

Background: Gain of function (GOF) mutations enhance the vWF-GPIbα interaction.
Results: GOF mutations induce destabilization of the N-terminal arm and increase mobility of the α2-helix.
Conclusion: Dynamics-driven up-regulation of A1 affinity to GPIbα serves as a GOF mechanism of type 2B mutations.
Significance: These results are helpful in understanding the structural basis of GOF mutants and in developing allosteric drugs against the activated A1 domain.
Keywords: Biophysics, Molecular Dynamics, Platelets, Protein Dynamics, Structural Biology, Thrombosis, von Willebrand Factor, GPIbα, von Willebrand Disease
Abstract
Binding of the A1 domain of von Willebrand factor (vWF) to glycoprotein Ibα (GPIbα) results in platelet adhesion, activation, and aggregation that initiates primary hemostasis. Both the elevated shear stress and the mutations associated with type 2B von Willebrand disease enhance the interaction between A1 and GPIbα. Through molecular dynamics simulations for wild-type vWF-A1 and its eight gain of function mutants (R543Q, I546V, ΔSS, etc.), we found that the gain of function mutations destabilize the N-terminal arm, increase a clock pendulum-like movement of the α2-helix, and turn a closed A1 conformation into a partially open one favoring binding to GPIbα. The residue Arg578 at the α2-helix behaves as a pivot in the destabilization of the N-terminal arm and a consequent dynamic change of the α2-helix. These results suggest a localized dynamics-driven affinity regulation mechanism for vWF-GPIbα interaction. Allosteric drugs controlling this intrinsic protein dynamics may be effective in blocking the GPIb-vWF interaction.
Introduction
In primary hemostasis, the initiation of platelet deposition at sites of vascular injury relies exclusively on the interaction between von Willebrand factor (vWF)3 and platelet glycoprotein Ibα (GPIbα) under the high flow conditions present in circulating blood (1). The intrinsic adhesive properties of vWF are tightly regulated such that plasma vWF and GPIbα could coexist without interaction under normal conditions. However, vWF could be activated to bind platelets under pathophysiological conditions, causing such conditions as microthrombosis for patients with thrombotic thrombocytopenic purpura (2) and bleeding for patients with type 2B von Willebrand disease (3). Therefore, understanding the regulatory mechanism of vWF activity is important for the diagnosis, treatment, and prevention of vWF-related diseases.
In circulation, the vWF presents as multimers of different molecular masses. Each vWF monomer consists of five types of repeat domains in the order of D1-D2-D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK, of which the A1 domain contains the known GPIbα-binding site (4). The crystal structure of the A1 domain comprises one globular body region and the N- and C-terminal arms connected by a disulfide bridge between the residues Cys509 and Cys695 (5) (Fig. 1, A and B). The body region consists of a six-stranded hydrophobic β-sheet flanked by three amphipathic α-helices on each side. The N-terminal arm folds over the body region and contacts with the bottom face of the body region, whereas the C-terminal arm extends downwards from the body. Crystallographic studies reveal that the concave face of GPIbα wraps around the body region in a pincer-like grip with the β-switch and β-finger regions (Fig. 1C). Both arms are not involved in the interaction with GPIbα but approach the β-finger-binding site (6).
FIGURE 1.
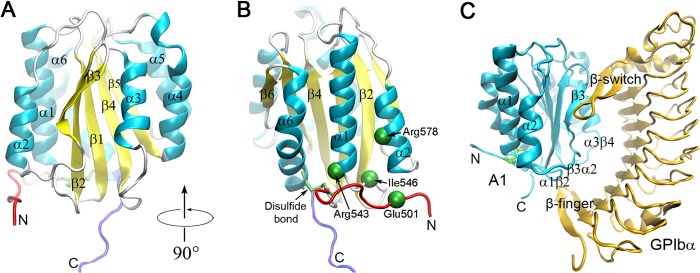
Structures of the wild-type vWF A1 domain and its complex with platelet GPIbα. All the structures were shown in Newcartoon representation. A, the A1 domain (Protein Data Bank code 1AUQ) (5), in which the colored secondary structure elements, such as the N-terminal arm (red), C-terminal arm (blue), six α-helixes (cyan), and six β-strands (yellow) as well as the loops (white), are labeled. B, the left side view of A, where the Cys509–Cys695 disulfide bond (green) and the four gain of function mutation sites (green spheres) are marked. C, the complex of A1 (cyan) bound GPIbα (orange) (Protein Data Bank code 1SQ0) (20), where all the labeled secondary structure elements, such as the β3-sheet, α2- and α3-helixes, and α1β2-, β3α2-, and α3β4-loops, are involved in the GPIbα binding surface.
Several lines of evidence indicate that plasma vWF is autoinhibited by internal domain-domain interactions between A2 and A1 (7), D′D3 and A1 (8), as well as the N-terminal flanking region of A1 and A1A2A3 tridomain (9). An isolated recombinant A1 domain binds GPIbα spontaneously (10). Hence, the exposure of the GPIbα-binding site in the A1 domain through a conformational change of vWF serves as an underlying mechanism of activating plasma vWF multimers. This activation process can be induced by high fluid shear forces detected at a site of arterial stenosis (11, 12).
The bond strength for the interaction between the exposed A1 and GPIbα is essential for efficient platelet adherence because this physical bonding must withstand strong hydrodynamic forces acting on cells. It has been demonstrated that the A1-GPIbα bond is a “flex bond” having two states with different lifetimes and force resistances (13); a tensile force changes the equilibrium as well as the switching rate between the two states (13). The gain-of-function (GOF) mutation R543Q associated with type 2B von Willebrand disease reduces the force that is required by the switch (14). In contrast, the WT A1 has been shown to form a catch bond with GPIbα, where the bond lifetime is prolonged by increasing force (15). A type 2B mutation can result in a left shift or a complete loss of the catch bond characteristics by increasing the lifetime at low force (15, 16). In addition, the binding affinity of an isolated A1 domain is increased ∼2.5-fold for the mutant R543Q (6) and 20-fold for the mutant R545A (17), independent of fluid shear stress. A similar increase in binding affinity can be induced in the isolated WT A1 domain by reduction and alkylation of the Cys509–Cys695 disulfide bond and/or by exposure to acidic pH (18, 19). These data suggest that the A1 domain itself may take either a low or a high affinity conformation, and tensile forces and GOF mutations favor the high affinity conformation.
However, the superposition of 12 A1 crystal structures (the WT A1 or its GOF mutants, unliganded or liganded with GPIbα, activators, or inhibitors) in the Protein Data Bank shows no significant structural heterogeneity to interpret these affinity differences (supplemental Fig. S1) (5, 6, 20–26). Because a crystal structure at a moderate resolution only provides time- and crystal lattice-averaged conformation present for more than 20% of the time (27), it is unknown whether the high affinity conformation is dynamically transient or reflects one of the known crystallized structures (3). Recently, the A1-GPIbα bond stability was correlated inversely with the intrinsic thermodynamic stability of the A1 domain (16). This suggests that apart from static structures, dynamic properties associated with the backbone and side chain mobilities responsible for the conformational stability may also play a key role in regulating the ligand-receptor interaction.
Molecular dynamics (MD) simulation has a unique ability to describe protein dynamics by tracking the precise position of each atom at any instant in time, and most importantly, can not only determine what is changing upon perturbations but can also tell us how and why the change happens (28). To better understand the molecular basis of GOF mutants, we herein examined the dynamic properties of the wild-type vWF A1 and its eight GOF mutants through MD simulations. The results depict a localized dynamics-driven affinity regulation mechanism for binding of vWF to GPIbα, provide a novel insight into the structural basis of GOF mutants, and may assist in developing allosteric drugs against the activated A1 domain.
EXPERIMENTAL PROCEDURES
System Setup
Two groups of molecular systems of vWF A1 structures were set up for MD simulations. The first group has four structures, namely the unliganded WT vWF A1 (Protein Data Bank code 1AUQ) (5) (Fig. 1A), its two gain-of-function mutants I546V (Protein Data Bank code 1IJB) (21) and R543Q (Protein Data Bank code 1M10) (6), and a modeled mutant labeled with “ΔSS” (Fig. 1B). Among the 25 type 2B mutants, R543Q and I546V were selected, because the thermodynamics and kinetics properties of these two mutants were better demonstrated than others (14–16, 19, 29). The mutant ΔSS was modeled by reducing the Cys509–Cys695 disulfide bond in the WT A1 via the AUTOPSF plug-in of VMD (30) (Fig. 1B). Both R543Q and I546V were reconstructed by replacing their N-terminal tails (residues 505–508 in R543Q and residues 500–508 in I546V) with that (residues 498–508) of the WT A1, through aligning with the body region of the WT A1 and making the N-terminal tails have an initial conformation the same as that of the WT A1. Missing atoms (all hydrogen atoms, side chain of Lys660 in WT A1, and side chain of Arg571 in I546V A1) in the partially resolved residues were added via Swiss-Pdb Viewer (31).
The reconstructed models of both R543Q and I546V were minimized along the protocol such that all atoms except those in the junction residues Tyr508 and Cys509 were fixed in the first 1000 minimization steps, the followed 2000 minimization steps were run for the free N-terminal tails (residues 498–508) joined to the fixed body, and the last 5000 minimization steps were performed for all atoms unconstrained. In each of the minimized models of R543Q and I546V, the irrational bond length of the peptide bond 508–509 had reverted to a normal value of 1.33 Å, and the 508–509 peptide plane had recovered too. The key contacts between the N-terminal arm and the rest of A1 were examined to be preserved (supplemental Fig. S2 and Table S1).
The other group includes six structures, of which, one, labeled as “WT-NC,” was a WT A1 reconstructed through taking the body of A1 from the crystal structure of the complex WT-A1/GPIbα (Protein Data Bank code 1SQ0) (20) and adding the N-terminal arm with the same protocol used in R543Q and I546V, and others, including a GOF mutant E501A (32) and four type 2B mutants (R578Q, R578W, R578P, and R578L) (33–35), were modeled per homology by replacing the corresponding side chain in WT A1 and subsequently performing 1000 steps of minimization in vacuo while all atoms except the mutated residue were fixed. The WT-NC A1 was used for a negative test to verify whether the reconstructed R543Q and I546V mutants were rational or not, and each GOF mutation (E501A, R578Q, R578W, R578P, and R578L) was selected to further examine whether it would cause a change of the dynamic properties of A1.
The protonation state of each titrable protein residue at neutral pH was determined with the software PROPKA (36). The terminal patches ACE and CT3 were added to the N terminus and the C terminus, respectively, to mimic the continuation of the protein chain. The crystallographic water molecules were retained. Each structure was soaked with TIP3P water molecules in a rectangular box with walls at least 15 Å away from any protein atom. Na+ and Cl− counter ions at physiological concentration of 150 mm were added into the water boxes to achieve charge neutrality and to mimic the real physiological environment.
Molecular Dynamics Simulations
Two software packages, visual molecular dynamics (VMD) for visualization and modeling (30) and NAMD 2.9 program for free MD simulations (37), were used in the simulations. The CHARMM22 all-atom force field (38), along with cMAP correction for backbone, particle mesh Ewald algorithm for electrostatic interaction and a 12 Å cutoff for electrostatic and van der Waals interaction, was used to perform MD simulations with periodical boundary condition and time step of 2 fs. All systems were subjected to energy minimizations for 5,000 steps with heavy atoms fixed and for another 10,000 steps with all atoms free. The energy-minimized systems were heated gradually from 0 to 310 K in 0.1 ns first and then equilibrated for 5 ns with pressure and temperature control. The temperature was held at 310 K using Langevin dynamics, and the pressure was held at 1 atmosphere by the Langevin piston method. From each system, three different structures in equilibrium were chosen as the initial conformations for free dynamics simulations to better capture the dynamics of the protein (28). The free dynamics simulations were run three times on each equilibrated system over 100 ns in a microcanonical ensemble (constant number of atoms, constant volume, and constant energy). The atomic coordinates were recorded and analyzed every other picosecond. All simulations were run on the Dell PowerEdge M910 supercomputer at the School of Bioscience and Bioengineering of South China University of Technology.
Data Analysis
All analyses were performed with VMD tools (30). The time courses of Cα root mean square deviation (RMSD) and radius of gyration (Rgyr) illustrated the conformational changes and the stabilities of the structures. The Cα root mean square fluctuation (RMSF) patterns marked the local structural flexibilities. The interhelical angle between the α2- and α1-helix in A1 was quantified by the cross-angle of two straight lines, which connected the N termini (residue 575 for α2 and 528 for α1) of the helixes to their respective C termini (residue 582 for α2 and 542 for α1) for simplicity. A hydrogen bond was defined if the donor-acceptor distance was <3.5 Å and the donor-hydrogen-acceptor angle was >150°. To define a salt bridge, the distance between any of the oxygen atoms of acidic residues (Asp or Glu) and the nitrogen atoms of basic residues (Lys or Arg) must be within 4 Å. A hydrogen bond or salt bridge occupancy was defined as the percentage of bond survival time, during which a hydrogen bond or salt bridge formed in the period of simulation. The residue hydration frequency was defined as the ratio of the hydration events, in which at least one water molecule appeared at a position less than 4 Å apart from a hydrophobic residue, to all events with or without hydration. Both the residue hydration frequency and the solvent-accessible surface area (SASA) with a 1.4 Å probe radius were measured to examine whether the hydrophobic core was exposed or not. All molecular images were generated using VMD (30).
Statistical Analysis
The p values of unpaired two-tailed Student's t test were used to indicate the statistical difference significance of the data (p < 0.05) or lack thereof (p > 0.05).
RESULTS
GOF Mutation Triggers the Switch from a Stable Conformation to a Localized Unstable One for the A1 Domain
Thermodynamic experiments demonstrated that type 2B mutations destabilize A1 (16). To reveal the structural changes caused by the mutations, we first performed free dynamics simulations three times over 100 ns with a time step of 2 fs for each of the WT A1 and its three mutants (R543Q, I546V, and ΔSS) (see “Experimental Procedures”). We observed this mutation-induced destabilization from the time courses of the Cα root mean square deviation (RMSD) (Fig. 2A and supplemental Fig. S3) and the gyration radius Rgyr (supplemental Fig. S4) of global A1 (residues 498–700), regardless of the C-terminal arm for its very flexible residue composition beyond Ala701. The type 2B mutations did remarkably raise the levels of both Cα RMSD and Rgyr or induce the destabilization, which occurred more severely in ΔSS than in both R543Q and I546V and was contributed mainly by the N-terminal arm (residues 498–508) rather than by the body region (residues 509–695) (Fig. 2, B and C). The major structural change occurred in the N-terminal flanking peptide (Fig. 2C) and was reflected by the increasing distance between the mass center (DMC) of the N-terminal arm and the body region for each mutant in comparison with the WT A1 (Fig. 2D), but the minor structural change in the body region was observable too (Fig. 2B). These structural changes were intuitively illustrated by a superimposition of four structures (Fig. 3A), each of which was an average over all conformational snapshots simulated one by one in duration of 100 ns in three runs for each of the WT A1 and its mutants.
FIGURE 2.
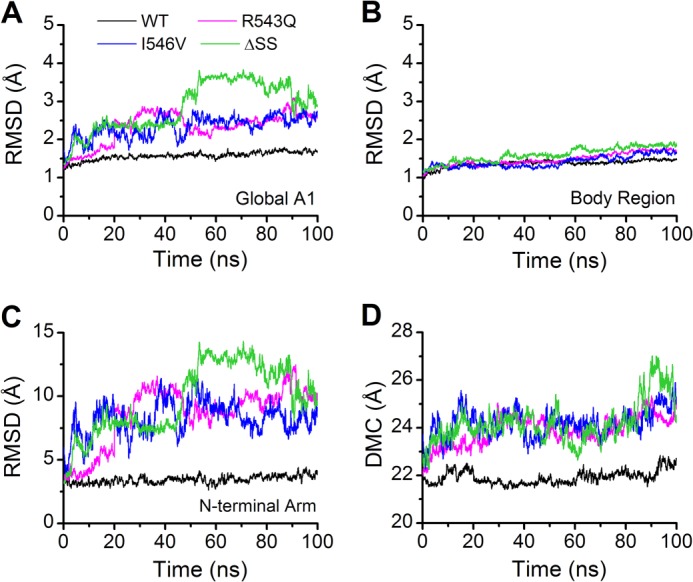
The mean time curves of RMSD and DMC for WT A1 (black) and its three mutants (R543Q, magenta; I546V, blue; ΔSS, green). RMSD stands for the Cα root mean square deviation, and DMC expresses the distance between the mass centers of the N-terminal arm and the A1 body region. Each of the time-RMSD profiles, which are shown with a moving average over 20 ps, for the global A1 (residues 498–700) (A) and the body region (residues 509–695) (B) as well as the N-terminal arm (residues 498–508) (C), presents an average of data from three independent runs and so do the time-DMC profiles (D). The time-RMSD profiles of the global WT A1 (A) and the N-terminal arm (C) are smoother and remain at a lower level in comparison with R543Q, I546V, and ΔSS, and so do the time curves of DMC (D), suggesting that the structural stability lies in the wild-type A1 but not in the mutants.
FIGURE 3.
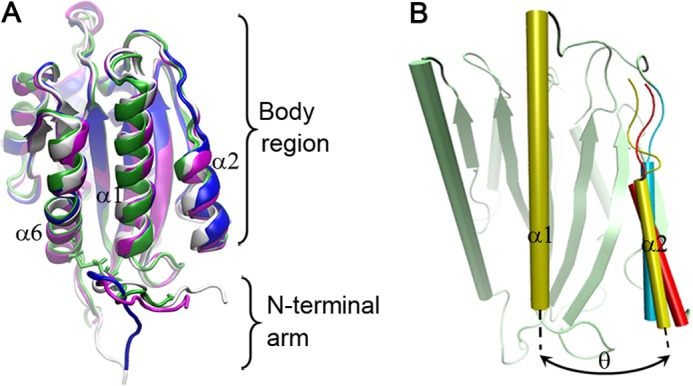
The mutation-induced change and evolution of the wild-type A1 structure. A, superposition of the structures of WT A1 (white), R543Q (magenta), I546V (blue), and ΔSS (green). Each conformation was averaged over 3 × 100 ns involved in three runs with simulation time of 100 ns. The mutation-induced change of A1 was small but observable, especially for N-terminal arm and the α2-helix. B, three representative poses of the α2-helix relative to the α1-helix for the wild-type A1 at three different moments such as 2.95 ns (yellow), 40.42 ns (red), and 84.09 ns (cyan) in a run, where three α1-helices and β-sheets were aligned very well and shown in cartoon representation. The α2-α1 interhelical angle θ, which was quantified by the cross-angle of the α1-helix axis (from N termini residue 528 to C termini residue 542) and the α2-helix axis (from N termini residue 575 to C termini residue 582) (B), was read, respectively, to be 25.61° (yellow), 40.17° (red), and 5.23° (cyan) at the moments of 2.95, 40.42, and 84.09 ns, showing a clock pendulum-like movement of the α2-helix relative to the α1-helix.
These results suggest that the GOF mutations trigger the switch from a stable conformation of A1 to a localized unstable one, and the region largely responsible for the destabilization is the N-terminal arm rather than the body region (Fig. 2, B and C), which remains stable even without the protection of the N-C-terminal disulfide bond. This localized destabilization of A1 may make the N-terminal arm either move away from or extend toward the β-finger-binding site of GPIbα (Figs. 2C and 3A), showing a structural heterogeneity of the N-terminal arm. This structural heterogeneity may be reflected by the various conformations of the N-terminal arm in the 12 A1 crystal structures (supplemental Fig. S1). The significant displacements of the N-terminal arm away from the body region in the GOF mutants (Figs. 3A and 2D) would weaken the interaction between the N-terminal arm and the body region. It demonstrates that an extensive N-terminal arm can shield the binding site for GPIbα, but the type 2B mutations are most likely to induce a conformational change in this peptide and make the binding site accessible, as suggested previously (5, 6, 39).
GOF Mutation Changes the Localized Dynamic Property of the A1 Domain
Because these GOF mutations do not lead to notable structural change in the GPIbα binding interface, an alternative explanation for the affinity modulation may lie in the mutation-induced change of the dynamic property of A1. To examine this possibility, we analyzed the Cα RMSF per residue in A1 and found that these GOF mutations enhanced the mobility of backbone in A1 (Fig. 4A and supplemental Fig. S5). The most significant fluctuation occurred consistently in the α2-helix (residues 574–583) and a part of the conjoint β3α2-loop (residues 569–573) rather than in the rest of A1 except the N-terminal arm, and the GOF mutations enhanced the fluctuation of the region spanning residues 569–583 (Fig. 4A). This mutation-induced change of the intrinsic dynamic property may regulate the binding of A1 to GPIbα, because both the α2-helix and the β3α2-loop make up part of the GPIbα binding interface (Fig. 1C).
FIGURE 4.
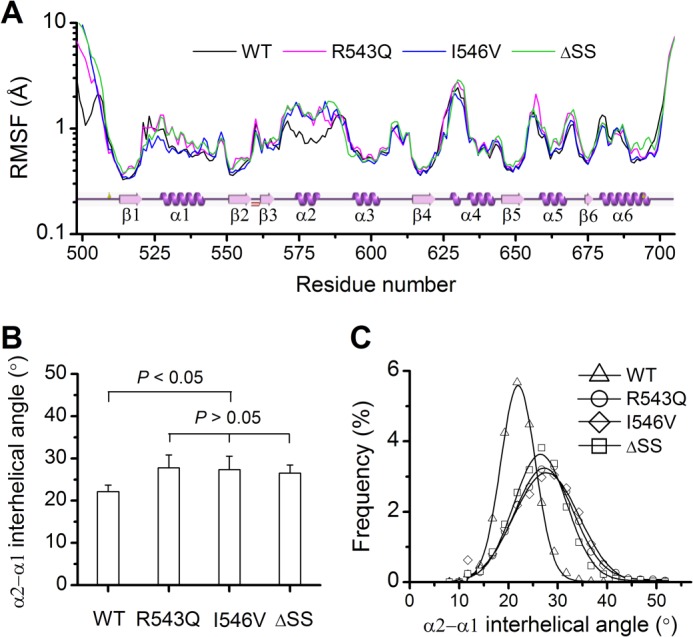
Dynamic properties of the WT A1 domain and its three GOF mutants. A, the distribution of RMSF (WT A1, black; R543Q, magenta; I546V, blue; ΔSS, green) on the residue chain of A1. Plot of the secondary structure versus the residue chain came from the website PDBsum, and the data shown were averaged over all three runs. B, the α2-α1 interhelical angle (θ) averaged over the entire simulation duration of 100 ns for each the three runs. The data shown are means ± S.E. The p values of the unpaired two-tailed Student's t test are shown to indicate the statistical difference significance, or lack thereof. C, distribution of the α2-α1 interhelical angle θ. The frequency (WT A1, ▵; R543Q, ○; I546V, ♢; ΔSS, □) is the ratio of the number of the clustered conformations with the α2-α1 interhelical angle variation of 0.25 degree above and below a given θ value to the sum of all conformations simulated within duration of 100 ns. The data of the frequency were fitted to the Gaussian distribution (solid black line) with the values given as the means ± S.D. such 21.96 ± 3.51 for WT, 27.23 ± 5.95 for R543Q, 27.69 ± 6.68 for I546V, and 26.47 ± 5.38 for ΔSS. All data are means of those in three runs.
In addition to the mutation-induced increase of local flexibility, we also observed a so-called clock pendulum-like movement of the α2-helix swinging inward and outward of the A1 body during simulations (Fig. 3B). This movement caused the variation of the α2-α1 interhelical angle, whose mean for the WT A1 was smaller than those for the GOF mutants (Fig. 4B and supplemental Fig. S6). The increased α2-α1 interhelical angle in each mutant demonstrated a localized dynamics-driven process such that a GOF mutation would turn the closed conformation of the WT A1 into an open one (Fig. 3, A and B). The distribution of the α2-α1 interhelical angle (Fig. 4C) showed that a GOF mutation distinctly widened the conformational space sampled in the thermodynamic equilibrium. The conformations with an α2-α1 interhelical angle over 30° accounted for 34.0% for R543Q, 35.6% for I546V, and 25.5% for ΔSS but only 2.3% for WT A1. The newly increased populations in the sampled space might include the “activated” A1 conformation with high affinity to GPIbα. As a result, a mutation may enhance the transition from a closed conformation to a partly open one in favor of binding to GPIbα, consistent with the reports that a partial unfolding conformation of A1 has high affinity to GPIbα (16, 29, 40), but a combination of two type 2B mutations in a single construct does nothing for the affinity of binding to platelets (41).
Mutations Induce the Exposure of the Hydrophobic Core in the A1 Domain
We found from free dynamics simulations that the above mentioned mutation-induced change of the localized dynamic property was accompanied with a partial exposure of the hydrophobic core in the A1 domain. The hydrophobic junction between the α1- and α2-helix is zipper-like, and the zipper teeth are constituted by the side chains of the hydrophobic residues (Phe530, Leu533, Val537, Val538, and Met541 in the α1-helix and Leu577, Ala581, and Val584 in the α2-helix) (Fig. 5A). As the α2-helix swung like a clock pendulum, the zipper opened and closed repeatedly with various frequencies and amplitudes, leading to partially exposing the hydrophobic residues (Val553, Ala554, Val555, and Val556) in the β2-strand beneath the zipper teeth to the bulk water. For example, the zipper was closed when the α2-α1 interhelical angle took a value of 17° (Fig. 5B) but partially opened when the angle increased to 37° (Fig. 5C) because of the swinging out of the α2-helix.
FIGURE 5.
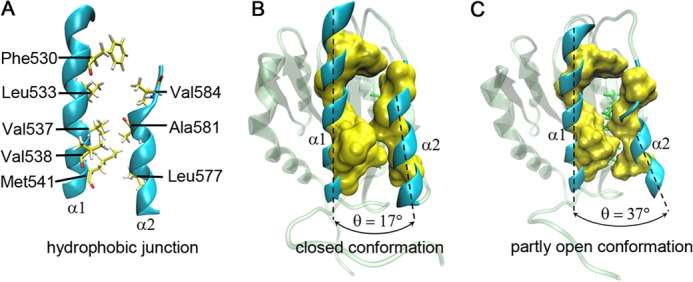
The representative closed and open conformations of the hydrophobic core in A1 domain. A, the zipper-like hydrophobic junction between the α1- and α2-helices in Newcartoon representation. All involved hydrophobic residues (Phe530, Leu533, Val537, Val538, Met541, Leu577, Ala581, and Val584), functioned as the “zipper teeth,” in the α1- or α2-helix are represented as yellow licorices. B, a closed state of the hydrophobic core in the mutant R543Q with the α2-α1 interhelical angle of 17°. C, this closed conformation becomes an open one; as such, the partly open conformation with the α2-α1 interhelical angle of 37°. The solvent-accessible surface (yellow) is displayed around the hydrophobic residues with a probe radius of 1.4 Å (B and C). The partly exposed hydrophobic residues (green licorices) (C) in the β2-strand beneath the hydrophobic junction between the α1- and α2-helices are shown in the open conformation too.
The hydrophobic core in the WT A1 should be less solvent-exposed than those in the mutants, because the GOF mutations could enhance the clock pendulum-like movement of the α2-helix relative to the α1-helix (Fig. 4C). To examine it, we calculated the average SASA of hydrophobic residues 553–556 (Fig. 6A) and the hydration frequency for each of residues 553–556 (Fig. 6B). As expected, a GOF mutation resulted in a marked increase of both the SASA in the hydrophobic region of the β2-strand and the hydration frequencies of the hydrophobic residues 553–556 in comparison with the WT A1 (Fig. 6, A and B) and thereby destabilized the hydrophobic core through weakening the hydrophobic contact between the α1- and α2-helices (Fig. 5), indicating that the mutation-induced weakening of the hydrophobic junction between the α2- and α1-helix increased the clock pendulum-like movement of the α2-helix.
FIGURE 6.

The mutation-mediated variation of the SASA and the residue hydration frequency. A and B, the SASA (A) and the residue hydration frequency (B), which are averaged over stimulation duration of 100 ns, of the hydrophobic core (residues 553–556) located at the β2-strand in the WT A1 and its three GOF mutants. The residue hydration frequency is the ratio of the hydration events, in which at least one water molecule appears at the position being less 4 Å apart from a hydrophobic residue, to all events with or without hydration. The data shown are means ± S.E. of three run results. The p values of the unpaired two-tailed Student's t test are shown to indicate the statistical difference significance, or lack thereof.
Residue Arg578 Serves as a Pivot in the Conformational Switch of the A1 Domain
A GOF mutation could change the dynamic properties of the N-terminal structure and the α2-helix, as mentioned above. However, it is unclear whether the localized structural change would correlate with the dynamic stabilization of the intradomain network of hydrogen bonds and/or salt bridges. We therefore examined the involved internal hydrogen bonds and/or salt bridges at the bottom of A1 via the free dynamics simulations and found that the dynamic change of the α2-helix is a consequent event after the N-terminal arm destabilization. In this process, residue Arg578 at the α2-helix, together with its two partner residues Glu501 at the N-terminal arm and Glu542 at the C termini of the α1-helix, is indispensably involved (Fig. 7).
FIGURE 7.
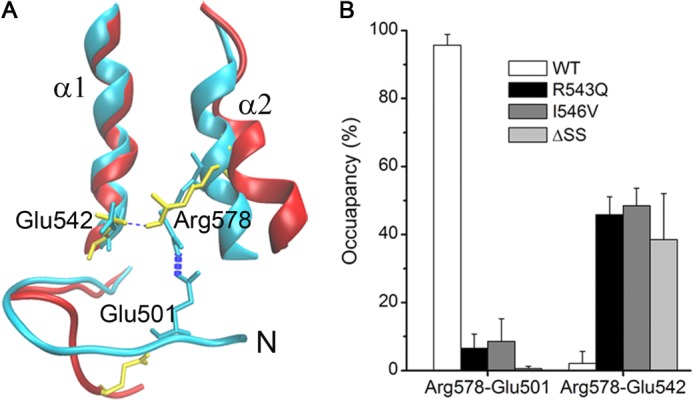
The two salt bridges between Arg578 at the α2-helix and its two partners (Glu501 at the N-terminal arm and Glu542 at the α1-helix) in A1. A, the superposition of the crystal structure pose (cyan) and a simulated pose (red) of the mutant I546V. In the crystal structure, the salt bridge Arg578–Glu501 has formed because the two residues (cyan licorices) are close to each other, but Arg578 fails to form another salt bridge with Glu542 (cyan licorices). In contrast, the salt bridge Arg578–Glu542 (yellow licorices) forms as the salt bridge Arg578–Glu501 (yellow licorices) breaks, as shown in an I546V pose (red) simulated for 9.72 ns in run 2. B, the occupancies of the above mentioned two salt bridges in the WT and its three GOF mutants. The data are presented as means ± S.D. of three run results.
Arg578 formed a firm salt bridge with Glu501 but a very weak one with Glu542 for the WT A1 (Fig. 7A). A mutation would make the firm salt bridge Arg578–Glu501 (occupancy > 95%) very weak (occupancy < 10%) and the weak salt bridge Arg578–Glu542 (occupancy < 5%) stronger (occupancy > 40%) (Fig. 7B). These results demonstrated that Arg578 serves as a pivot in the conformational switch of A1 from a stable state to a localized unstable one. As a firm linker between the N-terminal loop and the α2-helix in the WT A1, the salt bridge Arg578–Glu501 rather than the salt bridge Arg578–Glu542 maintains the structural stability of the N-terminal arm and restrains the movement of the α2-helix relative to the α1-helix; contrarily, a GOF mutation makes the salt bridge Arg578–Glu501 rarely formed so that the mutual restraint of the N-terminal loop and the α2-helix almost vanishes and subsequently reinforces the salt bridge Arg578-Glu542 to form a considerable restraint on the movement of the α2-helix (Fig. 7 and supplemental Fig. S7). However, the salt bridge Arg578–Glu542 (60% > occupancy > 40%) in the mutants should be a more flexible linker in comparison with the salt bridge Arg578–Glu501 (occupancy > 95%) in the WT A1, meaning that enhancing the clock pendulum-like movement of the α2-helix ensues from loosing of the salt bridge Arg578–Glu501.
Also, we also observed another seven hydrogen bonds and three salt bridges, which are intimately involved in the intradomain interactions at the bottom of the A1 body region (Fig. 8). Of these interactions, the hydrogen bond Lys569–Gln548 between the β3α2-loop and the β2α1-loop and the salt bridge Lys569–Asp610 between the β3α2-loop and the β4α3-loop are unstable (occupancy < 30%), but others are not (occupancy ≥ 50%), for each of the WT A1 and its three mutants (supplemental Table S2). It demonstrated the insubstantial effects of the GOF mutations on the stability of the hydrogen bond and/or salt bridge network in the intradomain of A1 except both the α2-helix and the N-terminal loop. This should provide another insight into why the mutations cannot change the structural stability of the A1 body.
FIGURE 8.

Hydrogen bonds and salt bridges at the A1 body bottom. Of all shown residues, Leu512 forms a stable hydrogen bond with Pro699 (occupancy > 50%), and others involved in hydrogen bonding contribute to three hydrogen bond networks, the unstable one such as Arg552-Asp610-Lys569-Gln548 for two Lys569-involved bonds with lower occupancies < 30% and the stable two, Ser547-Val551-Arg545-Cys509-Arg543 and Trp550-Arg611-Asp514 for their involved bonds with higher occupancies > 50% (the underlining means a salt bridge in the involved residue pair), not only in the wild-type A1 but also in the three GOF mutants (see supplemental Table S2). This demonstrates a weak linker of β3α2-loop to α3β4- and β3α2-loop and the strong linkers of C- to N-terminal loop, α3β4-loop to β1-strand, and α1β2-loop to α3β4-loop, N-terminal loop, and β2-strand.
Further Verification for the Type 2B GOF Mutation-induced Change of Dynamic Property of the A1 Domain
However, all of the analyses above were based on the assumption that the entire R543Q and I546V mutants, constructed by adding the N-terminal tail to the x-ray structures of the R543Q (Protein Data Bank code 1M10) and I546V (Protein Data Bank code 1IJB), were reliable, because the key contacts between the N-terminal arm and the rest of A1 were preserved (supplemental Fig. S2 and Table S1). A negative test for this assumption was performed therefore by examining the dynamic property of a reconstructed A1 domain named WT-NC (see “Experimental Procedures”). Also, all the type 2B mutations locate at the lower base of A1 and/or at the same side of the β-sheet (supplemental Fig. S8B), hinting that, as shown in R543Q, I546V, and ΔSS mutants (Figs. 2–4), the mutation-induced increase of flexibility in A1 may occur at other type 2B mutants too. To verify this hypothesis, we further examined the dynamic properties of other five mutants (E501A, R578Q, R578W, R578P, and R578L) via MD simulations (see “Experimental Procedures”). The mutant E501A has gain of function phenotype (32), and R578Q, R578W, R578P, and R578L are known as the type 2B mutations (33–35).
All results are summarized in Table 1, and we found that the Cα RMSD and DMC of the N-terminal arm remained at higher levels for all mutants and so did the α2-α1 interhelical angle and the RMSF (in the α2-helix and a part of the conjoint β3α2-loop) (supplemental Figs. S9 and S10) in comparison with the WT A1. The fluctuation of the N-terminal loop in each of the five newly examined mutants was stronger than those in both WT A1 and WT-NC A1, as observed in R543Q, I546V, and ΔSS mutants (Table 1 and supplemental Fig. S9). This observation suggests that a gain of function through destabilizing the N-terminal arm and increasing flexibility of the α2-helix may serve as a general mechanism for a type 2B mutation. In addition, the mutation-induced loss of the salt bridge between the α2-helix and the N-terminal tail for each mutant (E501A, R578Q, R578W, R578P, or R578L) further exhibited the pivot role of the residue Arg578 in maintaining stabilization of the N terminus.
TABLE 1.
Comparison of dynamic properties of the two wild-type A1 with those of the eight GOF mutants
| Number | Name | CαRMSDa |
DMCa | θa | Ratio of θ >30° | SASAa | Occupancyb |
||
|---|---|---|---|---|---|---|---|---|---|
| Body | N-terminal arm | Arg578–Glu501 | Arg578–Glu542 | ||||||
| Å | Å | ° | Å2 | % | |||||
| 1 | WT | 1.24 ± 0.16 | 3.18 ± 0.49 | 21.9 ± 0.2 | 22.6 ± 1.5 | 2.30 | 14.43 ± 0.95 | 95.7 ± 3.2 | 2.10 ± 3.55 |
| 2 | WT-NC | 1.11 ± 0.16 | 4.04 ± 0.52 | 22.2 ± 0.6 | 21.5 ± 1.6 | 0.50 | 12.07 ± 1.26 | 96.9 ± 1.0 | 3.07 ± 1.24 |
| 3 | R543Q | 1.47 ± 0.11 | 8.43 ± 2.28 | 23.9 ± 1.3 | 27.8 ± 3.4 | 34.0 | 16.85 ± 1.85 | 6.53 ± 4.2 | 45.9 ± 5.2 |
| 4 | I546V | 1.42 ± 0.03 | 8.27 ± 4.65 | 24.2 ± 0.7 | 27.3 ± 3.2 | 35.6 | 15.76 ± 1.23 | 8.61 ± 6.6 | 48.5 ± 5.2 |
| 5 | ΔSS | 1.61 ± 0.41 | 9.75 ± 5.23 | 24.2 ± 0.9 | 26.5 ± 1.9 | 25.5 | 18.88 ± 0.46 | 0.65 ± 0.7 | 38.5 ± 13.6 |
| 6 | E501A | 1.34 ± 0.12 | 8.53 ± 3.08 | 22.8 ± 0.8 | 24.1 ± 2.4 | 10.0 | 14.60 ± 1.34 | N/A | 50.2 ± 2.1 |
| 7 | R578Q | 1.53 ± 0.05 | 8.96 ± 4.67 | 24.0 ± 0.9 | 27.6 ± 2.4 | 36.4 | 15.57 ± 2.50 | N/A | N/A |
| 8 | R578W | 1.85 ± 0.33 | 7.25 ± 2.01 | 22.8 ± 0.2 | 30.1 ± 9.0 | 43.7 | 19.05 ± 1.16 | N/A | N/A |
| 9 | R578P | 1.37 ± 0.03 | 11.4 ± 2.08 | 24.3 ± 0.9 | 23.8 ± 0.8 | 11.1 | 14.73 ± 0.71 | N/A | N/A |
| 10 | R578L | 1.49 ± 0.08 | 6.29 ± 1.04 | 23.4 ± 1.3 | 24.0 ± 1.2 | 19.7 | 14.51 ± 0.45 | N/A | N/A |
a The data were averaged over the entire simulation duration of 100 ns for each of the three runs and shown as means ± S.E.
b The occupancy values were presented as means ± S.D. of three different occupancies measured from three independent simulations within simulation time of 100 ns for each A1. θ is the α2-α1 interhelical angle of A1, and SASA denotes the average solvent-accessible surface area of the hydrophobic residues 553–556 in the β2-strand.
The salt bridge Arg578–Glu501 between the α2-helix and the N-terminal tail in the WT-NC A1 was strong, making the N-terminal tail in the WT-NC stable, as in the WT A1. As a result, the difference in the dynamic properties of the WT A1 and the WT-NC A1 was not significant (Table 1 and supplemental Figs. S9 and S10). This result indicated that the higher fluctuations in both the entire I546V and R543Q mutants came from the type 2B mutations rather than the manually added N-terminal arms, suggesting these computer models of the I546V and R543Q mutants were available.
DISCUSSION
Less conformational change occurs in the vWF A1 domain, not like the homologous von Willebrand factor A domain, the integrin I domain, whose affinity is regulated by the obvious structural changes caused by a piston and connecting rod-like movement of the C-terminal α7-helix (42). This is why the structural basis of the GOF variants of the native vWF A1 domain remains an open question. From observation via free dynamics simulations on nanosecond scale, we demonstrated that a GOF mutation triggers the switch of A1 from a stable state to a localized unstable one. This switch is relevant to a localized dynamics-driven process, including a destabilization of the N-terminal arm and a consequent increase in flexibility of the α2-helix, and serving as a possible mechanism for vWF A1 activation and affinity modulation upon GOF mutation. In comparison with the WT A1, the partly open conformation of a GOF mutant should have higher compliance with the binding site of GPIbα, based on the conformational selection theory of molecular recognition (43, 44).
This GOF mutation effect is similar to those demonstrated in previous reports, such that the destabilization of A1 reduces concomitantly the interactions among A1-A2-A3 domains and thereby enhances the binding to GPIbα (45); the amino acid sequence 475–497, which stabilizes A1A2A3 tridomain (9), detaches from the body region together with the posterior N-terminal arm (residues 498–508) and induces a conformational change in the A1 domain to up-regulate A1 affinity, and as a linker between the A1 domain and the D′D3 domains, the N-terminal arm probably plays an important role in orientating the A1 domain with the neighboring domains and in exposing the GPIbα-binding site in arterial bleeding (42, 46).
Proteins are not static building blocks. Experimental and computational data suggest that dynamics and motions of protein structure are indispensable to protein activity modulation (47–51). We showed that each type 2B GOF mutation involved herein made A1 partially open. This is consistent with reports that a partial unfolding conformation of A1 has high affinity to GPIbα (16, 29, 40), because an open conformation of A1 may be in favor of unfolding, which may first occur at such weak regions as the β3α2-loop and the α2-helix (52, 53) (Figs. 7 and 8). The similar effects of the GOF mutations on the structure and dynamics of the A1 domain in the present results are also in good agreement with the experimental results such that a combination of two type 2B mutations in a single construct had no additive effect on the affinity of binding to platelets (41), possibly coming from the flexible motifs, such as the N-terminal arm, the α1-helix, the β3α2-loop, the β2-strand, and the α2-helix (Fig. 4A), which govern the conformational transition of A1, may be inherently sensitive to mutational events (54).
It was demonstrated that the allosteric communications can be transmitted solely by a variation in protein motions, which do not cause a macroscopic structural change at the backbone level (55, 56). This argument suggests that the mutation-induced change of dynamic property of A1 transmits the allosteric signals to the active site first and then makes A1 favoring binding to GPIbα. In signaling, the elaborate salt bridge network wrapping around the lower rim of A1 (supplemental Fig. S8A) should serve as the intramolecular allosteric signal pathway(s), because the type 2B mutation sites locate at or near the rim (5). In this region sensitive to mutagenesis, the motif that illustrated the largest mutation-induced deviation was observed to be around the hydrophobic cluster, which is comprised of the N-terminal arm, α1-helix, α1β2-loop, β2-strand, and α2-helix (Fig. 4A). Of the 16 type 2B mutation sites, 13 locate at and others are close to this cluster (supplemental Fig. S8B), which almost completely overlaps with the salt bridge network, showing that the hydrophobic interaction also participates in the allosteric signal transmission. Also, water molecules may be deeply involved in the allosteric signaling too, because water functions as a lubricant to ease rearrangements of necessary hydrogen bonds during conformational change (57). As the hydrophobic junction between the α1- and α2-helices is partly unzipped through the GOF mutations, a water channel toward the buried hydrophobic core forms (Fig. 5), and hydration ensues (Fig. 6). As a result, the local mobility of A1 increases (Fig. 4B) (58). The phenomenon of water molecules participating in the intramolecular interaction of A1 was observed with the x-ray crystallography (23). Therefore, water molecules should act as an effective medium for transmitting the allosteric signal at the mutation sites to the action site.
Unlike a gain of function through the type 2B mutation-induced increase of A1 flexibility, the fluid shear stress triggers the wild-type vWF activation in physiological conditions. Shear stress could greatly increase the oxidation of residues Met540 and Met541 at the α1-helix that are deeply buried by the adjacent α2-helix in the static structures (59). It suggests that shear stress may enhance A1 affinity through a similar mechanism as the GOF mutations, because A1 itself exposes the buried methionines through its conformational change under shear stress. Mechanically, in flows, stretching breaks the firm linker (the salt bridge Arg578–Glu501) between the N-terminal loop and the α2-helix first and then gradually unzips the hydrophobic junction of the α1- and α2-helices until the liganded A1 is partly unfolded. In this process, the liganded A1 may be more and more in favor of high affinity to GPIbα, suggesting another explanation for catch bond mechanism (15, 16). This is because a looser conjunction between the α1- and α2-helices in a GOF mutant up-regulates the affinity of A1 to GPIbα, as mentioned above, and the A1 structure at partly unfolding state takes a high affinity conformation in the urea unfolding process, as previously reported (16, 29, 40). Nevertheless, association of A1 with GPIbα should favor partly rather than overly unfolding of A1 (29). Thus, there should be a shear stress threshold, across which increasing shear stress will make the liganded A1 far away from its partial unfolding conformation ensemble in favor of association with its partner. Also, detachment of the N-terminal loop from the α2-helix requires stretching to be not too weak, meaning there is another shear stress threshold below which the phenomenon of the force-enhanced affinity does not occur. It follows that the dissociation of a liganded A1 from GPIbα may be tri- rather than bi-phase force-dependent, similar to the dissociation of E-selectin from its ligand (60, 61).
In the previous MD studies for the GOF phenomenon, one argued that conformational changes in the binding site of A1 are important for enhancing affinity. The present results showed that the mutation-induced GOF is relevant to the local mobility of A1. These two different arguments are complementary. The type 2B mutation R543Q was demonstrated to result in formation of an intermolecular salt bridge between Arg571 at the β3α2-loop of A1 and Glu14 at the β-finger of GPIbα (15). This could be demonstrated by the fact that the increased freedom of the α2-helix and the β3α2-loop in unliganded A1 caused by a GOF mutation would provide more chance for both Arg571 and Glu14 to meet and couple with each other. Through MD simulations for wild-type and mutant A1/GPIbα complexes, the bound A1 was observed to rotate away from the C terminus of GPIbα in the high force unbinding pathway but close to the GPIbα C terminus in thermal unbinding (62). In this process, the local mobility of A1 may be enhanced. This local mobility should be related to the reconstruction of the contact area between the two proteins under tensile force. The lower occupancies of the salt bridges in the mutant A1/GPIbα complex (62) might be related deeply to the mutation-enhanced fluctuations of the flexible α2-helix and β3α2-loop.
Indeed, the present work is limited not only by the small number of runs within the simulation time of 100 ns for each system, but also by the lack of experimental data and the reconstructed models of the mutants R543Q and I546V. However, the GOF phenotype of mutation E501A in an A1 variant had been obtained through the clustered charged to alanine scanning mutagenesis (32); the preservation of the key contacts between the N-terminal arm and the rest of A1 in the reconstructed models of R543Q and I546V (supplemental Fig. S2 and Table S1) should rationalize the present results; so did the negative test with the reconstructed model of the WT A1 (Table 1 and supplemental Figs. S9 and S10). Also, the flexibility in the flexible regions increases with each of the eight examined mutations (Table 1). Therefore, the present interpretation for the gain of function via the type 2B mutation-induced change of dynamic property of A1 should be persuasive, even without direct structural verification. This interpretation could be further tested through mutagenesis experiments such as introducing a disulfide bond between the N-terminal arm and the body of A1 for a GOF mutant or truncating the N-terminal loop of the WT A1. The present results provided a novel insight into the structural basis on the GOF variants of the native vWF A1 domain. The dynamics-driven regulation of function may serve as a mechanism not only for A1-GPIbα interaction but also for other receptor-ligand interactions.
This work was supported by National Natural Science Foundation of China Grants 10972081 (to Y. F.) and 11072080 and 31170887 (to J. W.) and by Guangdong Natural Science Foundation Grant S2011010005451 (to J. W.) and Specialized Research Fund for the Doctoral Program of Higher Education Grant 20110172110030 (to J. W.).

This article contains supplemental Tables S1 and S2 and Figs. S1–S10.
- vWF
- von Willebrand factor
- GPIbα
- glycoprotein Ibα
- GOF
- gain of function
- MD
- molecular dynamics
- DMC
- distance between the mass centers
- RMSD
- root mean square deviation
- RMSF
- root mean square fluctuation
- SASA
- solvent-accessible surface area.
REFERENCES
- 1. Savage B., Saldívar E., Ruggeri Z. M. (1996) Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 84, 289–297 [DOI] [PubMed] [Google Scholar]
- 2. Zhou Z., Dong J. F. (2012) Thrombotic thrombocytopenic purpura and anti-thrombotic therapy targeted to von Willebrand factor. Curr. Vasc. Pharmacol. 10, 762–766 [DOI] [PubMed] [Google Scholar]
- 3. Ruggeri Z. M. (2007) von Willebrand factor. Looking back and looking forward. Thromb. Haemost. 98, 55–62 [PubMed] [Google Scholar]
- 4. Sadler J. E. (1998) Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 67, 395–424 [DOI] [PubMed] [Google Scholar]
- 5. Emsley J., Cruz M., Handin R., Liddington R. (1998) Crystal structure of the von Willebrand factor A1 domain and implications for the binding of platelet glycoprotein Ib. J. Biol. Chem. 273, 10396–10401 [DOI] [PubMed] [Google Scholar]
- 6. Huizinga E. G., Tsuji S., Romijn R. A., Schiphorst M. E., de Groot P. G., Sixma J. J., Gros P. (2002) Structures of glycoprotein Ibα and its complex with von Willebrand factor A1 domain. Science 297, 1176–1179 [DOI] [PubMed] [Google Scholar]
- 7. Martin C., Morales L. D., Cruz M. A. (2007) Purified A2 domain of von Willebrand factor binds to the active conformation of von Willebrand factor and blocks the interaction with platelet glycoprotein Ibα. J. Thromb. Haemost. 5, 1363–1370 [DOI] [PubMed] [Google Scholar]
- 8. Ulrichts H., Udvardy M., Lenting P. J., Pareyn I., Vandeputte N., Vanhoorelbeke K., Deckmyn H. (2006) Shielding of the A1 domain by the D′D3 domains of von Willebrand factor modulates its interaction with platelet glycoprotein Ib-IX-V. J. Biol. Chem. 281, 4699–4707 [DOI] [PubMed] [Google Scholar]
- 9. Auton M., Sowa K. E., Behymer M., Cruz M. A. (2012) The N-terminal flanking region of A1 domain in von Willebrand factor stabilizes structure of A1A2A3 complex and modulates platelet activation under shear stress. J. Biol. Chem. 287, 14579–14585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arya M., Anvari B., Romo G. M., Cruz M. A., Dong J. F., McIntire L. V., Moake J. L., López J. A. (2002) Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex. Studies using optical tweezers. Blood 99, 3971–3977 [DOI] [PubMed] [Google Scholar]
- 11. Schneider S. W., Nuschele S., Wixforth A., Gorzelanny C., Alexander-Katz A., Netz R. R., Schneider M. F. (2007) Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc. Natl. Acad. Sci. U.S.A. 104, 7899–7903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Siedlecki C. A., Lestini B. J., Kottke-Marchant K. K., Eppell S. J., Wilson D. L., Marchant R. E. (1996) Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood 88, 2939–2950 [PubMed] [Google Scholar]
- 13. Kim J., Zhang C. Z., Zhang X., Springer T. A. (2010) A mechanically stabilized receptor-ligand flex-bond important in the vasculature. Nature 466, 992–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Springer T. A. (2011) Biology and physics of von Willebrand factor concatamers. J. Thromb. Haemost. 9, 130–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yago T., Lou J., Wu T., Yang J., Miner J. J., Coburn L., López J. A., Cruz M. A., Dong J. F., McIntire L. V., McEver R. P., Zhu C. (2008) Platelet glycoprotein Ibα forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J. Clin. Invest. 118, 3195–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Auton M., Sedlák E., Marek J., Wu T., Zhu C., Cruz M. A. (2009) Changes in thermodynamic stability of von Willebrand factor differentially affect the force-dependent binding to platelet GPIbα. Biophys. J. 97, 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miura S., Li C. Q., Cao Z., Wang H., Wardell M. R., Sadler J. E. (2000) Interaction of von Willebrand factor domain A1 with platelet glycoprotein Ibα-(1–289). Slow intrinsic binding kinetics mediate rapid platelet adhesion. J. Biol. Chem. 275, 7539–7546 [DOI] [PubMed] [Google Scholar]
- 18. Miyata S., Ruggeri Z. M. (1999) Distinct structural attributes regulating von Willebrand factor A1 domain interaction with platelet glycoprotein Ibα under flow. J. Biol. Chem. 274, 6586–6593 [DOI] [PubMed] [Google Scholar]
- 19. Miyata S., Goto S., Federici A. B., Ware J., Ruggeri Z. M. (1996) Conformational changes in the A1 domain of von Willebrand factor modulating the interaction with platelet glycoprotein Ibα. J. Biol. Chem. 271, 9046–9053 [DOI] [PubMed] [Google Scholar]
- 20. Dumas J. J., Kumar R., McDonagh T., Sullivan F., Stahl M. L., Somers W. S., Mosyak L. (2004) Crystal structure of the wild-type von Willebrand factor A1-glycoprotein Ibα complex reveals conformation differences with a complex bearing von Willebrand disease mutations. J. Biol. Chem. 279, 23327–23334 [DOI] [PubMed] [Google Scholar]
- 21. Fukuda K., Doggett T. A., Bankston L. A., Cruz M. A., Diacovo T. G., Liddington R. C. (2002) Structural basis of von Willebrand factor activation by the snake toxin botrocetin. Structure 10, 943–950 [DOI] [PubMed] [Google Scholar]
- 22. Celikel R., Varughese K. I., Madhusudan, Yoshioka A., Ware J., Ruggeri Z. M. (1998) Crystal structure of the von Willebrand factor A1 domain in complex with the function blocking NMC-4 Fab. Nat. Struct. Biol. 5, 189–194 [DOI] [PubMed] [Google Scholar]
- 23. Celikel R., Ruggeri Z. M., Varughese K. I. (2000) von Willebrand factor conformation and adhesive function is modulated by an internalized water molecule. Nat. Struct. Biol. 7, 881–884 [DOI] [PubMed] [Google Scholar]
- 24. Huang R. H., Fremont D. H., Diener J. L., Schaub R. G., Sadler J. E. (2009) A structural explanation for the antithrombotic activity of ARC1172, a DNA aptamer that binds von Willebrand factor domain A1. Structure 17, 1476–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maita N., Nishio K., Nishimoto E., Matsui T., Shikamoto Y., Morita T., Sadler J. E., Mizuno H. (2003) Crystal structure of von Willebrand factor A1 domain complexed with snake venom, bitiscetin. Insight into glycoprotein Ibα binding mechanism induced by snake venom proteins. J. Biol. Chem. 278, 37777–37781 [DOI] [PubMed] [Google Scholar]
- 26. Fukuda K., Doggett T., Laurenzi I. J., Liddington R. C., Diacovo T. G. (2005) The snake venom protein botrocetin acts as a biological brace to promote dysfunctional platelet aggregation. Nat. Struct. Mol. Biol. 12, 152–159 [DOI] [PubMed] [Google Scholar]
- 27. Bruice T. C. (2006) Computational approaches. Reaction trajectories, structures, and atomic motions. Enzyme reactions and proficiency. Chem. Rev. 106, 3119–3139 [DOI] [PubMed] [Google Scholar]
- 28. Wang Y., McCammon J. A. (2012) Introduction to molecular dynamics. Theory and applications in biomolecular modeling, in Computational Modeling of Biological Systems, pp. 3–30, Springer US, New York [Google Scholar]
- 29. Auton M., Zhu C., Cruz M. A. (2010) The mechanism of VWF-mediated platelet GPIbα binding. Biophys. J. 99, 1192–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Humphrey W., Dalke A., Schulten K. (1996) VMD. Visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–38 [DOI] [PubMed] [Google Scholar]
- 31. Guex N., Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer. An environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 32. Matsushita T., Sadler J. E. (1995) Identification of amino acid residues essential for von Willebrand factor binding to platelet glycoprotein Ib. Charged-to-alanine scanning mutagenesis of the A1 domain of human von Willebrand factor. J. Biol. Chem. 270, 13406–13414 [DOI] [PubMed] [Google Scholar]
- 33. Randi A. M., Jorieux S., Tuley E. A., Mazurier C., Sadler J. E. (1992) Recombinant von Willebrand factor Arg578 → Gln. A type IIB von Willebrand disease mutation affects binding to glycoprotein Ib but not to collagen or heparin. J. Biol. Chem. 267, 21187–21192 [PubMed] [Google Scholar]
- 34. Casaña P., Martínez F., Espinós C., Haya S., Lorenzo J. I., Aznar J. A. (1998) Search for mutations in a segment of the exon 28 of the human von Willebrand factor gene. New mutations, R1315C and R1341W, associated with type 2M and 2B variants. Am. J. Hematol. 59, 57–63 [DOI] [PubMed] [Google Scholar]
- 35. Hilbert L., Gaucher C., Abgrall J. F., Parquet A., Trzeciak C., Mazurier C. (1998) Identification of new type 2B von Willebrand disease mutations. Arg543Gln, Arg545Pro and Arg578Leu. Br. J. Haematol. 103, 877–884 [DOI] [PubMed] [Google Scholar]
- 36. Bas D. C., Rogers D. M., Jensen J. H. (2008) Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 73, 765–783 [DOI] [PubMed] [Google Scholar]
- 37. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. MacKerell A. D., Jr., Bashford D., Bellott M., Dunbrack R. L., Jr., Evanseck J., Field M., Fischer S., Gao J., Guo H., Ha S. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 39. Romijn R. A. (2002) Structural studies on the von Willebrand factor A1 and A3 domains. Ph.D. thesis, Universiteit Utrecht [Google Scholar]
- 40. Auton M., Cruz M. A., Moake J. (2007) Conformational stability and domain unfolding of the Von Willebrand factor A domains. J. Mol. Biol. 366, 986–1000 [DOI] [PubMed] [Google Scholar]
- 41. Cooney K. A., Ginsburg D. (1996) Comparative analysis of type 2b von Willebrand disease mutations. Implications for the mechanism of von Willebrand factor binding to platelets. Blood 87, 2322–2328 [PubMed] [Google Scholar]
- 42. Springer T. A. (2006) Complement and the multifaceted functions of VWA and integrin I domains. Structure 14, 1611–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boehr D. D., Nussinov R., Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Whitley M. J., Lee A. L. (2009) Frameworks for understanding long-range intra-protein communication. Curr. Protein Pept. Sci. 10, 116–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Auton M., Sowa K. E., Smith S. M., Sedlák E., Vijayan K. V., Cruz M. A. (2010) Destabilization of the A1 domain in von Willebrand factor dissociates the A1A2A3 tri-domain and provokes spontaneous binding to glycoprotein Ibα and platelet activation under shear stress. J. Biol. Chem. 285, 22831–22839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ma B., Tsai C. J., Haliloglu T., Nussinov R. (2011) Dynamic allostery. Linkers are not merely flexible. Structure 19, 907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Henzler-Wildman K. A., Lei M., Thai V., Kerns S. J., Karplus M., Kern D. (2007) A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 450, 913–916 [DOI] [PubMed] [Google Scholar]
- 48. Tzeng S. R., Kalodimos C. G. (2009) Dynamic activation of an allosteric regulatory protein. Nature 462, 368–372 [DOI] [PubMed] [Google Scholar]
- 49. Popovych N., Sun S., Ebright R. H., Kalodimos C. G. (2006) Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 13, 831–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smock R. G., Gierasch L. M. (2009) Sending signals dynamically. Science's STKE 324, 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wallnoefer H. G., Lingott T., Gutiérrez J. M., Merfort I., Liedl K. R. (2010) Backbone flexibility controls the activity and specificity of a protein-protein interface. Specificity in snake venom metalloproteases. J. Am. Chem. Soc. 132, 10330–10337 [DOI] [PubMed] [Google Scholar]
- 52. Ishikawa H., Kwak K., Chung J. K., Kim S., Fayer M. D. (2008) Direct observation of fast protein conformational switching. Proc. Natl. Acad. Sci. U.S.A. 105, 8619–8624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Whitten S. T., García-Moreno E. B., Hilser V. J. (2005) Local conformational fluctuations can modulate the coupling between proton binding and global structural transitions in proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 4282–4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Csermely P., Palotai R., Nussinov R. (2010) Induced fit, conformational selection and independent dynamic segments. An extended view of binding events. Trends Biochem. Sci. 35, 539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cooper A., Dryden D. (1984) Allostery without conformational change. Eur. Biophys. J. 11, 103–109 [DOI] [PubMed] [Google Scholar]
- 56. Fuentes E. J., Der C. J., Lee A. L. (2004) Ligand-dependent dynamics and intramolecular signaling in a PDZ domain. J. Mol. Biol. 335, 1105–1115 [DOI] [PubMed] [Google Scholar]
- 57. Chaplin M. (2006) Do we underestimate the importance of water in cell biology? Nat. Rev. Mol. Cell Biol. 7, 861–866 [DOI] [PubMed] [Google Scholar]
- 58. Zhang H., Zhang T., Chen K., Shen S., Ruan J., Kurgan L. (2009) On the relation between residue flexibility and local solvent accessibility in proteins. Proteins 76, 617–636 [DOI] [PubMed] [Google Scholar]
- 59. Fu X., Chen J., Gallagher R., Zheng Y., Chung D. W., López J. A. (2011) Shear stress-induced unfolding of VWF accelerates oxidation of key methionine residues in the A1A2A3 region. Blood 118, 5283–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wayman A. M., Chen W., McEver R. P., Zhu C. (2010) Triphasic force dependence of E-selectin/ligand dissociation governs cell rolling under flow. Biophys. J. 99, 1166–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li Q., Fang Y., Ding X., Wu J. (2012) Force-dependent bond dissociation govern rolling of HL-60 cells through E-selectin. Exp. Cell Res. 318, 1649–1658 [DOI] [PubMed] [Google Scholar]
- 62. Interlandi G., Thomas W. (2010) The catch bond mechanism between von Willebrand factor and platelet surface receptors investigated by molecular dynamics simulations. Proteins 78, 2506–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]