

Background: Saturated fatty acids disrupt protein trafficking and promote endoplasmic reticulum (ER) stress in pancreatic β-cells.
Results: Chronic palmitate selectively reduces ER sphingomyelin and cholesterol and disrupts ER lipid rafts.
Conclusion: Altered ER lipid rafts contribute to defective ER protein export.
Significance: This provides novel insights into the mechanisms of β-cell death that underlie type 2 diabetes.
Keywords: Ceramide, Endoplasmic Reticulum Stress, Lipotoxicity, Pancreatic Islets, Trafficking, ER Lipid Raft, Lipidomics, Palmitate, Sphingomyelin, Type 2 Diabetes
Abstract
Chronic saturated fatty acid exposure causes β-cell apoptosis and, thus, contributes to type 2 diabetes. Although endoplasmic reticulum (ER) stress and reduced ER-to-Golgi protein trafficking have been implicated, the exact mechanisms whereby saturated fatty acids trigger β-cell death remain elusive. Using mass spectroscopic lipidomics and subcellular fractionation, we demonstrate that palmitate pretreatment of MIN6 β-cells promoted ER remodeling of both phospholipids and sphingolipids, but only the latter was causally linked to lipotoxic ER stress. Thus, overexpression of glucosylceramide synthase, previously shown to protect against defective protein trafficking and ER stress, partially reversed lipotoxic reductions in ER sphingomyelin (SM) content and aggregation of ER lipid rafts, as visualized using Erlin1-GFP. Using both lipidomics and a sterol response element reporter assay, we confirmed that free cholesterol in the ER was also reciprocally modulated by chronic palmitate and glucosylceramide synthase overexpression. This is consistent with the known coregulation and association of SM and free cholesterol in lipid rafts. Inhibition of SM hydrolysis partially protected against ATF4/C/EBP homology protein induction because of palmitate. Our results suggest that loss of SM in the ER is a key event for initiating β-cell lipotoxicity, which leads to disruption of ER lipid rafts, perturbation of protein trafficking, and initiation of ER stress.
Introduction
The consumption of the calorific Western diet, high in saturated fatty acid (FA),2 in combination with decreased whole-body energy expenditure, has been a strong driver of global obesity and associated metabolic diseases, such as type 2 diabetes (T2D). The latter is a whole-body disorder that causes hyperglycemia and is characterized by insulin resistance in liver, skeletal muscle, and adipose tissue in combination with pancreatic β-cell secretory dysfunction and death. Obese patients commonly have elevated serum FAs, and this is an independent risk factor for the development of T2D (1). Although the etiology of T2D remains elusive, current opinion proposes that deposition of these excess lipids in non-adipose tissues such as skeletal muscle, liver, and the pancreatic β-cell contributes to dysfunction in these organs (2). The dysfunction of the pancreatic β-cell follows many stages. Decreased glucose sensing and glucose-stimulated insulin secretion as well as an increased proinsulin-to-insulin ratio are early defects, whereas enhanced apoptosis occurs in late-stage decompensation (3). The resultant loss of pancreatic β-cells to a mass that is unable to control blood glucose homeostasis by adequate insulin secretion is a critical event preceding the advent of T2D.
The failure of the β-cells to successfully compensate for insulin resistance and prevent progression into T2D is in part due to the strong toxicity of FAs on the β-cell. Upon entry into the cell, FAs need to be metabolized to exert their effects, although the exact mechanisms through which lipid-induced β-cell death occur remain poorly understood and are likely to be multifactorial (4–9). However, apoptosis is particularly linked to saturated FAs (7–11), which has focused attention on pathways such as ceramide (Cer) generation and, more recently, endoplasmic reticulum (ER) stress, both of which also show specificity for saturated FAs, such as palmitate, over unsaturated FAs, such as oleate (7, 10–13).
Palmitate acts as a precursor in the de novo synthesis of Cer through the enzyme serine palmitoyltransferase 1. This sphingolipid (SL) is implicated in many forms of apoptosis, including those because of chronic lipid exposure in multiple cell types (14). In β-cells, the strongest evidence has arisen using obese Zucker diabetic fatty rats, a model of T2D characterized by gross obesity (4, 15). There is also more limited evidence implicating Cer in cellular models of β-cell lipotoxicity (7, 8, 16–19). The in vitro models are extremely powerful, however, because they allow a mechanistic focus on saturated FAs in isolation and, indeed, led to an appreciation of the role of ER stress in mediating β-cell apoptosis. Thus, chronic exposure to saturated FAs was shown to selectively enhance the unfolded protein response (UPR) (11, 12, 20). This response initially serves a protective function by promoting the folding and/or degradation of secretory protein in the lumen of the ER but also triggers apoptosis if ER stress remains unresolved by these means (21, 22). As a professional secretory cell, β-cells are particularly susceptible to ER stress. Activation of the UPR arm, comprising phosphorylation of PRKR-like endoplasmic reticulum kinase (PERK) and induction of the transcription factor C/EBP homology protein (CHOP), are especially important for the saturated FA-induced progression to apoptosis (23, 24). Indeed, ER stress has been shown to be essential for full apoptosis in β-cells in response to (especially mild) lipotoxicity (10, 11). Relevance of these in vitro models to human disease was confirmed by the enhanced expression of ER stress markers in β-cells of T2D patients (11, 21, 22) and the recent clinical trial of an ER stress-reducing drug, phenylbutyric acid, that diminished β-cell dysfunction caused by prolonged hyperlipidemia (25).
The mechanism by which saturated FAs cause ER stress is, thus, a key question but remains controversial. One hypothesis moots a disruption in the efficiency of protein folding because of down-regulation of the calcium pump SERCA2 and depletion of lumenal ER Ca2+ (10). But this depletion has not been universally observed and correlates poorly with the effectiveness of different FAs to trigger ER stress (12, 26). Moreover, when assessed directly, palmitate did not appear to promote misfolding of a reporter protein (27). An alternative, initially proposed by us (27) and now confirmed independently (28, 29), postulates that palmitate slows protein trafficking out of the ER, which would, therefore, enhance ER stress because of lumenal protein overload. Our work further linked this trafficking defect to alterations in SL metabolism, although both the exact metabolite and the underlying mechanism remained obscure (30).
In this study, by extensively characterizing SL modifications under various interventions in both pancreatic islets and whole cell lysates and subcellular fractions of MIN6 β-cells, we define localized reductions in sphingomyelin (SM) in the ER as key determinants of lipotoxic ER stress. We propose that the loss of SM disrupts ER lipid rafts that are essential for the correct packaging of secretory cargo into export vesicles and that this contributes to defective protein trafficking, ER stress, and apoptosis.
EXPERIMENTAL PROCEDURES
Reagents
All tissue culture media, supplements, and trypsin for MIN6 cells and islets were purchased from Invitrogen. The cell death ELISAPLUS kit, SYBR Green I, liberase, and protease inhibitor tablets were obtained from Roche Diagnostics. Sodium palmitate, sodium orthovanadate, fatty acid-free fraction V BSA, sucrose, sodium oleate, sphingolipid standards for TLC, high-performance TLC plates (catalog no. z22718-25EA), solid iodine, GW4869, z-nitraphenyl-β-D-galactopyranoside, and hexyl-β-D-glucopyranoside were from Sigma-Aldrich. The Dual-Luciferase reporter assay kit (catalog no. E1910) and Coomassie Plus protein assay reagent were from Promega (Alexandria, Australia). TLC plates (catalog no. 1.11798.0001), Nanojuice transfection reagents (catalog nos. 71900-3 Core and 719001-3 Boost) were from Merck. Ultima Gold scintillation fluid and EN3HANCE spray were from PerkinElmer Life Sciences. [3H]Sphinganine was from American Radiolabeled Chemicals (St. Louis, MO). The GCS construct (in pCMWSport6, clone no. BC050828.1) and SMS1 construct (in pCMWSport6, clone no. BC019443) were from the ATCC. The Smpd3 construct (in pCMWSport6, clone no. BC046980) and Smpd4 construct (in pCMWSport6, clone no. BC026767) were from Thermo Scientific (Scoresby, Australia). The pEGFP-C1 plasmid (catalog no. 6084-1) was from Clontech (Mountain View, CA). NBD-Cer (catalog no. N2261), precast NuPAGE gels, sample buffer, reducing agent, antioxidant, and the electrophoresis tank and transfer system for immunoblotting were from Invitrogen. The rat insulin radioimmunoassay kit (catalog no. RI-13K) was from Millipore (Kilsyth, Australia). The BCA protein assay kit was from Pierce. The black 96-well assay plates were from Nunc (Scoresby, Australia). The cell disruption bomb and nitrogen connector were from Preiser Scientific (St. Albans, WV).
Islet Isolation and Culture
Pancreatic islets from male 12- to 16-week-old C57Bl6 mice were isolated as described previously (31) by ductal perfusion of collagenase, followed by purification on Ficoll-Paque gradients (GE Healthcare). Islets were cultured for 48 h in RPMI 1640 medium (Rosewell Park Memorial Institute, 11 mm glucose) supplemented with BSA or palmitate/BSA (see below). For Erlin-GFP studies, islets were dispersed with trypsin and attached to poly-L-lysine-treated glass coverslips (32) and then cultured as above for whole islets after transfection.
Cell Culture and Chronic Cell Treatments
The mouse MIN6 insulinoma cell line was routinely passaged and cultured as described previously (9, 30, 33). In brief, chronic palmitate or oleate treatment involved culture for 48 h in DMEM (6 mm glucose) with either 0.4 mm lipid precoupled to 0.92 g/100 ml BSA or BSA-only controls. The lipid couplings were prepared at a 3:1 palmitate:BSA molar ratio (33). Cells for immunoblotting experiments were seeded in 6-well plates at 8 × 105 cells/well in 3 ml of growth medium. Apoptosis assays were performed in either a 96-well-plate or 24-well-plate format at 3 × 104 cells/well (0.2 ml) or 2 × 105 cells/well (0.5 ml), respectively. Cells for metabolic flux assays were also plated in 24-well plates at the above density. Cells for confocal imaging of ER rafts were plated in 6-well plates preloaded with two coverslips at 6 × 105 cells/well in 3 ml of growth medium. MIN6 cells were seeded in 15-cm2 tissue culture dishes at 1.6 × 107 cells/dish in 23 ml of growth medium for lipid profile studies and in 10-cm2 tissue culture dishes at 8 × 106 cells/dish in 15 ml of medium for lipid profile studies involving GCS overexpression. For overexpression studies, MIN6 cells were transfected with a control pEGFP-N1 construct (Clontech, 6085-1) or GCS, Erlin1-GFP, SMS1, Smpd3, or Smpd4 constructs with Nanojuice transfection reagents according to the instructions of the manufacturer, 16–24 h following plating in test dishes, as indicated above. Other chronic (48-h) treatments in DMEM (6 mm glucose) included the inhibitor GW4869.
Apoptosis Assay
MIN6 cells transfected with SMS1, Smpd3/4, or EGFP controls and then treated chronically (48 h) with palmitate, oleate ± SMase inhibitor, or GW4869 were harvested in lysis buffer provided in the cell death ELISAPLUS kit. Levels of histone-bound mono- and oligonucleosomes (DNA fragmentation) were quantified according to the kit instructions and corrected for total DNA as described previously (23, 30).
MIN6 Cell Subcellular Fractionation
One 15-cm2 tissue culture dish of MIN6 cells/condition (or 1 × 10 cm2 dish for GCS overexpression experiments) was lysed for subcellular fractionation following chronic palmitate or control treatment. After washes on ice with cold PBS, the cells were scraped in PBS and spun at 500 × g in a swinging bucket rotor at 4 °C for 10 min to pellet cells. Cells were resuspended in 1.3 ml of HES buffer (20 mm HEPES, 1 mm EDTA, 3g/100 ml sucrose, and protease inhibitors) and lysed gently using nitrogen cavitation (450 psi, 15 min). The lysate was layered on top of a discontinuous sucrose gradient comprised of 1.3-ml layers from 0.25 mm to 2 m in 0.25-mm steps. A gradient pouring machine (Auto Densi Flow, Labconco, Kansas City, MO) was utilized. The sucrose gradient was spun for 18 h in a swinging bucket rotor at 200,000 × g at 4 °C. Fractions (0.65 ml) were taken from the top, and each fraction was characterized for its organelle content via immunoblotting (the ER, Golgi, mitochondria, and plasma membrane were detected using antibodies for calnexin, Golgi matrix protein 130, COX-1 (BD Biosciences), and syntaxin 4). Insulin granules were detected via insulin RIA (Millipore), and lysosomes were detected by a β-galactosidase enzyme activity assay (34).
The mitochondrial sample was prepared separately as a pure, mitochondria-only fraction via lysis of MIN6 cells by passing them through a 27-gauge needle 10 times in isolation buffer (230 mm sucrose, 0.5 mm EGTA, and 5 mm HEPES). After centrifugation (18,000 × g, 25 min), the pellet was resuspended in 20 g/100 ml sucrose solution (10 mm Tris and 0.05 mm EDTA). This was then centrifuged (18,000 × g, 30 min), and the pellet was resuspended in 2 ml of 60 g/100 ml sucrose solution. This was loaded at the bottom of a thin-walled ultracentrifuge tube, layered with 3 ml of 53 g/100 ml sucrose and 7 ml of 44 g/100 ml sucrose and centrifuged at 141,000 × g for 2 h. The mitochondria at the interface of the 44 g/100 ml and 53 g/100 ml sucrose cushions were removed carefully, diluted into 5 ml of isolation buffer, and finally centrifuged at 18,000 × g for 30 min before the pellet was resuspended in the desired buffer for MS or immunoblotting analysis.
Lipid Profiling with MS
Following chronic palmitate treatment, lipids were extracted from MIN6 whole cell homogenates (1 × 15-cm2 dish/condition) or individual sucrose fractions using chloroform:methanol (2:1, v/v). Sucrose fractions from MIN6 cells pretransfected with GCS or GFP control construct and treated with palmitate (1 × 10-cm2 dish/condition) were extracted using chloroform:methanol (1:2, v/v) to increase yield. All samples were separated via liquid chromatography and spiked with internal sphingo-, phospho-, and neutral lipid standards before being analyzed by electrospray ionization-tandem MS as described previously (30). Lipid concentrations were calculated by relating the peak area of each species to the peak area of the corresponding internal standard and then correcting for cell content via BCA protein assay (Pierce) of a portion of the cell homogenate prior to lipid extraction. MIN6 cell samples with GCS overexpression were corrected internally by expressing the lipid concentration (pmol) as a percentage of total lipids measured to maximize yield for LC-MS/MS analysis.
Metabolic Flux Assays
MIN6 cells pretransfected with GCS or EGFP control constructs were assayed for GCS and SMS enzyme activity by acute labeling with fluorescent NBD-Cer substrate, as described previously (30). High-performance TLC plates of separated lipid extracts were imaged on an UV light box within a Bio-Rad ChemiDoc XRS imager, and GluCer and SM bands were quantified with ImageJ software.
ER Raft Staining
Following Erlin1-GFP transfection ± cotransfection with GCS or SMS1 constructs, MIN6 cells or islet cell monolayers were treated chronically with palmitate. Glass coverslips were washed, fixed, permeabilized, and stained for ER with a mouse anti-KDEL antibody (catalog no. ADI-SPA-827-D, Enzo Life Sciences, Farmingdale, NY) to be visualized by an anti-mouse Alexa Fluor 647-conjugated secondary antibody (Invitrogen) on a Leica SP2 confocal microscope using a ×100 oil objective. All transfected cells observed were imaged, and two to five images were taken per treatment condition per experiment, which contained one to three transfected cells per image. The Erlin1-GFP/KDEL costaining of every imaged cell was then analyzed utilizing the intensity correlation analysis plugin within MBF_Image J software (16). Visual representation of the intensity of positive red and green costained pixels was generated on a costaining heatmap by intensity correlation analysis plugin plotting areas of low-to-high (blue-to-white) intensity. The intensity correlation analysis of costaining intensity generated the intensity correlation quotient (ICQ), which measured the degree of covariance within the red (ER) and green (ER lipid raft) channels as a measure of codependent (0 < ICQ ≤ + 0.5) or segregated (0 > ICQ ≥ − 0.5) staining. The total lipid raft area was measured using the analyze particles function (ImageJ) of Erlin-GFP images that were calibrated to their micron scale bars, and the threshold was adjusted to reveal peak lipid raft-stained areas only. ICQ values and images are representative of mean ICQ values or raft area measurements from three to six independent experiments.
ER Vesicle Budding Assay
A microsomal (chiefly ER) fraction was isolated from MIN6 cells pretreated chronically with palmitate via an adaptation of the methodology utilized by Nohturfft et al. (35). Briefly, MIN6 cells from duplicate 15-cm2 dishes were washed with ice-cold PBS, centrifuged at 500 × g for 10 min at 4 °C, resuspended in PBS, centrifuged again at 500 × g for 10 min at 4 °C. Then, membrane pellets were snap-frozen in liquid nitrogen. Pellets were thawed at 37 °C, resuspended in 0.4 ml of buffer F (10 mm HEPES-KOH (pH 7.2), 250 mm sorbitol, 10 mm KOAc, 1.5 mm Mg(OAc)2, and protease inhibitors), and then passed through a 22-gauge needle 20 times. Suspensions were centrifuged at 1000 × g for 5 min at 4 °C, transferred to siliconized (low-retention) microcentrifuge tubes, and centrifuged at 16,000 × g for 3 min at 4 °C. Each pellet was resuspended in 0.5 ml of buffer E (50 mm HEPES-KOH (pH 7.2), 250 mm sorbitol, 70 mm KOAc, 5 mm potassium EGTA, 2.5 mm Mg(OAc)2, and protease inhibitors) and centrifuged again at 16,000 × g for 3 min at 4 °C. Each pellet was then resuspended in 60–100 μl of buffer E to obtain microsomes for use in the in vitro vesicle formation assay, which was adapted from procedures described by the laboratories of Schekman (36) and Balch (37). The protein content of microsomes was determined with a 5-μl sample added to 5 μl of a solution of 20% (w/v) of hexyl-β-D-glucopyranoside and assayed with Coomassie Plus protein assay reagent (Pierce), according to the instructions of the manufacturer, using BSA as a standard. These isolated microsomal membranes were incubated at 37 °C for 15 min with bulk cytosol extracted from mouse liver and ATP/GTP (35). To begin the in vitro vesicle formation assay, 1.5 mm ATP, 0.5 mm GTP, 10 mm creatine phosphate, 4 units/ml of creatine kinase, and 600 μg of liver cytosol (isolated according to Ref. 35) were added to 30–80 μg of protein of the MIN6 microsome preparations to a final volume of 80 μl. Vesicles budding during this reaction were separated from remaining membranes by differential centrifugation to obtain a membrane pellet (16,000 × g for 3 min at 4 °C) or vesicle pellet (supernatants from the previous spin at 131,558 × g in a Beckman TLA100 rotor for 3 min at 4 °C). Vesicle and membrane fractions were resuspended in the appropriate amounts of NuPAGE sample buffer + reducing agent, heated at 70 °C for 10 min prior to SDS-PAGE separation of proteins, and immunoblotting with carboxypeptidase E (packaged into ER vesicles, catalog no. 610758, BD Biosciences) and Grp78/BiP (excluded from ER vesicles, catalog no. ADI-SPA-827-D, Enzo Life Sciences) antibodies.
Dual-Luciferase Assay
Following cotransfection of the pGL-TK 6xSRE-luciferase and pRL-TK Renilla reporter constructs ± the GCS construct into MIN6 cells, the degree of sterol response element (SRE)-driven gene transcription following chronic palmitate treatment was quantified using the Dual-Luciferase reporter assay (Promega). The quantification of 6xSRE-luciferase or Renilla luminescence was performed by a Fluostar Omega plate reader (BMG Labtech). The 6xSRE-luciferase luminescence of each sample was corrected by the Renilla luminescence of the sample, which utilizes a different substrate, to correct for transfection efficiency and the nonspecific effects of CMV promoter (GCS construct) cross-regulation of the expression of the thymidine kinase promoter-led luciferase construct. Data were then expressed as fold control 6xSRE-luciferase activity (luminescence).
Immunoblotting
Protein lysates were prepared, and immunoblotting was performed as described previously (11, 30). Antibodies used include GADD 153 (CHOP, Santa Cruz Biotechnology), β-actin (Sigma-Aldrich), phospho-PERK (Cell Signaling Technology), and cleaved caspase 3 (Cell Signaling Technology). Quantification of immunoblot films was performed using ImageJ.
RNA Analysis
After extraction of total RNA using an RNeasy mini kit (Qiagen, Doncaster, Australia), cDNA was generated with the QuantiTect reverse transcription kit (Qiagen, Victoria, Australia). Real-time PCR was performed using Power SYBR Green PCR master mix (Applied Biosystems, Foster City, CA) on a 7900HT real-time PCR system (Applied Biosystems). Primer sequences are as provided (23, 38), with the addition of those for thioredoxin-interacting protein (TXNIP) cggctttcgtttttcttgaacc (forward) and tgacggctttgactcgggtaac (reverse). The value obtained for each specific product was normalized to a control gene (cyclophilin A) and expressed as a fold change of the value in control extracts.
Statistical Analysis
All results are presented as means ± S.E. of experimental means. Data sets were subject to paired or unpaired Student's t tests as suited the experimental design for individual data set comparisons. Analysis of variance analysis with Tukey's or Bonferroni's multiple comparison post-tests were employed for experiments with multifactorial design. The non-parametric Kruskal-Wallis test with Dunn's multiple comparisons was employed for confocal image analysis. All statistical tests were performed at a 95% confidence interval with Prism 6 software or Microsoft Excel.
RESULTS
Comparative Lipidomic Analysis of MIN6 Cells and Pancreatic Islets of Langerhans Cultured Chronically in the Presence of Palmitate
By lipidomic profiling of MIN6 cells chronically exposed to palmitate, we had previously identified SL, but not phospholipids or neutral lipids, as mediators of β-cell lipotoxicity (30). We therefore embarked on a more extensive investigation of SL metabolism in this model and also extended our analysis to primary mouse islets. Total Cer content was neither significantly affected by palmitate exposure in whole islets (Fig. 1B, 1.44 ± 0.18 compared with 1.24 ± 0.12 nmol/mg protein control treated, p = 0.126) nor in whole MIN6 cells (A, 5.66 ± 0.42 compared with 4.87 ± 0.67 nmol/mg protein control treated, p = 0.249), consistent with our earlier study (30). We did, however, observe small increases in Cer C18:0, C20:0, and C22:0 species within both palmitate-treated MIN6 cells and islets (Fig. 1, A and B). No increases in islet glycosylceramide (GluCer) content were observed (Fig. 1D), unlike in MIN6 cells (C and Ref. 30). Palmitate induced little change in overall SM species in either MIN6 cells or islets. However, total very long chain unsaturated SM species (C25:1 and C26:1 together) were decreased significantly in both MIN6 cells (Fig. 1E, to 112.7 ± 11.2 from 209.3 ± 31.6 pmol/mg protein, p = 0.015) and islets (F, to 38.3 ± 4.2 from 61.5 ± 5.1 pmol/mg protein, p = 0.0045) with palmitate treatment. Previous studies of INS-1 β-cells showed an increased content of the de novo Cer biosynthetic product dihydroceramide after 24 h of palmitate exposure (28). However, we did not observe this in either MIN6 cells (Fig. 1G) or islets (H) following 48-h incubation. These data suggest that either the increase in C18:0, C20:0, or C22:0 Cer species or, possibly, the decrease in very long chain unsaturated SM species may contribute to palmitate-induced ER stress (30).
FIGURE 1.

Chronic palmitate treatment (48 h) induces similar sphingolipid species changes within MIN6 cells and islets of Langerhans. MIN6 cells (left panel) or WT islets ex vivo (right panel) were cultured with 0.4 mm palmitate complexed to BSA (0.92%) for 48 h before total lysates were prepared for quantification of all major sphingolipid species via mass spectrometry. Each species was corrected for total protein content of lysates. Data represent mean ceramide (A and B), glycosylceramide (C and D), sphingomyelin (E and F), and dihydroceramide (G and H) species from six (MIN6) or four (islet) independent experiments + S.E. *, p < 0.05; **, p < 0.01, ***, p < 0.001; paired Student's t tests compared with control condition.
Palmitate Exposure Diminishes the Budding of Secretory Vesicles from the ER and Causes Ceramide Accumulation in the ER of MIN6 Cells
β-Cell lipotoxicity is associated with a delay in ER-to-Golgi vesicular trafficking (27, 28), which contributes to the induction of ER stress (27). Therefore, we postulated that the palmitate may alter ER membrane lipid species that may affect membrane curvature and, therefore, the biophysics of vesicle budding from the ER membrane, which may contribute to this delay. Consistent with this notion, palmitate pretreatment inhibited the rate of vesicles budding from microsomes over 15 min, as detected using the endogenous cargo protein carboxypeptidase E (CPE) (Fig. 2A). We next addressed whether there were localized changes in subcellular SL content that may cause subcellular membrane disturbances, contributing to the defective trafficking that could initiate ER stress and toxicity (39). To investigate the accumulation of both Cer and GluCer more closely, we undertook subcellular fractionation of MIN6 cells (Fig. 2B) and measured these metabolites in peak fractions corresponding to Golgi, plasma membrane, lysosomes, and ER. MIN6 mitochondria were isolated in parallel using a separate sucrose gradient technique (see “Experimental Procedures”) and checked for purity by immunoblotting for subcellular protein markers (data not shown). In response to palmitate pretreatment, Cer increased mainly in the ER (Fig. 2C) and also the lysosome, whereas accumulation of GluCer was more widespread (D). The particular species of Cer accruing at the ER included C18:0 to C22:0 species (Fig. 2E), consistent with previous whole cell lysate data (Fig. 1A). All major species of GluCer detected accumulated at the ER (Fig. 2F).
FIGURE 2.

Subcellular changes in MIN6 cells because of palmitate treatment (48 h) include inhibition of ER vesicle budding and enhanced ceramide accrual. A, microsomes (chiefly ER) were prepared from MIN6 cells pretreated with 0.4 mm palmitate/0.92% BSA. Isolated microsomes underwent a budding assay (see “Experimental Procedures”) to probe the rate of carboxypeptidase E (CPE) incorporation into ER vesicle buds over 15 min. Grp78/BiP (excluded from buds) was used as a negative control. Results are presented as representative blots (each condition in duplicate) from a total of three independent experiments (i) or as densitometry relative to 0-min control (n = 3) (ii). B, MIN6 cells were pretreated with 0.4 mm palmitate/0.92% BSA (48 h) before lysis and fractionation. The position of subcellular peaks within the sucrose gradient corresponding to the ER, Golgi, plasma membrane (PM), lysosome (Lyso), and insulin granules as characterized by organelle markers (see “Experimental Procedures”). C, mass spectrometry of total ceramide and glycosylceramide (D) content from peak fractions corresponding to each compartment and expressed as total lipid per milliliter of sucrose fraction extract. Peak fractions are as follows: ER, 13–18; Golgi, 5–8; lysosome, 11–12; and plasma membrane, 9–10. Mitochondria (Mito) were isolated separately (see “Experimental Procedures”). Cont, control; Palm, palmitate. The specific ER ceramide (E) and glycosylceramide (F) species accumulating in response to palmitate are detailed. Data represent mean ± S.E. from three independent experiments. *, p < 0.05, unpaired Student's t test compared with control condition.
The Protective Effects of GCS Overexpression Do Not Appear to Be due to Alterations in Phospholipid Saturation
We then studied the effect of palmitate treatment on the ER content of phospholipids, whose loosely packed, flexible, unsaturated acyl chains facilitate insertion and folding of proteins in the lipid bilayer and efficient budding of vesicles from ER exit sites (40). Moreover, Fu et al. (67) recently reported an increase in the phosphatidylcholine (PC)/phosphatidylethanolamine (PE) ratio within the ER of liver from obese mice, which they implicated in the induction of lipotoxic ER stress. Although we observed a similar trend in ER fractions of MIN6 cells treated chronically with palmitate (Fig. 3A), this was not statistically significant. More importantly, but not surprisingly, the PC/PE ratio was unaltered by overexpression of the ceramide-catabolizing enzyme GCS, which we demonstrated previously to reverse the trafficking defect in our model (30). We also investigated the degree of PC saturation. Fractions from ER (and lysosomes) were enriched in unsaturated PC species compared with the plasma membrane and Golgi (not shown), and this was partially reversed by palmitate pretreatment (Fig. 3B). However, GCS overexpression did not alter PC saturation in the ER (or lysosome, not shown) regardless of the pretreatment conditions, suggesting that this effect of palmitate does not contribute to lipotoxic ER stress and defective protein trafficking in pancreatic β-cells.
FIGURE 3.
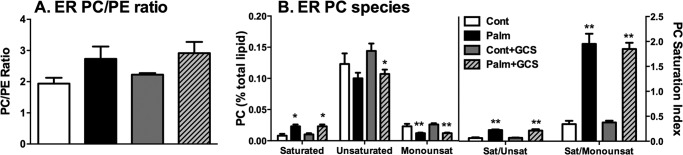
MIN6 cell ER PC species and saturation. A, MIN6 cells were pretransfected ± the GCS construct and then treated chronically (48 h) with 0.4 mm palmitate (Palm)/0.92% BSA and fractionated. Then, peak ER fractions were quantified via mass spectrometry. The ER PC/PE ratio represents the ratio of mean PC to PE lipid (percent of total lipid content) + S.E. from three independent experiments. B, saturated (Sat), unsaturated (Unsat) (≥ 1 double bond), and monounsaturated (Monounsat) (= 1 double bond) ER PC species (left panel) and those species represented as ratios (right panel). Data represents mean lipid (percent of total lipid content) + S.E. from three independent experiments. *, p < 0.05; **, p < 0.01; unpaired Student's t test compared with control. Con, control.
Reductions in the Sphingomyelin and Cholesterol Content of the ER because of Palmitate Pretreatment Are Partially Reversed by GCS Overexpression
Alterations in ceramide have also been implicated in regulating the entry of cargo protein into the secretory pathway (41–43). To address this, we reinvestigated ER ceramide accumulation to test the effects of GCS overexpression. Here we again observed a significant increase in C18:0, C20:0, and C22:0 species with palmitate treatment (Fig. 4A), although this was not reversed with GCS overexpression, as would be expected if these increases were mechanistically important in the protein trafficking defect (30).
FIGURE 4.

ER sphingolipid profiling implicates altered SM and FC content in the protection afforded by GCS overexpression. MIN6 cells were pretreated chronically (48 h) with 0.4 mm palmitate (Palm)/0.92% BSA and fractionated, and then peak ER fractions were quantified via mass spectrometry. A, ceramide species from the ER fraction ± GCS overexpression. *, p < 0.05; **, p < 0.01; paired Student's t test compared with control. Cont, control. B, total SM and FC content of the ER ± GCS overexpression. ** and #, p < 0.05; paired Student's t test compared with control (*) or palmitate (#) GCS conditions. Data in A and B represent mean lipid (percent of total lipid content) + S.E. from three independent experiments. C, fold 6xSRE-luciferase activity (luminescence) from MIN6 cells cotransfected with 6xSRE-tagged firefly luciferase reporter construct and Renilla luciferase (to correct for transfection efficiency) ± GCS construct prior to lipid treatment as a measure of 6xSRE-driven cholesterol gene expression. Data represent mean fold + S.E. from four independent experiments. D, GCS and SMS1 activity was measured via the acute (1-h) conversion of fluorescent NBD-labeled Cer to GluCer and SM in MIN6 cells ± GCS overexpression. #, p < 0.05; paired Student's t test, construct (GCS) effect versus GFP over both control and palmitate conditions.
The decrease in whole cell SM content, seen previously in both MIN6 cells and whole islets (Fig. 1), was most apparent at the ER and partially reversed by GCS overexpression (Fig. 4B). Further inspection of the ER lipidome revealed that free cholesterol (FC) was also modulated in a very similar manner (Fig. 4B). As an independent confirmation of these lipidomic studies, we also employed an SRE reporter assay to quantify the activation of sterol regulatory element-binding protein (SREBP), which is predominantly regulated by FC levels in the ER. Consistent with depletion of cholesterol in this compartment, palmitate pretreatment triggered enhanced SRE-driven gene expression, which was reversed by GCS overexpression (Fig. 4C). The apparently close coordination of ER SM and FC is to be expected, given the integrated regulation of these metabolites (44). However, it is more surprising that an enzyme-catalyzing conversion of ceramide to GluCer would also augment SM. Nevertheless, this was confirmed by assaying the catabolism of NBD-ceramide in MIN6 cells (Fig. 4D). This fluorescent ceramide analog targets the Golgi directly, bypassing the requirement of endogenous ceramide transport from the ER, and is, therefore, directly converted to SM or GluCer by the Golgi enzymes SMS1 or GCS, respectively. Thus, GCS overexpression increased conversion of NBD-ceramide to GluCer but also to SM, suggesting an unexpected coregulation of SMS1 and GCS in this system. Because palmitate failed to diminish conversion of NBD-ceramide to SM under control conditions (Fig. 4D), our results also suggest that inhibition of SMS1 by palmitate does not contribute to the loss of SM mass observed under these conditions (Fig. 4B), perhaps suggesting a role for enhanced sphingomyelinase (SMase) activation.
Palmitate Promotes Aggregation of ER Lipid Rafts Consistent with a Perturbation in ER Sphingomyelin and Cholesterol Content
Because SM and cholesterol physically associate in lipid microdomains (rafts), we reasoned that perturbation of these rafts at the ER via the effects of palmitate on SM and cholesterol might contribute to lipotoxic ER stress and defective vesicular trafficking. The function of lipid rafts at the ER, as opposed to the plasma membrane, is a relatively new area of cell biology (45) and has not been addressed previously in β-cells. To visualize these, we transfected the GFP-tagged version of the ER raft protein Erlin1 (45) into MIN6 cells. We firstly confirmed that the Erlin1-GFP construct did localize to the ER, as assessed by staining with a KDEL antibody (a marker of the ER-resident proteins Grp94 and Bip). Under control conditions, Erlin1 was distributed homogenously throughout the ER (Fig. 5A). Following palmitate treatment, Erlin1-GFP clearly aggregated within certain areas of the ER, and this aggregation was quantified using the ICQ (ImageJ intensity correlation analysis plugin (16)), which measured the degree of covariance within the red (ER) and green (ER lipid raft) channels as a measure of codependent (0 < ICQ ≤ + 0.5) or segregated (0 > ICQ ≥ − 0.5) staining. As shown in the representative images (Fig. 5), palmitate increases the codependent staining in MIN6 cells from ICQ 0.145 (control) to 0.215 (palmitate), indicating a significant aggregation of Erlin1 within the ER. Similar effects were seen in transfected monolayers derived from primary islets (Fig. 5A, lower panels). We next employed an overexpression strategy to determine the effects of repleting SM either directly with SMS1 (responsible for de novo SM synthesis) or indirectly with GCS. Both interventions reversed the palmitate-treated Erlin1 staining pattern to one more closely resembling control conditions, with the Erlin1 once again more evenly distributed throughout the ER (SMS1, ICQ 0.100; GCS, ICQ 0.118). Moreover, when a heatmap of only the positively colocalized pixel pairs from the ICQ analysis was generated, these areas of intense aggregation with palmitate and reversal with SMS1 and GCS are visualized more clearly (Fig. 5, right panels). This palmitate-induced aggregation caused an increase in total raft area within the cell from 3.83 ± 0.64 μm2 (control, Fig. 5B) to 20.6 ± 3.06 μm2 (palmitate). Again, both GCS and SMS overexpression significantly reversed this effect, generating rafts of a comparable size to control conditions (GCS, 5.87 ± 1.21 μm2; SMS, 5.3 ± 0.69 μm2). This argues in favor of the SM content of the ER being responsible for the changes in ER raft distribution seen during palmitate exposure.
FIGURE 5.

Palmitate-induced alterations in ER sphingomyelin and cholesterol are implicated in the disruption of ER lipid raft distribution. A, MIN6 cells or primary islet cell monolayers were transfected with the ER raft marker Erlin1-GFP (green) prior to 0.4 mm palmitate (Palm)/0.92% BSA treatment (48 h). Fixed cells were stained with anti-KDEL antibody (a marker of the ER-resident proteins Grp94 and Bip) and Cy3 secondary (red) to verify ER localization of Erlin-1. The visual representation of the intensity of positive red and green costaining areas was generated on a costaining heatmap by intensity correlation analysis plugin (16) (Image J), plotting areas of low-to-high (blue-to-white) intensity. Scale bar = 10 μm. B, total mean lipid raft area ± S.E. (μm2) of MIN6 Erlin-GFP images. ***, p < 0.001 versus control; ##, p < 0.01; ###, p < 0.001 versus palmitate; Kruskal-Wallis test with Dunn's multiple comparisons. Images are representative of mean ICQ and raft area values from three to six independent experiments (Cont, Palm, +GCS, and +SMS conditions).
SM Modulation Impacts Lipotoxic ER Stress and Apoptosis
We next investigated whether alterations in SM metabolism contribute to apoptosis and ER stress as well as ER lipid raft formation. MIN6 cell apoptosis (DNA fragmentation) because of palmitate was diminished by overexpression of SMS1 and enhanced by the ER-resident neutral SMase Smpd4 (Fig. 6A). Transfection of Smpd3, a plasma membrane-localized neutral SMase, did not significantly affect the apoptotic response in any condition. These data implicate the depletion of a pool of ER-localized SMs in triggering lipoapoptosis, perhaps via activation of an SMase. To investigate this more extensively, we treated MIN6 cells with the specific neutral SMase inhibitor GW4869 (46). This resulted in a significant and dose-dependent decrease in both palmitate-induced apoptosis (DNA fragmentation) (Fig. 6B) and CHOP expression (C). Although the latter is commonly used a marker of terminal ER stress, we also analyzed mRNA expression of a broader cohort of ER stress genes (Fig. 7). This revealed that not all arms of the UPR that are triggered by lipotoxicity are reversed by inhibition of SMase. Induction of quality control proteins such as Fkbp11, Dnajb9, and Herpud1 was not sensitive to GW4869, nor was TXNIP, a terminal effector of the IRE1 pathway (47). In contrast, inhibition of SMase impacted strongly on ATF4 and its target genes ATF3 and Trib3 (48) and, to a lesser extent, on Grp94 and Sel1l, thought to be downstream of ATF6 (49). ATF6 itself, as well as CHOP, showed trends toward reversal with the inhibitor, although induction of these genes with palmitate did not quite reach statistical significance (Fig. 7).
FIGURE 6.

SM content and SMase activity play a significant role in palmitate-induced ER stress and apoptosis. A, MIN6 cells were transfected with GFP, SMS1, Smpd3, or Smpd4 constructs prior to 48-h palmitate (Palm) or oleate (0.4 mm/0.92% BSA) treatment and then quantified for the level of apoptosis. #, p < 0.05; ###, p < 0.001; two-way analysis of variance with Bonferroni's multiple comparisons of Smpd4 to SMS1 where indicated; *, p < 0.05; ***, p < 0.001; unpaired Student's t tests compared with GFP control unless indicated. Data represent mean apoptosis (fold control) + S.E. from three to four independent experiments. Cont, control. B, MIN6 cells were cultured with 0.4 mm palmitate complexed to BSA (0.92%) ± SMase inhibitor, GW4869, for 48 h before total lysates were prepared for the quantification of apoptosis (DNA fragmentation via cell death ELISA, Roche) or CHOP induction (immunoblotting) (C). *, p < 0.05; **, p < 0.01; ***, p < 0.001; two-way analysis of variance with Bonferroni's multiple comparisons to control (0 μm) or where indicated.
FIGURE 7.

Inhibition of SMase activity differentially regulates UPR gene expression. MIN6 cells were cultured for 48 h with 0.4 mm palmitate complexed to BSA (0.92%) ± SMase inhibitor, GW4869 (5 μm). mRNA was isolated, converted to cDNA, and analyzed by real-time PCR. *, p < 0.05; **, p < 0.01 for effect of inhibitor versus control or palmitate alone group or as indicated using one-way analysis of variance with Tukey's multiple comparisons. Data are mean ± S.E. from three to four independent experiments.
DISCUSSION
There is growing evidence of a role for ER stress as a mediator of β-cell apoptosis in T2D, arising from both animal models and studies with humans. Saturated FAs, either through oversupply or inappropriate metabolism in the β-cell, appear to be the primary triggers, although the molecular mechanisms remain poorly understood. This study provides several important and unexpected advances in this context.
Our most novel finding was the appreciation that palmitate pretreatment alters ER lipid raft composition and distribution. This was shown in a number of ways. First, although comprehensive screening of different subcellular fractions revealed that palmitate altered several features of the ER lipidome, including the PC/PE ratio and PC saturation, the only changes that were counterregulated by GCS overexpression were the observed reductions in SM and FC (Fig. 4B). This overexpression strategy has been shown previously to ameliorate lipotoxic ER stress, apoptosis, and defective protein trafficking (30) and, thus, supports a causative role for the reduction in ER SM/FC in β-cell failure. Moreover, the metabolism of these two lipids is tightly coregulated, which reinforces the significance of their coordinated changes during treatment with palmitate and/or GCS overexpression. Second, we independently confirmed loss of FC in the ER by taking advantage of its key role in regulating SREBP activation. Consistent with a loss of ER FC because of palmitate, we observed enhanced activation of an SRE reporter following palmitate treatment (Fig. 4C). Conversely, overexpression of GCS (and reversal of ER FC depletion) led to diminished SRE reporter activation. Third, we visualized Erlin1-GFP as a specific marker of ER lipid rafts (45) to demonstrate that these were also reciprocally modulated by palmitate and GCS overexpression (Fig. 5). This is also consistent with SM and FC being the key structural components of lipid rafts. Although cholesterol overload inhibits protein trafficking and causes ER stress in macrophages (50, 51), a separate body of work demonstrates that cholesterol depletion also specifically blocks cargo export from the ER via disruption of ER exit sites (52, 53).
Lipid rafts are abundant in the plasma membrane, where their function has been studied extensively, but more recent research has also identified them in the ER. Here they appear to play a role in regulating the entry of certain types of cargo protein into the secretory pathway, consistent with the requirement for cholesterol, as discussed above (45, 54–56). The bending of the membrane at ER exit sites is thought to influence the rate of budding vesicle formation and the rate at which cargo proteins enter the vesicle (40). We now present new evidence that palmitate-induced modulations of ER lipid raft distribution may also impact similar processes in β-cells, consistent with our observation of reduced ER vesicle budding (Fig. 2A). By thus diminishing the vesicular trafficking rate, this would be expected to contribute to ER stress via protein overload (27). It is also possible that ER lipid rafts influence processes other than protein trafficking. Indeed, Erlin-1 itself contributes to the maintenance of ER protein folding capacity and the regulation of ER-associated degradation (45), both of which would impact more directly on the induction of the UPR.
To our knowledge, this study is also the first to demonstrate a primary role for SM in regulating lipid rafts in the ER, although they are disrupted at other cellular sites by activation of SMase, for example. This is potentially explained both by the loss of SM partners for cholesterol and displacement of the sterol from its interaction with saturated phospholipids by ceramide (57). Strong ER stress, however, may in turn further activate SMase and amplify the response. Indeed, Cer generated specifically from a neutral SMase in INS-1 β-cells was promoted by the pharmacological ER stressor thapsigargin via the activation of Ca2+-independent phospholipase A2 (58). Likewise, there have been reports of similar increases in minor species of ceramide within INS-1 cells secondary to ER stress induction by reducing agents and high glucose, perhaps consistent with activation of SMase (59). ER stress can also activate de novo cholesterol synthesis in β-cells following high-glucose treatment and via the induction of SREBP1 (60). Although these secondary events might contribute to overall β-cell failure, particularly under strong ER stress, the lipid remodeling we observed in our milder lipotoxic model occurs upstream and not downstream of ER stress and is causally implicated in its induction (27).
Our data highlight the importance of reductions in SM in the ER with particular reference to ER-to-Golgi protein trafficking. However, SM has also been shown to regulate vesicular trafficking from post-Golgi compartments within insulin-secreting INS-1 cells (61), so palmitate might also impact at this site. In general, however, most prior studies have focused on increases in ceramide rather than loss of SM. Although we believe that the latter is more important for regulating ER stress, we do not rule out other signaling roles of Cer acting in parallel or distal to ER stress, especially under strong lipotoxic conditions, such as in the presence of high glucose to drive partitioning. Cer has been a candidate mediator of FA-induced β-cell apoptosis because of the seminal work of the Unger laboratory (4) using the Zucker diabetic fatty rat model and reduction of apoptosis following treatment with serine palmitoyltransferase inhibitors. Cer formation in response to chronic FA overload has been implicated in β-cell apoptosis in subsequent studies (5–7, 13) via activation of the JNK/SAPK stress signaling pathways (8). Our previous work had suggested that de novo synthesis of Cer at the ER in response to palmitate treatment contributed to caspase-dependent apoptotic signaling and terminal ER stress (30). Here we highlight the specific accrual of Cer in the ER (Fig. 2C), which, when blocked previously with the de novo Cer synthesis inhibitor myriocin, completely prevented caspase 3 cleavage (30). We further report that the C18:0–22:0 Cer species were increased with palmitate treatment (Fig. 1 and 2), confirming prior observations in an INS-1 lipotoxicity model (28). Differences in the time course (24 versus 48 h) and mode of FA exposure (28) to our MIN6 model may explain the differences in dihydroceramide content and the severity of Cer accumulation between our two studies. Most importantly, we observed an excellent concordance between MIN6 cells and islets for alterations in ceramide and potentially very long chain unsaturated SM. The exception was GluCer. GCS activity is known to be important for cancer cell multidrug resistance to chemotherapeutics such as paclitaxel (62). Therefore, a greater accumulation of GluCer may be expected in the transformed MIN6 cell line when compared with primary, normal β-cells in islet tissue.
By now providing a mechanistic framework to explain how palmitate disrupts lipid rafts in β-cells, we further highlight the importance of protein overload, or disruption in ER-to-Golgi protein trafficking, in lipotoxic ER stress. This mechanism has now been demonstrated in three independent laboratories (27–30). An alternative theory postulates a primary role for protein misfolding secondary to depletion of ER lumenal Ca2+ as a result of down-regulation of SERCA2 activity (10, 20). A link to lipid remodeling has been described recently, at least in liver, where lipotoxicity results in an enhanced PC/PE ratio in the ER, which, in turn, impairs SERCA2 and promotes ER stress (67). In β-cells, however, we observed only a small and statistically insignificant increase in this ratio (Fig. 3A) that was not reversed by GCS overexpression, which would argue against a causal involvement in ER stress. Other work has suggested that saturated phospholipids might activate ER stress either directly or indirectly in yeast and fibroblasts (63–65). A similar increase in PL saturation is seen in the ER from lipotoxic β-cells (Fig. 3B), but this was not counteracted by GCS overexpression. Although we do not rule out a modulatory role for this mechanism in mediating ER stress in β-cells, we believe that milder lipotoxicity in a professional secretory cell is more likely to involve disrupted protein trafficking. Interestingly, it has been speculated that ER stress arising in this manner preferentially augments CHOP and ATF4 rather than the other UPR arms that are triggered by accumulation of misfolded protein (49). This is perhaps consistent with our observations that inhibition of neutral SMase also impacted predominately on CHOP and ATF4 (Figs. 6 and 7). Recent work suggests that ATF4 is a key transducer of terminal ER stress in β-cells through up-regulation of protein synthesis and oxidative stress (47, 66). The extent to which this contributes during lipotoxic ER stress and how it interacts with protein trafficking is likely to be complex but will be important to evaluate in future studies.
In conclusion, this study highlights that specific alterations in subcellular SL might contribute to ER stress and apoptosis during lipotoxic exposure of MIN6 β-cells and mouse islets. In particular, we implicate a primary and novel role for palmitate exposure in diminishing ER SM and disrupting lipid rafts in that compartment. Thus, our data thus further support the concept that protein overload as a consequence of defective ER-to-Golgi trafficking contributes to the induction of ER stress under these conditions.
Acknowledgments
We thank S. M. Robbins (University of Calgary, Canada) for the Erlin1-GFP construct, D. James (Garvan Institute, Australia) for the anti-syntaxin-4 antibody, A. Brown (University of New South Wales, Australia) for the 6xSRE-luciferase construct, P. Whitworth (Garvan Institute, Australia) for pancreatic islet isolation expertise, and A. Cooper (Garvan Institute, Australia) for use of the Auto Densi Flow machine.
This work was supported by a National Health and Medical Research Council project grant (to T. J. B.), by research fellowships (to T. J. B. and P. J. M.), and by the Young Garvan Fellowship (to E. B.).
- FA
- fatty acid
- T2D
- type 2 diabetes
- Cer
- ceramide
- CHOP
- C/EBP homology protein
- ER
- endoplasmic reticulum
- SL
- sphingolipid
- UPR
- unfolded protein response
- SM
- sphingomyelin
- GCS
- glucosylceramide synthase
- NBD
- 12-(N-methyl-N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl))
- ICQ
- intensity correlation quotient
- SRE
- sterol response element
- GluCer
- glycosylceramide
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- FC
- free cholesterol
- PERK
- PRKR-like endoplasmic reticulum kinase
- SMase
- sphingomyelinase.
REFERENCES
- 1. Paolisso G., Tataranni P. A., Foley J. E., Bogardus C., Howard B. V., Ravussin E. (1995) A high concentration of fasting plasma non-esterified fatty acids is a risk factor for the development of NIDDM. Diabetologia 38, 1213–1217 [DOI] [PubMed] [Google Scholar]
- 2. Kahn S. E., Hull R. L., Utzschneider K. M. (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846 [DOI] [PubMed] [Google Scholar]
- 3. Weir G. C., Bonner-Weir S. (2004) Five stages of evolving β-cell dysfunction during progression to diabetes. Diabetes 53, S16–21 [DOI] [PubMed] [Google Scholar]
- 4. Shimabukuro M., Higa M., Zhou Y. T., Wang M. Y., Newgard C. B., Unger R. H. (1998) Lipoapoptosis in β-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J. Biol. Chem. 273, 32487–32490 [DOI] [PubMed] [Google Scholar]
- 5. El-Assaad W., Joly E., Barbeau A., Sladek R., Buteau J., Maestre I., Pepin E., Zhao S., Iglesias J., Roche E., Prentki M. (2010) Glucolipotoxicity alters lipid partitioning and causes mitochondrial dysfunction, cholesterol, and ceramide deposition and reactive oxygen species production in INS832/13 ss-cells. Endocrinology 151, 3061–3073 [DOI] [PubMed] [Google Scholar]
- 6. Cnop M., Hannaert J. C., Hoorens A., Eizirik D. L., Pipeleers D. G. (2001) Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes 50, 1771–1777 [DOI] [PubMed] [Google Scholar]
- 7. Lupi R., Dotta F., Marselli L., Del Guerra S., Masini M., Santangelo C., Patané G., Boggi U., Piro S., Anello M., Bergamini E., Mosca F., Di Mario U., Del Prato S., Marchetti P. (2002) Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets. Evidence that β-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 51, 1437–1442 [DOI] [PubMed] [Google Scholar]
- 8. Maedler K., Spinas G. A., Dyntar D., Moritz W., Kaiser N., Donath M. Y. (2001) Distinct effects of saturated and monounsaturated fatty acids on β-cell turnover and function. Diabetes 50, 69–76 [DOI] [PubMed] [Google Scholar]
- 9. Busch A. K., Gurisik E., Cordery D. V., Sudlow M., Denyer G. S., Laybutt D. R., Hughes W. E., Biden T. J. (2005) Increased fatty acid desaturation and enhanced expression of stearoyl coenzyme A desaturase protects pancreatic β-cells from lipoapoptosis. Diabetes 54, 2917–2924 [DOI] [PubMed] [Google Scholar]
- 10. Cunha D. A., Hekerman P., Ladrière L., Bazarra-Castro A., Ortis F., Wakeham M. C., Moore F., Rasschaert J., Cardozo A. K., Bellomo E., Overbergh L., Mathieu C., Lupi R., Hai T., Herchuelz A., Marchetti P., Rutter G. A., Eizirik D. L., Cnop M. (2008) Initiation and execution of lipotoxic ER stress in pancreatic β-cells. J. Cell Sci. 121, 2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Laybutt D. R., Preston A. M., Akerfeldt M. C., Kench J. G., Busch A. K., Biankin A. V., Biden T. J. (2007) Endoplasmic reticulum stress contributes to β cell apoptosis in type 2 diabetes. Diabetologia 50, 752–763 [DOI] [PubMed] [Google Scholar]
- 12. Karaskov E., Scott C., Zhang L., Teodoro T., Ravazzola M., Volchuk A. (2006) Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic β-cell apoptosis. Endocrinology 147, 3398–3407 [DOI] [PubMed] [Google Scholar]
- 13. Maedler K., Oberholzer J., Bucher P., Spinas G. A., Donath M. Y. (2003) Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic β-cell turnover and function. Diabetes 52, 726–733 [DOI] [PubMed] [Google Scholar]
- 14. Pettus B. J., Chalfant C. E., Hannun Y. A. (2002) Ceramide in apoptosis. An overview and current perspectives. Biochim. Biophys. Acta 1585, 114–125 [DOI] [PubMed] [Google Scholar]
- 15. Shimabukuro M., Zhou Y. T., Levi M., Unger R. H. (1998) Fatty acid-induced β cell apoptosis. A link between obesity and diabetes. Proc. Natl. Acad. Sci. U.S.A. 95, 2498–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beeharry N., Chambers J. A., Green I. C. (2004) Fatty acid protection from palmitic acid-induced apoptosis is lost following PI3-kinase inhibition. Apoptosis 9, 599–607 [DOI] [PubMed] [Google Scholar]
- 17. González-Pertusa J. A., Dubé J., Valle S. R., Rosa T. C., Takane K. K., Mellado-Gil J. M., Perdomo G., Vasavada R. C., García-Ocaña A. (2010) Novel proapoptotic effect of hepatocyte growth factor: synergy with palmitate to cause pancreatic β-cell apoptosis. Endocrinology 151, 1487–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thörn K., Bergsten P. (2010) Fatty acid-induced oxidation and triglyceride formation is higher in insulin-producing MIN6 cells exposed to oleate compared to palmitate. J. Cell. Biochem. 111, 497–507 [DOI] [PubMed] [Google Scholar]
- 19. Boslem E., Meikle P. J., Biden T. J. (2012) Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets 4, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kharroubi I., Ladrière L., Cardozo A. K., Dogusan Z., Cnop M., Eizirik D. L. (2004) Free fatty acids and cytokines induce pancreatic β-cell apoptosis by different mechanisms. Role of nuclear factor-κB and endoplasmic reticulum stress. Endocrinology 145, 5087–5096 [DOI] [PubMed] [Google Scholar]
- 21. Back S. H., Kaufman R. J. (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 81, 767–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eizirik D. L., Cardozo A. K., Cnop M. (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 29, 42–61 [DOI] [PubMed] [Google Scholar]
- 23. Akerfeldt M. C., Howes J., Chan J. Y., Stevens V. A., Boubenna N., McGuire H. M., King C., Biden T. J., Laybutt D. R. (2008) Cytokine-induced β-cell death is independent of endoplasmic reticulum stress signaling. Diabetes 57, 3034–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song B., Scheuner D., Ron D., Pennathur S., Kaufman R. J. (2008) Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Invest. 118, 3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xiao C., Giacca A., Lewis G. F. (2011) Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and β-cell dysfunction in humans. Diabetes 60, 918–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gwiazda K. S., Yang T. L., Lin Y., Johnson J. D. (2009) Effects of palmitate on ER and cytosolic Ca2+ homeostasis in β-cells. Am. J. Physiol. Endocrinol. Metab. 296, E690–701 [DOI] [PubMed] [Google Scholar]
- 27. Preston A. M., Gurisik E., Bartley C., Laybutt D. R., Biden T. J. (2009) Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse beta cells by promoting protein overload. Diabetologia, 52, 2369–2373 [DOI] [PubMed] [Google Scholar]
- 28. Véret J., Coant N., Berdyshev E. V., Skobeleva A., Therville N., Bailbé D., Gorshkova I., Natarajan V., Portha B., Le Stunff H. (2011) Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 β-cells. Biochem. J. 438, 177–189 [DOI] [PubMed] [Google Scholar]
- 29. Pétremand J., Puyal J., Chatton J. Y., Duprez J., Allagnat F., Frias M., James R. W., Waeber G., Jonas J. C., Widmann C. (2012) HDLs protect pancreatic β-cells against ER stress by restoring protein folding and trafficking. Diabetes 61, 1100–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boslem E., MacIntosh G., Preston A. M., Bartley C., Busch A. K., Fuller M., Laybutt D. R., Meikle P. J., Biden T. J. (2011) A lipidomic screen of palmitate-treated MIN6 β-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem. J. 435, 267–276 [DOI] [PubMed] [Google Scholar]
- 31. Cantley J., Burchfield J. G., Pearson G. L., Schmitz-Peiffer C., Leitges M., Biden T. J. (2009) Deletion of PKCϵ selectively enhances the amplifying pathways of glucose-stimulated insulin secretion via increased lipolysis in mouse β-cells. Diabetes 58, 1826–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cantley J., Choudhury A. I., Asare-Anane H., Selman C., Lingard S., Heffron H., Herrera P., Persaud S. J., Withers D. J. (2007) Pancreatic deletion of insulin receptor substrate 2 reduces β and α cell mass and impairs glucose homeostasis in mice. Diabetologia 50, 1248–1256 [DOI] [PubMed] [Google Scholar]
- 33. Busch A. K., Cordery D., Denyer G. S., Biden T. J. (2002) Expression profiling of palmitate- and oleate-regulated genes provides novel insights into the effects of chronic lipid exposure on pancreatic β-cell function. Diabetes 51, 977–987 [DOI] [PubMed] [Google Scholar]
- 34. Bonifacino J. S. (2003) Current Protocols in Cell Biology, pp. 3.0.1–3.0.8, John Wiley and Sons, New York [Google Scholar]
- 35. Nohturfft A., Yabe D., Goldstein J. L., Brown M. S., Espenshade P. J. (2000) Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell 102, 315–323 [DOI] [PubMed] [Google Scholar]
- 36. Rexach M. F., Schekman R. W. (1991) Distinct biochemical requirements for the budding, targeting, and fusion of ER-derived transport vesicles. J. Cell Biol. 114, 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rowe T., Aridor M., McCaffery J. M., Plutner H., Nuoffer C., Balch W. E. (1996) COPII vesicles derived from mammalian endoplasmic reticulum microsomes recruit COPI. J. Cell Biol. 135, 895–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chan J. Y., Luzuriaga J., Bensellam M., Biden T. J., Laybutt D. R. (2013) Failure of the adaptive unfolded protein response in islets of obese mice is linked with abnormalities in β-cell gene expression and progression to diabetes. Diabetes 62, 1557–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hannun Y. A., Obeid L. M. (2008) Principles of bioactive lipid signalling. Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 40. Spang A. (2009) On vesicle formation and tethering in the ER-Golgi shuttle. Curr. Opin Cell Biol. 21, 531–536 [DOI] [PubMed] [Google Scholar]
- 41. Giussani P., Maceyka M., Le Stunff H., Mikami A., Lépine S., Wang E., Kelly S., Merrill A. H., Jr., Milstien S., Spiegel S. (2006) Sphingosine-1-phosphate phosphohydrolase regulates endoplasmic reticulum-to-Golgi trafficking of ceramide. Mol. Cell. Biol. 26, 5055–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rosenwald A. G., Machamer C. E., Pagano R. E. (1992) Effects of a sphingolipid synthesis inhibitor on membrane transport through the secretory pathway. Biochemistry 31, 3581–3590 [DOI] [PubMed] [Google Scholar]
- 43. Maceyka M., Machamer C. E. (1997) Ceramide accumulation uncovers a cycling pathway for the cis-Golgi network marker, infectious bronchitis virus M protein. J. Cell Biol. 139, 1411–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Worgall T. S. (2011) Sphingolipid synthetic pathways are major regulators of lipid homeostasis. Adv. Exp. Med. Biol. 721, 139–148 [DOI] [PubMed] [Google Scholar]
- 45. Browman D. T., Resek M. E., Zajchowski L. D., Robbins S. M. (2006) Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J. Cell Sci. 119, 3149–3160 [DOI] [PubMed] [Google Scholar]
- 46. Luberto C., Hassler D. F., Signorelli P., Okamoto Y., Sawai H., Boros E., Hazen-Martin D. J., Obeid L. M., Hannun Y. A., Smith G. K. (2002) Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J. Biol. Chem. 277, 41128–41139 [DOI] [PubMed] [Google Scholar]
- 47. Lerner A. G., Upton J. P., Praveen P. V., Ghosh R., Nakagawa Y., Igbaria A., Shen S., Nguyen V., Backes B. J., Heiman M., Heintz N., Greengard P., Hui S., Tang Q., Trusina A., Oakes S. A., Papa F. R. (2012) IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Han J., Back S. H., Hur J., Lin Y. H., Gildersleeve R., Shan J., Yuan C. L., Krokowski D., Wang S., Hatzoglou M., Kilberg M. S., Sartor M. A., Kaufman R. J. (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Adachi Y., Yamamoto K., Okada T., Yoshida H., Harada A., Mori K. (2008) ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 33, 75–89 [DOI] [PubMed] [Google Scholar]
- 50. Feng B., Yao P. M., Li Y., Devlin C. M., Zhang D., Harding H. P., Sweeney M., Rong J. X., Kuriakose G., Fisher E. A., Marks A. R., Ron D., Tabas I. (2003) The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 5, 781–792 [DOI] [PubMed] [Google Scholar]
- 51. Kockx M., Dinnes D. L., Huang K. Y., Sharpe L. J., Jessup W., Brown A. J., Kritharides L. (2012) Cholesterol accumulation inhibits ER to Golgi transport and protein secretion. Studies of apolipoprotein E and VSVGt. Biochem. J. 447, 51–60 [DOI] [PubMed] [Google Scholar]
- 52. Runz H., Miura K., Weiss M., Pepperkok R. (2006) Sterols regulate ER-export dynamics of secretory cargo protein ts-O45-G. EMBO J. 25, 2953–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ridsdale A., Denis M., Gougeon P. Y., Ngsee J. K., Presley J. F., Zha X. (2006) Cholesterol is required for efficient endoplasmic reticulum-to-Golgi transport of secretory membrane proteins. Mol. Biol. Cell 17, 1593–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bagnat M., Keränen S., Shevchenko A., Shevchenko A., Simons K. (2000) Lipid rafts function in biosynthetic delivery of proteins to the cell surface in yeast. Proc. Natl. Acad. Sci. U.S.A. 97, 3254–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Campana V., Sarnataro D., Fasano C., Casanova P., Paladino S., Zurzolo C. (2006) Detergent-resistant membrane domains but not the proteasome are involved in the misfolding of a PrP mutant retained in the endoplasmic reticulum. J. Cell Sci. 119, 433–442 [DOI] [PubMed] [Google Scholar]
- 56. Hayashi T., Su T. P. (2003) σ-1 receptors (σ(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum. Roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Ther. 306, 718–725 [DOI] [PubMed] [Google Scholar]
- 57. Maxfield F. R., Menon A. K. (2006) Intracellular sterol transport and distribution. Curr. Opin. Cell Biol. 18, 379–385 [DOI] [PubMed] [Google Scholar]
- 58. Lei X., Zhang S., Bohrer A., Bao S., Song H., Ramanadham S. (2007) The group VIA calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry 46, 10170–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Epstein S., Kirkpatrick C. L., Castillon G. A., Muñiz M., Riezman I., David F. P., Wollheim C. B., Riezman H. (2012) Activation of the unfolded protein response pathway causes ceramide accumulation in yeast and INS-1E insulinoma cells. J. Lipid Res. 53, 412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang H., Kouri G., Wollheim C. B. (2005) ER stress and SREBP-1 activation are implicated in β-cell glucolipotoxicity. J. Cell Sci. 118, 3905–3915 [DOI] [PubMed] [Google Scholar]
- 61. Subathra M., Qureshi A., Luberto C. (2011) Sphingomyelin synthases regulate protein trafficking and secretion. PLoS ONE 6, e23644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gouazé-Andersson V., Yu J. Y., Kreitenberg A. J., Bielawska A., Giuliano A. E., Cabot M. C. (2007) Ceramide and glucosylceramide upregulate expression of the multidrug resistance gene MDR1 in cancer cells. Biochim. Biophys. Acta 1771, 1407–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Volmer R., van der Ploeg K., Ron D. (2013) Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U.S.A. 110, 4628–4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pineau L., Colas J., Dupont S., Beney L., Fleurat-Lessard P., Berjeaud J. M., Bergès T., Ferreira T. (2009) Lipid-induced ER stress. Synergistic effects of sterols and saturated fatty acids. Traffic 10, 673–690 [DOI] [PubMed] [Google Scholar]
- 65. Ariyama H., Kono N., Matsuda S., Inoue T., Arai H. (2010) Decrease in membrane phospholipid unsaturation induces unfolded protein response. J. Biol. Chem. 285, 22027–22035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Krokowski D., Han J., Saikia M., Majumder M., Yuan C. L., Guan B. J., Bevilacqua E., Bussolati O., Bröer S., Arvan P., Tchórzewski M., Snider M. D., Puchowicz M., Croniger C. M., Kimball S. R., Pan T., Koromilas A. E., Kaufman R. J., Hatzoglou M. (2013) A self-defeating anabolic program leads to β-cell apoptosis in endoplasmic reticulum stress-induced diabetes via regulation of amino acid flux. J. Biol. Chem. 288, 17202–17213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fu S., Yang L., Hofmann O., Dicker L., Hide W., Lin X., Watkins S. M., Ivanov A. R., Hotamisligil G. S. (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]