

Background: Listeria monocytogenes has two putative agmatine deiminase homologs, AguA1 and AguA2.
Results: Only AguA1, but not AguA2, acts as functional agmatine deiminase and mediates acid tolerance in L. monocytogenes.
Conclusion: Provided is the first biological insight into the roles of AgDI in acid tolerance of L. monocytogenes.
Significance: We have discovered a novel residue Gly-157 other than the known catalytic triad (Cys-His-Glu/Asp) critical for L. monocytogenes AgDI activity.
Keywords: Enzyme Kinetics, Enzyme Mechanisms, Microbiology, Site-directed Mutagenesis, Stress Response, Listeria monocytogenes, Acid Tolerance, Agmatine Deiminase, Biochemical Characterization, Kinetic Properties
Abstract
Listeria monocytogenes is adaptable to low pH environments and therefore crosses the intestinal barrier to establish systemic infections. L. monocytogenes aguA1 and aguA2 encode putative agmatine deiminases (AgDIs) AguA1 and AguA2. Transcription of aguA1 and aguA2 was significantly induced at pH 5.0. Deletion of aguA1 significantly impaired its survival both in gastric fluid at pH 2.5 and in mouse stomach, whereas aguA2 deletion did not show significant defect of survival in gastric fluid. With agmatine as the sole substrate, AguA1 expressed in Escherichia coli was optimal at 25 °C and over a wide range of pH from 3.5 to 10.5. Recombinant AguA2 showed no deiminase activity. Site-directed mutagenesis revealed that all nine AguA1 mutants completely lost enzymatic activity. AguA2 acquired AgDI activity only when Cys-157 was mutated to glycine. AguA1 mutation at the same site, G157C, also inactivated the enzyme. Thus, we have discovered Gly-157 as a novel residue other than the known catalytic triad (Cys-His-Glu/Asp) in L. monocytogenes that is critical for enzyme activity. Of the two putative AgDIs, we conclude that only AguA1 functionally participates in the AgDI pathway and mediates acid tolerance in L. monocytogenes.
Introduction
Listeria monocytogenes is a Gram-positive food-borne pathogen that causes listeriosis with high mortality (1, 2). The bacterium is resistant to acidic environments where it encounters acidic food, such as processing environments, the stomach, and phagosomes of macrophages (3, 4). Agmatine deiminase (AgDI)2 pathway is an acid tolerance mechanism described in bacterial species like Enterococcus faecalis or Streptococcus mutans (5, 6). AgDI converts agmatine into putrescine and ammonia upon catalyzation (5, 7, 8). Agmatine enters bacterial cells via an agmatine-putrescine antiporter (aguD), where it is hydrolyzed to N-carbamoylputrescine by agmatine deiminase (EC 3.5.3.12) (9). Putrescine carbamoyltransferase (EC 2.1.3.6), encoded by aguB, mediates phosphorolysis of N-carbamoylputrescine, producing putrescine and carbamoylphosphate (10). Finally, a carbamate kinase (EC 2.7.2.2) transfers the phosphate group from carbamoylphosphate to ADP (Fig. 1A) (11). This pathway is thought to play an important role in acid resistance and competitive survival of Lactobacillus brevis (8).
FIGURE 1.
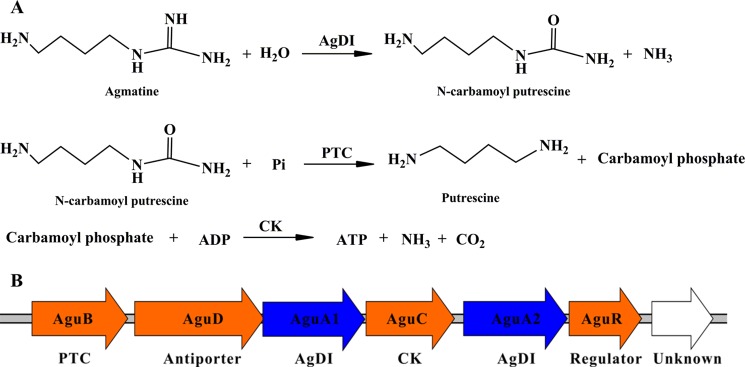
The AgDI pathway and organization of the gene cluster in L. monocytogenes. A, the AgDI pathway. Agmatine is first hydrolyzed to form N-carbamoylputrescine by AgDI. N-Carbamoylputrescine is then converted into putrescine and carbamoylphosphate by putrescine transcarbamoylase. Carbamate kinase (CK) then catalyzes the phosphate moiety in carbamoylphosphate to ADP, forming ATP as well as ammonia and CO2. B, genetic organization of the L. monocytogenes agmatine deiminase gene cluster.
AgDI is a member of the guanidinium-modifying enzyme family that hydrolyzes the guanidinium groups of agmatine, arginine, or methylarginine to form the ureido-containing derivatives (12). All enzymes of the guanidinium-modifying enzyme family contain a highly conserved catalytic triad (Cys-His-Glu/Asp), which plays a significant role in catalysis and substrate binding (12, 13).
The putative AgDI pathway in L. monocytogenes is encoded by the agu operon (14). There are two homologs, AguA1 and AguA2, coded for by aguA1 and aguA2, respectively (Fig. 1B). Sequence alignment of L. monocytogenes AgDI homologs to those from other species indicates that Asp-94, Glu-155, His-216, Asp-218, and Cys-356 might be the corresponding catalytic residues in both AguA1 and AguA2. Listerial AguA1 was initially annotated as a putative peptidyl-arginine deiminase (14). Homology analysis reveals that AguA1 is actually an AgDI of the guanidinium-modifying enzyme family. Our previous study showed that this molecule was involved in acid tolerance because deletion of aguA1 (lmo0038 in strain EGD) led to reduced survival in acidic brain heart infusion broth at pH 4.0 (15).
This study further characterized the biochemical and catalytic properties of two AgDI homologs of L. monocytogenes, AguA1 and AguA2, and their roles in acid stress response. We conclude that aguA1, but not aguA2 of L. monocytogenes, encodes a functional agmatine deiminase that contributes to acid tolerance, thus adding an additional acid stress response mechanism of L. monocytogenes other than the two well known glutamate decarboxylase and arginine deiminase (ADI) systems (10, 16–19).
MATERIALS AND METHODS
Chemicals
Agmatine sulfate, l-arginine monohydrochloride, l-citrulline, diacetyl-monoxime, and thiosemicarbazid were purchased from Sigma-Aldrich. N-Carbamoyl-putrescine was synthesized by the Institute of Pesticide and Environmental Toxicology or Zhejiang University. All other chemicals of analytical reagent grade were obtained from commercial sources.
Bacterial Strains, Plasmids, and Culture Conditions
L. monocytogenes 10403S was used as the wild-type strain. Escherichia coli DH5α was employed as the host strain for plasmids pMD18-T (Takara, Dalian, China), pET30a(+) (Merck), pERL3 (10) and pKSV7 (15, 20). L. monocytogenes was cultured in brain-heart infusion (BHI) medium (Oxoid, Hampshire, England). E. coli DH5α and Rosetta (DE3) (Merck) were grown at 37 °C in LB broth (Oxoid). Stock solutions of ampicillin (50 mg/ml), erythromycin (50 mg/ml), kanamycin (50 mg/ml), and chloramphenicol (50 mg/ml) were added to the medium, where appropriate, at required levels.
Bioinformatic Analysis
The amino acid sequences of AguA1 and AguA2 of L. monocytogenes and its homologs in other microbial species were obtained from the National Centre for Biotechnology Information database. The sequences were aligned with Clustal X. Phylogenetic tree was constructed using the neighbor-joining method. The known crystal structure of E. faecalis agmatine deiminase (Protein Data Bank code 2JER) was acquired from the Protein Data Bank. Putative models of AguA1 and AguA2 were constructed using SWISS-MODEL Workspace (21–23).
Transcriptional Analysis Using Quantitative PCR
L. monocytogenes 10403S was grown to the stationary phase (A600 = 0.6) in BHI broth at 37 °C and then exposed to acidic conditions (pH 5.0) and neutral conditions (pH 7.0), respectively, for 60 min. Total RNA was extracted using the TRIzol method, and cDNA was synthesized with reverse transcriptase (TOYOBO, Osaka, Japan). Quantitative PCR was then performed in 20-μl reaction mixtures containing SYBR quantitative PCR mix (TOYOBO) to measure transcriptional levels of aguA1 and aguA2 using an iCycler iQ5 real time PCR detection system (Bio-Rad) with specific primer pairs (supplemental Table S1). The housekeeping gene gyrB was used as an internal control for normalization as previously described (10). Relative transcription levels were quantified by the 2−ΔΔCT method and shown as relative fold changes in comparison with that at pH 7.0 (24). Transcriptional analysis was repeated for three times on each test condition.
Construction of Deletion Mutants
Genomic DNA of L. monocytogenes 10403S was extracted as described previously (25, 26). A homologous recombination strategy with SOE-PCR procedure was used for in-frame deletion to construct aguA1 and aguA2 single and double deletion mutants as described previously (15, 27). The DNA fragments containing homologous arms upstream and downstream of aguA1 or aguA2 were obtained by PCR amplification of 10403S DNA templates using the SOE primers (supplemental Table S1). Each of the SOE-PCR products with deletion of aguA1 or aguA2 was cloned into the temperature-sensitive shuttle vector pKSV7, which was then transformed into E. coli DH5α. The recombinant vectors containing the target gene deletion cassettes were confirmed by sequencing. A previous protocol was followed for deletion of the targeted genes via allelic exchange (27). Briefly, the competent L. monocytogenes 10403S cells were electroporated with one of the vector constructs. Transformants were grown at a nonpermissive temperature (41 °C) in BHI medium containing chloramphenicol (10 μg/ml) to promote chromosomal integration, and the homologous recombinants were passaged successively in BHI medium without antibiotic at a permissive temperature (30 °C) to enable plasmid excision and curing (28). The recombinants were identified as chloramphenicol-sensitive colonies and confirmed by PCR and DNA sequencing. The single deletion mutants ΔaguA1 and ΔaguA2 were generated initially. The mutant strain ΔaguA1 was used in a second round of mutagenesis to construct the ΔaguA1ΔaguA2 double deletion mutants.
Complementation of aguA1 Deletion
To complement the L. monocytogenes ΔaguA1 strain, the aguA1 ORF with its promoter region was amplified from 10403S using primer pairs aguA1-w/x and aguA1-y/z by SOE-PCR (27) (supplemental Table S1). After restriction with appropriate enzymes, the PCR fragment was cloned into pERL3. The resulting plasmid was then electroporated into L. monocytogenes ΔaguA1. The regenerated cells were plated on BHI agar containing erythromycin (5 μg/ml). The complemented strain was designated as L. monocytogenes ΔaguA1+aguA1.
Survival in Synthetic Gastric Fluid
Stationary phase cultures of L. monocytogenes 10403S, mutants (ΔaguA1, ΔaguA2, and ΔaguA1ΔaguA2), and complemented strain ΔaguA1+aguA1 were harvested, washed in PBS, and resuspended in synthetic human gastric fluid (8.3 g/liter of proteose peptone, 3.5 g/liter of d-glucose, 2.05 g/liter of NaCl, 0.6 g/liter of KH2PO4, 0.11 g/liter of CaCl2, 0.37 g/liter of KCl, 0.05 g/liter of bile, 0.1 g/liter of lysozyme, and 13.3 mg/liter of pepsin; adjusted to pH 2.5 with HCl) as described previously (16). After 30 min of incubation at 37 °C, the survival bacterial cells were plated onto BHI agar after appropriate dilutions. The plates were incubated at 37 °C for 24 h, and survival rates are reported as the means of three independent experiments, each performed in duplicate.
Survival in Mouse Stomach
ICR mice (20 ± 2 g, female) were divided into five groups (eight mice per group) and acclimated for 3 days before experiments. Feed was removed 12 h prior to intragastric inoculation. Two-hundred μl of listerial cells containing 4–6 × 108 CFU of wild-type, mutant, and complemented strains grown overnight in BHI medium at pH 7.0 was administered to mice of corresponding groups by intragastric inoculation using a ball-tipped gavage needle. At 1 h after inoculation, the stomach samples were separated and homogenized in sterile PBS (10 mm, pH 7.4). Serial dilutions of the homogenates were plated on PALCAM (Listeria selective medium) (Luqiao, Beijing, China) agar (29). The plates were incubated at 37 °C for 24 h for colony counting. A group of eight mice from the same batch was left uninoculated as controls that did not show visible colonies on PALCAM agar. The results were expressed as means ± S.E. of log10 CFU per stomach for each group. The animal experiments were approved by the Laboratory Animal Management Committee of Zhejiang University (approval no. 20111025).
Prokaryotic Expression and Purification of AguA1 and AguA2
AguA1 and AguA2 were expressed as fusion proteins to the N-terminal His tag using pET30a(+) as the expression vector. E. coli Rosetta (DE3) was used as the expression host. The full-length ORFs of aguA1 and aguA2 were amplified with primer pairs aguA1-exp-F/R and aguA2-exp-F/R (supplemental Table S1), respectively, and then inserted into the pET30a(+) vector after restriction digestion. The resulting plasmids were designated as pET-aguA1 and pET-aguA2. E. coli cells harboring the recombinant plasmids were grown in 500 ml of LB medium supplemented with 50 μg/ml kanamycin at 37 °C until the cultures reached 0.6–0.8 at A600. Isopropyl β-d-1-thiogalactopyranoside was then added to a final concentration of 0.4 mm to induce expression of AguA1 and AguA2 for 12 h at 15 °C.
The His-tagged fusion proteins AguA1 and AguA2 were purified using the nickel-chelated affinity column chromatography. Briefly, isopropyl β-d-1-thiogalactopyranoside-induced cell pellets were collected, resuspended in 50 mm PBS (pH 7.4), and disrupted with 100 cycles of sonication at 300 W for 5 s with intermittent cooling on ice for 10 s (25 min in total). After centrifugation at 12,000 × g for 20 min, the supernatant samples were collected and loaded onto a 2-ml prepacked nickel-chelated agarose gel column (Weishi-Bohui Chromtotech Co., Beijing, China). The columns were washed with PBS containing 500 mm NaCl and 30 mm imidazole, and the bound proteins were eluted with a linear gradient of 25–500 mm imidazole prepared in the same buffer. Expression and purification of recombinant proteins was analyzed by 12% SDS-PAGE followed by Coomassie Brilliant Blue staining. Protein concentration was quantified using the Bradford method for AgDI activity and kinetic assays. The purified enzyme AguA1 was stored in 50% glycerol at −80 °C.
Site-directed Mutagenesis
To identify the predicted active sites of AguA1 and AguA2, single site-directed mutants D94A, D94E, E155A, E155D, H216A, H216R, D218A, D218E, G157C, and C356A mutants for AguA1 and Y47F, C157G, F196L, N236D, and H360Q for AguA2 were generated on the vector template pET-aguA1 or pET-aguA2 using the QuikChange site-directed mutagenesis kit (Agilent, Santa Clara, CA) and the oligonucleotide primers (supplemental Table S2). The template DNA was removed by digestion with DpnI (TOYOBO) for 2 h at 37 °C. All mutants were sequenced to ensure that only the desired single mutations had been incorporated correctly into the wild-type expression constructs. The mutant proteins were expressed and purified as described above.
Enzyme Kinetic Assays of AguA1 and AguA2
The AgDI activity was assayed by monitoring production of ureido-containing compounds (30–33). Ureido groups (i.e., citrulline and N-carbamoylputrescine) could react with diacetyl-monooxime under strong acidic conditions to generate a colorimetric chemical that absorbs at 530 nm. The coloring reagent consists of 1 volume of solution A and 3 volumes of solution B (solution A: 80 mm diacetyl-monoxime (Sigma) and 2.0 mm thiosemicarbazide (Sigma); and solution B: 3 m H3PO4, 6 m H2SO4, and 2 mm NH4Fe(SO4)2) (32). N-Carbamoylputrescine or citrulline formation was quantified from the calibration curves of reference chemicals at A530 nm.
The Michaelis-Menten kinetic curve was determined by incubating 2.5 μm purified enzyme with varying amounts of agmatine (0–10 mm) in the assay buffer (100 mm potassium phosphate buffer, pH 7.5; final reaction volume, 500 μl). The reaction was initiated by adding the enzyme and terminated by the addition of 100 μl of 50% trichloroacetic acid (Sigma) after incubation at 25 °C for 30 min. Volumes of 120-μl reaction samples were then mixed with 400 μl of the coloring reagent. The mixtures were boiled at 100 °C for 5 min and then cooled down to room temperature for absorbance measurement at A530 nm. Product formation was quantified from the calibration curves as described above. Enzymatic reaction rates (v0) versus substrate concentrations were fitted to a Michaelis-Menten equation (Equation 1) by a nonlinear regression method, and the values Km, Vmax, and kcat were determined by fitting the data to the equation using the program GraphPad Prism (GraphPad Software, San Diego, CA),
where [S] is the substrate concentration, v0 is the initial velocity, Vmax is the maximum velocity, and Km is the Michaelis-Menten constant for the substrate. The kcat value was calculated from Vmax, and the enzyme concentration [E] was calculated using the equation kcat = Vmax/[E]. One unit of enzyme activity corresponds to production of 1 μm N-carbamoylputrescine min−1 or citrulline under the assay conditions.
Metal Inhibition and IC50 Determination
The effects of metal ions Co2+, Cu2+, Fe3+, Mg2+, Zn2+, and Ni2+ on AguA1 activity were determined by preincubating the enzyme with varying concentrations (0–10 mm) of each metal ion for 30 min at 4 °C. The remaining procedures were the same as above. The 50% inhibitory concentration (IC50) is defined as the concentration of metal ions required for 50% decrease of maximum enzyme activity. The percentage of inhibition versus the logarithm of inhibitor concentrations was plotted, and the IC50 values were determined by using the program GraphPad Prism.
Substrate Specificity and Competitive Assay
To determine the substrate specificity of AguA1, arginine was used as another guanidinium-containing compound to test for its ability to act as the substrate or competitive binding inhibitor. The reaction was initiated by adding 10 mm agmatine and varying concentrations (0–10 mm) of arginine into the assay buffer (100 mm potassium phosphate buffer, pH 7.5; 500 μl of final volume) containing 2.5 μm enzyme. The remaining procedures were the same as above.
Statistical Analysis
All data comparisons were analyzed using the two-tailed homoscedastic Student's t test. In all cases, differences with p values of <0.05 were considered as statistically significant. The GraphPad Prism 5 program was used for nonlinear fitting to the Michaelis-Menten equation.
RESULTS
Amino Acid Sequence Analysis of AguA1 and AguA2
The two putative agmatine deiminases AguA1 and AguA2 of L. monocytogenes share a high level of amino acid sequence identity (67.7%) and are clustered together in the same branch. However, they form a sister branch to AgDIs from other species with 23.9–73.6% amino acid sequence identity (Fig. 2A).
FIGURE 2.

Bioinformatic analysis of AguA1 and AguA2. A, phylogenetic tree of AguA1/AguA2 and homologs from other bacterial species. The tree was constructed by the neighbor-joining program, and a bootstrap test of 100 replicates was used to estimate the confidence of branching patterns, where the numbers on internal nodes are the support values. B, amino acid sequence alignment of AguA1/AguA2 against homologs from A. thaliana, H. pylori, S. mutans, E. faecalis, L. lactis, and P. aeruginosa. The conserved motifs and residues are shaded. The key amino acid residues noted with asterisks are involved in substrate binding and enzyme activity. The boxed residues are different between AguA2 and AguA1 or other AgDIs.
Agmatine deiminases are members of the guanidinium-modifying family enzymes, a family that includes enzymes that hydrolyze the guanidinium groups of arginine, peptidyl-arginine, methylarginine, and agmatine to form the ureido-containing derivatives (12). A catalytic triad (Cys-His-Glu/Asp), which plays a significant role in substrate binding and catalytic activity, has been identified from the crystal structures of known ADIs (34–36). Although AgDIs show homology to ADIs with similar structures and reaction mechanisms, they differ in their substrates (5, 8, 30). Alignment with AgDIs from other species indicates that Asp-94, Glu-155, His-216, Asp-218, and Cys-356 are the corresponding catalytic residues of L. monocytogenes AguA1 and AguA2 (Fig. 2B). However, there are five specific residues (Tyr-47, Cys-157, Phe-196, Asn-236, and His-360) of AguA2 differing from other AgDIs including AguA1, which might affect its agmatine deiminase activity (Fig. 2B). We modeled the structures of AguA1 and AguA2 using the crystal structure of E. faecalis AgDI (Protein Data Bank code 2JER) as the template (supplemental Fig. S1) (5). The predicted protein structures of AguA1 and AguA2 show high similarity to E. faecalis AgDI (supplemental Fig. S1), including a fanlike structure with five blades that result from a 5-fold pseudosymmetric structure in which each repeating element consists of a three-stranded mixed β sheet and a helix in a ββαβ arrangement (5, 30). These results suggest that AguA1 and AguA2 of L. monocytogenes are typical AgDIs that might exhibit agmatine deimination activity.
AguA1, but Not AguA2, Mediates Acid Tolerance of L. monocytogenes
To investigate whether aguA1 and aguA2 were induced under acidic conditions, quantitative RT-PCR was conducted to assess relative gene transcription levels in response to acidic stress at pH 5.0. Transcription of both aguA1 and aguA2 was about 13- and 15-fold, higher at acidic pH 5.0 than at pH 7.0 with the statistical p values of 0.018 and 0.016, respectively (Fig. 3A).
FIGURE 3.
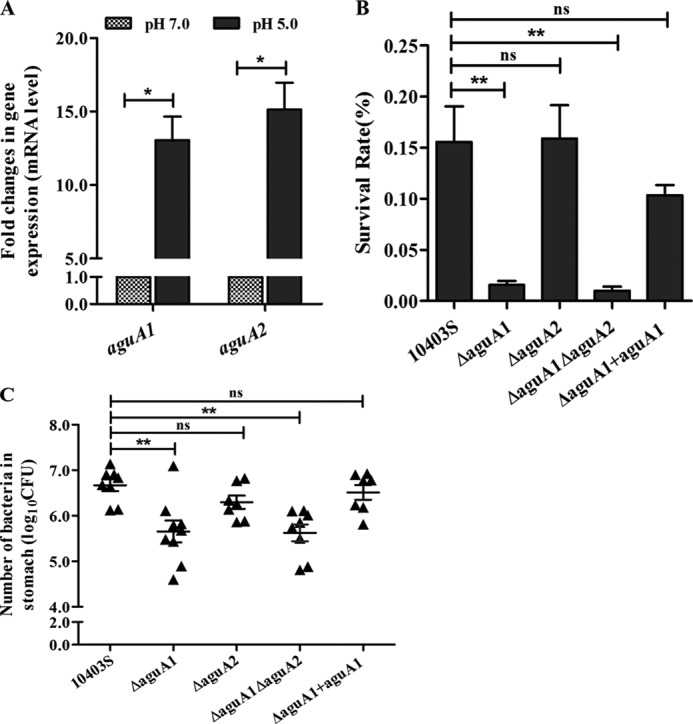
Responses or roles of aguA1 and aguA2 under acidic stresses. A, relative quantification of aguA1 and aguA2 mRNA levels in L. monocytogenes 10403S exposed to BHI medium at pH 5.0 and 7.0. Values are expressed as means ± S.D. B, survival of L. monocytogenes 10403S, its mutant, and complement strains in synthetic human gastric fluid at pH 2.5. Values are expressed as means ± S.D. C, survival of L. monocytogenes 10403S, its mutant, and complement strains in the stomach of mice receiving intragastric inoculation. Values are expressed as means ±S.E. *, p < 0.05; **, p < 0.01; ns, p > 0.05.
To further determine whether aguA1 and aguA2 contribute to survival in acidic environments, L. monocytogenes 10403S, aguA1/aguA2 single and double mutants and aguA1 complement strain were exposed to synthetic gastric fluid at pH 2.5. Only the mutants ΔaguA1 and ΔaguA1ΔaguA2 exhibited significant reduction in the survival rate as compared with the parent and complemented strains (p < 0.01). Deletion of aguA2 did not significantly impair its survival (Fig. 3B). Furthermore, we found that only the mutants ΔaguA1 and ΔaguA1ΔaguA2 showed significantly lower survival in mice stomach than the parent strain 10403S (Log10 CFU: 5.7 and 5.5 versus 6.6, p < 0.01). Complementation of aguA1 restored its survival to a level (Log10 CFU: 6.5) similar to that of the parent strain (Fig. 3C). These findings suggest that only AguA1, but not AguA2, is involved in listerial resistance to acid stress, although both were induced at the mRNA level under the acidic condition.
Expression and Purification of AguA1 and AguA2
The two putative agmatine deiminase homologs AguA1 and AguA2 from L. monocytogenes were expressed in E. coli Rosetta (DE3) with apparent bands at expected molecular masses (46.8 kDa for AguA1 and 47.9 kDa for AguA2). His-tagged AguA1 and AguA2 were purified to homogeneity by nickel-chelated affinity column chromatography, yielding pure proteins (∼20 and ∼40 mg/liter culture for AguA1 and AguA2, respectively) seen as single bands on the SDS-PAGE gel (Fig. 4A).
FIGURE 4.

Gel electrophoresis of AguA1 and AguA2 expressed in E. coli and enzymatic characterization of AguA1. A, SDS-PAGE analysis of the recombinant AguA1 and AguA2 proteins. Lane M, molecular mass standards; lane 1, supernatant of E. coli harboring pET30a; lanes 2 and 3, purification of AguA1 and AguA2, respectively. B, time course analysis of AguA1 and AguA2 activity with 10 mm agmatine. C, effect of pH on AguA1 activity. D, effect of temperature on AguA1 activity. E, Michaelis-Menten plot of AguA1 determined by measuring the enzymatic velocity. F, substrate specificity and competitive assay of AguA1 in the presence of 10 mm of agmatine and varying concentration (0–10 mm) of arginine. ND, not detectable.
Optimal Conditions for Enzyme Activity
Enzymatic activities were tested by incubation of 2.5 μm of each enzyme in the reaction mixtures containing 10 mm agmatine or arginine as a substrate, and the product N-carbamoylputrescine or citrulline was measured at the indicated time points. AguA1 produced increasing amounts of N-carbamoylputrescine that was linear until 20 min and reached the highest level after 30 min of incubation (Fig. 4B). Specific activity was 2050 units/mg when calculated from the linear range in the presence of agmatine. No citrulline was formed when arginine was used as the substrate for AguA1. However, AguA2 showed no deiminase activity either on agmatine (Fig. 4B) or on arginine (data not shown) as the substrate, suggesting that only AguA1, but not AguA2, acts as the functional agmatine deiminase in L. monocytogenes.
AguA1 exhibited high agmatine deiminase activity in a wide range of pH conditions from 3.5 to 10.5 with optimal pH at 7.5 (Fig. 4C), suggesting that the activity of AguA1 is not sensitive to pH, which is consistent with its contribution to acid tolerance under low pH conditions (Fig. 3, B and C). However, AguA1 showed varying activities at different temperatures, optimal at 25 °C and the residual activity was 39 and 12% at 37 and 45 °C, respectively (Fig. 4D).
Kinetic Properties of AguA1
AguA1 was tested for its kinetic properties under the optimal conditions at 25 °C and pH 7.5 by incubating 2.5 μm of the enzyme in the reaction mixtures containing 10 mm agmatine as the substrate. Plotting of enzymatic velocity against substrate concentrations yielded a curve that fits the classical Michaelis-Menten model: Km, Vmax, kcat, and kcat/Km values of the enzyme were 0.65 ± 0.23 mm, 85.69 ± 7.58 μm/min, 34.28 ± 3.03 min−1, and 5.30 × 104 min−1 m−1, respectively (Table 1 and Fig. 4E). The Km value of AguA1 is similar to that for P. aeruginosa AgDI (0.6 mm) (37) but much higher than those for maize shoots (12 μm) (38), cucumber (16 μm) (39), Arabidopsis thaliana (112 μm) (40), E. faecalis (35 μm) (5), and Helicobacter pylori (33 μm) (30). However, the kcat/Km value we obtained was quite close to that of P. aeruginosa AgDI (4.2 × 105 min−1 m−1) (37), H. pylori AgDI (1.47 × 105 min−1 m−1) (30), and peptidyl-arginine deiminase 4 (3.0 × 105 min−1 m−1) (31), indicating that AguA1 is a functional AgDI enzyme with reasonable kinetic properties.
TABLE 1.
Steady-state kinetic constants measured for agmatine with wild-type and mutant AguA1 enzymes
| Enzyme | Vmax | kcat | Km | kcat/Km |
|---|---|---|---|---|
| μm/min | min−1 | mm | min−1 m−1 | |
| Wild-type | 85.69 ± 7.58 | 34.28 ± 3.03 | 0.65 ± 0.23 | 5.30 × 104 |
| D94A | Inactive | |||
| E155A | Inactive | |||
| H216A | Inactive | |||
| D218A | Inactive | |||
| C356A | Inactive | |||
| D94E | Inactive | |||
| E155D | Inactive | |||
| H216R | Inactive | |||
| D218E | Inactive | |||
| G157C | Inactive |
Substrate Specificity and Competitive Assay of AguA1
Agmatine has been identified as the sole and preferred substrate for AgDIs of soybean (41), H. pylori (30), P. aeruginosa (37), E. faecalis (5), and L. brevis (8). A variety of guanidinium-containing small molecules (agmatine analogs) fail to substitute for agmatine as substrates for this enzyme. To determine whether AguA1 also has similar substrate specificity, l-arginine, another guanidinium-containing compound, was used to see whether it could be utilized by AguA1. Fig. 4F shows that there was no formation of citrulline from l-arginine in the presence of AguA1, indicating that l-arginine does not act as the substrate for AguA1. Moreover, arginine showed no competitive effect on agmatine even at high concentrations up to 10 mm (Fig. 4F). Thus, AguA1 demonstrates distinct preference for agmatine, as is the case with other AgDIs.
Metal Inhibition of AguA1
Metal ions Cu2+, Zn2+, and Co2+ inhibited AguA1 with varying degrees of potency with respective IC50 of 0.034 ± 0.008 mm (Fig. 5A), 0.25 ± 0.05 mm (Fig. 5B), and 2.89 ± 0.82 mm (Fig. 5C), indicating that copper ion is the most inhibitory. There was no inhibitory effect on AguA1 activity with Fe3+, Mg2+, and Ni2+ ions, even at highest concentrations (10 mm) (data not shown). These results suggest that the active sites of AguA1 could be susceptible to such metal ions as Cu2+, Zn2+, and Co2+.
FIGURE 5.

Enzymatic stability of AguA1 in the presence of metal ions and at room temperature. A–C, effects of metal ions Cu2+ (A), Zn2+ (B), and Co2+ (C) up to 10 mm on AguA1 activity. The dotted lines represent 50% inhibition of the enzyme activity (IC50). D, effect of room temperature at 25 °C on stability of AguA1.
Thermostability of AguA1
The purified enzyme preparation, when stored at 4 °C, maintained activity for at least 1 month (85% activity retained). However, 80% of the activity was lost when it was exposed to 25 °C for 6 h (Fig. 5D), indicating that AguA1 is sensitive to temperature, and its purification should proceed under 4 °C.
Effect of Site-directed Mutagenesis on the Activities of AguA1 and AguA2
Bioinformatic analysis suggests that Asp-94, Glu-155, His-216, Asp-218, and Cys-356 could be the corresponding catalytic residues for AguA1 and AguA2 (Fig. 2B). These residues were substituted by site-directed mutagenesis. The mutant proteins were expressed and purified, and their enzymatic activity was characterized in a way identical to that of the wild-type enzyme. Mutant proteins at D94A, E155A, H216A, D218A, and C356A completely lost their ability to degrade agmatine (Fig. 6A). Mutants substituted with amino acids of similar biochemical properties at the same sites (D94E, E155D, H216R, and D218E) also lost the enzyme activity (Fig. 6A). Therefore, kinetic analysis was not applied to these mutant proteins (Table 1). These results indicate that the corresponding residues in L. monocytogenes AguA1 play a critical and irreplaceable role in its catalytic activity, consistent with those obtained for other AgDIs and ADIs (30, 34, 42, 43).
FIGURE 6.

Enzymatic activity of wild-type AguA1 and AguA2 and their mutant proteins. A, relative activity of wild-type AguA1 and its mutants (D94A/D94E, E155A/E155D, H216A/H216R, D218A/D218E, and C356A). B, enzymatic activity of wild-type AguA2 and its mutants (Y47F, C157G, F196L, N236D, and H360Q). C, enzymatic activity of wild-type AguA1 and its mutant G157C. ND, not detectable.
AguA2 did not show any deiminase activity (Fig. 4B), although it has high homology and similar crystal model to AguA1, including the five critical residues (Fig. 2B and supplemental Fig. S1). However, we found that there are five specific residues (Tyr-47, Cys-157, Phe-196, Asn-236, and His-360) of AguA2 that differ from AguA1 and other AgDIs (corresponding residues: Phe, Gly, Leu, Asp, and Gln) (Fig. 2B), which might account for the loss of its deiminase activity. To verify our hypothesis, site-directed mutant proteins at Y47F, C157G, F196L, N236D, and H360Q for AguA2 were generated, and their enzymatic activity was determined. Interestingly, we found that AguA2 acquired agmatine deiminase activity only when Cys-157 was mutated to Gly, whereas the other mutants were still inactive (Fig. 6B). We also confirmed that AguA1 with G157C mutation led to the complete loss of the enzyme activity (Fig. 6C). This is the first time that Gly-157 has been discovered as a key residue in addition to those known to be essential for AgDI activity. Protein BLAST did not show any mutation with Gly-157 from the available UniProt Protein database for L. monocytogenes, further confirming that AguA1 is the only functional AgDI in this bacterium.
DISCUSSION
The ability of Listeria monocytogenes to tolerate low pH environments is of importance to its pathogenicity because the pathogen encounters acidic environments in the nature and food processing industry in vitro and during passage through the stomach or even within macrophage phagosomes in vivo (44). A number of acid resistance mechanisms have been identified in L. monocytogenes, such as glutamate decarboxylase and ADI systems, and F1F0-ATPase (2, 37, 45). The AgDI pathway generally contains a panel of genes coding for AgDI, putrescine transcarbamoylase, carbamate kinase, and agmatine-putrescine exchanger (8). However, there are two putative AgDI homologs, AguA1 and AguA2, encoded by aguA1 and aguA2, respectively, in L. monocytogenes. This is also seen in L. brevis IOEB 9809, Lactobacillus sakei, and Pediococcus pentosaceus (8). AgDI is one of the three crucial enzymes constituting the AgDI pathway, which deiminates the substrate agmatine to produce N-carbamoylputrescine and NH3. Ammonia can combine with intracellular cytoplasmic protons to yield ammonium ions (NH4+), thereby alleviating acidification of the cytoplasm and contributing to pH homeostasis (6, 7, 46).
Initially, we found that both aguA1 and aguA2 were induced at the mRNA level when L. monocytogenes was subjected to low pH at 5.0, suggesting that both AguA1 and AguA2 could be involved in acid tolerance (Fig. 3A). However, only the ΔaguA1 mutant, but not the ΔaguA2 mutant, exhibited significant reduction of its survival both in synthetic human gastric fluid (pH 2.5) and mice stomach, as compared with its parent and complemented strains (Fig. 3, B and C). Enzyme assays revealed that L. monocytogenes AguA1 had typical substrate specificity of AgDI, whereas AguA2 showed no deiminase activity. However, we found a specific residue, Cys-157, of AguA2 differing from other AgDIs including AguA1 that could be involved in AgDI activity because the mutant AguA2 at C157G converted agmatine to N-carbamoylputrescine. To our surprise, single mutation of G157C of AguA1 led to the loss of its enzyme activity. Further bioinformatic search reveals that Gly-157 for AguA1 and Cys-157 for AguA2 are conserved in all L. monocytogenes sequences (total 68 sequences) available in UniProt protein database. Therefore, we come to the conclusion that AguA1 is the only functional AgDI that contributes to acid tolerance of L. monocytogenes. To our knowledge, This is the first time that Gly-157 has been uncovered as another key residue other than those previously found essential for AgDI activity.
The AgDI pathway closely resembles the ADI pathway. The two pathways consist of similar enzymes performing similar reactions but only differ in their substrates. ADI catalyzes deimination of l-arginine but not l-agmatine, whereas AgDI catalyzes deimination of agmatine but not l-arginine. Although ADI and AgDI are structurally divergent in the region of the active site that interacts with the CαH(NH3+)(COO−) region of the substrate, the regions that interact with the guanidino group of the substrate are quite similar (43). Despite their unique substrate specificity, both ADIs and AgDIs contain a highly conserved catalytic triad (Cys-His-Glu/Asp). In this study, site-directed mutagenesis indicates that Asp-94, Glu-155, His-216, Asp-218, and Cys-356 are the corresponding catalytic residues in L. monocytogenes AguA1 because single mutation at any one of these residues completely abolished the enzymatic activity (Table 1 and Fig. 6). Although these residues are also present in AguA2, there was no enzyme activity unless the Cys-157 residue was mutated to glycine as discussed above. Our findings expand the current understanding of microbial AgDIs including L. monocytogenes with a novel key residue uncovered as having catalytic activity (30, 34, 42, 43). The AguA1 of L. monocytogenes most likely employs similar catalytic mechanisms of the guanidinium-modifying enzyme family previously described (30, 34, 35).
In conclusion, L. monocytogenes harbors two putative agmatine deiminases, but only AguA1 functionally participates in the AgDI pathway and contributes to its acid tolerance. AguA1 is specific for agmatine catalysis, sensitive to some metal ions such as Cu2+, Zn2+, and Co2+, and optimal at 25 °C and over a wide range of pH from 3.5 to 10.5. We have also discovered a novel residue Gly-157 other than the known catalytic triad (Cys-His-Glu/Asp) critical for L. monocytogenes AgDI activity. Further research is required to examine the role of aguD, as the agmatine-putrescine antiporter, in the proposed AgDI system of L. monocytogenes and in acid tolerance.
Acknowledgment
We thank Dr. Martin Wiedmann at Cornell University for kindly providing the shuttle plasmid pKSV7.
This study was supported by National Natural Science Foundation of China Grants 31101829 and 31272570 and Natural Science Foundation of Zhejiang Province Grant Q12C190019.

This article contains supplemental Tables S1 and S2 and Fig. S1.
- AgDI
- agmatine deiminase
- ADI
- arginine deiminase
- BHI
- brain-heart infusion
- SOE
- splice overlap extension.
REFERENCES
- 1. Vázquez-Boland J. A., Kuhn M., Berche P., Chakraborty T., Domínguez-Bernal G., Goebel W., González-Zorn B., Wehland J., Kreft J. (2001) Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14, 584–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Corr S. C., O'Neill L. A. (2009) Listeria monocytogenes infection in the face of innate immunity. Cell Microbiol. 11, 703–709 [DOI] [PubMed] [Google Scholar]
- 3. Cotter P. D., Hill C. (2003) Surviving the acid test. Responses of gram-positive bacteria to low pH. Microbiol. Mol. Biol. Rev. 67, 429–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gray M. J., Freitag N. E., Boor K. J. (2006) How the bacterial pathogen Listeria monocytogenes mediates the switch from environmental Dr. Jekyll to pathogenic Mr. Hyde. Infect. Immun. 74, 2505–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Llácer J. L., Polo L. M., Tavárez S., Alarcón B., Hilario R., Rubio V. (2007) The gene cluster for agmatine catabolism of Enterococcus faecalis. Study of recombinant putrescine transcarbamylase and agmatine deiminase and a snapshot of agmatine deiminase catalyzing its reaction. J. Bacteriol. 189, 1254–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Griswold A. R., Jameson-Lee M., Burne R. A. (2006) Regulation and physiologic significance of the agmatine deiminase system of Streptococcus mutans UA159. J. Bacteriol. 188, 834–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Griswold A. R., Chen Y. Y., Burne R. A. (2004) Analysis of an agmatine deiminase gene cluster in Streptococcus mutans UA159. J. Bacteriol. 186, 1902–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lucas P. M., Blancato V. S., Claisse O., Magni C., Lolkema J. S., Lonvaud-Funel A. (2007) Agmatine deiminase pathway genes in Lactobacillus brevis are linked to the tyrosine decarboxylation operon in a putative acid resistance locus. Microbiology 153, 2221–2230 [DOI] [PubMed] [Google Scholar]
- 9. Driessen A. J., Smid E. J., Konings W. N. (1988) Transport of diamines by Enterococcus faecalis is mediated by an agmatine-putrescine antiporter. J. Bacteriol. 170, 4522–4527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen J., Cheng C., Xia Y., Zhao H., Fang C., Shan Y., Wu B., Fang W. (2011) Lmo0036, an ornithine and putrescine carbamoyltransferase in Listeria monocytogenes, participates in arginine deiminase and agmatine deiminase pathways and mediates acid tolerance. Microbiology 157, 3150–3161 [DOI] [PubMed] [Google Scholar]
- 11. Liu Y., Zeng L., Burne R. A. (2009) AguR is required for induction of the Streptococcus mutans agmatine deiminase system by low pH and agmatine. Appl. Environ. Microbiol. 75, 2629–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shirai H., Blundell T. L., Mizuguchi K. (2001) A novel superfamily of enzymes that catalyze the modification of guanidino groups. Trends Biochem. Sci. 26, 465–468 [DOI] [PubMed] [Google Scholar]
- 13. Shirai H., Mokrab Y., Mizuguchi K. (2006) The guanidino-group modifying enzymes. Structural basis for their diversity and commonality. Proteins 64, 1010–1023 [DOI] [PubMed] [Google Scholar]
- 14. Glaser P., Frangeul L., Buchrieser C., Rusniok C., Amend A., Baquero F., Berche P., Bloecker H., Brandt P., Chakraborty T., Charbit A., Chetouani F., Couvé E., de Daruvar A., Dehoux P., Domann E., Domínguez-Bernal G., Duchaud E., Durant L., Dussurget O., Entian K. D., Fsihi H., García-del Portillo F., Garrido P., Gautier L., Goebel W., Gómez-López N., Hain T., Hauf J., Jackson D., Jones L. M., Kaerst U., Kreft J., Kuhn M., Kunst F., Kurapkat G., Madueno E., Maitournam A., Vicente J. M., Ng E., Nedjari H., Nordsiek G., Novella S., de Pablos B., Pérez-Diaz J. C., Purcell R., Remmel B., Rose M., Schlueter T., Simoes N., Tierrez A., Vazquez-Boland J. A., Voss H., Wehland J., Cossart P. (2001) Comparative genomics of Listeria species. Science 294, 849–852 [DOI] [PubMed] [Google Scholar]
- 15. Chen J., Jiang L., Chen Q., Zhao H., Luo X., Chen X., Fang W. (2009) lmo0038 is involved in acid and heat stress responses and specific for Listeria monocytogenes lineages I and II, and Listeria ivanovii. Foodborne Pathog. Dis. 6, 365–376 [DOI] [PubMed] [Google Scholar]
- 16. Cotter P. D., Gahan C. G., Hill C. (2001) A glutamate decarboxylase system protects Listeria monocytogenes in gastric fluid. Mol. Microbiol. 40, 465–475 [DOI] [PubMed] [Google Scholar]
- 17. Cotter P. D., O'Reilly K., Hill C. (2001) Role of the glutamate decarboxylase acid resistance system in the survival of Listeria monocytogenes LO28 in low pH foods. J. Food Prot. 64, 1362–1368 [DOI] [PubMed] [Google Scholar]
- 18. Ryan S., Begley M., Gahan C. G., Hill C. (2009) Molecular characterization of the arginine deiminase system in Listeria monocytogenes. Regulation and role in acid tolerance. Environ. Microbiol. 11, 432–445 [DOI] [PubMed] [Google Scholar]
- 19. Cheng C., Chen J., Shan Y., Fang C., Liu Y., Xia Y., Song H., Fang W. (2013) Listeria monocytogenes ArcA contributes to acid tolerance. J. Med. Microbiol. 62, 813–821 [DOI] [PubMed] [Google Scholar]
- 20. Wiedmann M., Arvik T. J., Hurley R. J., Boor K. J. (1998) General stress transcription factor sigmaB and its role in acid tolerance and virulence of Listeria monocytogenes. J. Bacteriol. 180, 3650–3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace. A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 22. Bordoli L., Kiefer F., Arnold K., Benkert P., Battey J., Schwede T. (2009) Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4, 1–13 [DOI] [PubMed] [Google Scholar]
- 23. Bordoli L., Schwede T. (2012) Automated protein structure modeling with SWISS-MODEL workspace and the protein model portal. Methods Mol. Biol. 857, 107–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 25. Chen J., Jiang L., Chen X., Luo X., Chen Y., Yu Y., Tian G., Liu D., Fang W. (2009) Listeria monocytogenes serovar 4a is a possible evolutionary intermediate between L. monocytogenes serovars 1/2a and 4b and L. innocua. J. Microbiol. Biotechnol. 19, 238–249 [PubMed] [Google Scholar]
- 26. Jiang L., Chen J., Xu J., Zhang X., Wang S., Zhao H., Vongxay K., Fang W. (2008) Virulence characterization and genotypic analyses of Listeria monocytogenes isolates from food and processing environments in eastern China. Int. J. Food Microbiol. 121, 53–59 [DOI] [PubMed] [Google Scholar]
- 27. Monk I. R., Gahan C. G., Hill C. (2008) Tools for functional postgenomic analysis of listeria monocytogenes. Appl. Environ. Microbiol. 74, 3921–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Camilli A., Tilney L. G., Portnoy D. A. (1993) Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol. Microbiol. 8, 143–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Netten P., Perales I., van de Moosdijk A., Curtis G. D., Mossel D. A. (1989) Liquid and solid selective differential media for the detection and enumeration of L. monocytogenes and other Listeria spp. Int. J. Food Microbiol. 8, 299–316 [DOI] [PubMed] [Google Scholar]
- 30. Jones J. E., Causey C. P., Lovelace L., Knuckley B., Flick H., Lebioda L., Thompson P. R. (2010) Characterization and inactivation of an agmatine deiminase from Helicobacter pylori. Bioorg. Chem. 38, 62–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kearney P. L., Bhatia M., Jones N. G., Yuan L., Glascock M. C., Catchings K. L., Yamada M., Thompson P. R. (2005) Kinetic characterization of protein arginine deiminase 4. A transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 44, 10570–10582 [DOI] [PubMed] [Google Scholar]
- 32. Knipp M., Vasák M. (2000) A colorimetric 96-well microtiter plate assay for the determination of enzymatically formed citrulline. Anal. Biochem. 286, 257–264 [DOI] [PubMed] [Google Scholar]
- 33. Jones J. E., Dreyton C. J., Flick H., Causey C. P., Thompson P. R. (2010) Mechanistic studies of agmatine deiminase from multiple bacterial species. Biochemistry 49, 9413–9423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galkin A., Lu X., Dunaway-Mariano D., Herzberg O. (2005) Crystal structures representing the Michaelis complex and the thiouronium reaction intermediate of Pseudomonas aeruginosa arginine deiminase. J. Biol. Chem. 280, 34080–34087 [DOI] [PubMed] [Google Scholar]
- 35. Das K., Butler G. H., Kwiatkowski V., Clark A. D., Jr., Yadav P., Arnold E. (2004) Crystal structures of arginine deiminase with covalent reaction intermediates. Implications for catalytic mechanism. Structure 12, 657–667 [DOI] [PubMed] [Google Scholar]
- 36. Galkin A., Kulakova L., Sarikaya E., Lim K., Howard A., Herzberg O. (2004) Structural insight into arginine degradation by arginine deiminase, an antibacterial and parasite drug target. J. Biol. Chem. 279, 14001–14008 [DOI] [PubMed] [Google Scholar]
- 37. Nakada Y., Itoh Y. (2003) Identification of the putrescine biosynthetic genes in Pseudomonas aeruginosa and characterization of agmatine deiminase and N-carbamoylputrescine amidohydrolase of the arginine decarboxylase pathway. Microbiology 149, 707–714 [DOI] [PubMed] [Google Scholar]
- 38. Yanagisawa H. (2001) Agmatine deiminase from maize shoots. Purification and properties. Phytochemistry 56, 643–647 [DOI] [PubMed] [Google Scholar]
- 39. Sakakibara Y., Yanagisawa H. (2003) Agmatine deiminase from cucumber seedlings is a mono-specific enzyme. Purification and characteristics. Protein Expr. Purif. 30, 88–93 [DOI] [PubMed] [Google Scholar]
- 40. Janowitz T., Kneifel H., Piotrowski M. (2003) Identification and characterization of plant agmatine iminohydrolase, the last missing link in polyamine biosynthesis of plants. FEBS Lett. 544, 258–261 [DOI] [PubMed] [Google Scholar]
- 41. Park K. H., Cho Y. D. (1991) Purification of monomeric agmatine iminohydrolase from soybean. Biochem. Biophys. Res. Commun. 174, 32–36 [DOI] [PubMed] [Google Scholar]
- 42. Wei Y., Zhou H., Sun Y., He Y., Luo Y. (2007) Insight into the catalytic mechanism of arginine deiminase. Functional studies on the crucial sites. Proteins 66, 740–750 [DOI] [PubMed] [Google Scholar]
- 43. Lu X., Li L., Wu R., Feng X., Li Z., Yang H., Wang C., Guo H., Galkin A., Herzberg O., Mariano P. S., Martin B. M., Dunaway-Mariano D. (2006) Kinetic analysis of Pseudomonas aeruginosa arginine deiminase mutants and alternate substrates provides insight into structural determinants of function. Biochemistry 45, 1162–1172 [DOI] [PubMed] [Google Scholar]
- 44. O'Driscoll B., Gahan C. G., Hill C. (1996) Adaptive acid tolerance response in Listeria monocytogenes. Isolation of an acid-tolerant mutant which demonstrates increased virulence. Appl. Environ. Microbiol. 62, 1693–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cotter P. D., Gahan C. G., Hill C. (2000) Analysis of the role of the Listeria monocytogenes F0F1-AtPase operon in the acid tolerance response. Int. J. Food Microbiol. 60, 137–146 [DOI] [PubMed] [Google Scholar]
- 46. Cunin R., Glansdorff N., Piérard A., Stalon V. (1986) Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 50, 314–352 [DOI] [PMC free article] [PubMed] [Google Scholar]