

Background: The bacterial type VI secretion system (T6SS) translocates toxic effector proteins into target cells.
Results: Novel T6S peptidoglycan glycoside hydrolase effector-immunity families are identified and a representative pair is structurally and functionally characterized.
Conclusion: Peptidoglycan glycoside hydrolase effectors are an important component of the T6S effector arsenal.
Significance: This work expands the repertoire of known T6S substrates and reports the first structure of a peptidoglycan glycoside hydrolase effector.
Keywords: Glycoside Hydrolases, Microbiology, Peptidoglycan, Protein Crystallization, Protein Secretion, Effector-Immunity Pair, Interbacterial Interactions, Type VI Secretion
Abstract
Bacteria employ type VI secretion systems (T6SSs) to facilitate interactions with prokaryotic and eukaryotic cells. Despite the widespread identification of T6SSs among Gram-negative bacteria, the number of experimentally validated substrate effector proteins mediating these interactions remains small. Here, employing an informatics approach, we define novel families of T6S peptidoglycan glycoside hydrolase effectors. Consistent with the known intercellular self-intoxication exhibited by the T6S pathway, we observe that each effector gene is located adjacent to a hypothetical open reading frame encoding a putative periplasmically localized immunity determinant. To validate our sequence-based approach, we functionally investigate a representative family member from the soil-dwelling bacterium Pseudomonas protegens. We demonstrate that this protein is secreted in a T6SS-dependent manner and that it confers a fitness advantage in growth competition assays with Pseudomonas putida. In addition, we determined the 1.4 Å x-ray crystal structure of this effector in complex with its cognate immunity protein. The structure reveals the effector shares highest overall structural similarity to a glycoside hydrolase family associated with peptidoglycan N-acetylglucosaminidase activity, suggesting that T6S peptidoglycan glycoside hydrolase effector families may comprise significant enzymatic diversity. Our structural analyses also demonstrate that self-intoxication is prevented by the immunity protein through direct occlusion of the effector active site. This work significantly expands our current understanding of T6S effector diversity.
Introduction
Bacteria utilize specialized protein secretion systems to mediate interactions with organisms in their environment. The bacterial type VI secretion system (T6SS)6 is a multi-protein apparatus that translocates substrate effector proteins into neighboring Gram-negative bacterial cells (1–3). Many of these proteins act as toxins; thus, their delivery grants the donor bacterium fitness in co-culture with susceptible competitors. Although the precise mechanism of effector translocation by the T6SS remains unclear, data indicate that effectors are delivered in single step from the cytoplasm of a donor bacterium to the periplasm of a recipient (3). Structural similarity between secreted T6S apparatus components and bacteriophage proteins involved in cell puncturing suggest that the translocation process may resemble phage infection (4, 5).
In recent years, significant progress has been made toward the identification and functional characterization of antibacterial effectors secreted by the T6SS. Bacteria-targeting effectors associated with T6S have been shown to elicit toxicity through the disruption of a variety of cellular structures including nucleic acids (6), membranes (7, 8), and the bacterial cell wall (3, 9–11). For example, the RhsA (recombination hot spot A) and RhsB proteins of Dickeya dadantii contain C-terminal toxin domains that degrade chromosomal DNA (6), and Tle (type VI secretion lipase effector) proteins from Vibrio cholerae, Burkholderia thailandensis, and Pseudomonas aeruginosa are lipases that catalyze the breakdown of membrane phospholipids (7). Cell wall-degrading T6S effectors distribute into two groups: those that act as amidases, cleaving the peptidoglycan molecule within its peptide stems and cross-links and those that act as glycoside hydrolases, cleaving the glycan backbone of the molecule. T6S amidase effectors have been studied extensively (3, 9, 10, 12–15). The enzymes are broadly distributed among Proteobacteria and form four phylogenetically distinct families that constitute the Tae (type IV secretion amidase effector) superfamily. Interestingly, the preferred cleavage site within peptidoglycan can vary between Tae families, suggesting the possibility that optimal effector specificity is dependent on the organism(s) targeted and/or the precise structure of the peptidoglycan found in those organisms.
In contrast to the amidases, there are few identified glycoside hydrolase cell wall-targeting effectors. Moreover, the general utilization of this effector activity by T6SS+ organisms remains uncertain. P. aeruginosa Tse3, the sole biochemically characterized glycoside hydrolase effector, acts as a muramidase, cleaving the β-(1,4) linkage between N-acetylmuramic acid (MurNAc) and N-acetylglucosamine (GlcNAc) (3). The only other glycoside hydrolase effector identified to date is VgrG-3 from V. cholerae. This protein carries a C-terminal lysozyme-like effector domain that degrades peptidoglycan (3, 11, 16).
A commonality among all identified effectors with antibacterial activity is that their corresponding open reading frames are found adjacent to genes encoding cognate immunity proteins. These proteins confer immunity by specifically binding to and inactivating their associated effector (3, 13–15). As effectors do not access the donor cell periplasm in transit to the recipient cell, the immunity proteins of cell wall-targeting effectors, which reside in the periplasm, serve the exclusive purpose of preventing intercellular self-intoxication.
The sequence divergence and sporadic distribution of T6S effectors present a challenge for the identification of these important mediators of interbacterial interactions. Owing to their frequent horizontal inheritance, bacteria related at the genus, or even the species level, often contain a completely unique repertoire of these proteins. For example, the plant commensal bacterium Pseudomonas protegens does not contain homologs of the three established effectors of the P. aeruginosa Hcp secretion island I-encoded T6SS (H1-T6SS), Tse1–3; however, this organism possesses a T6SS orthologous to the H1-T6SS (17).
One way in which the challenge of identifying T6SS effectors has been overcome is by exploiting the tendency of their corresponding genes to reside within or in close proximity to T6SS-encoding gene clusters. This approach was used for the identification of Tae4 family members from Serratia marcescens (12). Alternatively, mass spectrometry-based methodologies have been successful in the identification of T6S effectors from P. aeruginosa, Burkholderia thailandensis, and S. marcescens (2, 9, 18). Finally, our group utilized a sequence homology-independent informatic search based on common properties found within effector-immunity (E-I) pairs to identify the Tae superfamily (9). These properties, applied independently to the candidate effector and immunity protein, included size, isoelectric point, predicted subcellular localization, and the presence of a cysteine-histidine catalytic dyad.
In this study, we performed an informatic search for T6SS substrates and found previously unidentified families of peptidoglycan glycoside hydrolase effectors, herein named Tge proteins (type VI secretion glycoside hydrolase effectors). Characterization of a representative Tge from P. protegens showed that the protein displays periplasmic toxicity, is secreted in a T6-dependent manner, and confers a fitness advantage when P. protegens is grown in competition against P. putida, a co-occurring soil bacterium. Additionally, we solved the 1.4 Å crystal structure of P. protegens Tge in complex with its cognate immunity protein. Together, our findings show a broader distribution of T6S glycoside hydrolase effectors than was previously appreciated and offer insights into the molecular basis for glycoside hydrolase activity and inhibition.
EXPERIMENTAL PROCEDURES
Bioinformatic Screen
Putative effector-immunity candidates were identified using a similar informatic search protocol as described previously (9). Briefly, a custom Perl script was used to search 115 T6SS+ genomes for bicistronic genes with the following criteria for the encoded effector protein: 1) no predicted signal sequence, 2) a predicted pI greater than 8.0, and 3) fewer than 200 amino acids. The criteria for the immunity protein included the presence of a predicted signal sequence and fewer than 200 amino acids. Protein sequences obtained from this screen were submitted in batch mode to the Phyre2 server and examined manually for the presence of lysozyme-like folds (19). Candidate peptidoglycan glycoside hydrolases and associated immunity proteins were then used as Blastp search queries to identify all unique family members in the NCBI database.
Bacterial Strains and Growth Conditions
All P. protegens strains generated in this study were derived from the sequenced strain Pf-5 (20). P. protegens strains were grown in Luria-Bertani (LB) media at 30 °C supplemented with 15 μg ml−1 gentamycin and 25 μg ml−1 irgasan where appropriate. The pEXG2 suicide vector was used for in-frame chromosomal deletions in P. protegens as described previously for P. aeruginosa (21). Similar to P. aeruginosa, deletion of retS is required for activation of T6S in P. protegens (22, 23). Locus tags for retS, tge2PP, tgi2PP, and clpV are PFL_0664, PFL_3037, PFL_3036, and PFL_6093, respectively. The P. putida strain used for competition assays was derived from the sequenced strain KT2440 (24) and grown in LB media at 30 °C. E. coli strains used included DH5α for cloning, SM10 for conjugal transfer of plasmids into P. protegens, BL21 pLysS for toxicity assays, growth curves, and phase contrast microscopy, and Shuffle® T7 Express lysY (New England Biolabs) for expression of proteins for purification. E. coli strains were either grown in LB or LB low salt (LB-LS) at 37 °C supplemented with 50 μg ml−1 kanamycin, 150 μg ml−1 carbenicillin, 30 μg ml−1 chloramphenicol, 200 μg ml−1 trimethoprim, 0.1% (w/v) l-rhamnose and the indicated concentrations of IPTG as required.
E. coli Toxicity Assays
Tge2PP was cloned into pET-29b(+) and pET-22b(+) using the BamHI/HindIII and NdeI/HindIII restriction sites, respectively. Tgi2PP and tgi1PA (PA3485) were cloned into pSCrhaB2-CV using the NdeI/XbaI restriction sites. The tge2PP E69Q mutation was introduced using SOE PCR. Overnight cultures of E. coli BL21 pLysS pET-29b(+), pET-29b(+)::tge2PP, pET-22b(+), pET-22b(+)::tge2PP, pET-22b(+)::tge2PP E69Q, pET-22b(+) + pSCrhaB2-CV, pET-22b(+)::tge2PP + pSCrhaB2-CV, pET-22b(+)::tge2PP + pSCrhaB2-CV::tgi2PP and pET-22b(+)::tge2PP + pSCrhaB2-CV::tgi1PA were diluted 106 in 10-fold increments and stamp plated onto LB-LS 3% agar plates containing the appropriate antibiotics. For comparison of cytoplasmic versus periplasmic toxicity of Tge2PP, cells were induced with 100 μm IPTG. For immunity rescue experiments, expression was induced with 40 μm IPTG for Tge2PP and 0.1% (w/v) l-rhamnose for Tgi2PP/Tgi1PA. Western blot analysis of Tge2PP and Tge2PP E69Q expression levels was performed using an anti-His HRP conjugate according to the manufacturer's instructions (Qiagen). The E. coli BL21 pLysS expression strain was used in these experiments to prevent leaky expression prior to IPTG induction.
Interbacterial Competitions
Competition experiments were performed in a similar manner as described previously for B. thailandensis (9). Briefly, P. protegens and P. putida strains were mixed in a 1:1 ratio, and 10 μl of the mixture was spotted on 0.2-μm nitrocellulose membrane overlaid on LB-LS 3% agar plates. Plate counts were taken of the initial innoculum and after 18 h of competition at 30 °C. The recipient P. putida strain contained a GFP expression construct integrated into the attTn7 site allowing for differentiation of the donor and recipient cells using fluorescence imaging as described previously (1, 25, 26). Statistical analyses were carried out using a two-tailed Student's t test.
P. protegens Secretion Assay and Western Blot Analysis
Tge2PP, including its native ribosome binding site, were cloned into pPSV35-CV using the SacI/XbaI restriction sites. The resulting construct encodes Tge2PP fused to a C-terminal vesicular stomatitis virus G (VSV-G) epitope tag to facilitate detection by immunoblotting. Overnight cultures of P. protegens ΔretS and ΔretS ΔclpV harboring pPSV35-CV::tge2PP were subcultured 1:500 into LB containing 100 μm IPTG and grown to mid-log phase. Cell and supernatant fractions were separated and analyzed for the presence of Tge2PP by Western blot using an anti-VSV-G antibody as described previously (21).
Growth Curves
Overnight cultures of E. coli BL21 pLysS containing pET-22b(+), pET-22b(+)::tge2PP and pET-22b(+)::tge2PP E69Q were subinoculated to an optical density at 600 nm (A600) of 0.01 in LB-LS and grown at 37 °C. A600 measurements were taken every 30 min until the cultures reached an A600 of ∼0.2, at which point protein expression was induced with 100 μm IPTG. Following IPTG induction, A600 measurements were taken every 15 min for the remainder of the growth curve.
Protein Expression and Purification
For the isothermal titration calorimetry (ITC) experiment, full-length tge2PP and tgi2PP lacking its encoded signal sequence (Tgi2PP(24–156) were cloned into pET-28b(+) and pET-15b using the NdeI/XhoI and NdeI/BamHI restriction sites, respectively, to generate N-terminal His6 tags on both proteins. For crystallization, full-length tge2PP was cloned into pET-24a(+) using the NdeI/XhoI restriction sites to generate an untagged construct suitable for coexpression with the Tgi2PP(24–156) construct described above. All protein constructs were expressed in Shuffle® T7 Express lysY cells, which allow for cytoplasmic disulfide bond formation, and grown in LB broth supplemented with the appropriate antibiotics. For each construct, cells were grown at 37 °C to an A600 of 0.6 before protein expression was induced with 1 mm IPTG at room temperature for 18 h. For the protein samples used in the ITC experiment, cells were resuspended in 50 mm sodium phosphate, pH 7.2, 300 mm NaCl, 5 mm imidazole, and lysed by sonication. Following centrifugation, the cleared cell lysates were purified by Ni2+-nitrilotriacetic acid affinity chromatography using a linear gradient of 5–400 mm imidazole. The purified proteins were then dialyzed against 20 mm sodium phosphate, pH 7.2, 150 mm NaCl prior to use in downstream experiments. For crystallization, cells containing coexpressed Tge2PP/Tgi2PP(24–156) were resuspended in 50 mm Hepes, pH 7.5, 300 mm NaCl, 5 mm imidazole, and lysed by sonication. The complex was then purified using Ni2+-nitrilotriacetic acid affinity chromatography in 20 mm Hepes, pH 7.5, 300 mm NaCl, 5–400 mm imidazole followed by size exclusion chromatography in 20 mm Hepes, pH 7.5, 150 mm NaCl.
Isothermal Titration Calorimetry
Purified Tge2PP and Tgi2PP(24–156) were degassed before experimentation. ITC measurements were performed with a VP-ITC microcalorimeter (MicroCal Inc., Northampton, MA). Titrations were carried out with 250 μm Tgi2PP(24–156) in the syringe and 16 μm Tge2PP in the cell. The titration experiment consisted of one 2-μl injection followed by 29 5-μl injections with 300-s intervals between each injection. The ITC data were analyzed using the Origin software (version 5.0, MicroCal, Inc.) and fit using a single-site binding model.
Phase Contrast Microscopy
Phase contrast microscopy images were acquired with a Nikon Ti-E inverted microscope fitted with a 60× oil objective, a xenon light source (Sutter Instruments), and a CCD camera (Clara series, Andor). Overnight cultures of E. coli BL21 pLysS pET-22b(+)::tge2PP and pET-22b(+)::tge2PP E69Q were subcultured 1:300 into LB broth containing the appropriate antibiotics and grown to an A600 of 0.2. IPTG was then added to a concentration of 200 μm, and after a 45-min incubation, the bacterial suspension (1 μl) was spotted onto growth pads made with LB-LS medium and 1% (w/v) agarose, and cells were imaged immediately.
Crystallization and Structure Determination
Selenomethionyl-incorporated Tge2PP·Tgi2PP(24–156) complex was concentrated to 10 mg ml−1 by spin ultrafiltration (10 kDa molecular mass cut-off, Millipore) and screened against commercially available sparse matrix crystal screens (Crystal Screens 1 and 2, Hampton Research). Crystal trials were setup in 48-well plates using the sitting drop vapor diffusion technique. Protein and crystallization solutions were mixed in a 1:1 ratio with a final drop size of 2 μl suspended over 250 μl of crystallization solution and stored at room temperature.
After 3–4 days, diffraction quality crystals grew in 0.1 m Tris-HCl, pH 8.5, 0.2 m CaCl2, and 30% (w/v) PEG 4000 and were flash frozen without any added cryoprotectant. X-ray diffraction data were collected on beamline 5.0.3 at the Lawrence Berkeley National Laboratory Advanced Light Source (27). The data were merged, integrated, and scaled using the xia2 system (28). Experimental phases were obtained using the Phenix AutoSol Wizard (29) resulting in density modified selenium single-wavelength anomalous diffraction phased maps of excellent quality, which allowed for automated model building with the Phenix AutoBuild Wizard (30). Subsequent model adjustments were made manually in COOT (31) between iterative rounds of refinement carried out using PHENIX.REFINE (32). The final model was refined to an Rwork/Rfree of 14.9% and 17.5% (Table 1).
TABLE 1.
X-ray data collection and refinement statistics
| Tge2PP/Tgi2PP | |
|---|---|
| Data collection | |
| Beamline | ALS 5.0.3 |
| Wavelength (Å) | 0.976 |
| Space group | P212121 |
| Cell dimensions | a = 40.1, b = 81.5, and c = 86.4 Å; α = 90.0, β = 90.0, and γ=90.0° |
| Resolution (Å) | 59.31–1.44 (1.47–1.44)a |
| Total no. of reflections | 348,034 |
| Total no. of unique reflections | 52,547 |
| Rsym (%)b | 5.7 (53.0)a |
| I/σI | 15.1 (2.2)a |
| Completeness (%) | 99.9 (99.9)a |
| Redundancy | 6.6 (4.6)a |
| Refinement | |
| Rwork/Rfree (%)c | 14.9/17.5 |
| No. of atoms | |
| Protein | 2005 |
| Water | 353 |
| Average B-factors (Å2) | |
| Protein | 21.3 |
| Water | 32.7 |
| r.m.s.d.d | |
| Bond lengths (Å) | 0.007 |
| Bond angles | 1.105° |
| Ramachandran plot (%)e | |
| Total favored | 98.1 |
| Total allowed | 100.0 |
| Coordinate error (Å)f | 0.13 |
a Values in parentheses correspond to the highest resolution shell.
b Rsym = ΣΣ|I(k) − 〈I〉|/ΣI(k) where I(k) and 〈I〉 represent the diffraction intensity values of the individual measurements and the corresponding mean values. The summation is over all unique measurements.
c Rwork = Σ‖Fobs| − k|Fcalc‖/|Fobs| where Fobs and Fcalc are the observed and calculated structure factors, respectively. Rfree is the sum extended over a subset of reflections (5%) excluded from all stages of the refinement.
d r.m.s.d. indicates root mean square deviation.
e Values were calculated using MOLPROBITY (52).
f Shown is the maximum likelihood-based coordinate error, as determined by PHENIX (53).
RESULTS
Identification of Peptidoglycan Glycoside Hydrolase Effector/Immunity Families
Previously, we reported the development of a heuristic search method that was successfully employed for the identification of a widespread type VI peptidoglycan amidase effector superfamily (9). Using a customized Perl script, T6SS+ genomes were searched for bicistrons that encode protein products with a defined set of properties commonly associated with T6S effector-immunity pairs. For the effector protein, this included the absence of a signal peptide, an isoelectric point greater than 8.0, fewer than 200 amino acids, and the presence of conserved catalytic histidine and cysteine residues. Parameters used for the immunity protein included the presence of a signal peptide and primary sequence length limitation (<200 amino acids). These selection criteria yielded 418 total candidate E-I pairs, within which amidase effectors were identified by structure prediction algorithms and subsequent direct experimentation (19). To adapt this pipeline for the identification of glycoside hydrolase effectors, we excluded the histidine and cysteine constraints, generating a list of 831 candidate E-I pairs from 115 T6SS+ genomes. These were then examined by structure prediction servers to identify lysozyme-like folds and manually curated to remove systematic false-positives (see “Experimental Procedures”). This approach yielded two phylogenetically distinct families, which combine with P. aeruginosa Tse3 to form the basis for three distinct type VI peptidoglycan glycoside hydrolase effector and immunity groups (Tge1–3 and Tgi1–3) (Fig. 1 and supplemental Figs. S1 and S2). Although it likely functions as a muramidase, we have opted not to include V. cholerae VgrG-3 in this reclassification of T6S glycoside hydrolase effectors. This emphasizes the additional postulated structural role of VgrG-3 in the T6S apparatus, which has not been observed for non-VgrG-fused effectors similar to the Tge1–3 proteins (11, 16).
FIGURE 1.

Overview of the tge1–3 families. Genomic arrangement of tge1 (A), tge2 (B), and tge3 (C) families in representative organisms. Shading is used to indicate effector (light) and immunity (dark) open reading frames. Accession numbers and sequence alignments of Tge2 and Tge3 effector and immunity proteins are available in supplemental Figs. S1 and S2.
Many peptidoglycan glycoside hydrolases share a lysozyme-like fold; however, they can be divided into two enzymatic subgroups based on their cleavage mechanism. Lysozymes and lytic transglycosylases cleave the β-1,4-glycosidic bond between MurNAc and GlcNAc, the former hydrolyzing the bond forming a reducing MurNAc end, and the latter catalyzing the formation of 1,6-anhydroMurNAc (33). In contrast, N-acetylglucosaminidases cleave the β-1,4-glycosidic bond between GlcNAc and MurNAc, leaving GlcNAc as the reducing sugar. Irrespective of their substrate specificity, peptidoglycan glycoside hydrolases universally employ a conserved glutamate, acting as a catalytic acid, to initiate cleavage of the glycosidic bond (33). Consistent with our hypothesis that Tge2 and Tge3 proteins such as Tse3 function as peptidoglycan glycoside hydrolase enzymes, we identified a strictly conserved Glu within a putative active motif in both enzyme families (supplemental Figs. S1 and S2). Additionally, our sequence analyses indicated that Tge2 proteins likely hydrolyze peptidoglycan using a standard goose-type (G-type) single displacement mechanism (34), whereas the Tge3 family contains a Glu-Asp-Thr catalytic triad utilized by phage-type lysozymes (35, 36).
Tge2PP-Tgi2PP Are an Effector-immunity Pair Secreted by the T6SS
Tge1 from P. aeruginosa (Tge1PA, formerly Tse3) has been demonstrated previously to function as a peptidoglycan glycoside hydrolase effector. To validate a new family identified by our informatics search, we chose to characterize a Tge2-Tgi2 E-I pair from the soil-dwelling bacterium P. protegens (Tge2PP-Tgi2PP). To confirm that Tge2PP functions as a periplasmic effector, we performed toxicity assays in E. coli. As expected for a peptidoglycan-degrading enzyme, we observed a significant decrease in E. coli viability and optical density when Tge2PP was artificially targeted to the periplasm via a sec-dependent leader sequence (peri-Tge2PP, Fig. 2, A and B). Furthermore, these effects of Tge2PP periplasmic toxicity were specific to Tge2PP catalytic activity, as a conservative mutation of its predicted catalytic glutamate (E69Q) abolished toxicity. Consistent with its proposed role as a peptidoglycan glycoside hydrolase effector, peri-Tge2PP induced cell rounding and lysis compared with the catalytic point mutant (Fig. 2C). Importantly, Western blot analysis confirmed that the observed reduction in toxicity and cell rounding was not due to decreased expression levels of the E69Q protein relative to wild-type (Fig. 2D). Despite our ability to observe Tge2PP activity in a cellular context, we were unable to detect significant degradation of purified peptidoglycan sacculi by Tge2PP over a range of conditions, raising the intriguing possibility that the effector may require a yet unidentified periplasmic cofactor or binding partner.
FIGURE 2.

Tge2PP-Tgi2PP are an effector-immunity pair secreted by the T6SS. A, growth of E. coli on solid media harboring inducible plasmids expressing the indicated proteins. Empty vector controls are indicated by a dash. Error bars represent ± S.D. (n = 3). B, optical density of E. coli in liquid media harboring a vector control or inducible plasmids expressing the indicated proteins. The arrow indicates the time at which protein expression was induced. Error bars represent ± S.D. (n = 3). C, representative phase contrast micrographs of E. coli expressing peri-Tge2PP or peri-Tge2PP E69Q. Images were acquired 45 min after IPTG induction. Scale bar, 2 μm. D, Western blot analysis of E. coli expressing vector control, Tge2PP and Tge2PP E69Q. RNA polymerase (RNAP) was used as a loading control. E, growth of E. coli on solid media harboring inducible plasmids expressing the indicated proteins. Empty vector controls are indicated by a dash. Error bars represent ± S.D. (n = 3). F, ITC analysis showing the interaction between Tge2PP and Tgi2PP. The top panel displays the heats of injection, whereas the bottom panel shows the normalized integration data as a function of the molar syringe and cell concentrations. G, Tge2PP is secreted in a T6-dependent manner. Western blot analysis of the cell and supernatant (sup) fractions of the indicated P. protegens strains expressing Tge2PP fused to a C-terminal vesicular stomatitis viral glycoprotein (VSV-G) tag. H, Tge2PP provides a competitive growth advantage under cell contact-promoting conditions. Competitive outcome of the indicated P. protegens strains against P. putida. Asterisks denote competition outcomes significantly different than wild-type P. protegens (n = 4; *, p < 0.05). Error bars indicate ± S.D.
Next, we examined whether Tgi2PP could inhibit the toxicity exhibited by peri-Tge2PP. Tgi2PP co-expressed with peri-Tge2PP significantly rescued Tge2PP-dependent toxicity, indicating that Tgi2PP functions as an immunity protein (Fig. 2E). As predicted by its lack of sequence homology to Tgi2PP, co-expression of P. aeruginosa Tgi1 (Tgi1PA, formerly Tsi3) was not able to rescue E. coli viability. Next, we investigated the mechanism of inhibition of Tge2PP activity by Tgi2PP. To probe for a direct binding mechanism, we conducted ITC measurements on purified Tge2PP and Tgi2PP. The binding isotherm obtained by titration of the effector with Tgi2PP fit a single-site binding model with a stoichiometry of ∼1:1 (n = 0.93) and a dissociation constant of 0.26 ± 0.04 nm (Fig. 2F). Titration of Tgi2PP into buffer and buffer into Tge2PP confirmed that the observed saturation of the heats of injection was due to the Tge2PP-Tgi2PP interaction and not between either of the proteins and a component of the buffer solution (supplemental Fig. S3). The tight binding observed between Tge2PP and Tgi2PP is consistent with the affinities measured for other T6SS E-I pairs, including P. aeruginosa Tae1-Tai1, Enterobacter cloacae Tae4-Tai4, and two Tae4-Tai4 family members from S. marcescens, which all bind with low nanomolar to high picomolar dissociation constants (12, 13, 15).
P. aeruginosa possesses three functionally non-redundant T6SSs, encoded by HSI-I-III. Of these, the H1-encoded system is post-transcriptionally activated by the Gac/Rsm pathway (23). P. protegens possesses only a single T6SS. This system is orthologous to the H1-T6SS, and is also regulated by the Gac/Rsm pathway (22). The RetS hybrid sensor kinase functions within the Gac/Rsm pathway as a negative regulator of T6S (2, 22, 23). Given that T6SSs are largely quiescent under in vitro cultivation conditions, we used a P. protegens ΔretS strain to test the hypothesis that Tge2PP is a T6SS substrate. Robust secretion of Tge2PP was observed in the supernatant fraction of the parental strain; however, this was abrogated in a P. protegens strain containing the additional deletion of the gene encoding the essential type VI component, ClpV (Fig. 2G). Given that Tge2PP is secreted by the T6SS, we next sought to ascertain its contribution to interbacterial fitness. When P. protegens strains were competed under cell contact-promoting conditions against P. putida, a bacterium that inhabits similar environments (37, 38), those lacking tge2PP were ∼6-fold less fit compared with the wild-type (Fig. 2H). Although this difference is highly significant and confirms the role of Tge2PP as a T6 effector, it is also interesting to note that P. protegens strains with a non-functional T6SS display a ∼1000-fold decrease in competitive fitness against P. putida. This finding suggests that there are other currently uncharacterized effectors produced by P. protegens that mediate antagonistic interactions between these organisms. In total, these data indicate that Tge2PP is an antibacterial T6S effector.
Structure of the Tge2PP·Tgi2PP Complex
To gain further insight into Tge2PP function and its mode of inhibition by Tgi2PP, we determined the 1.4 Å crystal structure of the Tge2PP·Tgi2PP complex using the single-wavelength anomalous diffraction technique with selenomethionyl-incorporated protein (Table 1). Crystals of the Tge2PP·Tgi2PP complex grew in the orthorhombic space group P212121 and contained a single 1:1 complex in the asymmetric unit. Disordered residues not built in the final model include 1–29, 101–104, and 152–155 of Tge2PP, and residues 24–26, 155–156, and the N-terminal His6 tag of Tgi2PP. The final model was refined to an Rwork and Rfree of 14.9% and 17.5%, respectively.
Tge2PP Resembles Family 73 Glycoside Hydrolases
Tge2PP adopts a lysozyme-like fold consisting of large and small lobes that are positioned such that a substrate-binding groove is formed between them (Fig. 3A). The large lobe is made up of a short 310 helix, α1-α2 and α4-α6, whereas the small lobe consists of α3 and a β-hairpin formed by β1-β2. The termini are covalently linked by a Cys32-Cys177 disulfide bond, which connects the N-terminal 310 helix to the C-terminal loop. As determined by DALI (39), the overall structure resembles closely that of glycoside hydrolase family 73 members, which includes the peptidoglycan hydrolase FlgJ from Sphingomonas sp. strain A1 (Z-score, 16.4; Cα root mean square deviation of 1.6 Å over 124 equivalent positions) and Auto, a peptidoglycan N-acetylglucosaminidase from Listeria monocytogenes (Z-score, 13.9; Cα root mean square deviation of 2.6 Å over 126 equivalent positions) (Fig. 3B) (40, 41). It is proposed that members of this family catalyze the hydrolysis of peptidoglycan using a single displacement mechanism similar to that originally demonstrated for G-type egg white lysozyme (GEWL). In this mechanism, a catalytic glutamate donates a proton to the scissile glycosidic bond, resulting in its cleavage and formation of an oxocarbenium intermediate. This short-lived intermediate then undergoes nucleophilic attack by a water molecule, resulting in the hydrolyzed product with inverted stereochemistry at the anomeric position (34). In agreement with our functional studies, the catalytic glutamate in Tge2PP corresponds to Glu69, inferred from its nearly identical position to the corresponding glutamate residue in both FlgJ and Auto (Fig. 3B). Glu69 protrudes from the end of α2 and lies deep within the substrate-binding groove. In addition, the hydroxyl group of Tyr147 forms a hydrogen bond with Glu69, in a manner similar to the aforementioned peptidoglycan glucosaminidase and G-type lysozyme enzymes, perhaps to orient it for catalysis.
FIGURE 3.
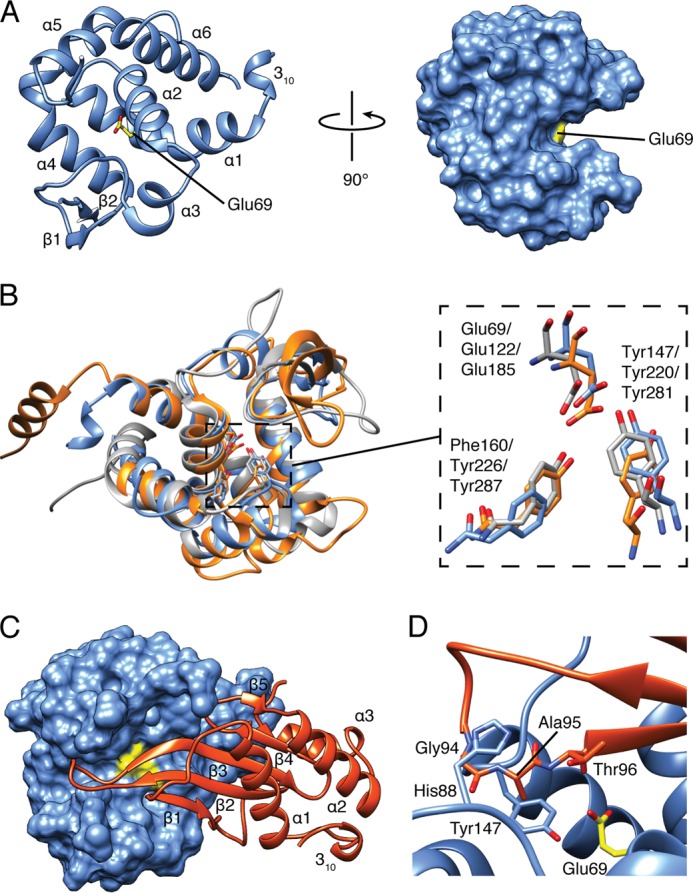
Overall structure of Tge2PP in complex with Tgi2PP. A, Tge2PP adopts a lysozyme-like fold. Ribbon (left) and surface (right) representations of Tge2PP shown at two orthogonal orientations. Secondary structure elements and the catalytic acid, Glu69, are labeled. Tgi2PP is omitted for clarity. B, structural overlay of Tge2PP (blue), Auto (orange), and FlgJ (gray) shown as schematic representations (left panel). Superposition of the Tge2PP, Auto and FlgJ active site residues are shown as stick representations (right panel). The labels indicate the residue numbering of Tge2PP followed by Auto and FlgJ. C, structure of the Tge2PP·Tgi2PP complex. Tge2PP and Tgi2PP are shown as surface and ribbon representations, respectively. Secondary structure elements of Tgi2PP are labeled, and Glu69 of Tge2PP is colored yellow. D, a protruding loop of Tgi2PP occludes the active site of Tge2PP. Shown is a close-up view of the β2-β3 loop of Tgi2PP interacting with the active site of Tge2PP. Invariant residues Gly94 and Ala95 of Tgi2PP interact with His88 and Tyr147 in the active site cleft of Tge2PP.
Tgi2PP Inhibits Tge2PP by Protruding into the Substrate-binding Groove
The structure of Tgi2PP consists of a short N-terminal 310 helix and a central five-stranded β-sheet flanked by three α-helices (Fig. 3C). A Cys117-Cys152 disulfide bond connects β5 to the C-terminal loop, anchoring α3 to the β-sheet core. Tgi2PP does not share strong structural similarity to other proteins of known structure; however, it does contain a similar topology to the periplasmic E. coli colicin M immunity protein (Z-score, 3.5; Cα root mean square deviation of 3.7 Å over 62 equivalent positions) (42). Colicin M is a bacteriocin with phosphatase activity that targets the undecaprenyl phosphate-linked peptidoglycan precursors lipid I and lipid II in the periplasm of recipient bacteria (43). The observation that the colicin M immunity protein resembles Tgi2PP suggests that these immunity proteins may have undergone diversifying selection to acquire effector specificity.
The interaction between Tge2PP and Tgi2PP involves the insertion of the β-sheet core of Tgi2PP into the substrate-binding groove of Tge2PP. Most notably, the elongated loop connecting β2 and β3 protrudes into the active site in an orientation predicted to prevent the catalytic glutamate from accessing its peptidoglycan substrate. Overall, the Tge2PP·Tgi2PP complex consists of a 1237 Å2 interface stabilized predominantly by hydrogen bonding and hydrophobic interactions. Conservation mapping of Tgi2PP homologs suggests that invariant residues Gly94 and Ala95, which are located in the β2-β3 loop, are important for direct interaction with the active site of Tge2PP (Fig. 3D). Binding site analysis using PDBePISA indicates that the amide nitrogen and oxygen atoms of Gly94 form hydrogen bonding interactions with the main chain of Tyr147 and His88 of Tge2PP, respectively, whereas the side chain of Ala95 becomes desolvated upon complex formation (44). Additional conserved interfacing residues include Tyr56, Asp66, Leu69, His78, Tyr88, Pro91, Trp99, Gly100, Leu101, Glu113, Cys117, Gly149, Leu150, Gly151, Cys152, and Asp154. The inhibition mode observed in the Tge2PP·Tgi2PP complex is reminiscent of bacterial proteinaceous inhibitors of eukaryotic lysozymes. The E. coli inhibitor of vertebrate lysozyme (Ivy), P. aeruginosa membrane-bound lysozyme inhibitor of C-type lysozyme (MliC), and E. coli PliG (periplasmic lysozyme inhibitor of G-type lysozyme) all use active site occlusion as a mechanism of inhibition (Fig. 4) (45–47). Moreover, in each case, the region involved in inhibition consists of a loop region connecting two β-strands. However, it is interesting to note that there is little fold similarity between these lysozyme inhibitors, suggesting they arose through convergent evolution.
FIGURE 4.
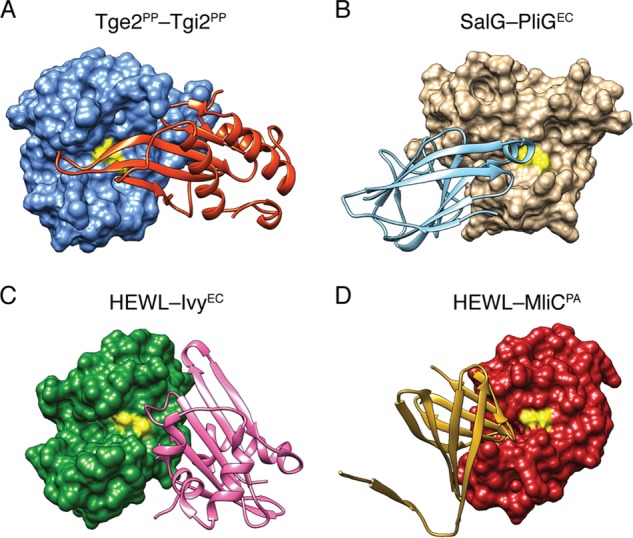
Structurally characterized lysozyme inhibitors display distinct folds but act via a common mechanism. Comparison of the Tge2PP·Tgi2PP complex to vertebrate lysozymes in complex with proteinaceous bacterial lysozyme inhibitors. Shown are the Tge2PP·Tgi2PP complex (A), E. coli periplasmic lysozyme inhibitor of G-type lysozyme (PliGEC) in complex with salmon G-type lysozyme (SalG) (B), E. coli proteinaceous inhibitor of vertebrate lysozyme (IvyEC) in complex with hen egg white lysozyme (HEWL) (C), and membrane-bound lysozyme inhibitor of C-type lysozyme (MliC) in complex with hen egg white lysozyme (D) (45–47). For each structural model, the lysozyme-like protein and its associated inhibitor are shown as surface and ribbon representations, respectively.
DISCUSSION
In this work, we identified two families of T6S peptidoglycan glycoside hydrolase effectors using informatic methods. Previously, we used homology-independent search criteria to identify a T6S peptidoglycan amidase effector superfamily. The identification of the peptidoglycan glycosyl hydrolase families described herein further exemplifies the utility of this method as a means to identify novel effector-immunity pairs. Prior characterization of Tge1PA has shown that this enzyme possesses β-(1,4)-N-acetylmuramidase (lysozyme) activity. Furthermore, Tge1PA contains a GLXQ motif found in both GEWL and E. coli soluble lytic transglycosylase (Slt70) in addition to its catalytic glutamate, suggesting it is likely structurally similar to these enzymes (3, 48). Interestingly, Tge3 family members contain a conserved Glu-Asp-Thr catalytic triad reminiscent of phage T4 lysozyme. Thus, similar to Tge1, we expect this family to exhibit β-(1,4)-N-acetylmuramidase activity. In contrast, we find that Tge2PP most closely resembles members of a peptidoglycan hydrolase family with ascribed N-acetylglucosaminidase activity. Members of this family contain a catalytic glutamate residue embedded in an AAXE(S/T) motif. This observation suggests that Tge2 enzymes could target a different chemical bond in the peptidoglycan backbone to facilitate breakdown of the cell wall in recipient bacteria.
Bacteria are known to chemically modify the glycan moieties of peptidoglycan. As this can affect their susceptibility to cell wall-targeting enzymes, it is intriguing to speculate that the different peptidoglycan glycoside hydrolase effectors delivered by the T6SS could harbor specificity for the presence or absence of these modifications. Modifications influencing muramidase activity include MurNAc and GlcNAc de-N-acetylation, as well as MurNAc O-acetylation (49). These generally result in lysozyme resistance, whereas N-acetylglucosaminidase enzymes can be unaffected by the acetylation state of peptidoglycan (50). Future studies will be needed to address the specificity of the Tge families, as well as the extent to which particular cell wall modifications influence their activity.
In light of the peptidoglycan glycoside hydrolases identified in this study, it is now apparent P. aeruginosa is not the only bacterium that secretes multiple T6 effectors targeting peptidoglycan. For example, in addition to Tge2 peptidoglycan glycoside hydrolases, Salmonella Typhi and P. protegens also possess Tae2 and Tae3 peptidoglycan amidases, respectively (9). The reason for secreting a multitude of peptidoglycan-targeting T6S effectors may be myriad. It is conceivable that the T6 donor bacterium requires complete digestion of recipient peptidoglycan sacculi to utilize it as a carbon source after lysis occurs. A non-mutually exclusive explanation may be the existence of enzymatic synergy between amidase and muramidase effectors, wherein the breakdown product of one enzyme may make the substrate of the second more accessible. The importance of this type of enzymatic synergy was recently demonstrated for cellulose-degrading enzymes (51). Alternatively, the various E-I loci within a given organism may be subject to differential regulation, such that the effector most beneficial in a particular environmental condition is expressed. The magnitude of this disproportionate benefit could be dependent upon the target organism(s) present, the physiological state of the target(s) (e.g. growing versus sessile), and the osmolarity of the surrounding milieu.
Acknowledgments
We thank Dr. P. Lynne Howell at the Hospital for Sick Children (Toronto, Canada) for providing reagents and laboratory equipment during the early stages of this work, Dr. William Catterall for use of the ITC instrument, and Michele LeRoux for microscopy expertise.
This work was supported by National Institutes of Health Grant AI080609.
The atomic coordinates and structure factors (code 4KT3) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Figs. S1–S3.
- T6SS
- bacterial type VI secretion system
- MurNAc
- N-acetylmuramic acid
- GlcNAc
- N-acetylglucosamine
- E-I
- effector-immunity
- LB-LS
- Luria-Bertani low salt
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- ITC
- isothermal titration calorimetry
- G-type
- goose-type.
REFERENCES
- 1. Schwarz S., West T. E., Boyer F., Chiang W. C., Carl M. A., Hood R. D., Rohmer L., Tolker-Nielsen T., Skerrett S. J., Mougous J. D. (2010) Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog. 6, e1001068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hood R. D., Singh P., Hsu F., Güvener T., Carl M. A., Trinidad R. R., Silverman J. M., Ohlson B. B., Hicks K. G., Plemel R. L., Li M., Schwarz S., Wang W. Y., Merz A. J., Goodlett D. R., Mougous J. D. (2010) A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7, 25–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Russell A. B., Hood R. D., Bui N. K., LeRoux M., Vollmer W., Mougous J. D. (2011) Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475, 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Leiman P. G., Basler M., Ramagopal U. A., Bonanno J. B., Sauder J. M., Pukatzki S., Burley S. K., Almo S. C., Mekalanos J. J. (2009) Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl. Acad. Sci. U.S.A. 106, 4154–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pell L. G., Kanelis V., Donaldson L. W., Howell P. L., Davidson A. R. (2009) The phage λ major tail protein structure reveals a common evolution for long-tailed phages and the type VI bacterial secretion system. Proc. Natl. Acad. Sci. U.S.A. 106, 4160–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koskiniemi S., Lamoureux J. G., Nikolakakis K. C., t'Kint de Roodenbeke C., Kaplan M. D., Low D. A., Hayes C. S. (2013) Rhs proteins from diverse bacteria mediate intercellular competition. Proc. Natl. Acad. Sci. U.S.A. 110, 7032–7037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Russell A. B., LeRoux M., Hathazi K., Agnello D. M., Ishikawa T., Wiggins P. A., Wai S. N., Mougous J. D. (2013) Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 496, 508–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miyata S. T., Kitaoka M., Brooks T. M., McAuley S. B., Pukatzki S. (2011) Vibrio cholerae requires the type VI secretion system virulence factor VasX to kill Dictyostelium discoideum. Infect. Immun. 79, 2941–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Russell A. B., Singh P., Brittnacher M., Bui N. K., Hood R. D., Carl M. A., Agnello D. M., Schwarz S., Goodlett D. R., Vollmer W., Mougous J. D. (2012) A widespread bacterial type VI secretion effector superfamily identified using a heuristic approach. Cell Host Microbe 11, 538–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chou S., Bui N. K., Russell A. B., Lexa K. W., Gardiner T. E., LeRoux M., Vollmer W., Mougous J. D. (2012) Structure of a peptidoglycan amidase effector targeted to Gram-negative bacteria by the type VI secretion system. Cell reports 1, 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brooks T. M., Unterweger D., Bachmann V., Kostiuk B., Pukatzki S. (2013) Lytic activity of the Vibrio cholerae type VI secretion toxin VgrG-3 is inhibited by the antitoxin TsaB. J. Biol. Chem. 288, 7618–7625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. English G., Trunk K., Rao V. A., Srikannathasan V., Hunter W. N., Coulthurst S. J. (2012) New secreted toxins and immunity proteins encoded within the type VI secretion system gene cluster of Serratia marcescens. Mol. Microbiol. 86, 921–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang H., Zhang H., Gao Z. Q., Wang W. J., Liu G. F., Xu J. H., Su X. D., Dong Y. H. (2013) Structure of the type VI effector-immunity complex (Tae4-Tai4) provides novel insights into the inhibition mechanism of the effector by its immunity protein. J. Biol. Chem. 288, 5928–5939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dong C., Zhang H., Gao Z. Q., Wang W. J., She Z., Liu G. F., Shen Y. Q., Su X. D., Dong Y. H. (2013) Structural insights into the inhibition of type VI effector Tae3 by its immunity protein Tai3. Biochem. J. 454, 59–68 [DOI] [PubMed] [Google Scholar]
- 15. Ding J., Wang W., Feng H., Zhang Y., Wang D. C. (2012) Structural insights into the Pseudomonas aeruginosa type VI virulence effector Tse1 bacteriolysis and self-protection mechanisms. J. Biol. Chem. 287, 26911–26920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dong T. G., Ho B. T., Yoder-Himes D. R., Mekalanos J. J. (2013) Identification of T6SS-dependent effector and immunity proteins by Tn-seq in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 110, 2623–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loper J. E., Hassan K. A., Mavrodi D. V., Davis E. W., 2nd, Lim C. K., Shaffer B. T., Elbourne L. D., Stockwell V. O., Hartney S. L., Breakwell K., Henkels M. D., Tetu S. G., Rangel L. I., Kidarsa T. A., Wilson N. L., van de Mortel J. E., Song C., Blumhagen R., Radune D., Hostetler J. B., Brinkac L. M., Durkin A. S., Kluepfel D. A., Wechter W. P., Anderson A. J., Kim Y. C., Pierson L. S., 3rd, Pierson E. A., Lindow S. E., Kobayashi D. Y., Raaijmakers J. M., Weller D. M., Thomashow L. S., Allen A. E., Paulsen I. T. (2012) Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet. 8, e1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fritsch M. J., Trunk K., Alcoforado Diniz J., Guo M., Trost M., Coulthurst S. J. (2013) Proteomic identification of novel secreted anti-bacterial toxins of the Serratia marcescens Type VI secretion system. Mol. Cell. Proteomics [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 20. Paulsen I. T., Press C. M., Ravel J., Kobayashi D. Y., Myers G. S., Mavrodi D. V., DeBoy R. T., Seshadri R., Ren Q., Madupu R., Dodson R. J., Durkin A. S., Brinkac L. M., Daugherty S. C., Sullivan S. A., Rosovitz M. J., Gwinn M. L., Zhou L., Schneider D. J., Cartinhour S. W., Nelson W. C., Weidman J., Watkins K., Tran K., Khouri H., Pierson E. A., Pierson L. S., 3rd, Thomashow L. S., Loper J. E. (2005) Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat. Biotechnol. 23, 873–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mougous J. D., Cuff M. E., Raunser S., Shen A., Zhou M., Gifford C. A., Goodman A. L., Joachimiak G., Ordoñez C. L., Lory S., Walz T., Joachimiak A., Mekalanos J. J. (2006) A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312, 1526–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hassan K. A., Johnson A., Shaffer B. T., Ren Q., Kidarsa T. A., Elbourne L. D., Hartney S., Duboy R., Goebel N. C., Zabriskie T. M., Paulsen I. T., Loper J. E. (2010) Inactivation of the GacA response regulator in Pseudomonas fluorescens Pf-5 has far-reaching transcriptomic consequences. Environ. Microbiol. 12, 899–915 [DOI] [PubMed] [Google Scholar]
- 23. Goodman A. L., Kulasekara B., Rietsch A., Boyd D., Smith R. S., Lory S. (2004) A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev. Cell 7, 745–754 [DOI] [PubMed] [Google Scholar]
- 24. Nelson K. E., Weinel C., Paulsen I. T., Dodson R. J., Hilbert H., Martins dos Santos V. A., Fouts D. E., Gill S. R., Pop M., Holmes M., Brinkac L., Beanan M., DeBoy R. T., Daugherty S., Kolonay J., Madupu R., Nelson W., White O., Peterson J., Khouri H., Hance I., Chris Lee P., Holtzapple E., Scanlan D., Tran K., Moazzez A., Utterback T., Rizzo M., Lee K., Kosack D., Moestl D., Wedler H., Lauber J., Stjepandic D., Hoheisel J., Straetz M., Heim S., Kiewitz C., Eisen J. A., Timmis K. N., Düsterhöft A., Tümmler B., Fraser C. M. (2002) Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 4, 799–808 [DOI] [PubMed] [Google Scholar]
- 25. Choi K. H., Gaynor J. B., White K. G., Lopez C., Bosio C. M., Karkhoff-Schweizer R. R., Schweizer H. P. (2005) A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2, 443–448 [DOI] [PubMed] [Google Scholar]
- 26. Lambertsen L., Sternberg C., Molin S. (2004) Mini-Tn7 transposons for site-specific tagging of bacteria with fluorescent proteins. Environ. Microbiol. 6, 726–732 [DOI] [PubMed] [Google Scholar]
- 27. MacDowell A. A., Celestre R. S., Howells M., McKinney W., Krupnick J., Cambie D., Domning E. E., Duarte R. M., Kelez N., Plate D. W., Cork C. W., Earnest T. N., Dickert J., Meigs G., Ralston C., Holton J. M., Alber T., Berger J. M., Agard D. A., Padmore H. A. (2004) Suite of three protein crystallography beamlines with single superconducting bend magnet as the source. J. Synchrotron Radiat. 11, 447–455 [DOI] [PubMed] [Google Scholar]
- 28. Winter G. (2010) xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 [Google Scholar]
- 29. Terwilliger T. C., Adams P. D., Read R. J., McCoy A. J., Moriarty N. W., Grosse-Kunstleve R. W., Afonine P. V., Zwart P. H., Hung L. W. (2009) Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 65, 582–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vollmer W., Joris B., Charlier P., Foster S. (2008) Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32, 259–286 [DOI] [PubMed] [Google Scholar]
- 34. Weaver L. H., Grütter M. G., Matthews B. W. (1995) The refined structures of goose lysozyme and its complex with a bound trisaccharide show that the “goose-type” lysozymes lack a catalytic aspartate residue. J. Mol. Biol. 245, 54–68 [DOI] [PubMed] [Google Scholar]
- 35. Kuroki R., Weaver L. H., Matthews B. W. (1993) A covalent enzyme-substrate intermediate with saccharide distortion in a mutant T4 lysozyme. Science 262, 2030–2033 [DOI] [PubMed] [Google Scholar]
- 36. Kuroki R., Weaver L. H., Matthews B. W. (1995) Structure-based design of a lysozyme with altered catalytic activity. Nat. Struct. Biol. 2, 1007–1011 [DOI] [PubMed] [Google Scholar]
- 37. Deflaun M. F., Marshall B. M., Kulle E. P., Levy S. B. (1994) Tn5 insertion mutants of Pseudomonas fluorescens defective in adhesion to soil and seeds. Appl. Environ. Microbiol. 60, 2637–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rodríguez-Herva J. J., Reniero D., Galli E., Ramos J. L. (1999) Cell envelope mutants of Pseudomonas putida: physiological characterization and analysis of their ability to survive in soil. Environ. Microbiol. 1, 479–488 [DOI] [PubMed] [Google Scholar]
- 39. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic acids research 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hashimoto W., Ochiai A., Momma K., Itoh T., Mikami B., Maruyama Y., Murata K. (2009) Crystal structure of the glycosidase family 73 peptidoglycan hydrolase FlgJ. Biochem. Biophys. Res. Commun. 381, 16–21 [DOI] [PubMed] [Google Scholar]
- 41. Bublitz M., Polle L., Holland C., Heinz D. W., Nimtz M., Schubert W. D. (2009) Structural basis for autoinhibition and activation of Auto, a virulence-associated peptidoglycan hydrolase of Listeria monocytogenes. Mol. Microbiol. 71, 1509–1522 [DOI] [PubMed] [Google Scholar]
- 42. Gérard F., Brooks M. A., Barreteau H., Touzé T., Graille M., Bouhss A., Blanot D., van Tilbeurgh H., Mengin-Lecreulx D. (2011) X-ray structure and site-directed mutagenesis analysis of the Escherichia coli colicin M immunity protein. J. Bacteriol. 193, 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. El Ghachi M., Bouhss A., Barreteau H., Touzé T., Auger G., Blanot D., Mengin-Lecreulx D. (2006) Colicin M exerts its bacteriolytic effect via enzymatic degradation of undecaprenyl phosphate-linked peptidoglycan precursors. J. Biol. Chem. 281, 22761–22772 [DOI] [PubMed] [Google Scholar]
- 44. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 45. Abergel C., Monchois V., Byrne D., Chenivesse S., Lembo F., Lazzaroni J. C., Claverie J. M. (2007) Structure and evolution of the Ivy protein family, unexpected lysozyme inhibitors in Gram-negative bacteria. Proc. Natl. Acad. Sci. U.S.A. 104, 6394–6399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yum S., Kim M. J., Xu Y., Jin X. L., Yoo H. Y., Park J. W., Gong J. H., Choe K. M., Lee B. L., Ha N. C. (2009) Structural basis for the recognition of lysozyme by MliC, a periplasmic lysozyme inhibitor in Gram-negative bacteria. Biochem. Biophys. Res. Commun. 378, 244–248 [DOI] [PubMed] [Google Scholar]
- 47. Leysen S., Vanderkelen L., Weeks S. D., Michiels C. W., Strelkov S. V. (2013) Structural basis of bacterial defense against g-type lysozyme-based innate immunity. Cell. Mol. Life Sci. 70, 1113–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thunnissen A. M., Rozeboom H. J., Kalk K. H., Dijkstra B. W. (1995) Structure of the 70-kDa soluble lytic transglycosylase complexed with bulgecin A. Implications for the enzymatic mechanism. Biochemistry 34, 12729–12737 [DOI] [PubMed] [Google Scholar]
- 49. Vollmer W. (2008) Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol. Rev. 32, 287–306 [DOI] [PubMed] [Google Scholar]
- 50. De Las Rivas B., García J. L., López R., García P. (2002) Purification and polar localization of pneumococcal LytB, a putative endo-β-N-acetylglucosaminidase: the chain-dispersing murein hydrolase. Journal of bacteriology 184, 4988–5000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fox J. M., Jess P., Jambusaria R. B., Moo G. M., Liphardt J., Clark D. S., Blanch H. W. (2013) A single-molecule analysis reveals morphological targets for cellulase synergy. Nat. Chem. Biol. 9, 356–361 [DOI] [PubMed] [Google Scholar]
- 52. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]