig. 7, Table 3). Incubation of MLA (1 μm for 30 s) alone followed by a 17-min wash to the (α4[D204C])3(β2)2 receptors under identical conditions to MLA-maleimide resulted in a 17% (I∞ = 1.05 ± 0.1, n = 3, p < 0.01) reduction of ACh-elicited currents after 120 s of cumulative incubation and did not fit a single exponential decay curve (Fig. 7, Table 3). The reduction in current observed when MLA-maleimide was applied to the wild type is most likely a nonspecific effect of the moderately high concentrations of ligand applied, as incubation of 1 μm MLA for a cumulative 120 s followed by a 17-min wash to the wild-type receptor led to a similar reduction in current (I∞ = 0.61 ± 0.06, n = 4, p > 0.05, Table 3).
TABLE 3.
Rate of reaction to MLA-maleimide parameters derived from curve-fitting
| Receptor/conditions | I∞±S.E. | k (95% CI) | n |
|---|---|---|---|
| s−1 | |||
| (α4)3(β2)2 MLA | 0.61 ± 0.06a | b | 3 |
| (α4)3(β2)2 MLA-maleimide | 0.62 ± 0.12a | b | 6 |
| (α4[D204C])3(β2)2 MLA | 1.05 ± 0.10a | b | 4 |
| (α4[D204C])3(β2)2 MLA-maleimide | 0.18 ± 0.04 | 0.03 (0.02–0.05) | 6 |
| (α4[D204C])2(β2)3 MLA-maleimide | 0.73 ± 0.04 | 0.02 (0.00–0.04) | 3 |
a Where data from individual oocytes did not fit to an exponential curve, the I∞ is defined as the I120.
b Individual oocytes could not be fitted to an exponential curve fit.
MLA-maleimide Does Not React to the (α4[D204C])2(β2)3 nAChR
The α4(D204C) mutation is present in the α4-α4 interface of the (α4)3(β2)2 nAChR stoichiometry but also in the two β2-α4 interfaces of both the (α4)3(β2)2 and (α4)2(β2)3 nAChR stoichiometries (Fig. 5). To determine if MLA is being trapped at the α4-α4 or β2-α4 interface, we altered the RNA injection ratios to reduce or eliminate the (α4)3(β2)2 nAChR stoichiometry containing the α4-α4 interface.
The reactive probe MLA-maleimide was applied to the (α4[D204C])2(β2)3 nAChR in oocytes derived from a 1:10 injection ratio (Fig. 6, Table 3). We applied 1 μm ACh to oocytes injected with α4(D204C) and wild-type β2 subunits in a 1:10 ratio, to predominantly express the (α4[D204C])2(β2)3 stoichiometry, and then re-applied ACh to ensure the current was reproducible. We then applied 1 μm MLA-maleimide for 30 s and washed with buffer for 17 min to remove any MLA within the perfusion system. We then applied 1 μm ACh again and measured the reduction in the ACh-elicited current before MLA-maleimide addition (Fig. 7). After 30 s of incubation with the MLA-maleimide, the ACh-elicited current was reduced by 11 ± 0.02% (mean ± S.E., n = 3) (Fig. 7). This was significantly different to the (α4[D204C])3(β2)2 stoichiometry (p < 0.01), demonstrating that MLA-maleimide was not reacting to the introduced cysteine in the (α4[D204C])2(β2)3.
We performed a rate of reaction experiment on the (α4[D204C])2(β2)3 nAChRs under identical conditions to those conducted on the (α4[D204C])3(β2)2 nAChRs derived from a 1:1 RNA injection ratio. Experiments from individual oocytes were fitted to single exponential curves, and the MLA-maleimide application and wash inhibited the ACh-elicited currents by a maximum of 27% (I∞ = 0.73 ± 0.04, n = 3), significantly lower than that observed for MLA-maleimide with the (α4[D204C])3(β2)2 nAChR of 82% (I∞ = 0.18 ± 0.04, n = 5, p < 0.001, Fig. 7, Table 3). The reduction in the ACh-elicited current was similar to that observed for the control studies of MLA with the (α4[D204C])3(β2)2 nAChR or MLA-maleimide and the (α4)3(β2)2 nAChR (Fig. 7, Table 3). Together, these data indicated that at the (α4[D204C])2(β2)3 nAChR, MLA-maleimide was not covalently trapped at an appreciable rate to irreversibly inhibit channel opening. We infer that with a 1:10 injection ratio, the population of receptors has altered to contain (α4[D204C])2(β2)3 receptors, and as such no α4-α4 interface is available. The Asp-204 residue is located on the complementary face of the α4-α4 and β2-α4 interfaces, and these data demonstrate that MLA-maleimide reacts rapidly with the α4(D204C) when it is located in the α4-α4 interface but not when it is located in the β2-α4 interfaces.
DISCUSSION
We have demonstrated that MLA-maleimide, a reactive analog of MLA, is covalently trapped at an introduced cysteine (D204C) on the complementary face of the α4 subunit only in the (α4)3(β2)2 stoichiometry where an α4-α4 interface is present, unequivocally demonstrating that MLA binds at the α4-α4 interface. Covalent trapping of a reactive analog at an introduced cysteine has previously been used to integrate computational techniques such as homology modeling and molecular dynamics, with functional analysis of the trapped ligand-receptor complex (29, 40). Here we have used this method for a different purpose, to reveal the interfacial binding site of a ligand in a heteropentameric nAChR.
There are four structurally distinct subunit interfaces within the two known stoichiometries of the α4β2 nAChR, two of which, α4-α4 and α4-β2 contain ACh binding sites (26–27). The reactive MLA-maleimide is only trapped by the (α4[D204C])3(β2)2 nAChR cysteine mutant where the receptor contains an α4-α4 interface, demonstrating unambiguously that MLA-maleimide and by extension MLA also binds at this site. No covalent trapping was observed for the (α4[D204C])2(β2)3 nAChR that contains two β2-α4 interfaces modified by cysteine but no α4-α4 site. Preincubation of the (α4)3(β2)2 nAChR with MLA results in insurmountable inhibition of Ach-induced currents. The covalent trapping experiments in this work may implicate the α4-α4 binding site in this mode of receptor antagonism. Indeed, the binding of MLA to the α4-α4 interface should not be surprising given the high affinity of the structurally related α7-α7 interface of the α7 nAChR for MLA. Furthermore, the elegant work of Palma et al. (41) has demonstrated that binding of a single MLA molecule to the α7 nAChR is sufficient to observe receptor inhibition despite the fact that five α7-α7 interfaces are available for MLA binding. In the current context, binding of a MLA molecule to the α4-α4 binding site displayed in the (α4)3(β2)2 may also be sufficient to observe insurmountable inhibition.
Recent investigations by Harpsøe et al. (26) and Mazzaferro et al. (27) have demonstrated that binding of ACh at the α4-α4 interface modulates the response to ACh in the (α4)3(β2)2 nAChR stoichiometry. The observation that MLA and ACh can both bind at the α4-α4 interface appears contradictory to our pharmacological data; MLA and ACh would be expected to compete at the α4-α4 binding site. Thus, the inhibition by MLA would be surmountable with high enough concentrations of ACh and parallel shifts of the ACh concentration-response curves in the presence of different concentrations for MLA would be expected (42). Under our experimental conditions, this was not observed (Fig. 2). Limitations in our experimental conditions, specifically ACh-induced desensitization of receptors and the osmotic effects of molar concentrations of ACh, prevent applying ACh in higher concentrations (>10 mm) to determine if the inhibitory effects of MLA are in fact surmountable. These complexities may contribute to the absence of a parallel shift in the concentration-response curves and/or the insurmountable inhibition.
Preincubation with MLA was performed to ensure the binding of MLA to the nAChRs was in equilibrium before ACh application, which along with a parallel shift in the concentration-response curves is a necessary precondition for the proper analysis of a Schild plot to characterize competitive inhibition (42). The preincubation was required to observe maximal insurmountable inhibition of the α4β2 nAChR in this study, which suggests a slow onset of MLA binding to the α4-α4 interface. Similar observations have been made for human α7 nAChRs where preincubation has been demonstrated to increase the potency of MLA inhibition of JN403-induced currents (43). In our covalent trapping study, the pseudo first order rate constant for binding and reaction of MLA-maleimide (1 μm) at the (α4[D204C])2(β2)3 was k = 0.038 ± 0.002 s−1, providing an estimated second-order rate constant of k = 3.8 ± 0.2 × 104 m−1 s−1. The pseudo first-order reaction rate constant for the binding and reaction of the same reactive probe MLA maleimide (10 nm) to the α7(S188C:L9′T) nAChR of k = 0.0058 ± 0.0014 s−1 provides an estimated second-order rate constant of k = 5.8 ± 1.4 × 105 m−1 s−1. This is comparable to the kon = 2.04 × 106 m−1 s−1 for binding of [3H]MLA to the α7 nAChR from rat membranes, which serves as an upper limit for the rate of the covalent trapping (44). The lower estimated second-order rate constant for covalent trapping at the (α4)3(β2)2 receptor subtype may in part reflect slower binding to the α4-α4 interface. The surmountable mode of inhibition that we demonstrate for the α4β2 nAChR in the absence of preincubation may also be a function of the relative time course of antagonist binding with respect to ACh binding. A slower time course of MLA binding would result in lower concentrations of ACh required to fully occupy the three agonist binding sites if the ligands were co-applied rather than the impractically high concentrations of ACh thought to be required if the binding of MLA was allowed to reach equilibrium before ACh application.
The covalent trapping experiment resulted in the α4(D204C)β2 receptor eliciting ∼14% of the current compared with the current before the reaction. It is tempting to speculate that this is a result of a mixed population of receptor stoichiometries, leading to some current elicited by (α4[D204C])2(β2)3 receptors that do not trap the inhibitor. However, we have previously demonstrated that when MLA-maleimide is trapped in the α7-α7 interface by the analogous α7(S188C:L9′T) receptor, we also do not see full inhibition, suggesting that the reaction does not go to completion or that bound MLA does not always covalently trap in the binding site (32). It is, therefore, not reliable to use this cysteine-trapping method as a proxy measure of different stoichiometric populations.
There is emerging evidence that ligands can exert pharmacological effects by binding to alternative subunit interfaces of heteromeric nAChRs at locales distinct from the α-β interface containing the consensus agonist binding site (28–30). These studies include an x-ray co-crystallographic study of the binding of non-competitive inhibitors and allosteric modulators to the AChBP coupled with radioligand binding studies of these inhibitors and modulators to AChBPs with mutations that resemble subunit interfaces that do not contain a principal α subunit (29), the application of the substituted cysteine accessibility method to identify the binding site of the binding of the positive allosteric modulator morantel at the β3-α2 interface of the α2β3 nAChR (30), and a photo-affinity labeling study of the positive allosteric modulators physostigmine and galantamine at the Torpedo californica muscle-type nAChR that identified three binding sites, including an γ-β subunit interfacial site (28). Furthermore, these non-agonist binding sites resemble the now well established modulatory benzodiazepine binding site on GABAA receptors.
In this study we have employed covalent trapping to directly reveal MLA binding to the α4-α4 interface in functional (α4)3(β2)2 nAChRs, the first time that ligand binding at the α4-α4 interface has been directly demonstrated. The identification of this new ligand binding site and the delineation of the pharmacological profile in functional receptors open new avenues for development of therapeutics by targeting this site.
There has been a concerted effort to identify modulators of nAChRs with therapeutic benefits. Agonists selectively targeting α4β2 or α7 nAChRs have been demonstrated to improve cognitive function in various learning and memory paradigms (45) as well as possess analgesic properties (46). Positive allosteric modulators only act in the areas of the brain where ACh was released, preserving the spatial and temporal activation by the endogenous neurotransmitter (46). The (α4)3(β2)2-selective positive allosteric modulator NS9283 increases the peak maximal current in response to ACh and augments ACh-evoked DhβE-sensitive currents in thalamocortical neurons and improves or alleviates pharmacological deficits in a wide variety of paradigms, including sensory gating, vigilance, attention, episodic, and reference memory (47). The selectivity exhibited by NS9283 for the (α4)3(β2)2 nAChR stoichiometry implicates binding to the α4-α4 interface. However, there is little understanding of the structural rearrangements after binding at the α4-α4 site that lead to positive or negative modulation, an understanding of which has the potential to lead to the identification of superior therapeutics. Our results suggest that MLA analogues may assist in addressing this question and also serve as lead compounds for α4-α4-targeted therapeutics, one that could be identified by a combination of molecular modeling, in silico docking, and functional electrophysiological techniques.
In conclusion, we have demonstrated for the first time that MLA binds to the α4-α4 subunit interface of the α4β2 nAChR through covalent-trapping of a modified MLA molecule. MLA binding at the α4-α4 interface leads to an apparently insurmountable mode of inhibition. We also demonstrated that trapping molecules to an introduced cysteine in individual subunits can identify otherwise ambiguous binding sites in ligand-gated ion channels. We propose that the α4-α4 binding site can be targeted to produce more selective pharmacological agents that can then be used to better understand the role of the different stoichiometries of the α4β2 nAChR.
Acknowledgments
We thank Professor Jim Boulter, University of California, Los Angeles, CA, and Professor Millet Treinin, Hebrew University of Jerusalem for the kind gifts of plasmid DNA. We are very grateful to the Department of Pharmacology, the University of Sydney, for managing and maintaining the X. laevis colony.
This work was supported by Australian Research Council Discovery Project DP0986469.
- AChBP
- acetylcholine (ACh)-binding protein
- nAChBP
- nicotinic AChBP
- M1–4
- transmembrane segments
- MLA
- methyllycaconitine
- nAChR
- nicotinic acetylcholine receptor.
REFERENCES
- 1. Gotti C., Clementi F. (2004) Neuronal nicotinic receptors. From structure to pathology. Prog. Neurobiol. 74, 363–396 [DOI] [PubMed] [Google Scholar]
- 2. Karlin A. (2002) Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci. 3, 102–114 [DOI] [PubMed] [Google Scholar]
- 3. Borovikova L. V., Ivanova S., Zhang M., Yang H., Botchkina G. I., Watkins L. R, Wang H., Abumrad N., Eaton J. W., Tracey K. J. (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458–462 [DOI] [PubMed] [Google Scholar]
- 4. Jensen A. A., Frølund B., Liljefors T., Krogsgaard-Larsen P. (2005) Neuronal nicotinic acetylcholine receptors. Structural revelations, target identifications, and therapeutic inspirations. J. Med. Chem. 48, 4705–4745 [DOI] [PubMed] [Google Scholar]
- 5. Grosman C., Zhou M., Auerbach A. (2000) Mapping the conformational wave of acetylcholine receptor channel gating. Nature 403, 773–776 [DOI] [PubMed] [Google Scholar]
- 6. Miyazawa A., Fujiyoshi Y., Unwin N. (2003) Structure and gating mechanism of the acetylcholine receptor pore. Nature 423, 949–955 [DOI] [PubMed] [Google Scholar]
- 7. Absalom N. L., Lewis T. M., Schofield P. R. (2004) Mechanisms of channel gating of the ligand-gated ion channel superfamily inferred from protein structure. Exp. Physiol. 89, 145–153 [DOI] [PubMed] [Google Scholar]
- 8. Bocquet N., Nury H., Baaden M., Le Poupon C., Changeux J. P., Delarue M., Corringer P. J. (2009) X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111–114 [DOI] [PubMed] [Google Scholar]
- 9. Hilf R. J., Dutzler R. (2009) Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 457, 115–118 [DOI] [PubMed] [Google Scholar]
- 10. Hilf R. J., Dutzler R. (2008) X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375–379 [DOI] [PubMed] [Google Scholar]
- 11. Hibbs R. E., Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brejc K., van Dijk W. J., Klaassen R. V., Schuurmans M., van Der Oost J., Smit A. B., Sixma T. K. (2001) Crystal structure of an ACh-binding protein reveals the ligand binding domain of nicotinic receptors. Nature 411, 269–276 [DOI] [PubMed] [Google Scholar]
- 13. Hansen S. B., Sulzenbacher G., Huxford T., Marchot P., Bourne Y., Taylor P. (2006) Structural characterization of agonist and antagonist-bound acetylcholine-binding protein from Aplysia californica. J. Mol. Neurosci. 30, 101–102 [DOI] [PubMed] [Google Scholar]
- 14. Unwin N. (1995) Acetylcholine receptor channel imaged in the open state. Nature 373, 37–43 [DOI] [PubMed] [Google Scholar]
- 15. Le Novère N., Grutter T., Changeux J. P. (2002) Models of the extracellular domain of the nicotinic receptors and of agonist and Ca2+ binding sites. Proc. Natl. Acad. Sci. U.S.A. 99, 3210–3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schapira M., Abagyan R., Totrov M. (2002) Structural model of nicotinic acetylcholine receptor isotypes bound to acetylcholine and nicotine. BMC Struct. Biol. 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bisson W. H., Scapozza L., Westera G., Mu L., Schubiger P. A. (2005) Ligand selectivity for the acetylcholine binding site of the rat α4β2 and α3β4 nicotinic subtypes investigated by molecular docking. J. Med. Chem. 48, 5123–5130 [DOI] [PubMed] [Google Scholar]
- 18. Huang X., Zheng F., Chen X., Crooks P. A., Dwoskin L. P., Zhan C. G. (2006) Modeling subtype-selective agonists binding with α4β2 and α7 nicotinic acetylcholine receptors. Effects of local binding and long-range electrostatic interactions. J. Med. Chem. 49, 7661–7674 [DOI] [PubMed] [Google Scholar]
- 19. Rohde L. A., Ahring P. K., Jensen M. L., Nielsen E. Ø., Peters D., Helgstrand C., Krintel C., Harpsøe K., Gajhede M., Kastrup J. S., Balle T. (2012) Intersubunit bridge formation governs agonist efficacy at nicotinic acetylcholine α4β2 receptors. Unique role of halogen bonding revealed. J. Biol. Chem. 287, 4248–4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Akdemir A., Rucktooa P., Jongejan A., Elk Rv., Bertrand S., Sixma T. K., Bertrand D., Smit A. B., Leurs R., de Graaf C., de Esch I. J. (2011) Acetylcholine binding protein (AChBP) as template for hierarchical in silico screening procedures to identify structurally novel ligands for the nicotinic receptors. Bioorg. Med. Chem. 19, 6107–6119 [DOI] [PubMed] [Google Scholar]
- 21. Nemecz A., Taylor P. (2011) Creating an α7 nicotinic acetylcholine recognition domain from the acetylcholine-binding protein. Crystallographic and ligand selectivity analyses. J. Biol. Chem. 286, 42555–42565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hansen S. B., Sulzenbacher G., Huxford T., Marchot P., Taylor P., Bourne Y. (2005) Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 24, 3635–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moroni M., Zwart R., Sher E., Cassels B. K., Bermudez I. (2006) α4β2 nicotinic receptors with high and low acetylcholine sensitivity. Pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol. Pharmacol. 70, 755–768 [DOI] [PubMed] [Google Scholar]
- 24. Zwart R., Broad L. M., Xi Q., Lee M., Moroni M., Bermudez I., Sher E. (2006) 5-I A-85380 and TC-2559 differentially activate heterologously expressed α4β2 nicotinic receptors. Eur. J. Pharmacol. 539, 10–17 [DOI] [PubMed] [Google Scholar]
- 25. Carbone A. L, Moroni M., Groot-Kormelink P. J., Bermudez I. (2009) Pentameric concatenated (α4)(2)(β2)(3) and (α4)(3)(β2)(2) nicotinic acetylcholine receptors. Subunit arrangement determines functional expression. Br. J. Pharmacol. 156, 970–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harpsøe K., Ahring P. K, Christensen J. K, Jensen M. L, Peters D., Balle T. (2011) Unraveling the high- and low-sensitivity agonist responses of nicotinic acetylcholine receptors. J. Neurosci. 31, 10759–10766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mazzaferro S., Benallegue N., Carbone A., Gasparri F., Vijayan R., Biggin P. C., Moroni M., Bermudez I. (2011) Additional acetylcholine (ACh) binding site at α4/α4 interface of (α4β2)2α4 nicotinic receptor influences agonist sensitivity. J. Biol. Chem. 286, 31043–31054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hamouda A. K., Kimm T., Cohen J. B. (2013) Physostigmine and galanthamine bind in the presence of agonist at the canonical and noncanonical subunit interfaces of a nicotinic acetylcholine receptor. J. Neurosci. 33, 485–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hansen S. B., Taylor P. (2007) Galanthamine and non-competitive inhibitor binding to ACh-binding protein. Evidence for a binding site on non-α-subunit interfaces of heteromeric neuronal nicotinic receptors. J. Mol. Biol. 369, 895–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seo S., Henry J. T., Lewis A. H., Wang N., Levandoski M. M. (2009) The positive allosteric modulator morantel binds at noncanonical subunit interfaces of neuronal nicotinic acetylcholine receptors. J. Neurosci. 29, 8734–8742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quek G. X., Lin D., Halliday J. I., Absalom N., Ambrus J. I., Thompson A. J., Lochner M., Lummis S. C., McLeod M. D., Chebib M. (2010) Identifying the binding site of novel methyllycaconitine (MLA) analogs at α4β2 nicotinic acetylcholine receptors. ACS Chem. Neurosci. 1, 796–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ambrus J. I., Halliday J.I., Kanizaj N., Absalom N., Harpsøe K., Balle T., Chebib M., McLeod M. (2012) Covalent attachment of antagonists to the α7 nicotinic acetylcholine receptor. Synthesis and recovery of substituted maleimides. Chem. Commun. (Camb) 48, 6699–6701 [DOI] [PubMed] [Google Scholar]
- 33. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 34. UniProt Consortium (2011) Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 39, D214–D219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Notredame C., Higgins D. G., Heringa J. (2000) T-Coffee. A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 36. Shen M. Y., Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 11, 2507–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alkondon M., Pereira E. F., Wonnacott S., Albuquerque E. X. (1992) Blockade of nicotinic currents in hippocampal neurons defines methyllycaconitine as a potent and specific receptor antagonist. Mol. Pharmacol. 41, 802–808 [PubMed] [Google Scholar]
- 38. Drasdo A., Caulfield M., Bertrand D., Bertrand S., Wonnacott S. (1992) Methyl lycaconitine. A novel nicotinic antagonist. Mol. Cell Neurosci. 3, 237–243 [DOI] [PubMed] [Google Scholar]
- 39. Chalker J. M., Bernardes G. J., Lin Y. A., Davis B. G. (2009) Conversion of cysteine into dehydroalanine enables access to synthetic histones bearing diverse post-translational modifications. Chem. Asian J. 4, 630–640 [DOI] [PubMed] [Google Scholar]
- 40. Wang J., Horenstein N. A., Stokes C., Papke R. L. (2010) Tethered agonist analogs as site-specific probes for domains of the human α7 nicotinic acetylcholine receptor that differentially regulate activation and desensitization. Mol. Pharmacol. 78, 1012–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palma E., Bertrand S., Binzoni T., Bertrand D. (1996) Neuronal nicotinic α 7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllycaconitine. J. Physiol. 491, 151–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wyllie D. J., Chen P. E. (2007) Taking the time to study competitive antagonism. Br. J. Pharmacol. 150, 541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arias H. R., Gu R. X., Feuerbach D., Wei D. Q. (2010) Different interaction between the agonist JN403 and the competitive antagonist methyllycaconitine with the human α7 nicotinic acetylcholine receptor. Biochemistry 49, 4169–4180 [DOI] [PubMed] [Google Scholar]
- 44. Davies A. R., Hardick D. J., Blagbrough I. S., Potter B. V., Wolstenholme A. J., Wonnacott S. (1999) Characterisation of the binding of [3H]methyllycaconitine. A new radioligand for labelling α7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology 38, 679–690 [DOI] [PubMed] [Google Scholar]
- 45. Taly A., Corringer P. J., Guedin D., Lestage P., Changeux J. P. (2009) Nicotinic receptors. Allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Discov. 8, 733–750 [DOI] [PubMed] [Google Scholar]
- 46. Rowbotham M. C., Duan W. R., Thomas J., Nothaft W., Backonja M. M. (2009) A randomized, double-blind, placebo-controlled trial evaluating the efficacy and safety of ABT-594 in patients with diabetic peripheral neuropathic pain. Pain 146, 245–252 [DOI] [PubMed] [Google Scholar]
- 47. Timmermann D. B., Sandager-Nielsen K., Dyhring T., Smith M., Jacobsen A. M., Nielsen E. O., Grunnet M., Christensen J. K., Peters D., Kohlhaas K., Olsen G. M., Ahring P. K. (2012) Augmentation of cognitive function by NS9283, a stoichiometry-dependent positive allosteric modulator of α2- and α4-containing nicotinic acetylcholine receptors. Br. J. Pharmacol. 167, 164–182 [DOI] [PMC free article] [PubMed] [Google Scholar]