

Background: Proteinase 3 is an abundant serine protease with high similarity to neutrophil elastase and a major autoimmune target in systemic vasculitis.
Results: We identified a monoclonal antibody that inhibits PR3 activity.
Conclusion: PR3-inhibiting antibodies can change its conformation and impair interactions with α1-proteinase inhibitor.
Significance: PR3-inhibiting antibodies may play a role in autoimmune vasculitis and could be exploited as highly selective inhibitors.
Keywords: Antibodies, Autoimmunity, Neutrophil, Protease, Protease Inhibitor, Allostery, α1-Proteinase Inhibitor, Proteinase 3, Thermophoresis, Zymogen
Abstract
Proteinase 3 (PR3) is an abundant serine protease of neutrophil granules and a major target of autoantibodies (PR3 anti-neutrophil cytoplasmic antibodies) in granulomatosis with polyangiitis. Some of the PR3 synthesized by promyelocytes in the bone marrow escapes the targeting to granules and occurs on the plasma membrane of naive and primed neutrophils. This membrane-associated PR3 antigen may represent pro-PR3, mature PR3, or both forms. To discriminate between mature PR3 and its inactive zymogen, which have different conformations, we generated and identified a monoclonal antibody called MCPR3-7. It bound much better to pro-PR3 than to mature PR3. This monoclonal antibody greatly reduced the catalytic activity of mature PR3 toward extended peptide substrates. Using diverse techniques and multiple recombinant PR3 variants, we characterized its binding properties and found that MCPR3-7 preferentially bound to the so-called activation domain of the zymogen and changed the conformation of mature PR3, resulting in impaired catalysis and inactivation by α1-proteinase inhibitor (α1-antitrypsin). Noncovalent as well as covalent complexation between PR3 and α1-proteinase inhibitor was delayed in the presence of MCPR3-7, but cleavage of certain thioester and paranitroanilide substrates with small residues in the P1 position was not inhibited. We conclude that MCPR3-7 reduces PR3 activity by an allosteric mechanism affecting the S1′ pocket and further prime side interactions with substrates. In addition, MCPR3-7 prevents binding of PR3 to cellular membranes. Inhibitory antibodies targeting the activation domain of PR3 could be exploited as highly selective inhibitors of PR3, scavengers, and clearers of the PR3 autoantigen in granulomatosis with polyangiitis.
Introduction
Proteinase 3 (PR33; EC 3.4.21.76) is one of four neutral serine proteases (elastase, cathepsin G, proteinase 3, and neutrophil serine protease 4) stored as fully processed mature enzymes in azurophil granules of human neutrophils (1–4). Small amounts of PR3 are also expressed on the plasma membrane of resting neutrophils (5, 6). The degree of this constitutive expression is genetically determined (7–9), but the surface exposure and pericellular activity of PR3 around neutrophils is further increased by priming and activation of neutrophils. Autoantibody responses to PR3 have been identified as a central pathogenic feature in patients suffering from granulomatosis with polyangiitis (GPA; formerly called Wegener granulomatosis). PR3-directed autoantibodies are capable of activating cytokine-primed neutrophils in vitro by binding to surface-exposed PR3 and Fcγ receptors (10). In its generalized form, a necrotizing vasculitic process affects and damages the endothelium of small vessels in the lungs and kidneys (11).
Although PR3 has been extensively studied for decades, its biological functions during immune defense responses are poorly understood. Likewise its interaction with anti-neutrophil cytoplasmic antibodies in patients with GPA and their pathogenic role for this relapsing-remitting disease have not been clarified. A large genome-wide association study recently confirmed the genetic association between anti-neutrophil cytoplasmic antibody formation and the PR3 locus on the one hand and the presence of the Z-variant of α1-proteinase inhibitor (α1PI) on the other hand in GPA (12). This finding suggests that PR3 activity and/or inactivation of PR3 by α1PI varies in the human population and contributes to the risk for GPA manifestations either at onset, during relapses, or during systemic progression.
Inhibition of neutrophil elastase and PR3 by α1PI is highly dependent on the proper conformation of an exposed reactive center loop, which serves as a pseudosubstrate. Single point mutations, even at distant sites within α1PI like a lysine substitution of Glu342 in the Z-variant, can affect the conformation of the reactive center loop and can decrease the association rates with target proteases (13). Once hydrolyzed after the methionine in position 358, the new carboxyl terminus of α1PI forms an irreversible covalent acyl·enzyme complex that undergoes a sophisticated conformational rearrangement. These enzyme·serpin complexes are quickly removed from neutrophil membranes, the interstitial fluids, and the circulation by a specific receptor-mediated uptake into endolysosomes (14). The question as to how antibodies can interfere with the activity of PR3 and impair its clearance by the natural plasma inhibitor α1PI, however, has not been addressed and answered.
Like other serine proteases of neutrophils, PR3 (15, 16) is synthesized as a proenzyme almost exclusively at the promyelocyte stage. Following cleavage of the signal peptide and translocation into the endoplasmic reticulum, the proenzyme (pro-PR3) egresses from the endoplasmic reticulum and migrates to the Golgi complex. At this stage, it carries a short amino-terminal extension, the dipeptide Ala-Glu. This dipeptide prevents the molecule from assuming its active enzyme conformation prematurely during biosynthesis but is cleaved off by the dipeptidyl aminopeptidase I (cathepsin C) just before storage in primary granules (17–20). After the removal of the amino-terminal dipeptide, the free positively charged amino terminus of Ile16 (chymotrypsinogen numbering) forms an internal salt bridge with the side chain carboxylate of Asp194. This rearrangement stabilizes the oxyanion hole and renders the active site cleft fully accessible to substrates. During biosynthesis, some catalytically inactive pro-PR3 escapes granule targeting and is transported to the cellular surface for secretion. As pro-PR3 is a catalytically inactive precursor, it is not cleared by α1PI and may be more easily accessible for autoantibodies in GPA.
Although the crystal structure of mature PR3 (without inhibitors bound to it) has been reported (21), inferences about the pro-PR3 structure can be drawn from comparisons with other closely related zymogen-enzyme pairs for which the structures are known. The best studied zymogen-enzyme pair, bovine cationic trypsinogen and its mature counterpart, bovine cationic trypsin (22), have identical structures for about 85% of the Cα chain, but four segments of the main chain are entirely different: the amino terminus (Ile16–Gly19), the so-called autolysis loop (Gly142–Ala152), the Val185–Gly193 loop, and the Val216–Leu223 loop (22). The latter three loops form the activation domain in the active enzyme in which the free amino terminus is inserted into the so-called activation pocket of the zymogen. All four segments are highly flexible in the zymogen but ordered in the active enzyme. Allosteric regulation of the two segments around residues 190 and 220 essentially switches the molecule from a functionally incompetent zymogen into a catalytically competent state and creates the S1 binding site and oxyanion hole (22, 23). This conformational switch between the proenzyme state and the catalytically active state of the mature enzyme is the well established structural basis for allosteric regulation of trypsin-/chymotrypsin-like enzyme activity (24).
The goal of our studies was to identify a new class of monoclonal antibodies (mAbs) that can discriminate between the catalytically competent state of the activation domain and the enzymatically inactive conformation of the zymogen. To develop such a monoclonal antibody with specificity close to the active site cleft and activity blocking properties, we immunized mice with a stable proform of PR3 carrying the tripeptide Ala-Glu-Pro at the mature amino terminus and selected hybridomas that showed preferential binding to the proform. We describe the binding specificity and activity blocking properties of the newly produced MCPR3-7 in comparison with the commercially available anti-PR3 mAb CLB-12.8. Besides the reduction of the PR3 activity, MCPR3-7 also interferes with the complexation of PR3 and α1PI and switches the active enzyme into an inactive, zymogen-like state by altering the autolysis and 190 loops. The antibody can be regarded as a starting point for the development of antibody-based allosteric inhibitors of PR3 that block activity and prevent the binding of neutrophil-activating, pathogenic anti-neutrophil cytoplasmic antibody.
EXPERIMENTAL PROCEDURES
Cells, Proteases, Antibodies, and Substrates
The human embryonic kidney (HEK) cell line 293 was purchased from the American Type Culture Collection (ATCC, Manassas, VA). The human embryonic kidney cell line 293 EBNA was received from Yves Durocher, National Research Council Canada, Montreal, Canada. Human neutrophil PR3 purified from neutrophils of peripheral blood was obtained from DIARECT AG, Freiburg, Germany. The anti-PR3 mAb MCPR3-2, which was used as control in most experiments, binds pro-PR3 and mature PR3 equally well and has been described previously (25). The anti-PR3 mAb CLB-12.8 was purchased from Sanquin, Amsterdam, The Netherlands. The FRET substrate Abz-Tyr-Tyr-Abu-ANB-NH2 was kindly provided by Adam Lesner, University of Gdansk, Gdansk, Poland and was described previously (26). The FRET substrate 5-TAMRA-VADnVADYQ-diaminopropionyl fluorescein (DAP(CF)) was ordered from EMC Microcollections, Tübingen, Germany. The thiobenzyl ester substrates Boc-Ala-Pro-nVal-SBzl and For-Ala-Ala-Pro-Abu-SBzl were purchased from Bachem AG, Bubendorf, Switzerland, and the substrate Boc-Ala-ONp was from Sigma-Aldrich. The substrate Ahx-PYFA-pNA was obtained from Dr. Francis Gauthier, Tours, France, and α1PI was from Athens Research and Technology, Athens, GA.
Expression of Recombinant Pro-PR3 and Mature PR3 Variants
The different recombinant PR3 variants used for the generation, selection, and characterization of mAbs with preferential binding to pro-PR3 are described schematically in Fig. 1A. The development of the cDNA construct coding for recombinant wild-type PR3 (pro-PR3ctp) and the cDNA construct of the active site mutant lacking the codons for the amino-terminal activation dipeptide (ΔPR3ctp-S195A) as well as their expression in HEK 293 cells have been described previously (18, 25). Similarly, the preparation and characterization of the active site mutant carrying a carboxyl-terminal c-myc-His tag (PR3-S195A-cmyc) has been described elsewhere (27). When expressed in HEK 293 cells and secreted into the cell culture supernatant, pro-PR3ctp and PR3ctp-S195A-cmyc have the conformation of pro-PR3, whereas ΔPR3ctp-S195A has the conformation of the mature enzyme (20, 27).
FIGURE 1.

Schematic diagram of cDNA constructs and identification of pro-PR3-binding monoclonal antibodies. A, the PR3 variants pro-PR3ctp, proP-PR3ctp, PR3ctp-S195A-cmyc, and pro(4DK)-PR3 are secreted by HEK 293 cells as an unprocessed zymogen (pro-PR3). ΔPR3ctp-S195A and ΔPR3-S195A do not contain the amino-terminal activation dipeptide AE and are secreted as mature but catalytically inactive PR3. Ile16 and Arg243 represent the amino-terminal and carboxyl-terminal residue, respectively, of mature PR3 and are numbered as in Protein Data Bank code 1FUJ according to chymotrypsinogen. Except proP-PR3ctp and ΔPR3-S195A, all constructs have been described previously (see references for more details). ProP-PR3ctp carries an additional proline residue between the natural dipeptide AE (light gray segment) and the amino terminus of mature PR3 (white bar). The resulting amino-terminal amino acid sequence of proP-PR3ctp is AEPIVGG. The insertion of this proline residue precludes the activation by cathepsin C and prevents any contamination by intracellularly processed (mature) PR3 in HEK 293 cells. The mutation S195A of the catalytic triad in mature PR3 eliminates the catalytic activity of the molecule, which was required to avoid autodigestion and damage of HEK 293 cells during biosynthesis of PR3. B, monoclonal antibodies from hybridoma supernatants were screened by capture ELISA with goat anti-mouse IgG. Subsequently, 0.025 μg of radiolabeled pro-PR3 (proP-PR3ctp) or mature PR3 (ΔPR3ctp-S195A) was added. The captured PR3 is quantified in counts/min (CPM). MCPR3-2 is known to recognize pro-PR3 and mature PR3 equally well and served as a control. MCPR3-7 displayed the largest difference in the binding to pro-PR3 and mature PR3. All other hybridomas (one example shown) discriminated less well between the two antigens. C, rabbit anti-mouse IgG bound to S. aureus protein A was incubated with various dilutions of MCPR3-7 (1.3 mg/ml stock solution) and used to immunoprecipitate radiolabeled pro-PR3 (proP-PR3ctp) or mature PR3 (ΔPR3ctp-S195A) from transfected HEK 293 cell supernatants. Immunoprecipitated proteins were then separated by SDS-PAGE (lanes 1–3, pro-PR3; lanes 4–6, mature PR3) and visualized by autoradiography (3 days of exposure). All dilutions of MCPR3-7 showed intense binding of pro-PR3 and weak recognition of mature PR3. Error bars represent S.E.
To obtain an immunogen that was not contaminated by traces of amino-terminally processed PR3 (mature PR3), we generated a pro-PR3 variant named proP-PR3ctp, which carries a three-residue-long propeptide ending with proline. This propeptide cannot be cleaved and removed by cathepsin C. The cDNA coding for the insertion of a proline residue between the natural activation dipeptide Ala-Glu and the Ile residue constituting the amino terminus of the mature enzyme was generated using the QuikChange site-directed mutagenesis kit (Stratagene Cloning Systems, La Jolla, CA) with the sense primer US127-5′-GCTGCGGAGCCAATCGTGGGC-3′, the antisense primer US128-5′-GCCCACGATTGGCTCCGCAGC-3′, and pro-PR3ctp as template. The underlined triplet of nucleotides encodes the inserted proline residue.
Stably transfected proP-PR3ctp-expressing HEK 293 cells were selected and cultured as described previously (20, 27), and proP-PR3ctp was purified from HEK 293 culture supernatant by immunoaffinity chromatography using MCPR3-2 coupled to a cyanogen bromide-activated Sepharose 4B column and 3 m KSCN in 1% NaHCO3 as elution buffer. ProP-PR3ctp in the eluted fractions was quantified by capture ELISA as described (25), pooled, and concentrated after dialysis.
Production of PR3 Variants in Flip-in HEK 293 and HEK 293 EBNA Cells
The constructs as well as the expression of human pro-PR3 (pro(4DK)-PR3), gibbon pro-PR3, and the two human/gibbon chimeras were produced as described (28). Furthermore, human pro-PR3 was expressed by transient transfection of HEK 293 EBNA cells as described elsewhere (29). The catalytically inactive PR3 variant ΔPR3-S195A (Fig. 1A) was also produced in the HEK 293 EBNA expression system. Purification of His-tagged proteins from cell supernatants was carried out as described previously (29). The PR3 variants, which were produced in their proform, were converted into an active state by enterokinase from calf intestine (Roche Applied Science). After dialysis against 20 mm Tris-HCl, 50 mm NaCl, and 2 mm CaCl2, pH 7.4, the proteins were cleaved at an enzyme to substrate ratio of 1:40 at room temperature overnight.
Generation of Antibodies and Selection of Hybridomas
For the generation of the mAb MCPR3-7, BALB/c mice were immunized with immunoaffinity-purified proP-PR3ctp following standard immunization protocols. Antibody titers of immunized mice were determined by ELISA. Spleen cells from immunized mice were fused with a FO myeloma cell line of BALB/c origin to generate hybridomas. Animal experimentation for this study was approved by the Institutional Animal Care and Use Committee. Handling and care of animals were in accordance with institutional guidelines.
For the identification and selection of hybridoma antibodies that bind to pro-PR3 but not to mature PR3, the following capture ELISA technique was applied. Immulon I strips were coated with rabbit anti-mouse IgG diluted 1:1000 in 0.5 m NaHCO3 buffer, pH 9.6 overnight. After blocking with BSA/immunoradiometric assay buffer (50 mm Tris, 0.1 m NaCl, and 2% BSA, pH 7.4), 0.1 μg of the monoclonal hybridoma antibodies or control antibody (MCPR3-2) was added and incubated at room temperature for 30 min while shaking. Supernatants from [35S]methionine-labeled proP-PR3ctp- or ΔPR3ctp-S195A-expressing HEK 293 cells diluted 1:20 in immunoradiometric assay buffer (50 mm Tris, 0.1 m NaCl, and 0.1% BSA, pH 7.4) were added and incubated at room temperature for 60 min. After removal of unbound PR3 by washing, antibody-bound radioactivity was measured in counts/min for each well. The hybridomas generating the highest signal difference between pro-PR3 and mature PR3 were chosen for all subsequent experiments (MCPR3-7).
Biosynthetic Labeling and Immunoprecipitation
ProP-PR3ctp- and ΔPR3ctp-S195A-expressing HEK 293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 μCi/ml [35S]methionine, penicillin, streptomycin, and 10% fetal bovine serum in a 5% CO2 atmosphere at 37 °C overnight. Unless otherwise stated, all immunoprecipitation steps were performed at 4 °C. Staphylococcal protein A was incubated with rabbit anti-mouse IgG and then with different concentrations of MCPR3-7 (1:100, 1:200, and 1:400 dilutions of a 1.3 mg/ml stock solution) in each case for 1 h. The cell culture supernatants of biosynthetically labeled cells were precleared by incubation with staphylococcal protein A for 30 min followed by centrifugation at 10,000 × g for 2 min. The precleared culture supernatants were then incubated for 10 min with the staphylococcal protein A/rabbit anti-mouse IgG/MCPR3-7 mixture and washed three times with radioimmune precipitation assay buffer (50 mm Tris, pH 8.0, 0.5% deoxycholate, 1% Triton X-100, 0.1% SDS, and 50 mm NaF in PBS to which 1 μg/ml leupeptin, 2 μg/ml aprotinin, and 75 μg/ml PMSF were freshly added prior to use). The precipitate was resuspended in SDS sample buffer, boiled for 5 min, and centrifuged at 10,000 × g for 2 min, and the supernatant was collected and stored at −20 °C until analysis by SDS-PAGE and Western blotting.
Bead-based Flow Cytometry
The possible competition between the two mAbs MCPR3-2 and MCPR3-7 in binding to pro-PR3 was analyzed by bead-based flow cytometry using the carboxyl-terminally tagged PR3 variant PR3ctp-S195A-cmyc (pro-PR3) (30). Stably transfected HEK 293 cells expressing the proforms were grown in serum-free medium for 2 days. Cell culture supernatants were concentrated using an Amicon Centriplus C-10 with a cutoff of 10,000 Da. Imidazole was added to a final concentration of 20 mm, and the concentrate was applied to a HiTrap chelating high performance column (GE Healthcare) according to the manufacturer's instructions. Proteins were eluted with 20 mm phosphate, 500 mm NaCl, and 200 mm imidazole. Imidazole was removed by centrifugation through a spin column equilibrated with 50 mm sodium phosphate, 300 mm NaCl, and 0.01% Tween 20, pH 8.0. Proteins were quantified by Coomassie Plus® (Pierce). A total of 2.8 × 106 beads coated with PR3 variants were incubated with 0.25 μg/100 μl MCPR3-2 or MCPR3-7 as described. After washing with PBS, 0.1% BSA, and 0.01% Tween 20, the beads were incubated with 100 μl of a 1:50 dilution of FITC-conjugated MCPR3-2 or FITC-conjugated MCPR3-7 (prepared in our laboratory) for 5 min, washed, and analyzed by FACScan (setting, fluorescence = 1 at 682 nm).
FRET-based Activity Assay
The activity of PR3 variants diluted to 50 nm in Tris-HCl buffer (100 mm Tris, 500 mm NaCl, and 0.01% Brij 39, pH 7.5) was measured over time using the FRET substrate Abz-Tyr-Tyr-Abu-ANB-NH2 (excitation at 325 nm, emission at 400 nm) at a concentration of 800 nm.
To locate the epitope of MCPR3-7, the activity of human PR3, gibbon PR3, and two gibbon/human chimeras (after activation of the respective pro(4DK) precursors) was determined. The activity of proteases was measured directly after adding a 2-fold molar excess of MCPR3-7. As controls, the emission of the FRET substrate without the enzyme and the activity of the enzyme without the antibody were measured. To detect the inhibitory effect of mAbs, the activity of PR3 was measured in the presence of increasing concentrations (0–750 nm) of MCPR3-7, CLB-12.8, and MCPR3-2. The activity was recorded over time directly after adding the mAbs. The effect of MCPR3-7 on PR3-mediated cleavage of the FRET substrate 5-TAMRA-VADnVADYQ-DAP(CF) (5 μm; excitation at 485 nm, emission at 520 nm) was measured over time. The activity of a 50 nm PR3 solution in activity buffer (50 mm Tris, 150 mm NaCl, and 0.01% Triton X-100, pH 7.4) was determined directly after adding a 3-fold molar excess of the antibody.
Absorbance-based Activity Assay
To search for conformational changes of PR3 in response to MCPR3-7 binding, the cleavage of different substrates was investigated. The activity of 50 nm PR3 in activity buffer at a 3-fold molar excess of MCPR3-7 was compared with its activity in the absence of antibodies using Ahx-PYFA-pNA, Boc-Ala-ONp, Boc-Ala-Pro-nVal-SBzl, and For-Ala-Ala-Pro-Abu-SBzl (1 mm each) as substrates. For the reaction with the two thiobenzyl ester substrates, 5,5′-dithiobis(2-nitrobenzoic acid) was added to the samples at a concentration of 500 μm.
ELISA
To investigate the differential interactions of mAbs with PR3·inhibitor complexes, two covalent and two canonical complexes were assembled. The covalent complexes were formed with active PR3 (converted pro(4DK)-PR3) and α1PI or AAPV-chloromethyl ketone (CMK) (American Peptide Co., Sunnyvale, CA), and the canonical complexes were formed with active PR3 and elafin or catalytically inactive ΔPR3-S195A in association with α1PI (PR3*·α1PI). The PR3·inhibitor complexes were coated on nickel plates via the His tag of the PR3, and a capture ELISA was performed as described elsewhere (31). The coating concentration of the PR3 complexes was 2 μg/ml. The dilution of the mAb was 1:1000 for CLB-12.8, 1:5000 for MCPR3-2, 1:200 for MCPR3-7, and 1:50 for the IgG1 control antibody (Mouse IgG1Pure, BD Biosciences). As secondary antibody, goat anti-mouse HRP antibody was used (dilution, 1:2500), and the reaction was developed with a peroxidase substrate. Recognition of PR3 variants was normalized to the signal obtained with pro(4DK)-PR3, which was set to 100%.
Thermophoresis
MCPR3-7 and α1PI were labeled with the red fluorescent dye NT647 using the Monolith NT Protein Labeling kit RED (NanoTemper Technologies, Munich, Germany) according to the supplied protocol. This approach is based on covalently attaching NT647 N-hydroxysuccinimide ester to primary amines of lysine residues. Prior to thermophoresis experiments, the free dye was removed via dialysis.
To quantify the affinity of MCPR3-7 to different PR3 conformations, a 1:1 dilution series of pro(4DK)-PR3, PR3-CMK, or PR3 was prepared starting at a maximum concentration of 5 μm each. To guarantee constant buffer conditions throughout the dilution, the proteinases were diluted in the exact same buffer as the stock solution: 20 mm sodium phosphate, 300 mm NaCl, and 0.02% Tween 20, pH 6.2 with (PR3-CMK) or without (pro(4DK)-PR3 and PR3) 1.75% DMSO. Each point of the dilution series was mixed 1:1 with MCPR3-7-NT647 in PBS with 0.05% Tween 20 and 2% BSA to yield a constant final antibody concentration of 50 nm and a maximum proteinase concentration of 2.5 μm.
In addition, thermophoresis was used to test whether the mAbs MCPR3-7 and CLB-12.8 have an influence on the formation of canonical α1PI·PR3 complexes. Separate 1:1 dilution series of ΔPR3-S195A in EB buffer (20 mm sodium phosphate and 500 mm NaCl, pH 7.4) were prepared, starting at a maximum concentration of 12 μm each. 5 μl of each dilution step were mixed with 1 μl of either PBS or 1 μl of a 4 μm stock of MCPR3-7 or CLB-12.8 in PBS, which results in an antibody concentration of 670 nm. After incubating the sample for 1 h at room temperature in the dark, 1 μl of a 6.15 μm α1PI-NT647 solution in PBS with 0.05% Tween 20 was added, resulting in a final α1PI concentration of 880 nm. To avoid adsorption to the capillary walls during the measurement, BSA was added at a final concentration of 1% (w/v).
Standard treated enhanced gradient capillaries (NanoTemper Technologies) were filled with the samples. Measurements were performed on a Monolith NT.115 system (NanoTemper Technologies) at a constant ambient temperature of 20 °C using 60% light-emitting diode (experiments with MCPR3-7-NT647) or 20% light-emitting diode (experiments with α1PI-NT647) and 20% infrared (IR) laser power with laser on and off times of 40 and 20 s.
After bleaching correction, the fluorescence after temperature jump and early thermophoresis was normalized to the fluorescence before laser heating with a LabVIEW routine. Mean Fnorm values of at least three technical repeats of each measurement were plotted on a linear y axis in per mil units (‰) against the proteinase concentration on a log10 x axis. The standard deviation of the repeats was calculated for each point. To get a better estimate of the error, the mean of all standard deviations of a binding curve was determined. This mean error is visualized as an error bar on all points of the corresponding graph. Using IGOR Pro, a weighted fit to the quadratic solution of the mass action law
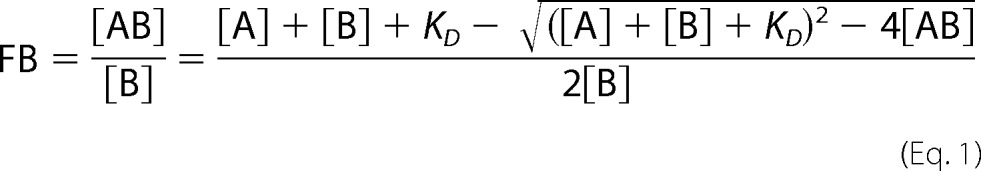 |
where FB is the fraction bound, [A] is the concentration of the titrated binding partner, [B] is the concentration of the labeled binding partner, [AB] is the concentration of bound complex of A and B, and KD is the equilibrium dissociation constant was performed with KD as the single free fit parameter (32). KD values are given together with an error estimation from the fit. The fitting procedure assumes a Gaussian, symmetric error distribution. Thus, the error can reach negative values if the sensitivity limit determined by the lowest detectable concentration of fluorescent partner is reached. In these cases, the KD values are presented as an upper limit. Fnorm of the unbound state as revealed by the fit was subtracted as a base-line value to yield ΔFnorm as depicted on the y axis of the figures.
Possible Interference of mAb with PR3 Complexation
The effect of mAbs on the covalent inactivation reaction of PR3 by α1PI was measured over time. A 3-fold molar excess of mAbs was added to an 800 nm solution of active PR3 in Tris-HCl buffer and incubated at room temperature for 1 h. After adding a 5-fold molar excess of α1PI, the sample was incubated at 37 °C, and 10-μl aliquots were taken after different time points. The reaction was stopped by adding Laemmli buffer and heating to 95 °C for 10 min. Proteins in the samples were then separated by SDS-PAGE under reducing conditions and visualized by silver staining.
Isolation and Priming of Neutrophils
The recognition of PR3 on the neutrophil membrane from one normal donor was evaluated without and after stimulation with TNFα. The neutrophils were isolated from EDTA-anticoagulated blood by centrifugation on PolymorphprepTM (Accurate Chemical and Scientific Corp., Westbury, NY) and hypotonic lysis of erythrocytes with distilled water. Cells were washed with cold Hanks' balanced salt solution without Ca2+/Mg2+ (Mediatech Inc., Herndon, VA) and resuspended in Hanks' balanced salt solution with Ca2+/Mg2+ (Mediatech Inc.) to obtain 1 × 107 cells/ml. One-half of the preparation was primed with 2 ng/ml recombinant TNFα (Roche Applied Science) for 15 min at 37 °C before analysis of membrane expression. Non-primed neutrophils were analyzed for membrane expression immediately after isolation.
PR3 on Neutrophil Membranes
The recognition of PR3 on the membrane was evaluated using flow cytometry. All steps were performed on ice. Samples containing 1 × 106 neutrophils were fixed with 0.5% paraformaldehyde for 10 min, washed with PBS and 1% BSA by centrifugation at 1200 × g at 4 °C for 3 min, and incubated with 0.5 mg/ml heat-aggregated goat immunoglobulins (IgG; Sigma) for 15 min to saturate Fcγ receptors. Next, cells were stained with a saturating dose of mouse monoclonal IgG1 directed against human PR3 (MCPR3-2 and MCPR3-7) and anti-CD32 (AbD Serotec, UK) or with an IgG1 control antibody for 30 min. Unbound antibodies were washed off with PBS and 1% BSA, and the cells were incubated for 30 min with FITC-conjugated goat anti-mouse antibody. After washing, the fluorescence intensity was analyzed on a FACScan flow cytometer (BD Biosciences Immunocytometry Systems).
RESULTS
Identification of New mAbs with Preferential Binding to Pro-PR3
To generate antibodies with preferred specificity for the inactive conformation of pro-PR3, we produced a pro-PR3 variant that was resistant to cathepsin C cleavage and other exopeptidases. Non-hematopoietic cells like HEK 293 cells were found to be devoid of cathepsin C and could not properly convert recombinant neutrophil serine protease precursors to mature enzymes. Nevertheless, minimal amounts of amino-terminally processed active recombinant PR3 have been noticed in cell lysates of HEK 293 cells transfected with wild-type PR3 cDNA.4 Therefore, a PR3 variant with an amino terminus that cannot be trimmed by exo- and endopeptidases was constructed and expressed in a stable HEK 293 cell line. The cDNA construct proP-PR3ctp codes for the insertion of a proline residue between the natural propeptide Ala-Glu and the amino terminus of mature PR3. The resulting pro-PR3 variant (Fig. 1A) with the amino-terminal sequence Ala-Glu-Pro-Ile-Val-Gly-Gly cannot be processed by cathepsin C or other exodipeptidases. This was verified by radiosequencing of supernatants and lysates from [3H]isoleucine pulse-labeled HEK 293 cells expressing proP-PR3ctp (data not shown). ProP-PR3ctp was purified via immunoaffinity chromatography from cell culture supernatants of HEK 293 cells.
Splenocytes from BALB/c mice immunized with this pro-PR3 variant were fused with FO myeloma cells. The resulting hybridoma cell clones were screened for anti-PR3 IgG secretion via capture ELISA as described under “Experimental Procedures.” Supernatants from 43 cell clones that reacted with pro-PR3 in the initial capture ELISA were retested in parallel for reactivity with pro-PR3 and mature PR3. Nineteen of these showed between 0 and 30% difference in reactivity (group A) with the two antigens, 13 showed a 31–60% difference (group B), and 11 displayed a 61–96% difference (group C). Four clones from group A, five from group B, and nine from group C were subjected to another round of dilution subcloning and retested for differential binding to pro-PR3 and mature PR3. Only six clones with persistent pro-PR3 reactivity emerged. Four of these showed similar reactivity with both antigens, and two had a 75% greater reactivity with pro-PR3 compared with mature PR3. Clone MCPR3-7, generating the highest signal difference between pro-PR3 and mature PR3 in the capture ELISA (Fig. 1B), was chosen for further characterization.
To demonstrate the differential binding of MCPR3-7 to pro-PR3 and mature PR3, rabbit anti-mouse IgG bound to Staphylococcus aureus protein A was incubated with dilutions (ranging from 1:100 to 1:400 of a 1.3 mg/ml stock solution) of MCPR3-7 and was used to precipitate 500 ng/ml radiolabeled pro-PR3 and mature PR3 from stably transfected HEK 293 cell culture supernatants. Immunoprecipitated proteins were separated by SDS-PAGE (12% gels) under reducing conditions (Fig. 1C). The preferential recognition of pro-PR3 by MCPR3-7 was observed at all mAb concentrations tested. These findings confirmed that MCPR3-7 preferentially bound pro-PR3 but that there was some cross-reactivity with mature PR3.
Characterization of the MCPR3-7 Binding Site
To determine whether MCPR3-7 and MCPR3-2, which is known to bind pro-PR3 and mature PR3 equally well, recognize non-overlapping PR3 surface structures, we modified the bead-based capture assay. Pro-PR3 (PR3ctp-S195A-cmyc) was attached to the beads as an antigen because both mAbs bound to it with similar affinity. Fig. 2A shows that each unlabeled mAb inhibited the binding of its FITC-labeled derivative by more than 90% (upper left and lower right panels). In contrast, unlabeled MCPR3-7 did not interfere with the binding of FITC-conjugated MCPR3-2 (lower left panel), and unlabeled MCPR3-2 inhibited the binding of FITC-conjugated MCPR3-7 by only 21% (upper right panel). These findings corroborated previous data (30), clearly indicating that MCPR3-7 recognized a unique epitope on pro-PR3 that is different from that of MCPR3-2 and all other mAbs. The precise binding site of MCPR3-7, however, remained uncertain as the epitope on human PR3 was not lost after replacing Ala146, Trp218, and Leu223 with the respective residues of murine PR3.
FIGURE 2.

Characterization of the MCPR3-7 binding site. A, pairwise competition between the unlabeled PR3-specific mAbs MCPR3-2 and MCPR3-7 and FITC-conjugated MCPR3-2 and MCPR3-7 using a bead-based FACS assay. Unlabeled MCPR3-2 and MCPR3-7 (on the left) served as inhibitors for the binding of the respective FITC-tagged mAbs (on the top) to immobilized pro-PR3. Pro-PR3 was chosen as it binds equally well to both mAbs. The continuous lines represent the binding of FITC-conjugated mAbs to pro-PR3-coated beads in the absence of a competing mAb; black reference histograms indicate the lack of binding to uncoated beads; the dashed lines refer to the binding of FITC-conjugated mAbs after preincubation of the pro-PR3-coated beads with unlabeled mAbs. The degree of inhibition of the FITC-conjugated mAb after preincubation with unlabeled antibodies is expressed in percent. Using the same mAb as unlabeled competitor, inhibition was 95% for MCPR3-2 (A, upper left panel) and 91% for MCPR3-7 (A, lower right panel). Unlabeled MCPR3-7 did not interfere with the binding of FITC-conjugated MCPR3-2 (A, lower left panel), and the binding of FITC-conjugated MCPR3-7 was only inhibited by 21% due to unlabeled MCPR3-2 (A, upper right panel). These findings indicate that the two mAbs recognize distinct and non-overlapping epitopes. B, PR3 activity was measured in the presence of three mAbs at different concentrations to study their differential inhibitory capacity (n = 3; ±S.E.). The activity of PR3 (50 nm) was measured with 800 nm Abz-Tyr-Tyr-Abu-ANB-NH2. MCPR3-2 and CLB-12.8 did not have any effect on the PR3 activity. By contrast, MCPR3-7 completely inhibited PR3 at a 3-fold molar excess and above. C, the activity of gibbon PR3, human PR3, and two gibbon/human chimeras was determined in the presence of a 2-fold molar excess of MCPR3-7. The h/gibPR3 variant consists of the human amino-terminal β-barrel and the gibbon carboxyl-terminal β-barrel, whereas the gib/hPR3 variant contains the human carboxyl-terminal β-barrel. The activity of PR3 variants in the presence or absence of MCPR3-7 was measured utilizing a highly sensitive FRET substrate, Abz-Tyr-Tyr-Abu-ANB-NH2, and is shown in relative fluorescence units (RFU) as an example of three technical repeats. MCPR3-7 strongly inhibited hPR3 and gib/hPR3 but not h/gibPR3. Accordingly, the epitope of MCPR3-7 is located on the carboxyl-terminal β-barrel. Error bars represent S.E.
To map the interaction of MCPR3-7 in closer detail, we studied its effects on the activity of mature human PR3 in a FRET-based activity assay. In contrast to CLB-12.8 and MCPR3-2, MCPR3-7 inhibited the activity of human PR3 (Fig. 2B). This inhibitory effect was already observed at a 1:1 ratio of PR3 and MCPR3-7. PR3 was completely inhibited at a 3-fold molar excess of MCPR3-7, whereas CLB-12.8 and MCPR3-2 did not show any effect on PR3 activity even at a 15-fold molar excess over PR3. This inhibitory effect of MCPR3-7 was sequence-dependent as it was much less pronounced for the closely related PR3 homolog of a gibbon species (Fig. 2C, second panel). The activity of a chimeric PR3 variant composed of the amino-terminal subdomain of gibbon PR3 and the carboxyl-terminal subdomain of human PR3 (gib/hPR3) was also suppressed by MCPR3-7 (Fig. 2C, right panel). Conversely, MCPR3-7 had a much smaller effect on the activity of an h/gibPR3 chimera with a humanized amino terminus and a carboxyl-terminal gibbon subdomain (Fig. 2C, third panel). As MCPR3-7 completely inhibited the activity of the gibbon/human PR3 but not that of the human/gibbon PR3 chimera, we conclude that its major binding region is located within the carboxyl-terminal subdomain (barrel) of human PR3.
Binding Affinity of MCPR3-7 for Different PR3 Conformations
To quantify the differences in binding affinities of MCPR3-7 to either pro(4DK)-PR3, active PR3, or PR3 in a complex with AAPV-CMK (see below for details), microscale thermophoresis (MST), the directed movement of molecules along a microscopic temperature gradient, was utilized. MST can be used to determine biomolecular binding affinities in free solution as it probes any binding-induced changes in size, charge, or conformation (33). In an all-optical, free solution-based approach, thermophoresis of the samples inside a glass capillary was induced via IR laser heating and observed via detection of a red fluorescent label covalently attached to MCPR3-7. When the IR laser is turned on, the fluorescence signal drops for two reasons: first, because of an intrinsic temperature effect on the fluorophore (100-ms time scale), the so-called temperature jump, and second, because of thermophoretic depletion (several seconds). Pro(4DK)-PR3, CMK-inhibited PR3, and PR3 were titrated against a fixed concentration of labeled MCPR3-7. Binding induced a change in both temperature jump and thermophoresis. As the thermophoretic depletion is linear to the bound fraction, the dissociation constant KD was determined by fitting the data points to the quadratic solution of the mass action law. MST revealed a high binding affinity of MCPR3-7 to the proform with a KD ≤10 nm. By contrast, we were not able to detect any binding of MCPR3-7 to the active conformation of PR3 stabilized by complexation with CMK, an irreversible, mechanism-based small molecule inhibitor of PR3 (Fig. 3A). This clearly indicated that MCPR3-7 selectively recognized the zymogen conformation of PR3. Using active PR3, however, we noticed that MCPR3-7 was indeed able to interact with mature PR3 but with a much lower affinity in comparison with pro(4DK)-PR3 (Fig. 3B). This is probably due to a reversible allosteric switch from the active form of PR3 to a more zymogen-like conformation in free solution that is recognized by MCPR3-7. A KD of 0.4 ± 0.2 μm could be inferred from the data. This means that the affinity of MCPR3-7 for the free active form is ∼40-fold lower than for its proform.
FIGURE 3.
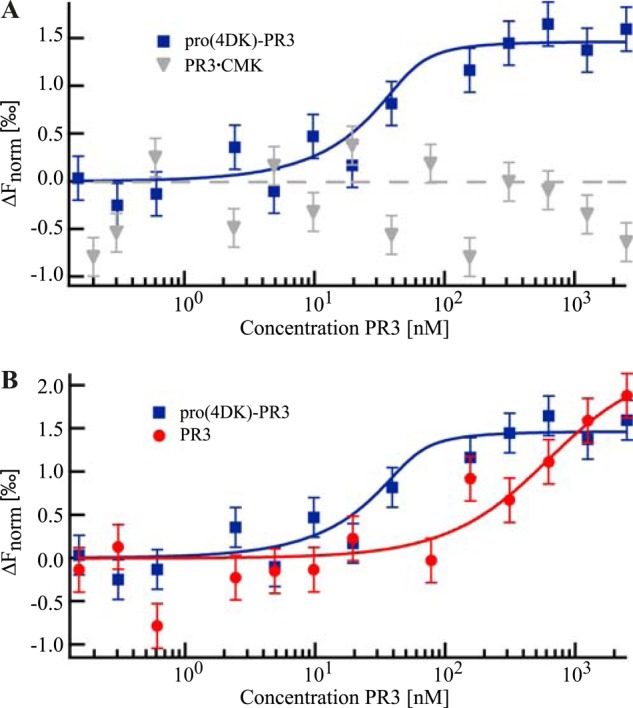
Thermophoretic quantification of MCPR3-7 binding affinity. Starting at a maximum concentration of 2.5 μm, pro(4DK)-PR3, active PR3, and PR3 complexed with AAPV-CMK (PR3.CMK) were titrated against a constant concentration of fluorescently labeled MCPR3-7 (50 nm). The biophysical method of MST allows an absolute affinity quantification by fitting the thermophoretic depletion to the quadratic solution of the mass action law with the dissociation constant KD as the single free fit parameter. Data points represent the mean value of at least three technical repeats; the error bars represent the mean of the S.D. KD values are given with an error estimation from the fit. A, MST revealed a strong binding affinity of MCPR3-7 to pro-PR3 (KD ≤ 10 nm; blue rectangles). In contrast, MCPR3-7 did not show binding to the mature conformation of the PR3·CMK complex (gray triangles). B, active PR3 was bound with a much lower affinity (KD = 0.4 ± 0.2 μm) corresponding to a 40-fold weaker binding compared with pro-PR3 (red circles).
Interaction of mAbs with Different PR3·Inhibitor Complexes
To test the differential binding capabilities of mAbs to PR3·inhibitor complexes, complexes were formed by adding a 10-fold molar excess of each inhibitor to PR3 and incubating these mixtures at 37 °C for 1 h. To generate covalently linked complexes, natural α1PI or AAPV-CMK was added to active His-tagged PR3 (activated pro(4DK)-PR3). In the case of α1PI, the resulting complexes were purified over a nickel column and checked for complete complexation on a polyacrylamide gel (data not shown). Free, uncomplexed PR3 was excluded in this way. The canonical PR3·elafin complexes were also made with active enzyme, whereas the catalytically inactive ΔPR3-S195A variant was used to assemble the so-called canonical encounter complex between PR3 and α1PI (PR3*·α1PI). These PR3 complexes were immobilized on nickel plates. MCPR3-7 (Fig. 4A) only showed weak binding to active PR3, PR3·elafin, CMK-inhibited PR3, and the encounter complex. By contrast, MCPR3-7 strongly bound to pro(4DK)-PR3. Most surprisingly, MCPR3-7 also showed strong binding to the covalent PR3·α1PI complex although, as mentioned before, not to the PR3·AAPV-CMK complex. The structural background for its differential binding to serpin-inactivated PR3 and canonical PR3 complexes is the precursor-like conformation of PR3 in the covalent serpin complex, whereas complexation of active PR3 with AAPV-CMK and elafin as well as complexation of catalytically inactive PR3 to α1PI does not alter the mature conformation of PR3. Conversely, CLB-12.8 was able to bind all PR3 variants and inhibitor complexes except for the covalent PR3·α1PI complex (Fig. 4B). The epitope of CLB-12.8 lies close to the active site cleft and may become inaccessible after binding and rearrangement of α1PI (30). MCPR3-2 was chosen as an appropriate coating control (Fig. 4C) as it could bind to all PR3 variants. Thus, equal coating of wells with different variants was ascertained. No interaction between PR3 complexes and an isotype control antibody was observed.
FIGURE 4.
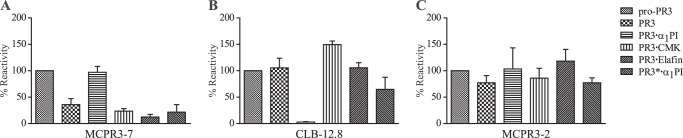
Interaction of monoclonal antibodies with different PR3·inhibitor complexes. Inhibitors were added to PR3 in solution at a 10-fold molar excess relative to PR3 and incubated at 37 °C for 1 h. His-tagged pro-PR3 (pro(4DK)-PR3), active PR3 (activated pro(4DK)-PR3), and PR3 complexes were immobilized on nickel plates. Binding of the monoclonal antibodies was analyzed via ELISA using a secondary anti-mouse HRP-conjugated polyclonal antibody. Recognition of PR3 variants of three technical repeats was normalized to the signal obtained with pro-PR3, which was set to 100% (n = 3; ±S.E.). A, MCPR3-7 was able to bind strongly to pro-PR3 and covalent PR3·α1PI complexes but failed to bind to active PR3 or other complexes. B, no binding of CLB-12.8 to the covalent PR3·α1PI complex was observed, whereas all other variants were bound similarly. C, binding of MCPR3-2 was used as a coating control and showed similar binding to all complexes. Error bars represent S.E.
Inhibitory Effect of mAbs on Serpin Complexation
To assess the impact of the antibodies on the interaction between PR3 and α1PI, we monitored the covalent complexation in the presence of a 3-fold molar excess of mAbs after different incubation times by SDS-PAGE. The reaction of PR3 with α1PI in the absence of antibodies at different time points is shown in Fig. 5A. The complexes occurred immediately after mixing the two components, and the reaction was already completed within 5 s of incubation. In contrast, preincubation of PR3 with MCPR3-7 led to a delay of complexation (Fig. 5B). At the early time points until 15 s, hardly any complexes could be detected. Complex formation was only completed within a time span of 3 min. By comparison, CLB-12.8 (Fig. 5C) and MCPR3-2 (Fig. 5D) did not show any effect on PR3·α1PI complexation. Under these assay conditions, complexes were formed within the first 5 s as seen without antibodies, and it took only 15 s until all PR3 was covalently attached to α1PI. These findings clearly indicate that the binding of MCPR3-7 to PR3 affects the covalent complexation with α1PI and delays the irreversible inhibition of PR3.
FIGURE 5.

Interference of monoclonal antibodies with PR3·serpin complexation. A, covalent complexation of PR3 (800 nm) with α1PI was measured over time by adding a 5-fold molar excess of α1PI at 37 °C. B–D, after preincubation of PR3 with a 3-fold molar excess of the different antibodies, the effects of MCPR3-7 (B), CLB-12.8 (C), and MCPR3-2 (D) on this reaction were measured. Examples of three technical repeats are shown. The covalent binding of α1PI to PR3 was delayed by MCPR3-7, whereas CLB-12.8 and MCPR3-2 did not affect this complexation. mAb, monoclonal antibody; IgGH, IgG heavy; IgGL, IgG light.
Effects of mAbs on Noncovalent Complex Formation via Thermophoresis
To find out whether the mAbs MCPR3-7 and CLB-12.8 interfere with the formation of the canonical PR3·α1PI complex, thermophoretic competition experiments were performed. To this end, the binding of NT647-labeled α1PI to ΔPR3-S195A was quantified in the presence or absence of mAbs (Fig. 6). To form the PR3·mAb complexes, the components were preincubated for 1 h before adding labeled α1PI and measuring the binding affinity.
FIGURE 6.
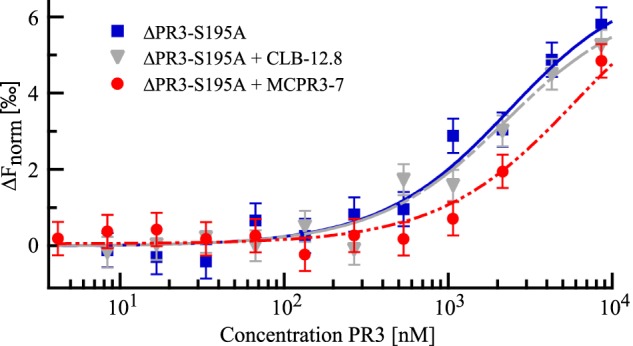
Evaluation of the interference of mAbs with the noncovalent PR3·α1PI complex formation via thermophoresis. Starting with a maximum concentration of 12 μm, dilution series of ΔPR3-S195A were prepared. Each sample of the dilution was mixed 6:1 with either PBS, MCPR3-7 in PBS, or CLB-12.8 in PBS to yield a mAb concentration of 670 nm. After preincubation for 1 h at room temperature, α1PI-NT647 was added to yield a final concentration of 880 nm. Thermophoresis revealed a KD of 1.9 ± 1.1 μm in the absence of mAbs (blue rectangles). Preincubation with CLB-12.8 did not have a significant effect on the affinity (KD = 1.8 ± 0.8 μm; gray triangles). In contrast, preincubation with MCPR3-7 led to a right shift of the binding curve corresponding to a 3-fold decrease in the apparent affinity to α1PI (KD = 5.6 ± 1.0 μm; red circles). Error bars represent S.D.
ΔPR3-S195A and α1PI bound with a KD of 1.9 ± 1.1 μm in the absence of mAbs. In the competition experiments, we were not able to use saturating amounts of MCPR3-7 as this antibody could not be concentrated to very high levels. We instead used a relatively low MCPR3-7 concentration of 570 nm. Thus, not only MCPR3-7·ΔPR3-S195A complexes but also free ΔPR3-S195A is present in solution. This reduces the putative overall effect of the antibody on the binding curve and leads to a more complex binding behavior. The thermophoresis signal corresponds to the binding of α1PI to free and MCPR3-7-bound ΔPR3-S195A, which is most likely characterized by two different KD constants. The data could not be fitted to a more complex binding model as saturation was not reached in the binding curve, and data points for higher MCPR3-7 concentrations were limited.
We instead chose to fit the data to the simple model according to the mass action law, which in this case revealed an apparent KD of 5.6 ± 1.0 μm. This apparent KD corresponds not only to the binding of α1PI to MCPR3-7·ΔPR3-S195A complexes but also to α1PI binding to free ΔPR3-S195A still present in solution. Taking into account the rather high affinity between α1PI and ΔPR3-S195A (1.9 μm), the actual binding affinity of α1PI for MCPR3-7·ΔPR3-S195A complexes can be expected to be lower than the measured apparent affinity (KD of 5.6 ± 1.0 μm). The competition experiment demonstrates the inhibiting impact of MCPR3-7 on PR3·α1PI noncovalent complex formation: preincubation with MCPR3-7 reduced the apparent affinity by a factor of 3. This inhibitory effect was already observed despite a relatively low MCPR3-7 concentration. Preincubation of ΔPR3-S195A with a higher MCPR3-7 concentration can be expected to lead to a much more pronounced decrease of the apparent affinity to α1PI. This finding clearly indicates that binding of MCPR3-7 leads to a conformational change of the free active PR3 and impairs the canonical interaction of PR3 and α1PI (Fig. 7).
FIGURE 7.
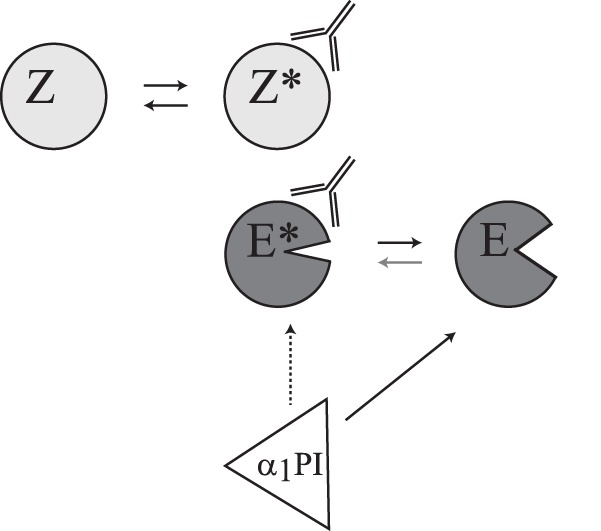
Impact of MCPR3-7 on PR3 activity and α1PI inhibition. Binding of MCPR3-7 to the proform of PR3 (Z) leads to slight conformational changes of the zymogen (Z*). The mature form of PR3 occurs in an equilibrium between a highly favored active (E) and an inactive (E*) conformation. Binding of the antibody shifts this equilibrium between E and E* toward the inactive E*. The antibody interaction stabilizes an altered conformation of E (modified substrate binding cleft; left shape) and changes activity and substrate specificity of PR3 (open substrate binding cleft; right shape). It also has an inhibitory effect (dotted arrow) on the complexation with α1PI.
In contrast to this pronounced impact of MCPR3-7, preincubation of ΔPR3-S195A with CLB-12.8 did not have an effect on its affinity to α1PI. The fitted binding curve depicted a nearly perfect overlay of the curve without mAbs, and the KD of 1.8 ± 0.8 μm did not significantly differ from the KD without mAbs (1.9 ± 1.1 μm). The affinity of CLB-12.8 to ΔPR3-S195A is expected to be much higher than the affinity of MCPR3-7 to ΔPR3-S195A. Thus, when adding the same concentration of CLB-12.8 and MCPR3-7, a much higher amount of CLB-12.8·PR3·α1PI complexes is present in solution. Nevertheless, CLB-12.8 did not have any effect on the canonical ΔPR3-S195A·α1PI complexation.
Activity Changes of PR3 Induced by MCPR3-7
To determine which subsites of PR3 are affected by conformational changes upon MCPR3-7 binding, the activity of PR3 toward different substrates was tested in the presence and absence of MCPR3-7. We used substrates with different amino acids at the P1 site and different leaving groups at the P1′ site. The cleavage of the substrate Abz-Tyr-Tyr-Abu-ANB-NH2 with the best fitting non-natural residue in P1 was strongly inhibited after binding of MCPR3-7 to PR3 (Fig. 8A). Likewise, the substrates 5-TAMRA-VADnVADYQ-DAP(CF) (Fig. 8B), an optimized extended peptide substrate, and Ahx-PYFA-pNA (Fig. 8C), a substrate with a relatively big leaving group in the P1′ position, could no longer be cleaved by PR3-MCPR3-7 complexes. In contrast, the substrates Boc-Ala-ONp (Fig. 8D) and Boc-Ala-Pro-nVal-SBzl (Fig. 8E), which carry residues of different sizes in their P1 position but have relatively small leaving groups in the P1′ position, were efficiently cleaved by PR3 even after binding of MCPR3-7. The cleavage of For-Ala-Ala-Pro-Abu-SBzl was partly inhibited by the binding of MCPR3-7 to PR3 (Fig. 8F), but PR3 still showed activity toward this substrate at a lower rate of about 50%. These findings strongly suggest that the conformation of the S1 pocket is not changed significantly after binding of MCPR3-7, but rather the S1′ subsite of PR3 is changed. Smaller leaving groups can still interact with PR3·MCPR3-7 complexes, whereas substrates with larger leaving groups could not be cleaved.
FIGURE 8.

Binding of MCPR3-7 leads to an alteration in the S1′ subsite of PR3. The effect of MCPR3-7 binding on the cleavage of different PR3 substrates was obtained in three technical repeats (±S.E.) with a 50 nm PR3 solution (A–E) or a 100 nm PR3 solution (F). A 3-fold molar excess of MCPR3-7 hindered the binding and cleavage of 800 nm Abz-Tyr-Tyr-Abu-ANB-NH2 (A), 5 μm 5-TAMRA-VADnVADYQ-DAP(CF) (B), and 1 mm Ahx-PYFA-pNA (C), whereas the cleavage of 1 mm Boc-Ala-ONp (D) and 1 mm Boc-Ala-Pro-nVal-SBzl (E) was not affected by the binding of MCPR3-7. For-Ala-Ala-Pro-Abu-SBzl (F) at a 1 mm concentration was still cleaved by PR3·MCPR3-7 complexes but at a lower rate of about 50%. The relative activity was determined by normalizing the activity of PR3 in the absence of antibodies to 1. Error bars represent S.E.
Detection of PR3 Expression on Neutrophil Membranes
After biochemical characterization of MCPR3-7 and MCPR3-2, we explored these as potential tools to determine PR3 expression on the membranes of non-stimulated neutrophils. A bimodal expression pattern of PR3 representing a large population of PR3-positive and a small subpopulation of PR3-negative neutrophils from peripheral blood was observed using MCPR3-2 for the recognition of membrane-bound PR3 (Fig. 9A, continuous black line). The majority of cells expressed PR3 on their surface, and a much smaller number of cells had no detectable membrane PR3 (Fig. 9A). After TNFα priming, the membrane-bound PR3 fraction increased, and essentially all PMNs expressed PR3 on their surface (Fig. 9B). CD32 staining served as a positive control and is indicated by the dotted line.
FIGURE 9.
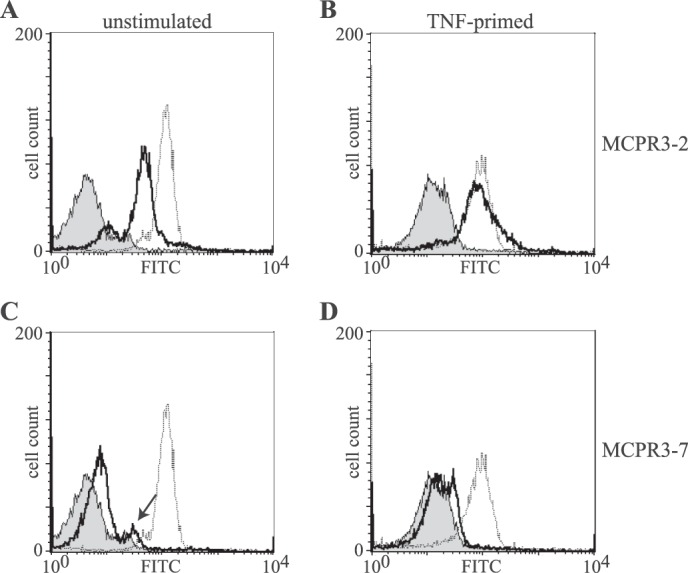
Differential recognition of PR3 on neutrophil membrane. Membrane expression of PR3 (continuous black line) in non-stimulated (A and C) and TNFα-primed neutrophils (B and D) was detected with MCPR3-2 (A and B) and MCPR3-7 (C and D). The gray peak represents an isotype control, and CD32 expression is represented by the dotted line. Non-stimulated neutrophils from this individual donor showed a bimodal distribution of PR3 membrane expression (A). PR3 membrane expression increased with priming of the neutrophils, and essentially all neutrophils expressed PR3 detectable with MCPR3-2 (B). By contrast, only a small fraction of unprimed neutrophils (arrow) expressed membrane PR3 using MCPR3-7 (C); PR3 expressed on primed neutrophils was essentially undetectable by MCPR3-7 (D).
When MCPR3-7 was used for the detection of membrane-bound PR3 on unstimulated neutrophils, the vast majority of cells did not display any membrane PR3, and only a tiny second population (arrow) with marginal MCPR3-7 reactivity was observed (Fig. 9C). After TNFα stimulation, a strong increase of membrane-bound PR3 was observed using MCPR3-2, and again this increase was not discernible with MCPR3-7; in fact, essentially none of the cells displayed PR3 binding to MCPR3-7 (Fig. 9D).
The likely reason for these disparate findings is that human PR3 is attached to membranes specifically and forms complexes with CD177 and/or lipid bilayers with its active conformation. Under these conditions, it cannot present the structural determinants recognized by MCPR3-7. These observations are fully consistent with the idea that MCPR3-7 interacts with hydrophobic patches of the activation domain that are not accessible in membrane-bound PR3.
DISCUSSION
The initial goal of this study was to identify a mAb that preferentially recognized the inactive proform of human PR3. As the proform sequence of PR3 is only two residues longer than the mature enzyme and the amino-terminal sequence of serine proteases is highly conserved, a unique amino-terminal peptide as a proform-specific immunogen cannot be designed. We therefore aimed at identifying a conformation-specific antibody that discriminates between the two principal conformations of mature and catalytically inactive PR3 (23). To prevent any spurious amino-terminal processing during synthesis of the recombinant pro-PR3 as well as subsequent in vivo modifications of the immunogen after injection, we inserted a proline residue between the natural propeptide and the amino terminus of mature PR3 (Fig. 1A). Most proteases including exoaminopeptidases even in combination with dipeptidyl aminopeptidase I cannot remove this tripeptide after a proline residue (34, 35). Moreover, endogenous PR3 inhibitors cannot associate with the proform of PR3, which would restrict the surface-accessible area for antibody responses.
The challenge of this strategy, however, was the high structural similarity between the proform (Z) and the mature form of PR3 (E) whose conformations only differ in one subregion, the so-called activation domain (22). The activation domain of mature PR3 can adopt a zymogen-like conformation (E*) even after propeptide removal although with low probability (24). Binding of an antibody could forcefully induce this zymogen-like, catalytically inactive state even in mature, amino-terminally processed PR3, resulting in a loss of the S1 binding site and oxyanion hole. In view of these circumstances, it was not too surprising that most mAbs generated failed to discriminate these two conformational states and showed very similar binding to both PR3 preparations.
Therefore, we selected a mAb on the basis of its differential affinities to both conformational states. To this end, we tested the direct binding of radioactively labeled pro-PR3 and PR3 to immobilized hybridoma IgG in an ELISA and to IgGs in solution followed by a single immunoprecipitation step (Fig. 1, B and C) and decided to use MCPR3-7, which showed the best discrimination between the two forms, for further analysis. To stabilize the activation domain in its active conformation, we used a mechanism-based small molecule inhibitor, Ala-Ala-Pro-Val-CMK, which forms a stable covalent complex with the transition state of PR3 (E) during catalysis, and tested the binding capability of this antibody to the active conformation. Indeed, this covalent complex displayed no detectable affinity toward MCPR3-7 as determined by ELISA (Fig. 4A) and by thermophoresis in free homogenous solution (Fig. 3A). In contrast, free active PR3 in solution showed some affinity for MCPR3-7 as revealed by thermophoresis, although it was ∼40-fold lower than that of pro-PR3. This weak interaction with free mature PR3 was also detectable by ELISA. Consistent with this finding, MCPR3-7 showed minimal binding to the so-called canonical complexes in which PR3 is constrained to adopt its active conformation (Fig. 4A). Covalently linked complexes between α1PI and PR3, however, exposed the epitope for MCPR3-7 (Fig. 4A). Two studies reporting covalent serpin structures between pancreatic elastase and α1PI (36) and trypsin with an arginine mutant of α1-antitrypsin (37) point out that the P1 residue of the reactive center loop is no longer buried in the S1 pocket of the protease and that the so-called autolysis loop 142–149, the 186–190 loop, and the amino-terminal four residues are disordered. Within this covalent complex, the protease adopts a zymogen-like conformation (E*) that carries the target epitope of MCPR3-7 on the accessible surface.
The MCPR3-7 epitope was mapped to the carboxyl-terminal half of PR3 using human/gibbon chimera (Fig. 2C). Non-conservative residue substitutions are found on all three loops of the so-called activation domain: a Lys to Gly change at position 187, a Trp to Arg substitution at 218, and an Ala to Thr switch at position 146 within the autolysis loop (31). MCPR3-7, which is able to interact with free active PR3 in solution, showed a strong inhibitory effect toward human PR3 and most significantly inhibited the activity of the chimera with the human sequence in the carboxyl-terminal half, which makes up the activation domain. This differential inhibitory effect on the gibbon/human versus the human/gibbon chimera (Fig. 2C) clearly indicates that the major binding site of MCPR3-7 is on the carboxyl-terminal half of PR3. These functional data are fully consistent with our previous observation that the epitope of MCPR3-7 could be reconstructed at least in part by a murine to human residue swap in the carboxyl-terminal half of murine PR3 (T146A,R218W,Q223L) (30).
Two other mAbs tested in this study (Fig. 2B) did not show any effect on the activity of human PR3 and also did not interfere with the inhibitory effect of MCPR3-7, indicating that the antibody binding site of MCPR3-7 is unique and does not overlap with the epitopes of the other mAbs (Fig. 2A). The strong inhibitory effect of MCPR3-7 was detected with an internally quenched substrate that occupies the S3 to S1 subsites of PR3 (Tyr-Tyr-Abu) and interacts with the S4 and S1′ pockets by virtue of its quencher ANB-NH2 and fluorogen Abz, respectively (Fig. 2B).
Substrates with other P1′ leaving groups, e.g. 5-amino-2-nitrobenozoic acid (ANB-OH), para-nitroaniline (pNA), the benzyl mercaptan from thiobenzyl esters, the para-nitrophenol from Boc-l-Ala-ONp, do not interact with the S1′ subsite of PR3 as efficiently as ANB-NH2 substrates (26). Indeed, we found that cleavage of the small ester substrate Boc-l-Ala-ONp and the thioester substrate Boc-Ala-Pro-nVal-SBzl (Fig. 8, D and E) was not significantly affected by MCPR3-7, suggesting that the shape and/or access to the S1′ pocket is primarily altered in the MCPR3-7·PR3 enzyme complex. Although the S1 pocket can accept P1 residues like Ala and nor-Val, which are shorter and longer than Abu, respectively, the S1′ pocket appears to accept only smaller and less polar leaving groups after MCPR3-7 binding. Extended peptide substrates that have to be optimally aligned with the S1, S1′, and S2′ pockets were most strongly affected by MCPR3-7 binding (Fig. 8B). Moreover, binding of this antibody to active PR3 reduced canonical interactions with the reactive center loop of α1PI (Fig. 6) and subsequent covalent complex formation (Fig. 5). Taken together, all these characteristics qualify MCPR3-7 as a first prototype of a PR3-directed conformation-specific mAb with inhibitory properties. Inhibition is most likely due to an allosteric effect changing the autolysis and 187–190 loops. Both loops shape the size and physical character of adjacent subsites around the cleaved peptide bond, namely S1, S1′, and S2′. The autolysis loop (Gly145–Ala152) shows the highest temperature factor of the main chain and is completely disordered in all four PR3 molecules of the asymmetric unit (21). A second region with a high temperature factor maps to the 187–190 loop (21). Such regions are in general preferred target sites for antibody interactions and are good targets for allosteric regulation of enzyme activities. Capture of partially altered loop conformations of PR3 by MCPR3-7 most likely changes substrate recognition and impairs the interaction of PR3 with the reactive center loop of α1PI.
Antibodies that can interfere with the catalytic activity of serine proteases are classified in two groups according to their mode of action as reviewed recently (38). Antibodies like MCPR3-7 induce an allosteric switch in the target antigen. This first group of antibodies does not directly compete with substrates for the active site cleft but rather interacts with a region at the periphery of the substrate binding region. Upon binding to the enzyme, antibodies of this group induce a significant conformational switch in surface loops that determine the shape and size of substrate binding pockets. The epitopes of these antibodies can be regarded as regulatory hot spots that are connected to mobile surface structures around the active site cleft. The altered conformation of loops surrounding the substrate binding pockets reduces, alters, or suppresses catalytic activity.
The second group can bind to the catalytic domain of a serine protease in a way that substrate access to the catalytic cleft is partially or fully occluded. Some complementarity-determining region loops of these antibodies directly insert into substrate binding pockets and occupy the substrate binding regions with high affinity. The latter type of antibodies, however, is rarely found in mammals and is hardly induced by immunizations as the antigen binding surface of mammalian antibodies mostly interacts with convex, protruding surfaces or a flat surface of an antigen. Directly inhibiting antibodies, however, have been produced in camelids. These antibodies represent unusual homodimers of a single heavy chain but are immunogenic in humans. Hence, camelid antibodies so far have found little application in preclinical (animal) and clinical studies.
Identification of monoclonal antibodies with similar properties and epitope specificity as MCPR3-7 but a higher affinity for mature PR3 should be of medical impact for two reasons. Antibodies of this type would not only block the binding of secreted soluble PR3 to neutrophils membranes but also could serve as selective PR3 inhibitors and clearers of PR3. Application of these antibodies in GPA patients may reduce the extent of neutrophil activation by anti-neutrophil cytoplasmic antibody via membrane-bound PR3 and Fcγ receptors and other tissue-damaging effects of secreted PR3.
Acknowledgments
The expert technical assistance of Heike Kittel is gratefully acknowledged. In addition, the authors thank Oliver Eickelberg for continuous interest in the project and Karl-Heinz Wiesmüller, EMC Microcollections for the design and synthesis of FRET substrates.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1-AR49806 and RC1-AR58303 (to U. S.) and U54-RR019497 from the Vasculitis Clinical Research Consortium (to F. S.). This work was also supported by European Union Seventh Framework Programme (FP7/2007–2013) under Grant Agreement 261382 (INTRICATE), the Alexander von Humboldt Foundation, a joint grant from the Deutsche Forschungsgemeinschaft (JE194/4-1; BR2152/2-1), the NanoSystems Initiative Munich, the Center for NanoScience, the Ludwig-Maximilians-Universität Munich Initiative Functional Nanosystems, and the Mayo Clinic Monoclonal Antibody Core Facility.
U. Specks, unpublished data.
- PR3
- proteinase 3
- GPA
- granulomatosis with polyangiitis
- α1PI
- α1-proteinase inhibitor
- ANB
- 5-amino-2-nitrobenzoic acid
- Abz
- γ-aminobenzoic acid
- EBNA
- Epstein-Barr nuclear antigen 1
- Abu
- ã-aminobutyric acid
- DAP(CF)
- diaminopropionyl fluorescein
- SBzl
- thiobenzyl ester
- nVal or nV
- norvaline
- For
- formyl
- Boc
- t-butoxycarbonyl
- TAMRA
- tetramethylrhodamine
- Ahx
- aminohexanoic acid
- ONp
- para-nitrophenyl ester
- pNA
- para-nitroaniline
- CMK
- chloromethyl ketone
- h
- human
- gib
- gibbon
- MST
- microscale thermophoresis.
REFERENCES
- 1. Korkmaz B., Horwitz M. S., Jenne D. E., Gauthier F. (2010) Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 62, 726–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perera N. C., Jenne D. E. (2012) Perspectives and potential roles for the newly discovered NSP4 in the immune system. Expert Rev. Clin. Immunol. 8, 501–503 [DOI] [PubMed] [Google Scholar]
- 3. Pham C. T. (2006) Neutrophil serine proteases: specific regulators of inflammation. Nat. Rev. Immunol. 6, 541–550 [DOI] [PubMed] [Google Scholar]
- 4. Heutinck K. M., ten Berge I. J., Hack C. E., Hamann J., Rowshani A. T. (2010) Serine proteases of the human immune system in health and disease. Mol. Immunol. 47, 1943–1955 [DOI] [PubMed] [Google Scholar]
- 5. Csernok E., Lüdemann J., Gross W. L., Bainton D. F. (1990) Ultrastructural localization of proteinase 3, the target antigen of anti-cytoplasmic antibodies circulating in Wegener's granulomatosis. Am. J. Pathol. 137, 1113–1120 [PMC free article] [PubMed] [Google Scholar]
- 6. Witko-Sarsat V., Cramer E. M., Hieblot C., Guichard J., Nusbaum P., Lopez S., Lesavre P., Halbwachs-Mecarelli L. (1999) Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood 94, 2487–2496 [PubMed] [Google Scholar]
- 7. Csernok E., Ernst M., Schmitt W., Bainton D. F., Gross W. L. (1994) Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin. Exp. Immunol. 95, 244–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Halbwachs-Mecarelli L., Bessou G., Lesavre P., Lopez S., Witko-Sarsat V. (1995) Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Lett. 374, 29–33 [DOI] [PubMed] [Google Scholar]
- 9. Schreiber A., Busjahn A., Luft F. C., Kettritz R. (2003) Membrane expression of proteinase 3 is genetically determined. J. Am. Soc. Nephrol. 14, 68–75 [DOI] [PubMed] [Google Scholar]
- 10. Kallenberg C. G. (2011) Pathogenesis of ANCA-associated vasculitis, an update. Clin. Rev. Allergy Immunol. 41, 224–231 [DOI] [PubMed] [Google Scholar]
- 11. Hoffman G. S., Specks U. (1998) Antineutrophil cytoplasmic antibodies. Arthritis Rheum. 41, 1521–1537 [DOI] [PubMed] [Google Scholar]
- 12. Lyons P. A., Rayner T. F., Trivedi S., Holle J. U., Watts R. A., Jayne D. R., Baslund B., Brenchley P., Bruchfeld A., Chaudhry A. N., Cohen Tervaert J. W., Deloukas P., Feighery C., Gross W. L., Guillevin L., Gunnarsson I., Harper L., Hrušková Z., Little M. A., Martorana D., Neumann T., Ohlsson S., Padmanabhan S., Pusey C. D., Salama A. D., Sanders J.-S., Savage C. O., Segelmark M., Stegeman C. A., Tesař V., Vaglio A., Wieczorek S., Wilde B., Zwerina J., Rees A. J., Clayton D. G., Smith K. G. (2012) Genetically distinct subsets within ANCA-associated vasculitis. N. Engl. J. Med. 367, 214–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ogushi F., Fells G. A., Hubbard R. C., Straus S. D., Crystal R. G. (1987) Z-type α1-antitrypsin is less competent than M1-type α1-antitrypsin as an inhibitor of neutrophil elastase. J. Clin. Investig. 80, 1366–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gettins P. G. (2002) Serpin structure, mechanism, and function. Chem. Rev. 102, 4751–4804 [DOI] [PubMed] [Google Scholar]
- 15. Campanelli D., Melchior M., Fu Y., Nakata M., Shuman H., Nathan C., Gabay J. E. (1990) Cloning of cDNA for proteinase 3: a serine protease, antibiotic, and autoantigen from human neutrophils. J. Exp. Med. 172, 1709–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zimmer M., Medcalf R. L., Fink T. M., Mattmann C., Lichter P., Jenne D. E. (1992) Three human elastase-like genes coordinately expressed in the myelomonocyte lineage are organized as a single genetic locus on 19pter. Proc. Natl. Acad. Sci. U.S.A. 89, 8215–8219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rao N. V., Rao G. V., Marshall B. C., Hoidal J. R. (1996) Biosynthesis and processing of proteinase 3 in U937cells. Processing pathways are distinct from those of cathepsin G. J. Biol. Chem. 271, 2972–2978 [DOI] [PubMed] [Google Scholar]
- 18. Specks U., Fass D. N., Fautsch M. P., Hummel A. M., Viss M. A. (1996) Recombinant human proteinase 3, the Wegener's autoantigen, expressed in HMC-1 cells is enzymatically active and recognized by c-ANCA. FEBS Lett. 390, 265–270 [DOI] [PubMed] [Google Scholar]
- 19. Garwicz D., Lindmark A., Hellmark T., Gladh M., Jögi J., Gullberg U. (1997) Characterization of the processing and granular targeting of human proteinase 3 after transfection to the rat RBL or the murine 32D leukemic cell lines. J. Leukoc. Biol. 61, 113–123 [DOI] [PubMed] [Google Scholar]
- 20. Sun J., Fass D. N., Viss M. A., Hummel A. M., Tang H., Homburger H. A., Specks U. (1998) A proportion of proteinase 3 (PR3)-specific anti-neutrophil cytoplasmic antibodies (ANCA) only react with PR3 after cleavage of its N-terminal activation dipeptide. Clin. Exp. Immunol. 114, 320–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujinaga M., Chernaia M. M., Halenbeck R., Koths K., James M. N. (1996) The crystal structure of PR3, a neutrophil serine proteinase antigen of Wegener's granulomatosis antibodies. J. Mol. Biol. 261, 267–278 [DOI] [PubMed] [Google Scholar]
- 22. Fehlhammer H., Bode W., Huber R. (1977) Crystal structure of bovine trypsinogen at 1.8 Å resolution. II. Crystallographic refinement, refined crystal structure and comparison with bovine trypsin. J. Mol. Biol. 111, 415–438 [DOI] [PubMed] [Google Scholar]
- 23. Jenne D. E., Kuhl A. (2006) Production and applications of recombinant proteinase 3, Wegener's autoantigen: problems and perspectives. Clin. Nephrol. 66, 153–159 [DOI] [PubMed] [Google Scholar]
- 24. Pozzi N., Vogt A. D., Gohara D. W., Di Cera E. (2012) Conformational selection in trypsin-like proteases. Curr. Opin. Struct. Biol. 22, 421–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun J., Fass D. N., Hudson J. A., Viss M. A., Wieslander J., Homburger H. A., Specks U. (1998) Capture-ELISA based on recombinant PR3 is sensitive for PR3-ANCA testing and allows detection of PR3 and PR3-ANCA/PR3 immune complexes. J. Immunol. Methods 211, 111–123 [DOI] [PubMed] [Google Scholar]
- 26. Wysocka M., Lesner A., Guzow K., Mackiewicz L., Legowska A., Wiczk W., Rolka K. (2008) Design of selective substrates of proteinase 3 using combinatorial chemistry methods. Anal. Biochem. 378, 208–215 [DOI] [PubMed] [Google Scholar]
- 27. Capizzi S. A., Viss M. A., Hummel A. M., Fass D. N., Specks U. (2003) Effects of carboxy-terminal modifications of proteinase 3 (PR3) on the recognition by PR3-ANCA. Kidney Int. 63, 756–760 [DOI] [PubMed] [Google Scholar]
- 28. Korkmaz B., Kuhl A., Bayat B., Santoso S., Jenne D. E. (2008) A hydrophobic patch on proteinase 3, the target of autoantibodies in Wegener granulomatosis, mediates membrane binding via NB1 receptors. J. Biol. Chem. 283, 35976–35982 [DOI] [PubMed] [Google Scholar]
- 29. Perera N. C., Schilling O., Kittel H., Back W., Kremmer E., Jenne D. E. (2012) NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc. Natl. Acad. Sci. U.S.A. 109, 6229–6234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Silva F., Hummel A. M., Jenne D. E., Specks U. (2010) Discrimination and variable impact of ANCA binding to different surface epitopes on proteinase 3, the Wegener's autoantigen. J. Autoimmun. 35, 299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuhl A., Korkmaz B., Utecht B., Kniepert A., Schönermarck U., Specks U., Jenne D. E. (2010) Mapping of conformational epitopes on human proteinase 3, the autoantigen of Wegener's granulomatosis. J. Immunol. 185, 387–399 [DOI] [PubMed] [Google Scholar]
- 32. Seidel S. A., Dijkman P. M., Lea W. A., van den Bogaart G., Jerabek-Willemsen M., Lazic A., Joseph J. S., Srinivasan P., Baaske P., Simeonov A., Katritch I., Melo F. A., Ladbury J. E., Schreiber G., Watts A., Braun D., Duhr S. (2013) Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seidel S. A., Wienken C. J., Geissler S., Jerabek-Willemsen M., Duhr S., Reiter A., Trauner D., Braun D., Baaske P. (2012) Label-free microscale thermophoresis discriminates sites and affinity of protein-ligand binding. Angew. Chem. Int. Ed. Engl. 51, 10656–10659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lindley H. (1972) The specificity of dipeptidyl aminopeptidase I (cathepsin C) and its use in peptide sequence studies. Biochem. J. 126, 683–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lone A. M., Nolte W. M., Tinoco A. D., Saghatelian A. (2010) Peptidomics of the prolyl peptidases. AAPS J. 12, 483–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huntington J. A., Read R. J., Carrell R. W. (2000) Structure of a serpin-protease complex shows inhibition by deformation. Nature 407, 923–926 [DOI] [PubMed] [Google Scholar]
- 37. Dementiev A., Dobó J., Gettins P. G. (2006) Active site distortion is sufficient for proteinase inhibition by serpins: structure of the covalent complex of α1-proteinase inhibitor with porcine pancreatic elastase. J. Biol. Chem. 281, 3452–3457 [DOI] [PubMed] [Google Scholar]
- 38. Ganesan R., Eigenbrot C., Kirchhofer D. (2010) Structural and mechanistic insight into how antibodies inhibit serine proteases. Biochem. J. 430, 179–189 [DOI] [PubMed] [Google Scholar]