

Background: Nonviability of cells lacking mitochondrial DNA ligase suggests essential function of this enzyme.
Results: We report the isolation of viable Lig3−/− cells, which lack mtDNA.
Conclusion: The lethality of the Lig3 knock-out is mediated by the ρ0 phenotype.
Significance: This is definitive proof that the essential function of LIG3 in mitochondria is limited to DNA transactions.
Keywords: DNA Damage, DNA Repair, DNA Replication, Mitochondria, Mitochondrial DNA, Reactive Oxygen Species, Respiration, Mitochondrial DNA Degradation, rho-0 Cells
Abstract
Multiple lines of evidence support the notion that DNA ligase III (LIG3), the only DNA ligase found in mitochondria, is essential for viability in both whole organisms and in cultured cells. Previous attempts to generate cells devoid of mitochondrial DNA ligase failed. Here, we report, for the first time, the derivation of viable LIG3-deficient mouse embryonic fibroblasts. These cells lack mtDNA and are auxotrophic for uridine and pyruvate, which may explain the apparent lethality of the Lig3 knock-out observed in cultured cells in previous studies. Cells with severely reduced expression of LIG3 maintain normal mtDNA copy number and respiration but show reduced viability in the face of alkylating and oxidative damage, increased mtDNA degradation in response to oxidative damage, and slow recovery from mtDNA depletion. Our findings clarify the cellular role of LIG3 and establish that the loss of viability in LIG3-deficient cells is conditional and secondary to the ρ0 phenotype.
Introduction
In mammalian cells, there are two DNA-containing organelles, the nucleus and the mitochondrion. Human mitochondrial DNA (mtDNA) is a 16,568-bp circular molecule (1) crucial for proper mitochondrial function and cellular ATP production. It encodes 13 proteins as follows: 11 polypeptide subunits of the mitochondrial electron transport chain complexes I, III, and IV and two subunits of the ATP synthase, complex V. These polypeptides are encoded using a genetic code distinct from that used in the nucleus and therefore require a separate translational apparatus, some components of which (22 tRNAs and 2 rRNAs) also are encoded in mtDNA (2). Therefore, it comes as no surprise that the loss of mtDNA integrity either through mutation or depletion may lead to serious, often fatal diseases (3, 4). The mitochondrial electron transport chain is a major cellular source for the production of reactive oxygen species. Although the exact rates of mitochondrial reactive oxygen species production in vivo remain controversial due to technical difficulties associated with their quantitation (5, 6), it is well established that reactive oxygen species can damage mtDNA (7). To deal with oxidative and other types of DNA damage, mitochondria possess an array of DNA repair mechanisms (8), of which the most active and the best studied is base excision repair (BER)2 (9–11).
DNA ligase activity is crucial for both DNA replication and repair (12). In BER, the ligase catalyzes the last step in the series, nick sealing (13). Of the three mammalian DNA ligases, Lig1, LIG3, and Lig4, only Lig3 has been documented to be present in mitochondria, where it functions in both DNA repair and replication (14, 15). The mitochondrial isoform is transcribed from the same gene as the nuclear isoform and is generated by alternative translation initiation using an upstream in-frame ATG codon. The resulting N-terminal extension encodes an amphipathic helix, which serves as the mitochondrial matrix targeting sequence (MTS) (16, 17).
Consistent with the essential role played by LIG3 in the maintenance of mtDNA, the whole body Lig3 knock-out is embryonically lethal, and Lig3-null mouse embryos die ∼8.5 days post-coitum (18). Mice with a tissue-specific Lig3 knock-out in the nervous system do not survive past postnatal day 20, and death of these animals is preceded by a reduction in mtDNA copy number (19). Similarly, mice with Lig3 ablation in muscle die abruptly between 3.5 and 4.5 weeks of age, due to defects in cardiac, but not skeletal, muscle (19). Repeated attempts to culture Lig3−/− fibroblasts from knock-out embryos proved unsuccessful, whereas fibroblast cultures from similarly aged wild type (WT) embryos were readily established (18). Similarly, Lig3-null mouse embryonic stem cells do not survive, unless cells are preemptively complemented with mitochondrially targeted DNA ligase (20). In the latter case, even DNA ligases as evolutionarily distant from LIG3 as the ATP-dependent Chlorella virus ligase and the NAD+-dependent ligA of Escherichia coli were able to substitute for LIG3. Finally, the loss of Lig3 was also reported to be lethal in chicken DT40 cells, unless preemptively complemented with mitochondrially targeted ligase (21). Provided that LIG3 is not involved in mitochondrial processes other than mtDNA replication and repair, the expected outcome of the Lig3 knock-out is a loss of mtDNA. This explains the lethality of the Lig3 knock-out in animal models but not in cultured cells. Cells lacking mtDNA (ρ0 cells) are viable in appropriately formulated media and can be obtained with reasonable ease through chronic treatment of cultures with DNA intercalators such as ethidium bromide (EtBr) (22, 23), ditercalinium chloride (24–26), or by targeting a restriction endonuclease to mitochondria (27). Therefore, the previously observed lethality of the Lig3 knock-out in cultured cells may suggest a possible novel role for the LIG3. Here, in contrast, we demonstrate that LIG3 does not play an indispensable role in cultured cells and that viable Lig3−/− mouse embryonic fibroblasts (MEFs) can be readily engineered and maintained in culture. Furthermore, we show that LIG3 is present in mitochondria in amounts exceeding those required for mtDNA maintenance under routine cell culture conditions.
EXPERIMENTAL PROCEDURES
Cell Growth and Treatment
Unless specified otherwise, all cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, 50 μg/ml gentamycin, 50 μg/ml uridine, and 1 mm sodium pyruvate in a humidified atmosphere containing 5% CO2 at 37 °C.
Mouse Embryonic Fibroblasts
Cre-TM mice (JAX 004682, B6.Cg-Tg(CAG-cre/Esr1)5Amc/J) were intercrossed with Lig3LoxP mice (19) to provide tamoxifen-inducible cre expression driven from the actin promoter. Lig3Cre-TM mouse embryonic fibroblast (MEF) cell cultures were prepared from E13.5 embryonic mesenchyme. Embryonic tissue was minced using dissection scissors, trypsinized, filtered through a sterile 40-μm sieve, and resuspended in DMEM supplemented with 10% fetal bovine serum (v/v), 1× GlutaMAX, 100 units/ml penicillin, 100 μg/ml streptomycin, and β-mercaptoethanol. Cells were allowed to proliferate in T-25 tissue culture flasks (Falcon) at 37 °C in a humidified CO2-regulated (5%) incubator and were split at a 1:4 dilution when the monolayer was confluent (24 h post-isolation). MEFs were immortalized with retroviral construct 3315, a derivative of pSF91 (28) which encodes SV40 large T antigen (Fig. 1A), and selected depending upon their ability to form colonies. One clone was selected for its superior growth characteristics and designated Cre4. This clone was further modified by introducing a Tet-On transactivator with retrovirus rv2641 (29), thus producing a clone designated 4B6. To a loss of mtDNA, Cre4 cells were treated with 5 μg/ml EtBr for 3 weeks and cloned. One of the clones (clone 11) demonstrated an absence of mtDNA as determined by both PCR and Southern blotting (data not shown) and was used in subsequent experiments.
FIGURE 1.
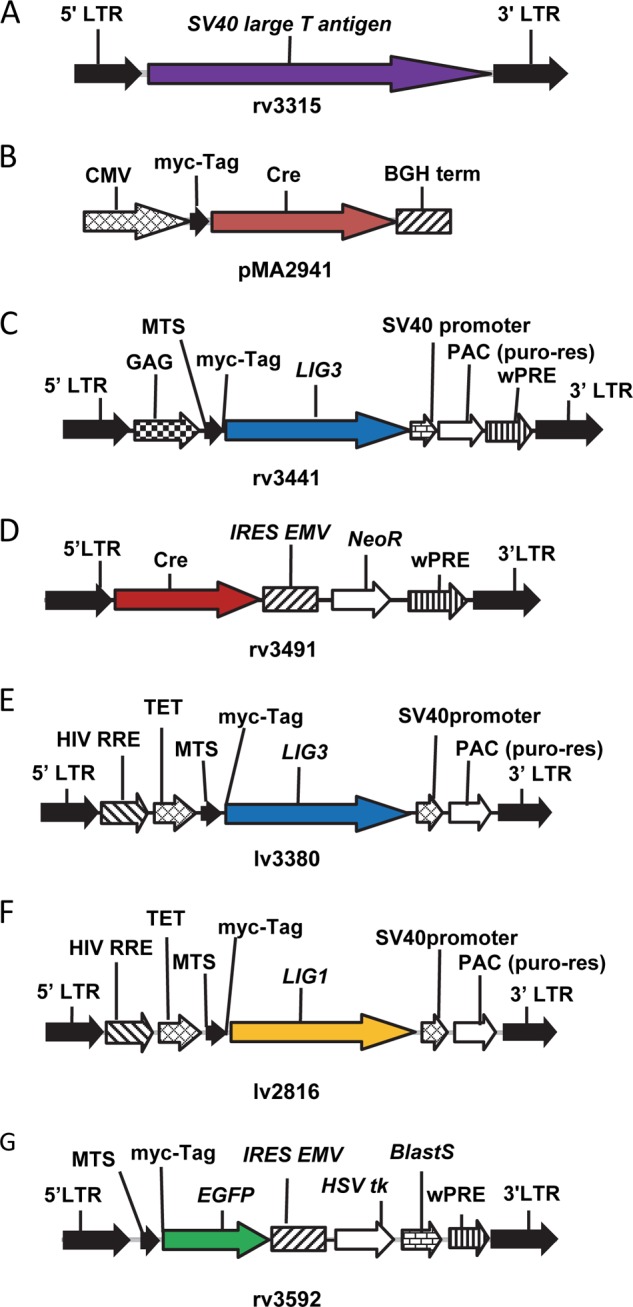
DNA constructs used in the study. A, retrovirus rv3315 encodes SV40 large T antigen driven by murine embryonic stem cell virus long terminal repeat (LTR) promoter. No antibiotic resistance marker is present. B, plasmid pMA2941 encodes CMV promoter-driven Myc-tagged cre recombinase gene. C, retrovirus rv3441 encodes Moloney murine leukemia virus LTR promoter-driven mitochondrially targeted LIG3. The gene's native MTS and an initiating codon were removed and replaced with MTS from human ornithine transcarbamylase followed by a Myc tag. D, retrovirus rv3491 encodes LTR promoter-driven cre recombinase whose expression is transcriptionally linked (through an internal ribosome entry site, IRES EMV) to the expression of the G418 resistance gene. E, lentivirus lv3380 encodes doxycycline-inducible promoter (TET)-driven mitochondrially targeted LIG3 as in C.
Lig3 knockdown in HeLa cells was performed using pSilencerTM 5.1-H1 Retro (Invitrogen). Six shRNA constructs were generated by cloning annealed complementary oligonucleotides (Table 1) into BamHI-HindIII digested vector. Retroviral supernatants were generated using Phoenix-Ampho cells as described earlier (29). HeLa cells were infected with retroviral supernatants overnight in the presence of 10 μg/ml Polybrene and selected with 3 μg/ml puromycin.
TABLE 1.
Oligonucleotides used to generate shRNA for hLig3
| Name | Sequence |
|---|---|
| hLig3–1F | GATCCGATCTTCTTTCCACAAACCTTCAAGAGAGGTTTGTGGAAAGAAGATCTTTTTTGGAAA |
| hLig3–1R | AGCTTTTCCAAAAAAGATCTTCTTTCCACAAACCTCTCTTGAAGGTTTGTGGAAAGAAGATCG |
| hLig3–2F | GATCCGAACTGTGCCTATTCCGAATTCAAGAGATTCGGAATAGGCACAGTTCTTTTTTGGAAA |
| hLig3–2R | AGCTTTTCCAAAAAAGAACTGTGCCTATTCCGAATCTCTTGAATTCGGAATAGGCACAGTTCG |
| hLig3–3F | GATCCGCATCACTGGCGTGATGTAATTCAAGAGATTACATCACGCCAGTGATGTTTTTTGGAAA |
| hLig3–3R | AGCTTTTCCAAAAAACATCACTGGCGTGATGTAATCTCTTGAATTACATCACGCCAGTGATGCG |
| hLig3–4F | GATCCGACAATTCAGCCAGTGGTCTTCAAGAGAGACCACTGGCTGAATTGTCTTTTTTGGAAA |
| hLig3–4R | AGCTTTTCCAAAAAAGACAATTCAGCCAGTGGTCTCTCTTGAAGACCACTGGCTGAATTGTCG |
| hLig3–5F | GATCCGTTCAGCCAGTGGTCAGAAATTCAAGAGATTTCTGACCACTGGCTGAATTTTTTGGAAA |
| hLig3–5R | AGCTTTTCCAAAAAATTCAGCCAGTGGTCAGAAATCTCTTGAATTTCTGACCACTGGCTGAACG |
| hLig3–6F | GATCCACAGATCTGCTTCATGGACTTCAAGAGAGTCCATGAAGCAGATCTGTTTTTTTGGAAA |
| hLig3–6R | AGCTTTTCCAAAAAAACAGATCTGCTTCATGGACTCTCTTGAAGTCCATGAAGCAGATCTGTG |
Quantitative Southern Blotting
Quantitative Southern blotting under alkaline (denaturing) conditions (QSBA) was performed essentially as described earlier (30). Quantitative Southern Blotting under the nondenaturing conditions (QSBN) was performed similarly, except that there was no alkaline pretreatment of DNA samples, and no NaOH was included in the loading dye, agarose gel, or the electrophoresis buffer (electrophoresis was performed using TBE buffer). BamHI-digested total human DNA was separated in appropriately formulated 0.6% agarose gels overnight. After blotting, the membrane was cut at the level of the 9-kb band of λ/HindIII marker. The top portion was then hybridized with the mtDNA probe (detects a 16,569-bp fragment), and the lower portion was hybridized with the 18 S rDNA probe (5,102-bp fragment). After hybridization, membranes were exposed to an imaging screen to determine band intensity. The number of pixels per band was determined by encompassing bands with identical rectangular regions of interest and subtracting the background.
The DNA break frequency per analyzed DNA fragment (16,569 bp for mtDNA and 5,102 bp for nDNA) was determined using the Poisson expression (s = −ln P0, where s is the number of breaks per fragment, and P0 is the fraction of fragments free of breaks). Break frequency was expressed as a number of breaks per 10,000 bp. The percent mtDNA degradation was determined by means of QSBN as % = (C − 6h)/C·100%, where C is intensity of the band in control and 6h is intensity of the band at 6 h.
The frequency of double strand breaks (DSB) was determined using QSB; the combined frequency of DSB and single-strand breaks (SSB) was determined using QSBA, and the frequency of SSB was determined by subtracting the frequency of DSB from the combined frequency of SSB and DSB.
Determination of mtDNA Copy Number
mtDNA copy number was estimated by QSBN as the ratio of the intensities of the bands corresponding to mtDNA and 18 S DNA. Precise determination of mtDNA copy number was performed using TaqMan qPCR with the primers and probes listed in the Table 2. For human cells, simplex qPCR was employed, and for mouse cells duplex qPCR was employed. To generate a standard curve for both human and mouse DNA, a separate calibrator plasmid containing cloned nuclear and mitochondrial targets was used.
TABLE 2.
Primers and probes used for quantitation of mtDNA copy number by TaqMan qPCR
| Human mtDNA | |
| F2198 | AAAGCGTTCAAGCTCAACACCCAC |
| R2353 | TAATCTGACGCAGGCTTATGCGGA |
| hMito probe3: | 6-FAM/TGAACTCCT/ZEN/CACACCCAATTGGACCA/IABkFQ |
| Human nDNA | |
| hTertTaqManF | CAAGCACTTCCTCTACTCCTC |
| hTertTaqManR | TGGAACCCAGAAAGATGGTC |
| hTertTaqManProbe | TEX615/CACGAGCCTCCGAGCGCCAG/IABkRQ |
| Mouse mtDNA | |
| rtF-mtDNA | ACTTCTAACTAAAAGAATTACAGC |
| rtR-mtDNA | TAGACGAGTTGATTCATAAAATTG |
| mtDNA-probe | 6-FAM/CCCGAAACC/ZEN/AAACGAGCTACCT/IAbFQ |
| Mouse nDNA | |
| rtF-mTert | CCTCAAGCATTCACCTCTTCTTTG |
| rtR-mTert | CCAAGGACCTGCTCGATGAC |
| mTret-probe | TEX613-Y/ACCACCCTCTCTGACCTCCAGCCA/IAbRQ |
Isolation of mitochondrion-enriched fractions was performed according to Pallotti and Lenaz (31), with some modifications. Briefly, cells were collected by centrifugation, washed with PBS, and frozen until isolation. For isolation, cells were thawed, resuspended in buffer containing 0.21 m mannitol, 70 mm sucrose, 10 mm KH2PO4, 5 mm HEPES, 0.5 mm EGTA, 1× Halt protease inhibitor mixture (Thermo Fisher Scientific, Waltham, MA), pH 7.4, and disrupted by five passages through a 27-gauge needle. Cell debris was removed by centrifugation for 5 min at 1,200 × g, 4 °C, and mitochondria were recovered from the supernatant by centrifugation for 5 min at 13,000 × g, 4 °C.
Clonogenic Assays
For clonogenic assays, cells were collected by trypsinization, counted using a Coulter counter, plated at a density of 250 cells per 60-mm dish, and allowed to attach overnight. The next day medium was removed; cells were washed once with PBS and exposed to various concentrations of either H2O2 or MMS in HBSS for 30 min at 37 °C in an atmosphere of 5% CO2. After treatment, the medium was replaced, and plates were left in the incubator to allow for the formation of colonies. Upon formation of colonies, they were washed with PBS, fixed with methanol/acetic acid (3:1), stained with hematoxylin, washed with deionized water, dried, and counted. Plating efficiency was expressed as percentage of untreated control. A survival and proliferation assay was performed essentially as the clonogenic assays, except experimental plates were trypsinized and cells counted, instead of being fixed and stained.
Mitochondrial membrane potential was measured with JC-1. The cells were plated in 12-well plates, allowed to attach overnight, and incubated with 5 μm JC-1 in DMEM for 10 min at 37 °C in an atmosphere of 5% CO2. After treatment, cells were trypsinized and immediately subjected to flow cytometry on FACSCantoII (BD Biosciences). Membrane potential was expressed as a ratio of fluorescence intensities in the phycoerythrin and FITC channels.
Mitochondrial respiration in whole attached cells was measured with the help of an XF-24 extracellular flux analyzer (Seahorse Biosciences, Billerica, MA) according to the manufacturer's recommendations and expressed as picomoles/min/μg of protein. ATP-linked respiration was determined with the help of oligomycin (5 μm); maximal respiration was induced with carbonyl cyanide m-chlorophenyl hydrazine;(1 μm), and nonmitochondrial respiration was determined after injection of rotenone and antimycin A (1 μm each). Transient mitochondrial DNA depletion was achieved by treating cells for 3 days with 5 μg/ml EtBr.
Genotyping ρ0 Cells
To test for a ρ0 genotype, total DNA was amplified with two pairs of primers (duplex reaction). Primers 4f (GCCTACACCCAGAAGATTTCAT) and 4r (AGACAGTTGGACCCTCGTTTAG) were used to amplify 1,000 bp of mouse mtDNA between coordinates 1,075 and 2074. Primers mPolGchrF4 (ACTGGATGGATATCAGCAGTGCCA) and mPolGchrR4 (ATGTGTTCTGTGCCTCTGTCAGGT) were used to amplify the 629-bp fragment encompassing intron 17 and parts of exons 17 and 18 of the PolG gene, which encodes mitochondrial DNA polymerase γ. PCR was performed using the following parameters: denaturation at 95 °C for 1 min and then 35 cycles of 94 °C (10 s), 55 °C for 20 s, 68 °C for 1 min, and a final extension of 68 °C for 3 min.
Genotyping Lig3 Allele
To genotype the Lig3 allele, the following primers were used: Lig3-Δ, CAGTCGACGAGATGGCTCAGTGGTTAAGAGC; Lig3–5, GATGCGGCCGCAGCCAAGTGTGAATATACAGC; and Lig3–8, CAGTCGACAGGGAGCTTGGGACGGATGC.
Lig3–5/Lig3–8 pair amplifies the 555-bp fragment of the Lig3loxP/loxP allele, and no amplification was observed with the Lig3−/−allele. Conversely, Lig3–5/Lig3-Δ pair amplifies a 560-bp fragment of the Lig3−/− allele, and no amplification was observed with Lig3loxP/loxP allele. PCR was performed using the following parameters: denaturation at 95 °C for1 min and then 35 cycles of 94 °C (10 s), 55 °C for 20 s, 68 °C for 1 min, and final extension of 68 °C for 3 min.
Detection of the transduced human Lig3 cDNA in mouse cells was performed with primers hLig3cDNAf (TGGCCACAAAGTCTTCTCCAGTGA) and hLig3cDNAr (CGTGCGTGGCTGAAGTCATATCAA). These primers amplify the 209-bp fragment using the same cycling parameters as above.
Western Blotting
For Western blot analysis, whole cell or mitochondrial lysates were prepared by lysis in a buffer containing 10 mm Tris, 0.5% SDS, 1× Halt protease inhibitor mixture, pH 8.0. Protein was quantitated by either the Bradford or BCA assay.
Confocal Microscopy
For confocal microscopy, cells were plated into 35-mm glass bottom MatTek dishes, allowed to attach overnight, and imaged with a Nikon A1R confocal microscope using a ×60 water immersion objective.
Statistical Analysis
Pairwise comparisons were performed using the two-tailed unpaired Student's t test assuming unequal variances.
RESULTS
Steady-state Levels of LIG3 Are Excessive for the Maintenance of mtDNA Copy Number
To evaluate the effect of reduced cellular LIG3 levels on mtDNA maintenance, six HeLa cell lines expressing shRNA constructs directed at different regions of the Lig3 mRNA were established (Fig. 1). Constructs 2842-1 and 2842-6 caused a modest knockdown, and construct 2842-2 initiated very efficient knockdown, as estimated by Western blotting. Little or no LIG3 signal was detected by Western blotting of whole cell extracts of WT and HeLa cells transduced with the 2842-2 construct (hereafter designated KD cells) (Fig. 2A). Therefore, to estimate the actual efficiency of the knockdown, Western blotting was performed on serial dilutions of the mitochondrial fraction of WT cells versus the undiluted mitochondrial fraction of KD cells. Using this approach, a 90% knockdown of LIG3 in the mitochondrial fraction of KD cells was estimated (Fig. 2B). Surprisingly, mtDNA copy number determined in KD cells by either QSBN (Fig. 2, C and D) or by TaqMan qPCR (Fig. 2E) failed to reveal any correlation between levels of Lig3 expression and mtDNA copy number indicating that as little as 10% of the actual LIG3 levels may be sufficient to maintain normal mtDNA copy number in HeLa cells.
FIGURE 2.

Effect of Lig3 knockdown on mtDNA copy number. Six shRNA constructs (2842-1 through 6) were generated, and their effects on Lig3 expression and mtDNA copy number were studied. A, Western blots of whole cell lysates from HeLa cells infected with shRNA constructs 2842-1 through 6. β-Actin, loading control. B, Western blotting of serial dilutions of the mitochondrial lysate of the WT HeLa cells and undiluted mitochondrial lysate of HeLa/2842-2. Mitochondrial HSP60, loading control. C, QSBN of the total DNA from HeLa cells and cells infected with knockdown constructs. Total DNA was digested with BamHI and processed as described under “Experimental Procedures.” Mito, mitochondria. D, quantitation of the QSBN in C. E, quantitation of mtDNA copy number in WT HeLa cells and HeLa/2842-2 by TaqMan qPCR. Error bars, S.E., n = 3.
Reduction of LIG3 Levels Increases Susceptibility of Cells to Alkylating and Oxidative Damage
To evaluate the effect of Lig3 knockdown on mitochondrial physiology, mitochondrial membrane potential and respiration were measured in WT and KD cells. Neither membrane potential nor basal, ATP-linked, maximal, or nonmitochondrial respiration differed significantly between the two cell types (Fig. 3, A and B). However, there was a slight decrease in the viability of the KD cells as compared with their WT counterparts in response to treatment with DNA-damaging agents, methyl methanesulfonate (MMS) and H2O2 (Fig. 3, C and D). This reduction was significant only at higher doses of MMS (Fig. 3C).
FIGURE 3.

Effect of Lig3 knockdown on physiological responses of the HeLa cells and DNA repair. A, effect of Lig3 knockdown on mitochondrial membrane potential as measured by JC-1 fluorescence. Val, 0.2 μm valinomycin (ionophore). B, effect of Lig3 knockdown on cellular respiration as measured by an XF-24 analyzer. Oxygen consumption is expressed in picomoles of O2/min/μg of protein. C and D, effect of Lig3 knockdown on susceptibility to DNA-damaging agents MMS (C) and hydrogen peroxide (H2O2, D) as determined by clonogenic assay. Two-tailed Student's t test assuming unequal variance; n = 5 or 6. ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Lig3 Knockdown Enhances mtDNA Degradation in Response to Oxidative Damage
To study the effect of reduced LIG3 levels on mtDNA damage and repair, WT and KD HeLa cells were exposed to the Sn2 alkylating agent MMS (32) or to H2O2, and mtDNA repair was followed with QSBA and QSBN as described under “Experimental Procedures.” At the concentrations used, MMS induced strand breaks with similar frequency in mtDNA of both WT and KD cells (Fig. 4, A and C). A vast majority of those breaks (>90%) were SSB. In contrast, H2O2 induced more strand breaks in mtDNA of KD cells. A majority of those were also SSBs (Fig. 4, B and C). Of note, H2O2, but not MMS, stimulated mtDNA degradation in KD cells, even though MMS induced twice as many strand breaks (Fig. 4D).
FIGURE 4.

Effect of LIG3 knockdown on mtDNA repair. A and B, WT and KD cells were treated with either 2 mm MMS (A) or 100 μm H2O2 (B) for 30 min at 37 °C in the humidified atmosphere of 5% CO2. Total cellular DNA was extracted either immediately after the treatment (0 h) or after 6 h of recovery in fresh DMEM (6 h), digested with BamHI, and subjected to QSBA and QSBN. C, frequencies of SSB and DSB induced in mtDNA by MMS and H2O2 as measured immediately after treatment (0-h time point). D, % mtDNA degradation as determined by QSBN at the 6-h time point. Error bars, S.E., n = 4. C and D, two-tailed Student's t test assuming unequal variance, WT versus KD. ns, not significant; Mito, mitochondria; **, p < 0.01; ***, p < 0.001.
LIG3 Is Dispensable for the Viability of MEFs
Previous studies reported that mitochondrial, but not nuclear, LIG3 is required for viability of MEFs (18), mouse embryonic stem cells (20), and chicken DT40 cells (21). The lethality of the Lig3−/− phenotype observed in those studies leaves open the possibility that mitochondrial DNA ligase activity was involved in an essential process other than mtDNA transactions. To resolve this issue, Cre4 cells were transfected with plasmid pMA2941 (Fig. 1B), which encodes cre recombinase, and selected for resistance to G418 in medium supplemented with uridine and pyruvate. Some of the resulting clones grew poorly and demonstrated the expected deletion in Lig3 and a concomitant loss of mtDNA (Fig. 5A). To see whether the loss of mtDNA can be rescued by expressing LIG3 in mitochondria, Cre4 cells were simultaneously infected with two retroviruses, rv3441 and rv3491 (Fig. 1, C and D). rv3441 encodes LIG3 from which the native MTS and initiating codon for the nuclear isoform were removed and replaced with the MTS from human ornithine transcarbamylase (33) followed by a Myc tag. rv3491 encodes cre recombinase. Co-infection with these two viruses allowed simultaneous cre-mediated excision and complementation of the deletion with LIG3 encoded by the second virus. As expected, mitochondrial LIG3 prevented the loss of mtDNA in Cre4/3441/3491 cells. Additional experiments demonstrated that in agreement with a previous report (20), neither nuclear LIG3 nor catalytically inactive mitochondrial LIG3 was able to rescue the loss of mtDNA, although mitochondrially targeted LIG1 prevented mtDNA loss (Fig. 5, B and D, and results not shown). Western blotting with monoclonal antibodies directed against LIG3 protein confirmed the loss of LIG3 expression in Cre4/2941 ρ0, but not in Cre4#11 ρ0 cells, which were derived by treatment with EtBr rather than by introducing a deletion into the Lig3 allele. However, no LIG3 expression was detected in whole cell lysates from the Cre4/3441/3491 cells, even though these cells did not exhibit any reduction in mtDNA copy number as compared with parental Cre4 cells (Fig. 5, C and E). Nevertheless, LIG3 was detectable in the mitochondrial fraction from these cells, although its level of expression was only ∼3% of that found in WT mitochondria (Fig. 5F). The loss of mtDNA was accompanied by shortening and swelling of mitochondria, which was not observed in Cre4/3441/3491 cells, in which loss of mtDNA was prevented by complementation with hLIG3 (Fig. 5G).
FIGURE 5.

Lig3 is dispensable for the viability of MEFs. A, PCR genotyping demonstrates the presence of the WT Lig3 allele in Cre4 and Cre4 ρ-0#11 (EtBr-derived ρ0) cell lines, whereas a deleted allele is present in the Cre4/2941 and Cre4/3441/3491 cell lines due to Cre-mediated floxing out. Deleted Lig3 allele in Cre4/2941 leads to a loss of mtDNA. However, mtDNA is present in Cre4/3441/3491 despite deletion in the Lig3 allele due to complementation with human Lig3 encoded by rv3441. This complementing hLig3 gene is detectable by PCR. WT Lig3 primers amplify the loxP/loxP allele but not the deleted allele. Conversely, ΔLig3 primers amplify deleted Lig3−/− allele but not the WT allele. mtDNA and nDNA primers amplify, in the same tube, mitochondrial and nuclear DNA, respectively. hLig3 primers amplify cDNA for the human Lig3 but not the endogenous mouse gene. B, PCR genotyping of the original 4B6 cell line, 4B6 cells in which deletion of the Lig3 allele was induced with Cre recombinase (4B6/2941), and 4B6 cells complemented with doxycycline-inducible mitochondrially targeted human Lig1 gene prior to excision of the endogenous Lig3 allele (4B6/2816/2941). See Fig. 1F for the map of lv2816. C and D, quantitation of mtDNA copy number in the same cell lines by real time TaqMan qPCR. Mitochondrially targeted hLig1 supports normal levels of mtDNA in 4B6/2816/2941 even without doxycycline induction due to the “leaky” expression from the Tet promoter. Error bars, S.E., n ≥3. E, Western blotting of the whole cell lysates of the same cell lines. Note that Lig3 expression is not detectable in the Cre4/3441/3491 cell line even though antibodies can detect both mouse (Cre4) and human (HeLa) proteins and even though this cell line normally maintains mtDNA, which suggests low expression. F, reduction in the Lig3 expression levels in Cre4/3441/3491 cells was estimated by Western blotting of the mitochondrial fractions of the Cre4/3441/3491 cells versus dilutions of the mitochondrial fraction of the Cre4 cells. G, the loss of mtDNA induces morphological changes in mitochondria. Cre4 cells (WT), Cre4/2941(ρ0, ΔLig3), and Cre4/3441/3491 (ΔLig3, complemented) were infected with rv3591 (Fig. 1G), which encodes mitochondrially targeted enhanced GFP and imaged by confocal microscopy.
Lig3−/− Cells Are Dependent on Uridine and Pyruvate for Their Survival and Growth
It has been demonstrated that loss of mitochondrial DNA induced by means other than deletion of the Lig3 (34) leads to the dependence of the resulting cell lines on media supplementation with uridine and pyruvate for survival and growth. Therefore, the Cre4 cell line as well as its derivatives Cre4/2941 (Lig3−/−), Cre4 ρ0#11 (obtained by EtBr treatment), and Cre4/3441/3491 were exposed to DMEM with and without uridine and pyruvate. Within 24 h of exposure to the medium without uridine and pyruvate, Cre4/2941 cells stopped their growth, and cell death was clearly detectable within 48 h (Fig. 6A). Cre4 ρ0#11 cells also died in media without uridine or pyruvate, although with somewhat slower kinetics. Both cell lines survived and grew in media containing uridine and pyruvate. Cre4 and Cre4/3441/3491, cell lines that contained mtDNA, grew regardless of supplementation with uridine and pyruvate (Fig. 6B).
FIGURE 6.
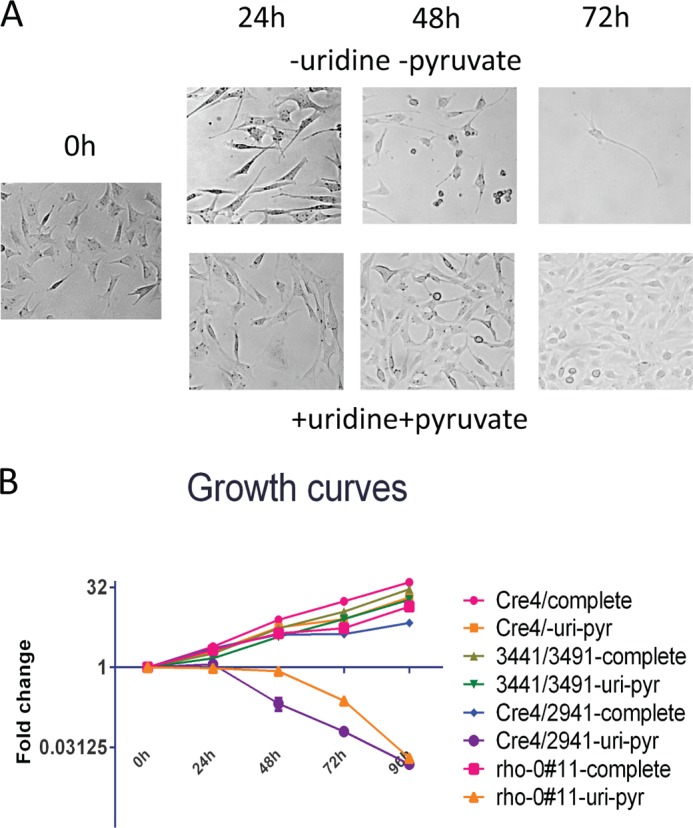
Lig3−/− cells are dependent on uridine and pyruvate for their survival and growth. A, representative microscopic field views of Cre4/2941 cells exposed to media with and without uridine and pyruvate for 0 h (immediately after media change) and 24, 48, and 72 h after media change. Top panels, DMEM without uridine or pyruvate supplementation. Bottom panels, DMEM supplemented with uridine and pyruvate. Cells proliferate in media supplemented with uridine and pyruvate and die in unsupplemented media. B, growth curves of Cre4, Cre4/2941, Cre4 ρ0#11, and Cre4/3441/3491 in media with or without uridine and pyruvate. Note that neither Cre4 ρ0#11 (ρ0 due to EtBr treatment) nor Cre4/2941 (ρ0 due to Lig3 deletion) survive in media without uridine or pyruvate.
Low Levels of Lig3 Support Normal mtDNA Replication and Cellular Respiration
To exclude positional effects of gene integration and to better evaluate cellular requirements for LIG3 under different conditions, we deleted Lig3 in MEFs that were engineered for doxycycline-inducible expression of the LIG3 with a retrovirally encoded reverse tetracycline transactivator (4B6, see under “Experimental Procedures”) and lentivirus 3380, which encodes Myc-tagged human LIG3 (Fig. 1E). After confirming proper deletion in the endogenous LIG3 gene and introduction of the LIG3, we observed that mtDNA was present in cells even in the absence of LIG3 induction (Fig. 7A). Moreover, induction did not alter mtDNA copy number (Fig. 7B), even though LIG3 was undetectable in whole cell lysates from uninduced cells, as determined by Western blotting with antibodies directed against either LIG3 or the Myc tag (Fig. 7C). Apparently, low levels of “leaky” LIG3 expression were sufficient to support mtDNA replication. Indeed, Western blotting of serial dilutions of the mitochondrial fraction from the induced cells side-by-side with the undiluted mitochondrial fraction from uninduced cells revealed that LIG3 content of mitochondria in uninduced cells was less than 1% of that in induced cells (Fig. 7D). These dramatically reduced levels of Lig3 supported normal mtDNA replication and respiration (Fig. 7, B and F). Similarly, mitochondrial levels of LIG3 in the WT 4B6 cells were more than 100-fold higher than those in uninduced 4B6/3380/2941 cells (Fig. 7E). Even though mtDNA copy number in uninduced 4B6/3380/2941 cells was lower than that in the WT 4B6 cells, this difference is not likely to be due to the difference in the Lig3 expression. Indeed, both induced and uninduced 4B6/3380/2941 cells have reduced mtDNA copy number as compared with the WT 4B6 cells, even though LIG3 levels in the WT 4B6 and induced 4B6/3380/2941 cells are comparable (Fig. 7, B–E).
FIGURE 7.

Low levels of Lig3 are sufficient to support normal mtDNA replication and cellular respiration. A, PCR genotyping of the 4B6 cells and their derivative obtained by simultaneous deletion and complementation of the Lig3. Note the presence of mtDNA in both induced (Ind) and uninduced (Unind) cells. B, quantitation of the mtDNA copy number in the 4B6 cell line and its deleted/complemented derivative (4B6/3380/2941) grown in the presence (Ind) or absence (Unind) of doxycycline (inducer). C, Lig3 expression in the 4B6 cell line and its deleted/complemented derivative 4B6/3380/2941, induced (Ind) and uninduced (Unind). Note that Lig3 in rv3380 is tagged with Myc tag. D, estimation of the relative levels of the Lig3 in induced and uninduced 4B6 cells with deletion/complementation. MnSOD, loading control. E, relative levels of the mitochondrial Lig3 in the WT 4B6 cells versus uninduced deleted/complemented (4B6/3380/2941) cells. F, mitochondrial respiratory profiles of the induced and uninduced Lig3 deleted/complemented 4B6 cells. Labels as in Fig. 3B. Oxygen consumption expressed in picomoles of O2/min/μg of protein. Error bars, S.E.; OCR, oxygen consumption rate.
Low Levels of LIG3 Increase Susceptibility to Alkylating and Oxidative Damage, Increase mtDNA Degradation in Response to Oxidative Stress, and Delay Recovery of mtDNA Levels after Depletion
LIG3, the only mitochondrial DNA ligase, catalyzes the final step in both SSB repair and BER in mitochondria. Because mtDNA integrity has been implicated in cell viability (35–42), it is conceivable that reduced LIG3 levels may sensitize cells to agents like MMS and H2O2, which produce lesions in mtDNA that are repaired by either BER or SSB repair pathways. Indeed, uninduced cells, which expressed less than 1% of LIG3 as compared with induced cells, were more sensitive to both MMS and H2O2 (Fig. 8, A and B). Moreover, they demonstrated more DSB and mtDNA degradation in response to oxidative stress (Fig. 8, C and D). LIG3 is also the only ligase available for the replication of mtDNA. Therefore, we examined the effect of reduced LIG3 expression on the recovery of mtDNA copy number after depletion with EtBr. After 3 days of exposure to 5 μg/ml EtBr, mtDNA copy number declined to similar levels in both induced and uninduced cells. However, after the removal of EtBr, the dynamics of mtDNA copy number per cell differed significantly between cells with high and low LIG3 levels. In cells with high LIG3 expression, mtDNA copy number remained stable for the first 24 h after the removal of EtBr, and then began to rise quickly, essentially restoring mtDNA copy number within 4 days. In contrast, in uninduced cells, mtDNA copy number continued to drop for the first 24 h after EtBr withdrawal, and then remained stable for another 48 h. Only 3 days after EtBr removal did mtDNA copy number began to rise in these cells, and even then at a slower rate than in induced cells (Fig. 8E). Cells with low levels of LIG3 grew somewhat faster than their induced counterparts during depletion. This trend continued for the first 24 h after EtBr removal, but then the growth rate of induced cells surpassed that of uninduced cells (Fig. 8F). Examination of the total mtDNA copy number in the population (an extrapolated product of cell number and mtDNA copy number per cell) revealed that for the first 24 h after the removal of EtBr, the total mtDNA copy number in the population of uninduced cells continued to decline, indicating mtDNA degradation. In contrast, the level of mtDNA in the population of induced cells remained stable during this period. Beyond 24 h of EtBr removal, the total mtDNA copy number in both populations grew, although it grew faster in the population of induced cells (Fig. 8, G and H).
FIGURE 8.

Low levels of Lig3 increase susceptibility to alkylating and oxidative damage, increase mtDNA degradation in response to oxidative stress, and delay recovery of mtDNA levels after depletion. A and B, survival and growth of induced and uninduced Lig3 deleted/complemented 4B6 cells (4B6/3380/2941) in response to alkylating and oxidative damage, respectively. Error bars, S.E., n = 6, two-tailed Student's t test assuming unequal variance. Cont, control. C and D, frequency of strand breaks and extent of mtDNA degradation, respectively, in induced and uninduced 4B6/3380/2941 cells in response to treatment with H2O2. Error bars, S.E., n = 3 or 4, two-tailed Student's t test assuming unequal variance. ns, not significant, *, p < 0.05; **, p < 0.01; ***, p < 0.001. E–H, dynamics of mtDNA copy number per cell, cell growth, dynamics of the total mtDNA copy number in the population, and the ratios of the total mtDNA copy numbers in the populations of induced and uninduced cells at different time points during depletion-repletion, respectively. Induced (Ind) and uninduced (Unind) cells were subjected to 3 days of mtDNA depletion with 5 μg/ml EtBr (grayed areas on the graphs) followed by release. Error bars, S.E.; n = 3 for 0-h time point and n = 5 for remaining time points. Aliquots of cells were withdrawn at different time points, followed by determination of the total cell number as well as mtDNA copy number per cell. Note that induced and uninduced cells have similar starting mtDNA copy numbers and deplete with similar kinetics, yet show different repopulation kinetics upon release.
DISCUSSION
Experimental evidence collected so far points to the possibility that LIG3 is essential for cellular viability due to the role it plays in mitochondria (12). Indeed, previous attempts to isolate viable cells from whole body knock-out embryos (18), as well as attempts to inactivate both Lig3 alleles in mouse embryonic stem cells (20) or in DT40 cells (21), all failed. Here, we report the generation, in vitro, of viable MEFs that carry a deletion in both alleles of Lig3. These cells are completely devoid of mtDNA and die, as do other ρ0 cells (34), unless the medium is supplemented with uridine and pyruvate. This may explain why previous attempts to isolate viable Lig3−/− cells were unsuccessful (18–21). Consistent with previous findings (20), the loss of mtDNA was readily prevented by introducing, simultaneously with Lig3 deletion, a retrovirus-encoded LIG3 or LIG1 that was engineered to localize exclusively to mitochondria, but not by introducing an enzymatically inactive mitochondrial or a nuclear LIG3.
Unexpectedly, both HeLa and MEF cells severely depleted of LIG3 were able maintain normal mtDNA copy number, membrane potential, and respiration. This indicates that steady-state levels of LIG3 are excessive for routine mtDNA maintenance and contradicts a previous report that the decrease in LIG3 level leads to a reduction in mtDNA copy number (43). The reason for this discrepancy is not clear, but it is worth noting that in a previous study an observed molecular weight of LIG3 (approximately 80 kDa) was considerably less than predicted (113 kDa, UniProtKB P49916) and that when quantitating, no attempt was made to normalize mtDNA to nDNA.
After depletion with EtBr, cells with reduced LIG3 expression demonstrated slow growth, an extended period of mtDNA degradation, a delayed recovery of mtDNA copy number, and a slower mtDNA replication. Taken together, these findings independently factor in on the debate pertaining to the mechanism of mtDNA replication, which has remained the subject of controversy for over a decade. Two models have been proposed as follows: the strand-asynchronous “Clayton model” in which replication is initiated at the origin of the heavy strand and proceeds ⅔ of the mtDNA circle prior to exposing the origin of the light strand and initiating replication of the second strand (44). The strand-asynchronous model does not involve synthesis (and hence ligation) of Okazaki fragments and therefore predicts a minimum requirement for LIG3. The alternative strand-coupled model (45) involves a traditional replication fork with continuously synthesized leading strand and lagging strand synthesized through Okazaki fragments. This model has a greater reliance on LIG3 activity because it necessitates ligation of Okazaki fragments. Our observations favor both strand-asynchronous and bimodal mechanisms for mtDNA replication over the strand-coupled model. The bimodal mechanism was first proposed (46) and later abandoned by the authors in favor of the strand-coupled model (45). It postulates that during routine maintenance, mtDNA replicates predominantly according to the strand-asynchronous mechanism, while during periods of recovery of mtDNA copy number from depletion, replication occurs predominantly according to the strand-coupled mechanism. Therefore, the bimodal model predicts only a minimal requirement for LIG3 to circularize mtDNA after the completion of replication during routine maintenance and a higher reliance for mtDNA replication on LIG3 during recovery from depletion due to the need to ligate Okazaki fragments in the lagging strand. We propose that the apparently excessive steady-state levels of LIG3 observed in cells are maintained to efficiently cope with various eventualities, such as degradation of mtDNA due to oxidative damage (47). Such a loss would have to be compensated for by mtDNA replication, which requires higher levels of LIG3.
HeLa cells with reduced expression of LIG3 showed increased mtDNA degradation in response to H2O2 but not to MMS. Similarly, reduced levels of LIG3 were associated with increased H2O2-induced mtDNA degradation in MEFs. This may reflect the fact that H2O2 can induce DNA strand breaks directly, whereas MMS-induced strand breaks are predominantly indirect and are intermediates of BER. Also, oxidative damage to deoxyribose generates a distinct lesion, deoxyribonolactone, which may inhibit BER by suicide trapping of the DNA polymerase γ and require processing by long patch BER (8, 48). Overwhelming mitochondrial BER machinery with such unprocessed lesions in cells with reduced LIG3 expression may lead to mtDNA degradation (49). In both HeLa cells and MEF, reduced levels of LIG3 were associated with increased susceptibility to MMS and H2O2, agents that induce lesions repaired by BER. This observation is consistent with the essential role played by LIG3 in the mitochondrial BER.
Overall, our observations indicate that mitochondrial levels of LIG3 are excessive for routine mtDNA maintenance in cultured cells and that even though LIG3 is essential in vivo, Lig3−/− cells can be successfully generated and propagated in vitro. Also, they suggest that LIG3 inhibitors can be used to sensitize cancer cells to chemotherapy with DNA-alkylating drugs or drugs that induce oxidative stress.
Acknowledgments
We acknowledge the excellent technical assistance by Victoriya Pastukh, Luanne Oliveira, and Larysa Yuzefovich. The purchase and operation of the Nikon A1R confocal system was supported by National Institutes of Health Grant S10RR027535.
This work was supported, in whole or in part, by National Institutes of Health Grants ES03456, PO1 HL66299, OD010944, NS37956, CA21765, and CCSG (P30 CA21765).
- BER
- base excision repair
- DSB
- double strand break
- EtBr
- ethidium bromide
- Lig3
- DNA ligase III
- MEF
- mouse embryonic fibroblasts
- MMS
- methyl methanesulfonate
- MTS
- matrix targeting sequence
- QSBN
- quantitative Southern blotting under the nondenaturing conditions
- QSBA
- quantitative Southern blotting under alkaline (denaturing) conditions
- SSB
- single strand break
- qPCR
- quantitative PCR
- h
- human.
REFERENCES
- 1. Anderson S., Bankier A. T., Barrell B. G., de Bruijn M. H., Coulson A. R., Drouin J., Eperon I. C., Nierlich D. P., Roe B. A., Sanger F., Schreier P. H., Smith A. J., Staden R., Young I. G. (1981) Sequence and organization of the human mitochondrial genome. Nature 290, 457–465 [DOI] [PubMed] [Google Scholar]
- 2. Herrmann J. M., Longen S., Weckbecker D., Depuydt M. (2012) Biogenesis of mitochondrial proteins. Adv. Exp. Med. Biol. 748, 41–64 [DOI] [PubMed] [Google Scholar]
- 3. Poulton J., Holt I. J. (2009) 163rd ENMC International Workshop: Nucleoid and Nucleotide Biology in Syndromes of Mitochondrial DNA Depletion Myopathy, December 12–14, 2008, Naarden, The Netherlands Neuromuscul. Disord. 19, 439–443 [DOI] [PubMed] [Google Scholar]
- 4. Wallace D. C. (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alexeyev M. F. (2009) Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J. 276, 5768–5787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yakes F. M., Van Houten B. (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 94, 514–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu P., Demple B. (2010) DNA repair in mammalian mitochondria: Much more than we thought? Environ. Mol. Mutagen. 51, 417–426 [DOI] [PubMed] [Google Scholar]
- 9. Pettepher C. C., LeDoux S. P., Bohr V. A., Wilson G. L. (1991) Repair of alkali-labile sites within the mitochondrial DNA of RINr 38 cells after exposure to the nitrosourea streptozotocin. J. Biol. Chem. 266, 3113–3117 [PubMed] [Google Scholar]
- 10. LeDoux S. P., Wilson G. L., Beecham E. J., Stevnsner T., Wassermann K., Bohr V. A. (1992) Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis 13, 1967–1973 [DOI] [PubMed] [Google Scholar]
- 11. Driggers W. J., LeDoux S. P., Wilson G. L. (1993) Repair of oxidative damage within the mitochondrial DNA of RINr 38 cells. J. Biol. Chem. 268, 22042–22045 [PubMed] [Google Scholar]
- 12. Simsek D., Jasin M. (2011) DNA ligase III: a spotty presence in eukaryotes, but an essential function where tested. Cell Cycle 10, 3636–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Svilar D., Goellner E. M., Almeida K. H., Sobol R. W. (2011) Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 14, 2491–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cotner-Gohara E., Kim I. K., Hammel M., Tainer J. A., Tomkinson A. E., Ellenberger T. (2010) Human DNA ligase III recognizes DNA ends by dynamic switching between two DNA-bound states. Biochemistry 49, 6165–6176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katyal S., McKinnon P. J. (2011) Disconnecting XRCC1 and DNA ligase III. Cell Cycle 10, 2269–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lakshmipathy U., Campbell C. (1999) The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol. Cell. Biol. 19, 3869–3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perez-Jannotti R. M., Klein S. M., Bogenhagen D. F. (2001) Two forms of mitochondrial DNA ligase III are produced in Xenopus laevis oocytes. J. Biol. Chem. 276, 48978–48987 [DOI] [PubMed] [Google Scholar]
- 18. Puebla-Osorio N., Lacey D. B., Alt F. W., Zhu C. (2006) Early embryonic lethality due to targeted inactivation of DNA ligase III. Mol. Cell. Biol. 26, 3935–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao Y., Katyal S., Lee Y., Zhao J., Rehg J. E., Russell H. R., McKinnon P. J. (2011) DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 471, 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simsek D., Furda A., Gao Y., Artus J., Brunet E., Hadjantonakis A. K., Van Houten B., Shuman S., McKinnon P. J., Jasin M. (2011) Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 471, 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arakawa H., Bednar T., Wang M., Paul K., Mladenov E., Bencsik-Theilen A. A., Iliakis G. (2012) Functional redundancy between DNA ligases I and III in DNA replication in vertebrate cells. Nucleic Acids Res. 40, 2599–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Desjardins P., de Muys J. M., Morais R. (1986) An established avian fibroblast cell line without mitochondrial DNA. Somat. Cell Mol. Genet. 12, 133–139 [DOI] [PubMed] [Google Scholar]
- 23. King M. P., Attardi G. (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503 [DOI] [PubMed] [Google Scholar]
- 24. Inoue K., Ito S., Takai D., Soejima A., Shisa H., LePecq J. B., Segal-Bendirdjian E., Kagawa Y., Hayashi J. I. (1997) Isolation of mitochondrial DNA-less mouse cell lines and their application for trapping mouse synaptosomal mitochondrial DNA with deletion mutations. J. Biol. Chem. 272, 15510–15515 [DOI] [PubMed] [Google Scholar]
- 25. Okamaoto M., Ohsato T., Nakada K., Isobe K., Spelbrink J. N., Hayashi J., Hamasaki N., Kang D. (2003) Ditercalinium chloride, a pro-anticancer drug, intimately associates with mammalian mitochondrial DNA and inhibits its replication. Curr. Genet. 43, 364–370 [DOI] [PubMed] [Google Scholar]
- 26. Segal-Bendirdjian E., Coulaud D., Roques B. P., Le Pecq J. B. (1988) Selective loss of mitochondrial DNA after treatment of cells with ditercalinium (NSC 335153), an antitumor bis-intercalating agent. Cancer Res. 48, 4982–4992 [PubMed] [Google Scholar]
- 27. Kukat A., Kukat C., Brocher J., Schäfer I., Krohne G., Trounce I. A., Villani G., Seibel P. (2008) Generation of rho0 cells utilizing a mitochondrially targeted restriction endonuclease and comparative analyses. Nucleic Acids Res. 36, e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rappa G., Anzanello F., Alexeyev M., Fodstad O., Lorico A. (2007) γ-Glutamylcysteine synthetase-based selection strategy for gene therapy of chronic granulomatous disease and graft-versus-host disease. Eur. J. Haematol. 78, 440–448 [DOI] [PubMed] [Google Scholar]
- 29. Alexeyev M. F., Fayzulin R., Shokolenko I. N., Pastukh V. (2010) A retro-lentiviral system for doxycycline-inducible gene expression and gene knockdown in cells with limited proliferative capacity. Mol. Biol. Rep. 37, 1987–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Driggers W. J., Holmquist G. P., LeDoux S. P., Wilson G. L. (1997) Mapping frequencies of endogenous oxidative damage and the kinetic response to oxidative stress in a region of rat mtDNA. Nucleic Acids Res. 25, 4362–4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pallotti F., Lenaz G. (2007) in Mitochondria (Pon L. A., Scon E. A., eds) 2nd Ed., pp. 4–44, Academic Press, San Diego [Google Scholar]
- 32. Wyatt M. D., Pittman D. L. (2006) Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 19, 1580–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pastukh V., Shokolenko I. N., Wilson G. L., Alexeyev M. F. (2008) Mutations in the passenger polypeptide can affect its partitioning between mitochondria and cytoplasm: Mutations can impair the mitochondrial import of DsRed. Mol. Biol. Rep. 35, 215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hashiguchi K., Zhang-Akiyama Q. M. (2009) Establishment of human cell lines lacking mitochondrial DNA. Methods Mol. Biol. 554, 383–391 [DOI] [PubMed] [Google Scholar]
- 35. Shokolenko I. N., Alexeyev M. F., Robertson F. M., LeDoux S. P., Wilson G. L. (2003) The expression of exonuclease III from E. coli in mitochondria of breast cancer cells diminishes mitochondrial DNA repair capacity and cell survival after oxidative stress. DNA Repair 2, 471–482 [DOI] [PubMed] [Google Scholar]
- 36. Dobson A. W., Grishko V., LeDoux S. P., Kelley M. R., Wilson G. L., Gillespie M. N. (2002) Enhanced mtDNA repair capacity protects pulmonary artery endothelial cells from oxidant-mediated death. Am. J. Physiol. Lung Cell. Mol. Physiol. 283, L205–L210 [DOI] [PubMed] [Google Scholar]
- 37. Dobson A. W., Xu Y., Kelley M. R., LeDoux S. P., Wilson G. L. (2000) Enhanced mitochondrial DNA repair and cellular survival after oxidative stress by targeting the human 8-oxoguanine glycosylase repair enzyme to mitochondria. J. Biol. Chem. 275, 37518–37523 [DOI] [PubMed] [Google Scholar]
- 38. Cai S., Xu Y., Cooper R. J., Ferkowicz M. J., Hartwell J. R., Pollok K. E., Kelley M. R. (2005) Mitochondrial targeting of human O6-methylguanine DNA methyltransferase protects against cell killing by chemotherapeutic alkylating agents. Cancer Res. 65, 3319–3327 [DOI] [PubMed] [Google Scholar]
- 39. Druzhyna N. M., Hollensworth S. B., Kelley M. R., Wilson G. L., Ledoux S. P. (2003) Targeting human 8-oxoguanine glycosylase to mitochondria of oligodendrocytes protects against menadione-induced oxidative stress. Glia 42, 370–378 [DOI] [PubMed] [Google Scholar]
- 40. Rinne M., Caldwell D., Kelley M. R. (2004) Transient adenoviral N-methylpurine DNA glycosylase overexpression imparts chemotherapeutic sensitivity to human breast cancer cells. Mol. Cancer Ther. 3, 955–967 [PubMed] [Google Scholar]
- 41. Rinne M. L., He Y., Pachkowski B. F., Nakamura J., Kelley M. R. (2005) N-Methylpurine DNA glycosylase overexpression increases alkylation sensitivity by rapidly removing non-toxic 7-methylguanine adducts. Nucleic Acids Res. 33, 2859–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tann A. W., Boldogh I., Meiss G., Qian W., Van Houten B., Mitra S., Szczesny B. (2011) Apoptosis induced by persistent single-strand breaks in the mitochondrial genome: Critical role of EXOG (5′ EXO/Endonuclease) in their repair. J. Biol. Chem. 286, 31975–31983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lakshmipathy U., Campbell C. (2001) Antisense-mediated decrease in DNA ligase III expression results in reduced mitochondrial DNA integrity. Nucleic Acids Res. 29, 668–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tapper D. P., Clayton D. A. (1981) Mechanism of replication of human mitochondrial DNA. Localization of the 5′ ends of nascent daughter strands. J. Biol. Chem. 256, 5109–5115 [PubMed] [Google Scholar]
- 45. Bowmaker M., Yang M. Y., Yasukawa T., Reyes A., Jacobs H. T., Huberman J. A., Holt I. J. (2003) Mammalian mitochondrial DNA replicates bidirectionally from an initiation zone. J. Biol. Chem. 278, 50961–50969 [DOI] [PubMed] [Google Scholar]
- 46. Holt I. J., Lorimer H. E., Jacobs H. T. (2000) Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell 100, 515–524 [DOI] [PubMed] [Google Scholar]
- 47. Shokolenko I., Venediktova N., Bochkareva A., Wilson G. L., Alexeyev M. F. (2009) Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 37, 2539–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kazak L., Reyes A., Holt I. J. (2012) Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 13, 659–671 [DOI] [PubMed] [Google Scholar]
- 49. Shokolenko I., LeDoux S., Wilson G., Alexeyev M. (2011) in DNA Repair: On the Pathways to Fixing DNA Damage and Errors (Storici F., ed) pp. 339–356, InTech., New York [Google Scholar]