

Background: The regulatory mechanism of BLT2 is largely unknown.
Results: RanBPM interacts with BLT2 and inhibits BLT2-induced ROS generation and chemotaxis.
Conclusion: Our findings suggest that RanBPM acts as a negative regulator of BLT2.
Significance: Identification of regulators would provide better understanding of BLT2 signaling and potentially various BLT2-associated inflammatory pathogenesis.
Keywords: Chemotaxis, G Protein-coupled Receptors (GPCR), Leukotriene, Reactive Oxygen Species (ROS), Scaffold Proteins, BLT2, GPCR, RanBPM, Chemotaxis, Reactive Oxygen Species
Abstract
BLT2, a low affinity receptor for leukotriene B4 (LTB4), is a member of the G protein-coupled receptor family and is involved in many signal transduction pathways associated with various cellular phenotypes, including chemotactic motility. However, the regulatory mechanism for BLT2 has not yet been demonstrated. To understand the regulatory mechanism of BLT2, we screened and identified the proteins that bind to BLT2. Using a yeast two-hybrid assay with the BLT2 C-terminal domain as bait, we found that RanBPM, a previously proposed scaffold protein, interacts with BLT2. We demonstrated the specific interaction between BLT2 and RanBPM by GST pulldown assay and co-immunoprecipitation assay. To elucidate the biological function of the RanBPM-BLT2 interaction, we evaluated the effects of RanBPM overexpression or knockdown. We found that BLT2-mediated motility was severely attenuated by RanBPM overexpression and that knockdown of endogenous RanBPM by shRNA strongly promoted BLT2-mediated motility, suggesting a negative regulatory function of RanBPM toward BLT2. Furthermore, we observed that the addition of BLT2 ligands caused the dissociation of BLT2 and RanBPM, thus releasing the negative regulatory effect of RanBPM. Finally, we propose that Akt-induced BLT2 phosphorylation at residue Thr355, which occurs after the addition of BLT2 ligands, is a potential mechanism by which BLT2 dissociates from RanBPM, resulting in stimulation of BLT2 signaling. Taken together, our results suggest that RanBPM acts as a negative regulator of BLT2 signaling to attenuate BLT2-mediated cell motility.
Introduction
G protein-coupled receptors (GPCRs)2 are the largest family of cell surface receptors. They regulate various biological functions in mammals and are targets of therapeutic drugs (1, 2). Ligand-induced activation of GPCRs mediates cellular responses, and signal transduction through GPCRs is tightly coupled to heterotrimeric G proteins (2). In addition to G proteins, a variety of proteins named GPCR-interacting proteins (GIPs) have been identified that bind directly to GPCRs (3). Most GIP proteins act as adaptor or scaffolding proteins and are involved in various functions, such as modulating GPCR signaling, directing GPCR trafficking, regulating localization, and influencing GPCR pharmacology (4, 5). As important modulators of GPCRs in cellular physiology, the role of GPCR-GIP interactions has been observed in many GPCR regulatory mechanisms.
BLT2 is a member of the GPCR family and is a low affinity receptor for leukotriene B4 (LTB4), which is a lipid metabolite of arachidonic acid derived via the 5-lipoxygenase-dependent pathway. BLT2 is widely expressed in various tissues; however, its physiological function has not yet been identified (6, 7). Recent studies have demonstrated that activation of BLT2 by ligand stimulation is associated with chemotaxis in primary keratinocytes (8). Previously, we observed that the LTB4-BLT2 cascade is associated with the generation of reactive oxygen species (ROS) (9–12). Overexpression of BLT2 caused an LTB4-induced increase in chemotactic responses in an ROS-dependent manner (10). Recent studies have suggested that LTB4 and its receptor BLT2 play a role in the pathogenesis of several inflammatory diseases and also critically regulate tumor progression by promoting cell proliferation, survival, migration, and metastasis (13–17). For example, LTB4-related inflammatory signaling has been shown to stimulate cell proliferation by activating extracellular signal-regulated kinase (ERK) in several types of cancer cells, such as colon and pancreatic cancer cell lines (18, 19). In addition, elevated levels of LTB4 and BLT2 have been observed in pancreatic, colon, and ovarian tumors; neuroblastoma; and many other tumor types (13, 14, 20). Although these various functions of BLT2 have been demonstrated, the regulatory mechanism of BLT2 remains unknown. To identify the proteins that modulate BLT2, we performed a yeast two-hybrid screen in a human thymus cDNA library using the BLT2 C terminus as bait to identify BLT2-interacting proteins.
In this study, we found that RanBPM, a member of the Ran-GTPase-binding protein family, interacts with BLT2. We demonstrate the specific interaction between RanBPM and BLT2 by in vitro GST pulldown assay and in vivo co-immunoprecipitation studies. We also demonstrate that the C-terminal region of BLT2 was responsible for binding to RanBPM and that the BLT2-binding region of RanBPM is a SPRY domain. We show that RanBPM overexpression attenuates BLT2-meditated ROS generation and motility. In addition, knockdown of endogenous RanBPM by shRNA strongly promoted BLT2-mediated ROS generation and motility. Finally, we show that Akt-induced BLT2 phosphorylation at residue Thr355 following treatment with BLT2 ligands causes the dissociation of BLT2 and RanBPM. These findings indicate a potential mechanism by which BLT2 is dissociated from RanBPM. Taken together, our results suggest that RanBPM acts as a negative regulator of BLT2 signaling in cell motility.
EXPERIMENTAL PROCEDURES
Chemicals and Plasmids
Triton X-100 was obtained from Sigma-Aldrich. 2′,7′-Dichlorodihydrofluorescein diacetate was purchased from Molecular Probes (Eugene, OR). LTB4, 12(S)-hydroxyheptadeca-5Z, 8E, 10E-trienoic acid (12HHT), and CAY10583 were purchased from Cayman Chemical (Ann Arbor, MI). LY294002 was purchased from Sigma-Aldrich. Fetal bovine serum (FBS), RPMI 1640 medium, and phenol red-free RPMI 1640 medium were purchased from Invitrogen. Dulbecco's modified Eagle's medium (DMEM) was from obtained from Life Technologies, Inc. All other chemicals were obtained from standard sources and were of molecular biology grade or higher. The human BLT2 (GenBankTM accession number NM_019839.1) plasmid was cloned by PCR using a human genomic bacterial artificial chromosome library, as described previously for other chemoattractant receptors, with minor modifications to the PCR conditions (21). The bacterial artificial chromosome library was a kind gift from Dr. Young-Chul Choi (Kyung Hee University, Seoul, Korea). The epitope-tagged wild-type BLT2 and C-terminal domain-deleted BLT2 mutant (ΔC) were obtained using the following primers: forward primer for HA-BLT2 5′-CTGGATCCCACCATGTACCCCTACGACGTGCCCGACTACGCCGCACCTTCTCATCGG-3′ and reverse primer for HA-BLT2 5′-GGTGAATTCTCAAAGGTCCCATTCCGG-3′; forward primer for c-Myc-BLT2 5′-CGGGATCCCACCATGGAACAAAAACTCATCTCAGAAGAGGATCTGGCACCTTCTCATCGG-3′ and reverse primer for c-Myc-BLT2 5′-GGTGAATTCTCAAAGGTCCCATTCCGG-3′; forward primer for ΔC mutant 5′-CTGGATCCCACCATGTACCCCTACGACGTGCCCGACTACGCCGCACCTTCTCATCGG-3′ and reverse primer for ΔC mutant 5′-GGTGAATTCTCATCCAGCGGTGAAGACGTAG-3′. The pcDNA3.1 and pET22b vectors were purchased from Invitrogen and Novagen, respectively. The 3×FLAG vector and the plasmids containing the coding region of the full-length (FL), amino acids 1–333 (N3), or amino acids 207–492 (I) of RanBPM were kindly provided by Dr. Shim Sang-Ohk (Yale University School of Medicine, New Haven, CT). The deletion mutant C1, expressing amino acids 334–729, was generated by restriction digestion followed by filling with Pfu polymerase and religating in the p3×FLAG-CMV7.1 vector. The plasmid pSilencer-shRanBPM was kindly provided by Dr. Dane Winner (University of Florida, Gainesville, FL).
Cell Culture and DNA Transfection
HEK 293T and the immortalized human keratinocyte HaCaT cells were cultured in DMEM supplemented with 10% FBS and antibiotic-antimycotic solution (Life Technologies, Inc.) at 37 °C in a 5% CO2 humidified atmosphere. CHO-K1 cells were obtained from the Korean Cell Line Bank (KCLB, 10061), and the cells were grown in RPMI 1640 medium supplemented with 10% FBS, penicillin (50 units/ml), and streptomycin (50 μg/ml) at 37 °C in a 5% CO2 humidified atmosphere. The primary human keratinocyte (PHK) cells were obtained from Invitrogen and cultured in EpiLife® medium (Invitrogen) supplemented with human keratinocyte growth supplement (Invitrogen) at 37 °C in a 5% CO2 humidified atmosphere. Transient transfection was performed by plating 2 × 105 cells in 60-mm dishes for 24 h and then adding Lipofectamine (4 μl) (Invitrogen) and DNA (2 μg) to each dish. For the immunoprecipitation assays, 5 μg of DNA was used to transfect 2 × 106 cells in 100-mm dishes. The total transfected DNA quantities were equalized in each experiment with the pcDNA3.1 vector DNA and 3×FLAG vector DNA. In the shRNA knockdown system, 2 μg of each pSilencer vector and shRNA was used to transfect 2 × 105 cells in 60-mm dishes for 24 h.
Semiquantitative RT-PCR for RanBPM
RanBPM, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcripts were amplified using a PCR PreMix kit (Intron Biotechnology, Seongnam, Korea). The primers for human RanBPM were 5′-GGTGATGTCATTGGCTGTTG-3′ (forward) and 5′-AATTTGGCGGTAGGTCAGTG-3′ (reverse). The PCR protocol for human RanBPM involved 31 cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 30 s, and elongation at 72 °C for 30 s. These cycles were followed by an extension at 72 °C for 10 min. The amplified PCR products were subjected to electrophoresis on 1.5% agarose gels, and the bands were visualized by ethidium bromide staining and photographed with a Gel Doc system (Bio-Rad). The specificity of all primers was confirmed by sequencing of the PCR products. The RNA extraction products were tested in control reverse transcription reactions and found to be free of DNA contamination.
Yeast Two-hybrid Screen
Yeast two-hybrid screening with the GAL4 DNA-binding domain (BD)-fused C-terminal domain of BLT2 (amino acids 325–389) was performed with the human thymus cDNA activation domain (AD) library. Yeast strain PBN204 (Panbionet Inc., Pohang, Korea) was co-transformed using the polyethylene glycol-lithium acetate method with the following two hybrid plasmids: (i) the bait plasmid pBCT-BLT2 that encodes a GAL4 DNA BD-fused BLT2 C-terminal domain cDNA and the HIS3 marker gene and (ii) the pACT2 plasmid that encodes the human thymus cDNA fused to GAL4 AD and the TRP1 marker gene. In our screening, three reporter genes, URA3, ADE2, and lacZ, each under the control of different GAL4-binding sites, were used to minimize false positives (as shown in Fig. 1). First, the transformants were spread on selective medium lacking leucine, tryptophan, and uracil but containing 2% glucose (SD-LWU). On this medium, the transformants grow when the BD-BLT2 interacts with the AD prey proteins. The independent colonies grew on the selective SD-LWU medium. Second, the positive colonies grew on selective medium lacking leucine, tryptophan, and adenine but containing 2% glucose (SD-LWA). Third, the positive colonies were blue in response to X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside), which was confirmed by a filter assay to detect β-galactosidase expression (see Fig. 1).
FIGURE 1.
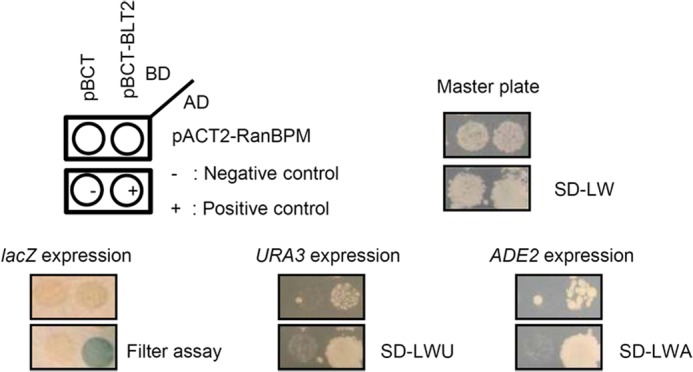
Identification of RanBPM as a BLT2-interacting protein, using a yeast two-hybrid screen. The PBN204 yeast strain was co-transformed with either the GAL4-BD fusion plasmid pBCT or pBCT-BLT2 and the GAL4-AD fusion plasmid pACT2-RanBPM isolated from the positive colonies. The yeast cells were spread on selective medium lacking leucine and tryptophan (SD-LW) to select for co-transformants. To select for interacting proteins, the isolated co-transformants were then replica-plated onto selective medium lacking leucine and tryptophan (SD-LW) for filter assays to detect lacZ expression. Selective medium lacking leucine, tryptophan, and uracil (SD-LWU) or selective medium lacking leucine, tryptophan, and adenine (SD-LWA) were used for filter assays. When two proteins interact with each other, the co-transformants express lacZ, URA3, and Ade2. pBCT-polypyrimidine tract-binding protein (PTB) and pACT2-PTB were used as the positive controls for the protein-protein interaction. pBCT and pACT2 were used as the negative controls.
GST Pulldown Assay
Interactions between the BLT2 C-terminal region (expressing amino acids 312–389) and each domain of RanBPM (FL, N3, I, and C1) were assessed by GST pulldown assay. Briefly, GST or GST fusion proteins were expressed in BL21 cells and purified using glutathione-SepharoseTM beads (Amersham Biosciences) in binding buffer (100 mm NaCl, 50 mm Tris-HCl, pH 7.5, 1 mm dithiothreitol, 2 μg/ml leupeptin, 2 μg/ml aprotinin, and 100 μg/ml phenylmethylsulfonyl fluoride). After 1 h at 4 °C, equal amounts of GST or GST fusion proteins were resuspended in reaction buffer (200 mm NaCl, 50 mm HEPES, pH 7.5, 1 mm MgCl2, and 0.2% Triton X-100) containing 0.2 mg/ml bovine serum albumin (BSA) (Fraction V, Sigma-Aldrich) and incubated for 2 h at 4 °C. Then, 1 mg of 3×FLAG-RanBPM and each mutant-transfected cell lysate was added to each mixture followed by rotation at room temperature for 1 h, centrifugation, and three washes. The beads were boiled in sodium dodecyl sulfate (SDS) sample buffer to elute the bound proteins, which were resolved by 10% SDS-polyacrylamide gel electrophoresis (PAGE) followed by Western blot analysis.
In Vivo Co-immunoprecipitation
The in vivo co-immunoprecipitation assay was performed as described (22). pcDNA3.1-HA-BLT2 or pcDNA3.1-HA-BLT2 and 3×FLAG-RanBPM were transfected into HEK 293T cells. After 24 h, the cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed with 0.5 ml of lysis buffer. The lysate was transferred to a 1.7-ml microcentrifuge tube and incubated on a rotating wheel for 1 h. Insoluble material was removed by centrifugation at 10,000 × g for 10 min, and the supernatant was precleared with protein G-agarose beads on a rotating wheel for 2 h. The precleared cell lysate was incubated with 4 μg of high affinity monoclonal anti-HA antibody (clone 12CA5, Roche Applied Science) for 2 h and incubated with protein G-agarose beads overnight to immunoprecipitate the epitope-tagged BLT2. The samples were washed twice with lysis buffer and twice with ice-cold PBS. Unless otherwise stated, all procedures were performed at 4 °C. SDS sample buffer (20 μl) was added to the pellet, which was then heated to 95 °C for 10 min. The proteins were separated by 10% SDS-PAGE and analyzed by immunoblotting.
Immunofluorescence Confocal Microscopy
HEK 293T cells that were transiently transfected with c-Myc-BLT2 and 3×FLAG-RanBPM were grown on coverslips, washed three times with ice-cold PBS, and then fixed with 2% paraformaldehyde for 20 min at room temperature. The cells were rinsed three times with ice-cold PBS and permeabilized with 0.1% Triton X-100 for 5 min. The cells were then washed two times with PBS containing 2% BSA (PBA) and incubated with either anti-c-Myc antibody (1:100 dilution, Santa Cruz Biotechnology) or anti-FLAG M2 antibody (1:100 dilution, Sigma-Aldrich) in 2% PBA for 40 min at room temperature followed by staining with fluorescein isothiocyanate-conjugated secondary anti-rabbit IgG (1:200 dilution, Molecular Probes) or tetramethylrhodamine isothiocyanate-conjugated secondary anti-mouse IgG antibody (1:200 dilution, Molecular Probes) for 30 min at room temperature. The integrity of the nuclei was confirmed by DAPI (Sigma-Aldrich) staining. The cells were washed twice with ice-cold PBS and imaged with a confocal laser-scanning microscope (510META, Carl Zeiss, Oberkochen, Germany) with ×630 oil immersion objective. Co-localization was quantitated using MetaMorph software version 6.0 (Molecular Devices). Additionally, co-localization of endogenous BLT2 and RanBPM was detected using anti-BLT2 antibody (1:50 dilution, Cayman Chemical) and anti-RanBPM antibody (1:100 dilution, Santa Cruz Biotechnology) in HaCaT cells.
Measurement of ROS
Intracellular H2O2 was measured as a function of dichlorodihydrofluorescein (DCF) fluorescence using the procedure described previously (9). Briefly, 2 × 105 CHO-K1 or HEK-293T cells were plated in 60-mm dishes and incubated in RPMI 1640 or DMEM medium supplemented with 10% FBS for 24 h prior to ROS measurement. The cells were then stabilized in serum-free RPMI 1640 or DMEM medium without phenol red for at least 2 h before exposure to the agonist. HaCaT or PHK cells were seeded onto 60-mm dishes and grown to 90% confluence. The cells were starved in serum-free DMEM or EpiLife® medium for 2 h before exposure to the agonist. To measure the intracellular H2O2 levels, the cells were incubated for 40 min in the dark at 37 °C in a humidified CO2 incubator with the H2O2-sensitive fluorophore 2′,7′-dichlorodihydrofluorescein diacetate (20 μm). DCF fluorescence was measured using a FACSCalibur flow cytometer (BD Biosciences) with excitation and emission wavelengths of 488 and 530 nm, respectively. The values represent the means ± S.D. of the DCF fluorescence from three independent experiments.
Chemotaxis Assay
Chemotactic motility was assayed using Transwell chambers with 6.5-mm diameter polycarbonate filters (8-μm pore size, Corning Costar) as described previously (23). Briefly, CHO-K1 or HEK-293T cells were transfected with pcDNA3.1, pcDNA3.1-HA-BLT2, or pcDNA3.1-HA-BLT2 and 3×FLAG-RanBPM. Twenty-four hours after transfection, the lower surfaces of the filters were coated with 10 μg/ml fibronectin (Roche Applied Science) or collagen type I (BD Bioscience) in serum-free RPMI 1640 or DMEM medium for 1 h at 37 °C. Dry, coated filters containing various amounts of LTB4 were placed in the lower wells of the Transwell chambers, and 100 μl of CHO-K1 or HEK-293T cells expressing either BLT2 or BLT2 and RanBPM in serum-free RPMI 1640 or DMEM medium was loaded into the top wells, resulting in a final concentration of 2 × 104 cells/ml. The Transwell chambers were then placed into 24-well companion plates (Corning Costar) containing serum-free RPMI 1640 or DMEM medium with ethanol, LTB4 (300 nm), 12HHT (100 nm), or CAY10583 (100 nm) and incubated at 37 °C in 5% CO2 for 2 h. HaCaT or PHK cells were transfected with either control shRNA or RanBPM shRNA. At 48 h after transfection, the transfected cells were exposed to LTB4 (300 nm) or 12HHT (100 nm) for 3 h. The cells that migrated through the membrane were fixed and stained with methanol for 3 min and stained with hematoxylin/eosin for 10 min. Chemotaxis was quantified by counting the cells on the lower sides of the filters under an optical microscope (magnification, ×200). Six fields were counted for each assay, and the values represent the means ± S.D. of the chemotactic motility from duplicate independent experiments.
Data Analysis and Statistics
All values are expressed as the means ± S.D. Statistical comparisons between experimental groups were performed using the Student's t test. Values of p < 0.01 were considered statistically significant. Values of p < 0.01, p < 0.001, and p < 0.0001 are designated by *, **, and ***, respectively, in Figs. 4–7.
FIGURE 4.

RanBPM overexpression inhibits BLT2-mediated ROS generation and chemotaxis in HEK-293T cells. A, HEK-293T cells were transiently co-transfected with HA-BLT2 and 3×FLAG control vector (−) or co-transfected with HA-BLT2 and 3×FLAG-RanBPM (+). At 48 h after transfection, the transfected cells were exposed to control buffer (ethanol) or 300 nm LTB4 for 2 h. The migrating cells were then fixed and stained with hematoxylin/eosin. Migratory activity was expressed as a percentage of the control. B and C, HEK-293T cells were transiently co-transfected with pcDNA3.1 (Mock) and 3×FLAG-RanBPM (+) or co-transfected with HA-BLT2 and 3×FLAG-RanBPM. Also, the cells were co-transfected with pcDNA3.1 (Mock) and 3×FLAG vector (−) for the control. B, HEK-293T cells expressing either HA-BLT2 or HA-BLT2 and 3×FLAG-RanBPM were exposed to control buffer (ethanol) or 100 nm 12HHT for 2 h and then assayed for chemotaxis as described above. C, HEK-293T cells expressing either HA-BLT2 or HA-BLT2 and 3×FLAG-RanBPM were exposed to control buffer (DMSO) or 100 nm CAY10583 for 2 h and then assayed for chemotaxis as described above. D, HEK-293T cells were transiently co-transfected with HA-BLT2 and 3×FLAG control vector (−) or co-transfected with HA-BLT2 and 3×FLAG-RanBPM (+). At 48 h after transfection, the transfected cells were stabilized in serum- and phenol red-free DMEM medium for at least 2 h prior to exposure to 300 nm LTB4 for 5 min. DCF fluorescence was then monitored by flow cytometry as described previously (11). DCF fluorescence was expressed as a -fold change from the control. E and F, HEK-293T cells were transiently co-transfected with pcDNA3.1 (Mock) and 3×FLAG-RanBPM (+) or co-transfected with HA-BLT2 and 3×FLAG-RanBPM. Also, the cells were co-transfected with pcDNA3.1 (Mock) and 3×FLAG vector (−) for the control. E, HEK-293T cells expressing either HA-BLT2 or HA-BLT2 and 3×FLAG-RanBPM were exposed to 100 nm 12HHT for 5 min and then assayed for ROS generation. F, HEK-293T cells expressing either HA-BLT2 or HA-BLT2 and 3×FLAG-RanBPM were exposed to 100 nm CAY10583 for 5 min and then assayed for ROS generation. The data are presented as the means ± S.D. of three independent experiments.
FIGURE 5.

RanBPM overexpression attenuates BLT2-mediated ROS generation and chemotaxis in CHO-K1 cells. A, CHO-K1 cells were transiently co-transfected with HA-BLT2 and 3×FLAG control vector (−) or co-transfected with HA-BLT2 and 3×FLAG-tagged full-length RanBPM (+) or each 3×FLAG-tagged RanBPM deletion mutant (N3, I, and C1). CHO-K1 cells transfected with HA-BLT2-ΔC were used as the negative control. At 48 h after transfection, the transfected cells were exposed to control buffer (ethanol) or 300 nm LTB4 for 2 h. The migrating cells were then fixed and stained with hematoxylin/eosin. Migratory activity was expressed as a percentage of the control. B–D, CHO-K1 cells were transiently co-transfected with pcDNA3.1 (Mock) and 3×FLAG-RanBPM (+) or co-transfected with HA-BLT2 and 3×FLAG-RanBPM. Also, the cells were co-transfected with pcDNA3.1 (Mock) and 3×FLAG vector (−) for the control. B, CHO-K1 cells expressing either HA-BLT2 or HA-BLT2 and 3×FLAG-RanBPM were exposed to control buffer (ethanol) or 100 nm 12HHT for 2 h and then assayed for chemotaxis as described above. C, CHO-K1 cells expressing either HA-BLT2 or HA-BLT2 and 3×FLAG-RanBPM were exposed to control buffer (DMSO) or 100 nm CAY10583 for 2 h and then assayed for chemotaxis as described above. D, CHO-K1 cells expressing either HA-BLT1 or HA-BLT1 and 3×FLAG-RanBPM were exposed to 300 nm LTB4 for 2 h and then assayed for chemotaxis as described above. E, CHO-K1 cells expressing either HA-BLT2 or HA-BLT2 and 3×FLAG-RanBPM were exposed to control buffer (ethanol) or 100 nm lysophosphatidic acid (LPA) for 2 h and then assayed for chemotaxis as described above. F, CHO-K1 cells were transiently co-transfected with HA-BLT2 and 3×FLAG control vector (−) or co-transfected with HA-BLT2 and 3×FLAG-tagged full-length RanBPM (+) or each 3×FLAG-tagged RanBPM deletion mutant (N3, I, and C1). CHO-K1 cells transfected with HA-BLT2-ΔC were used as the negative control. At 48 h after transfection, the transfected cells were stabilized in serum- and phenol red-free RPMI 1640 medium for at least 2 h prior to exposure to 300 nm LTB4 for 5 min. DCF fluorescence was then monitored by flow cytometry as described previously (11). DCF fluorescence was expressed as a -fold change from the control. G and H, CHO-K1 cells were transiently co-transfected with pcDNA3.1 (Mock) and 3×FLAG-RanBPM (+) or co-transfected with HA-BLT2 and 3×FLAG-RanBPM. Also, the cells were co-transfected with pcDNA3.1 (Mock) and 3×FLAG vector (−) for the control. G, CHO-K1 cells expressing either HA-BLT2 or HA-BLT2 and 3×FLAG-RanBPM were exposed to 100 nm 12HHT for 5 min and then assayed for ROS generation. H, CHO-K1 cells expressing either HA-BLT2 or HA-BLT2 and 3×FLAG-RanBPM were exposed to 100 nm CAY10583 for 5 min and then assayed for ROS generation. The data are presented as the means ± S.D. of three independent experiments. ns: not significant.
FIGURE 6.

Knockdown of endogenous RanBPM by shRNA promotes BLT2-mediated chemotaxis and ROS generation. A, HaCaT cells were transfected with either control shRNA (−) or RanBPM shRNA (+). At 48 h after transfection, the transfected cells were exposed to 300 nm LTB4 (left panel) or 100 nm 12HHT (right panel) for 3 h. The migrating cells were then fixed and stained with hematoxylin/eosin. The migratory activity was expressed as a percentage of the control. B, PHK cells expressing either control shRNA or RanBPM shRNA were exposed to 300 nm LTB4 (left panel) or 100 nm 12HHT (right panel) for 3 h and then assayed for chemotaxis as described above. C, HaCaT cells expressing either control shRNA or RanBPM shRNA were exposed to 300 nm LTB4 (left panel) or 100 nm 12HHT (right panel) for 5 min and then assayed for ROS generation. D, PHK cells expressing either control shRNA or RanBPM shRNA were exposed to 300 nm LTB4 (left panel) or 100 nm 12HHT (right panel) for 5 min and then assayed for ROS generation. E, 253J-BV bladder cancer cells were stained with primary antibodies against human BLT2 (Cayman Chemical) and RanBPM (Santa Cruz Biotechnology), and analyzed by FACS analysis. The results shown are representative of three independent experiments with similar results. F, dose dependence of LTB4-induced chemotactic motility was determined in 253J-BV bladder cancer cells, as described above. G, 253J-BV bladder cancer cells were transfected with either control shRNA (−) or RanBPM shRNA (+), and the amount of RanBPM mRNA was analyzed by semiquantitative RT-PCR. H, 253J-BV cells were transiently transfected with control shRNA or RanBPM shRNA. At 48 h after transfection, the transfected cells were exposed to 300 nm LTB4 (left panel) or 100 nm 12HHT (right panel) for 3 h and then assayed for chemotaxis as described above. The migratory activity was expressed as a percentage of the control. I, 253J-BV cells expressing either control shRNA or RanBPM shRNA were exposed to 300 nm LTB4 (left panel) or 100 nm 12HHT (right panel) for 5 min and then assayed for ROS generation. The data are presented as the means ± S.D. of three independent experiments.
FIGURE 7.
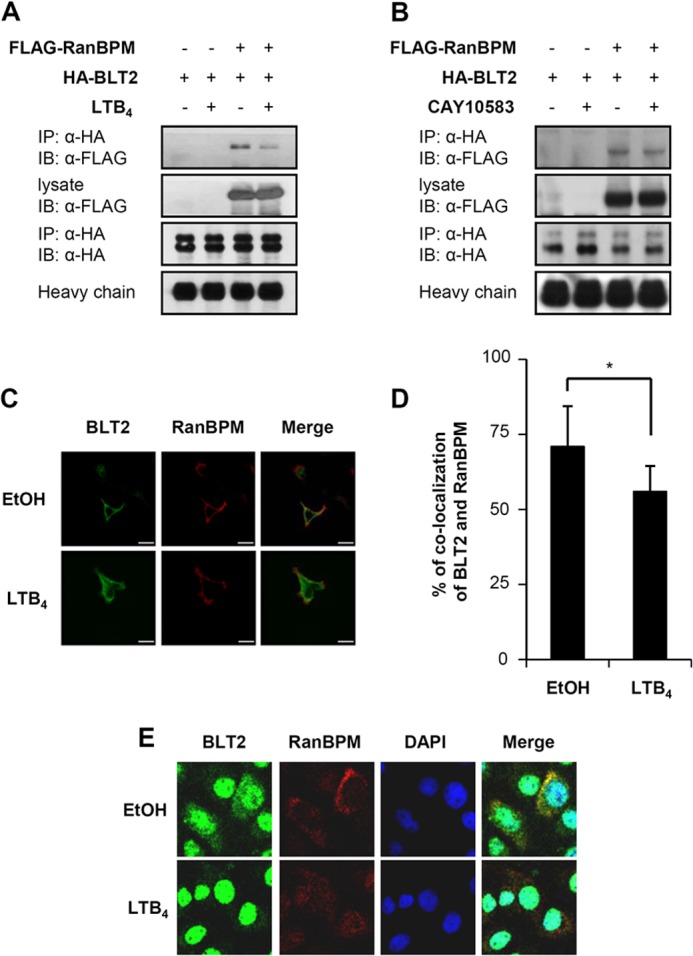
Ligand stimulation decreases the interaction between RanBPM and BLT2. A, 3×FLAG-RanBPM and HA-BLT2 were transiently co-transfected into HEK 293T cells. At 48 h after transfection, the transfected cells were exposed to control buffer (−) or 300 nm LTB4 (+) for 15 min, and immunoprecipitation (IP) was performed using an anti-HA antibody followed by Western blotting (IB) with an anti-FLAG antibody. B, 3×FLAG-RanBPM and HA-BLT2 were transiently co-transfected into HEK 293T cells. At 48 h after transfection, the transfected cells were exposed to control buffer (−) or 100 nm CAY10583 (+) for 15 min, and immunoprecipitation was performed using an anti-HA antibody followed by Western blotting with an anti-FLAG antibody. C, agonist-mediated dissociation of RanBPM and BLT2 was visualized by confocal microscopy of HEK 293T cells transiently transfected with either 3×FLAG-RanBPM or 3×FLAG-RanBPM and c-Myc-BLT2. The HEK 293T cells were treated with either ethanol or 300 nm LTB4 for 15 min. The data are representative of three independent experiments with similar results. Scale bars, 20 μm. D, the percentage of RanBPM and BLT2 co-localization was analyzed with the “co-localization” application of MetaMorph Software as described previously (47). RanBPM co-localization with BLT2 was observed in 73% of the HEK 293T cells, which decreased to 52% after LTB4 stimulation. The data are presented as the means ± S.D. of three independent experiments (20 cells/experiment). E, co-localization of endogenous RanBPM and BLT2 was visualized by confocal microscopy in HaCaT cells. The HaCaT cells were treated with either ethanol or 300 nm LTB4 for 15 min. The images are pseudocolored as follows: red, RanBPM; green, BLT2, with yellow areas indicating co-localization in the merged image and blue indicating DAPI staining of the nucleus. The data are representative of three independent experiments with similar results.
RESULTS
RanBPM Interacts with the BLT2 C-terminal Domain in Vitro and in Vivo
To identify BLT2-interacting proteins, we performed a yeast two-hybrid screen using the GAL4 AD-fused human thymus cDNA library and the GAL4 BD-fused human BLT2 C-terminal domain as the bait. Only one positive clone was identified after screening 1.2 × 107 clones with three reporter genes (Fig. 1). DNA sequencing revealed that the positive clone encoded a partial sequence (amino acids 140–443) of RanBPM/RanBP9 (GenBank accession number: NM_005493), corresponding to the SPRY and LISH domains of the protein. To confirm the interaction between RanBPM and BLT2, we performed a co-immunoprecipitation assay. HA-BLT2 and 3×FLAG-RanBPM were transiently transfected into HEK 293T cells. Whole-cell extracts were then immunoprecipitated with an anti-HA antibody followed by immunoblotting with an anti-FLAG antibody. The data clearly show that RanBPM co-immunoprecipitated with BLT2 (Fig. 2A). Also, the interaction was demonstrated by confocal microscopy analysis. HEK 293T cells were transiently co-transfected with c-Myc-BLT2 and 3×FLAG-RanBPM. Immunofluorescence analysis revealed that RanBPM was co-localized with BLT2 on the plasma membranes of HEK 293T cells (Fig. 2B).
FIGURE 2.

RanBPM interacts with the BLT2 C-terminal domain. A, 3×FLAG-RanBPM and HA-BLT2 were transiently co-transfected into HEK 293T cells, and immunoprecipitation (IP) was performed using an anti-HA antibody followed by Western blotting (IB) with an anti-FLAG antibody. co-IP, co-immunoprecipitation. B, confocal images show the co-localization of RanBPM with BLT2 in the plasma membrane when the two proteins were co-expressed in HEK 293T cells. The images are pseudocolored as follows: red, 3×FLAG-RanBPM; green, c-Myc-BLT2, with yellow areas indicating co-localization in the merged image and blue indicating DAPI staining of the nucleus. Scale bars, 20 μm. C, schematic of the domain organization of BLT2. The HA-tagged deletion construct is HA-BLT2-ΔC (amino acids 1–324), and the GST-tagged deletion construct is GST-BLT2-CT (amino acids 318–389). D, 3×FLAG-RanBPM and HA-BLT2 or 3×FLAG-RanBPM and HA-BLT2 C-terminal deletion mutant were co-transfected into HEK 293T cells, and immunoprecipitation was performed using an anti-HA antibody followed by Western blotting with an anti-FLAG antibody (left). Recombinant GST or GST-BLT2-CT fusion proteins purified by glutathione-SepharoseTM 4B were incubated with 3×FLAG-RanBPM-transfected cell lysates, and the bound proteins were analyzed by immunoblotting with an anti-FLAG antibody (right). The data are representative of three independent experiments with similar results.
Next, to identify the RanBPM-binding region of BLT2, we constructed a C-terminal domain-deleted mutant BLT2 (HA-BLT2-ΔC; Fig. 2C), and a co-immunoprecipitation assay was performed. The data clearly revealed that RanBPM bound to full-length BLT2 but not the C-terminal domain-deleted mutant BLT2 (Fig. 2D), suggesting that RanBPM interacts with the C-terminal domain of BLT2. To further identify the RanBPM-binding region of BLT2, an in vitro GST pulldown assay was performed using the purified recombinant C-terminal domain of BLT2 fused to GST (GST-BLT2-CT; Fig. 2C). Full-length 3×FLAG-RanBPM was transfected into HEK 293T cells. After 24 h, the cells were lysed, and the whole-cell lysates were incubated with either purified GST-BLT2-CT or GST alone. The data indicate that RanBPM was pulled down with GST-BLT2-CT but not GST alone (Fig. 2D). Together, these results suggest that RanBPM interacts directly with the C-terminal domain of BLT2.
The SPRY Domain of RanBPM Is Sufficient for Interaction with BLT2
RanBPM has four conserved domains, SPRY, LISH, CTLH, and CRA. The SPRY domain is involved in protein-protein interactions, and the LISH/CTLH domain is involved in dimerization. To identify which RanBPM domain is important for interaction with BLT2, we employed in vitro domain deletion analysis. Each domain of the N-terminal FLAG-tagged full-length RanBPM or mutant RanBPM (Fig. 3A) was transfected into HEK 293T cells, and the resulting cell lysates were pulled down with the GST-BLT2-CT protein. Full-length RanBPM, the N3 mutant, and the I mutant, which all contain the SPRY domain, strongly interacted with the BLT2 C-terminal domain. By contrast, the C1 mutant, which contains the LISH/CTLH and CRA domains but not the SPRY domain, failed to interact with the BLT2 C-terminal domain (Fig. 3B). These results suggest that the RanBPM SPRY domain contributes to the interaction with the BLT2 C-terminal domain.
FIGURE 3.
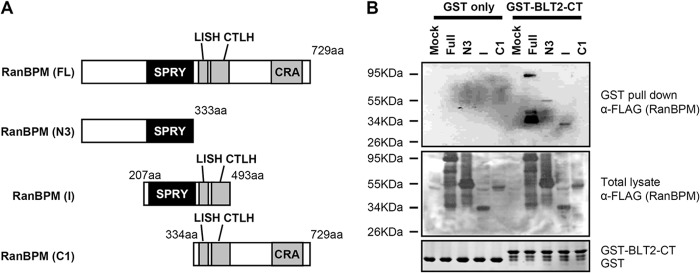
The RanBPM SPRY domain interacts with the BLT2 C-terminal domain. A, schematic of the domain organization of RanBPM and the FLAG-tagged deletion constructs, N3 (amino acids 1–333), I (amino acids 207–493), and C1 (amino acids 334–729). 729aa, 729 amino acids. B, recombinant GST or GST-BLT2 C-terminal fusion proteins purified by glutathione-SepharoseTM 4B were incubated with each RanBPM deletion mutant-transfected cell lysate, and the bound proteins were analyzed by immunoblotting with an anti-FLAG antibody. The data are representative of three independent experiments with similar results.
RanBPM Inhibits LTB4-BLT2-induced Chemotaxis and ROS Generation
Recent studies have shown that BLT2 regulates cell motility functions, such as chemotaxis (10); therefore, we tested whether RanBPM could regulate BLT2-mediated chemotaxis. To test the functional consequence of the interaction between BLT2 and RanBPM, HEK-293T cells were transiently transfected with expression plasmids for BLT2 and RanBPM. LTB4 elicited significantly increased chemotactic migration of the cells transfected with BLT2 alone; however, this effect was substantially blocked by co-transfection with RanBPM (Fig. 4A). In addition, an inhibitory effect of RanBPM on BLT2-mediated chemotaxis was similarly observed in response to a natural ligand of BLT2, 12HHT (Fig. 4B), and a specific BLT2-stimulating agonist, CAY10583, which is also known as Compound A (Fig. 4C). Recently, we reported that the LTB4-BLT2 cascade is associated with the generation of ROS and that the generated ROS play critical roles in cell motility (9–11). Therefore, we determined whether RanBPM could regulate BLT2-induced ROS generation, and we found that LTB4-BLT2-induced ROS generation was blocked by co-transfection with RanBPM (Fig. 4D). Similar inhibitory effects were observed in response to 12HHT (Fig. 4E) and CAY10583 (Fig. 4F).
To further demonstrate the negative regulatory function of RanBPM toward BLT2, we repeated the experiments using CHO-K1 cells, which were previously used for BLT2 functional studies (6, 10, 11). CHO-K1 cells were transiently transfected with expression plasmids for BLT2 and full-length RanBPM (FL) or RanBPM deletion variants N3, I, and C1. LTB4 elicited significantly increased chemotactic migration of the cells transfected with BLT2 alone; however, this effect was severely blocked by co-transfection with full-length RanBPM (FL) or the RanBPM mutants containing the SPRY domain (N3 and I) except the SPRY domain deletion mutant (C1) (Fig. 5A). Additionally, the inhibitory effect of RanBPM on BLT2-mediated chemotaxis was similarly observed in response to 12HHT (Fig. 5B) or CAY10583 (Fig. 5C). As shown in Fig. 5, D and E, overexpression of RanBPM did not inhibit BLT1- or lysophosphatidic acid-induced chemotactic migration, suggesting that RanBPM specifically inhibits BLT2-mediated chemotaxis. Also, we found that LTB4 elicited significantly increased ROS generation in the cells transfected with BLT2; however, this effect was blocked by co-transfection with full-length RanBPM (FL) or the RanBPM mutants containing the SPRY domain (N3 and I) except the SPRY domain deletion mutant (C1) (Fig. 5F). In addition, we observed similar inhibitory effects in response to 12HHT (Fig. 5G) or CAY10583 (Fig. 5H). Together, these results suggest that RanBPM interacts with BLT2 via its SPRY domain to regulate BLT2-induced chemotactic motility and ROS generation.
Knockdown of Endogenous RanBPM Enhanced BLT2-induced ROS Generation and Chemotaxis
To further identify the role of RanBPM in BLT2 inhibition in different cell types, we performed chemotaxis and ROS generation experiments using human keratinocyte HaCaT cells and PHK cells that exhibit endogenous expression of RanBPM and BLT2. Knockdown of endogenous RanBPM by shRNA significantly enhanced BLT2 agonist (LTB4 or 12HHT)-induced chemotaxis and ROS generation in HaCaT cells (Fig. 6, A and C) and PHK cells (Fig. 6, B and D). In addition, we performed similar shRNA knockdown experiments in 253J-BV bladder cancer cells. We observed that both BLT2 and RanBPM are highly expressed in 253J-BV cells and that LTB4 induces chemotaxis in a dose-dependent manner (Fig. 6, E and F). Knockdown of endogenous RanBPM (Fig. 6G) promoted BLT2 agonist (LTB4 or 12HHT)-induced chemotaxis and ROS generation in 253J-BV bladder cancer cells (Fig. 6, H and I). From these results, we are quite certain that RanBPM acts an endogenous brake for BLT2 activation in various cell types.
RanBPM and BLT2 Interaction Are Dependent on Ligand Stimulation
There have been studies indicating that interaction of the cognate receptor with RanBPM was affected by ligand stimulation (24). To test whether the binding of RanBPM and BLT2 was affected by ligand stimulation, a co-immunoprecipitation assay was performed. HA-BLT2 and 3×FLAG-RanBPM were transiently transfected into HEK 293T cells. The cells were stimulated with LTB4 for 15 min. The whole-cell extracts were then immunoprecipitated with anti-HA antibody followed by immunoblotting with an anti-FLAG antibody. Notably, we observed that the interaction between RanBPM and BLT2 was dissociated by stimulation with LTB4 (Fig. 7A). Moreover, we observed a dose-dependent dissociation effect in response to LTB4 (data not shown). As shown in Fig. 7B, CAY10583 treatment caused the dissociation between RanBPM and BLT2. Densitometric analysis showed that the relative amount of RanBPM that co-localized with BLT2 decreased following treatment with LTB4 (Fig. 7, C and D). Next, we performed confocal microscopy experiments to detect the co-localization of RanBPM and BLT2 in HaCaT cells that endogenously express RanBPM and BLT2. As shown in Fig. 7E, our immunofluorescence analysis clearly revealed that RanBPM (red) co-localized with BLT2 (green) in the absence of LTB4 (top panel). However, the co-localization of these proteins was abolished in the presence of LTB4 in HaCaT cells (bottom panel). Together, these results suggest that the interaction between RanBPM and BLT2 is dependent on ligand stimulation.
Ligand Stimulation Decreases the Interaction between RanBPM and BLT2 via an Akt-dependent Mechanism
Recently, we demonstrated that phosphorylation of BLT2 at Thr355 by PI3K-Akt signaling was critical for LTB4-evoked BLT2-mediated chemotaxis (11); therefore, to determine whether this signaling event is involved in the dissociation of RanBPM and BLT2 caused by ligand stimulation, a co-immunoprecipitation assay was performed. Wild-type HA-BLT2 and 3×FLAG-RanBPM or HA-BLT2 T355A mutant and 3×FLAG-RanBPM were transiently transfected into HEK 293T cells. Treatment with LTB4 caused the dissociation between RanBPM and BLT2 in cells transfected with wild-type BLT2; however, there was no change in the cells transfected with the BLT2 T355A mutant (Fig. 8A). Furthermore, RanBPM and BLT2 dissociation was abrogated by pretreatment with LY294002 and was rescued by co-transfection of active Akt (Fig. 8B). These results suggest that LTB4 caused the dissociation between RanBPM and BLT2 and that this dissociation requires the phosphorylation of BLT2 at Thr355 by Akt.
FIGURE 8.
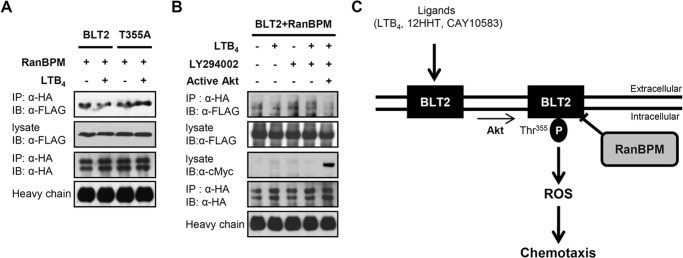
Ligand stimulation decreases the interaction between RanBPM and BLT2 via an Akt-dependent mechanism. A, 3×FLAG-RanBPM and HA-BLT2 or 3×FLAG-RanBPM and the HA-BLT2 T355A mutant were transiently co-transfected into HEK 293T cells. At 48 h after transfection, the transfected cells were exposed to control buffer (−) or 300 nm LTB4 (+) for 15 min, and immunoprecipitation (IP) was performed using an anti-HA antibody followed by Western blotting (IB) with an anti-FLAG antibody. B, 3×FLAG-RanBPM, HA-BLT2, and pUSEamp vector (control vector; −) or 3×FLAG-RanBPM, HA-BLT2, and active Akt (pUSEamp-Myr-Akt; +) (11) were transiently co-transfected into HEK 293T cells. At 48 h after transfection, the transfected cells were exposed to control buffer (−) or 300 nm LTB4 (+) for 15 min in the presence of DMSO (−) or 20 μm LY294002 (+), and immunoprecipitation was performed using an anti-HA antibody followed by Western blotting with an anti-FLAG antibody. Also, the cell lysates were subjected to immunoblot analysis with anti-c-Myc antibody to active Akt. The data are representative of three independent experiments with similar results. C, schematic model depicting the role of RanBPM in BLT2 signaling. P indicates phosphorylation.
DISCUSSION
In the present study, we identified RanBPM as a cellular binding protein for BLT2 using a yeast two-hybrid screen. Further characterization of the interaction between BLT2 and RanBPM via GST pulldown assay and co-immunoprecipitation studies revealed that the SPRY domain of RanBPM binds to the C-terminal domain of BLT2. We also showed that RanBPM inhibits BLT2-mediated ROS generation and chemotaxis, suggesting a negative regulatory function of RanBPM toward BLT2. In addition, our results suggest that Akt-induced phosphorylation of BLT2 at residue Thr355 is critical for ligand-induced dissociation of RanBPM and BLT2, thus releasing the negative regulatory effect of RanBPM and resulting in stimulation of BLT2 signaling.
LTB4 is a potent lipid mediator of inflammation that is biosynthesized from arachidonic acid via the 5-lipoxygenase pathway (25, 26). LTB4 exerts its biological functions via two types of GPCRs, BLT1 and BLT2. Like other GPCRs, BLT1 and BLT2 signal through the activation of guanine nucleotide-binding proteins (G proteins) (27). In addition to G proteins, GPCRs associate with many GIPs to form large functional complexes that induce different activities, from motility to transcriptional activation. Most GIPs interact with the C-terminal domains of GPCRs and modulate GPCRs (3). The C-terminal domain of the GPCR is called the “magic” C terminus because this domain contains many binding motifs that may be involved in protein-protein interactions; it also contains important posttranslational modifications, such as palmitoylation and phosphorylation sites. BLT1 couples with Gαi proteins to induce chemotaxis (28). Chen et al. (29) demonstrated that GRK2, a serine-threonine kinase, is associated with the C terminus of BLT1s and is necessary for receptor internalization. Unlike BLT1, the regulatory mechanism of BLT2 remains unknown. Here, we identified novel intracellular interactions between BLT2 and RanBPM. We showed the specific interaction between RanBPM and BLT2 by co-immunoprecipitation experiments. Moreover, immunofluorescence assays revealed that RanBPM was co-localized with BLT2 on the plasma membranes of HEK 293T cells. We also found that RanBPM directly interacted with the C-terminal region of BLT2 and that the C-terminal domain-deleted BLT2 showed no interaction with RanBPM in comparison with full-length BLT2 (Fig. 2).
RanBPM, a member of the Ran-GTPase-binding protein family, is ubiquitously expressed in a wide variety of tissues (30–32). Although the normal physiological functions of RanBPM are not fully understood, several lines of evidence suggest that it acts as a scaffolding protein by binding to the cytoplasmic domains of a diverse group of membrane receptors to regulate signal transduction (33–37). RanBPM contains multiple functional domains, including a SPRY domain, a LISH/CTLH domain, and a CRA domain. SPRY domains are involved in protein-protein interactions in various cellular functions (38). Our observations suggest that the SPRY domain of RanBPM strongly interacts with BLT2, whereas there is no interaction between the SPRY domain deletion mutant and BLT2 (Fig. 3B). Recently, studies have shown that RanBPM regulates cell migration by linking extracellular signals via plasma membrane receptors with the intracellular signal transduction machinery (39–41). For example, RanBPM interacts with the plasma membrane receptor integrin LFA-1 to regulate cell motility. Consistent with the proposed role of RanBPM in cell motility, our observations suggest that RanBPM inhibits BLT2-meditated chemotaxis (Figs. 4, A–C, and 5, A–C) but not BLT1, a high affinity LTB4 receptor (Fig. 5D), or lysophosphatidic acid, a major serum lysophospholipid that stimulates CHO-K1 cell migration via lysophosphatidic acid-mediated (42) chemotaxis in CHO-K1 cells (Fig. 5E), suggesting that RanBPM specifically inhibits BLT2-mediated chemotaxis. The regulatory mechanism of RanBPM in the LTB4-BLT2-induced signaling pathway is not understood, and recently, it was shown that RanBPM binds to the mu opioid receptor to alter agonist-induced internalization (43, 44). Interestingly, we also observed that overexpression of RanBPM significantly inhibited LTB4-triggered BLT2 internalization (supplemental Fig. S1). Thus, one possibility is that RanBPM-inhibited BLT2 signaling occurs through its blocking of BLT2 internalization. Future studies are necessary to understand the detailed mechanisms by which RanBPM inhibits BLT2 signaling pathway.
Recently, we reported that BLT2 signaling is associated with aggressive 253J-BV bladder cancer cells and promotes chemotactic migration through ROS generation (45). The role of RanBPM in cancer cells remains unknown, although RanBPM is highly expressed in breast cancer cells and prostate cancer (32, 46). Interestingly, BLT2 is also highly expressed in many cancer cell types (13–17). Therefore, it has been speculated that RanBPM reduces the cancer-promoting effect of BLT2. In accordance with this idea, we observed that knockdown of endogenous RanBPM by shRNA further promoted LTB4-BLT2-induced ROS generation and chemotaxis in 253J-BV bladder cancer cells (Fig. 6, H and I). In addition, in aggressive MDA-MB-231 breast cancer cells, BLT2-mediated invasion was dramatically reduced by overexpression of RanBPM, and knockdown of endogenous RanBPM strongly promoted BLT2-mediated invasion (data not shown). These results demonstrate the negative regulatory function of RanBPM toward the cancer-promoting effects of BLT2. Further studies are necessary to clarify how RanBPM reduces the effect of BLT2 in cancer cells.
The interaction between RanBPM and BLT2 is direct and occurs in the absence of ligand. Notably, in the presence of ligand, we found that ligand treatment caused the dissociation of BLT2 and RanBPM (Fig. 7). It is possible that conformational changes in the C terminus of the receptor following activation inhibit binding and induce the release of RanBPM. Previously, we showed that activation of Akt by LTB4, which phosphorylates BLT2 at Thr355, is critical for BLT2-mediated chemotactic responses (11). In addition, we found that blockade of Akt signaling or the BLT2 T355A mutant inhibited the LTB4-triggered dissociation of BLT2 and RanBPM (Fig. 8). Therefore, we postulate that Akt-induced phosphorylation of BLT2 at Thr355 may be a regulatory mechanism by which the ligand stimulates the dissociation of BLT2 and RanBPM. On the basis of our observations, we propose a model that describes the mechanism by which the interaction between RanBPM and BLT2 contributes to BLT2-mediated cell migration. In this model, RanBPM interacts with BLT2 to suppress cell migration by inhibiting ROS generation. Furthermore, the addition of BLT2 ligands causes the dissociation of BLT2 and RanBPM, thus releasing the negative regulatory effect of RanBPM and resulting in stimulation of BLT2 signaling. We found that Akt-induced BLT2 phosphorylation at residue Thr355 is a potential mechanism by which BLT2 is dissociated from RanBPM. This novel role for RanBPM in the regulation of BLT2 should contribute to a better understanding of the regulatory mechanisms of BLT2-mediated cellular phenotypes, including chemotactic motility.
This work was supported by grants from the Bio and Medical Technology Development Program (Grant 2012M3A9C5048709), the Basic Science Research Program (Grant 2012R1A2A2A01044526) through the National Research Foundation (NRF) funded by the Ministry of Science, ICT and Future Planning, Republic of Korea, and the National Research and Development Program for Cancer Control (Grant 1220020), Ministry for Health and Welfare, Republic of Korea. This work was also supported by a Korea University grant (Grant G1300090).

This article contains supplemental Fig. S1.
- GPCR
- G protein-coupled receptor
- GIP
- GPCR-interacting protein
- BLT
- leukotriene B4 receptor
- LTB4
- leukotriene B4
- 12HHT
- 12(S)-hydroxyheptadeca-5Z, 8E, 10E-trienoic acid
- ROS
- reactive oxygen species
- DCF
- dichlorofluorescein
- PHK
- primary human keratinocyte
- AD
- activation domain
- BD
- binding domain
- SD
- synthetic defined
- CT
- C-terminal domain
- DMSO
- dimethyl sulfoxide
- SPRY
- repeats in splA and RyR
- LISH
- Lis1-homologous
- CTLH
- C-terminal to LisH
- CRA
- CT11-RanBPM.
REFERENCES
- 1. Bridges T. M., Lindsley C. W. (2008) G-protein-coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem. Biol. 3, 530–541 [DOI] [PubMed] [Google Scholar]
- 2. Cotton M., Claing A. (2009) G protein-coupled receptors stimulation and the control of cell migration. Cell. Signal. 21, 1045–1053 [DOI] [PubMed] [Google Scholar]
- 3. Bockaert J., Fagni L., Dumuis A., Marin P. (2004) GPCR interacting proteins (GIP). Pharmacol. Ther. 103, 203–221 [DOI] [PubMed] [Google Scholar]
- 4. Ritter S. L., Hall R. A. (2009) Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol. 10, 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bockaert J., Perroy J., Bécamel C., Marin P., Fagni L. (2010) GPCR interacting proteins (GIPs) in the nervous system: Roles in physiology and pathologies. Annu. Rev. Pharmacol. Toxicol. 50, 89–109 [DOI] [PubMed] [Google Scholar]
- 6. Yokomizo T., Kato K., Terawaki K., Izumi T., Shimizu T. (2000) A second leukotriene B4 receptor, BLT2. A new therapeutic target in inflammation and immunological disorders. J. Exp. Med. 192, 421–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamohara M., Takasaki J., Matsumoto M., Saito T., Ohishi T., Ishii H., Furuichi K. (2000) Molecular cloning and characterization of another leukotriene B4 receptor. J. Biol. Chem. 275, 27000–27004 [DOI] [PubMed] [Google Scholar]
- 8. Iizuka Y., Yokomizo T., Terawaki K., Komine M., Tamaki K., Shimizu T. (2005) Characterization of a mouse second leukotriene B4 receptor, mBLT2: BLT2-dependent ERK activation and cell migration of primary mouse keratinocytes. J. Biol. Chem. 280, 24816–24823 [DOI] [PubMed] [Google Scholar]
- 9. Choi J. A., Kim E. Y., Song H., Kim C., Kim J. H. (2008) Reactive oxygen species are generated through a BLT2-linked cascade in Ras-transformed cells. Free Radic. Biol. Med. 44, 624–634 [DOI] [PubMed] [Google Scholar]
- 10. Kim J. Y., Lee W. K., Yu Y. G., Kim J. H. (2010) Blockade of LTB4-induced chemotaxis by bioactive molecules interfering with the BLT2-Gαi interaction. Biochem. Pharmacol. 79, 1506–1515 [DOI] [PubMed] [Google Scholar]
- 11. Wei J. D., Kim J. Y., Kim J. H. (2011) BLT2 phosphorylation at Thr355 by Akt is necessary for BLT2-mediated chemotaxis. FEBS Lett. 585, 3501–3506 [DOI] [PubMed] [Google Scholar]
- 12. Kim C., Kim J. Y., Kim J. H. (2008) Cytosolic phospholipase A2, lipoxygenase metabolites, and reactive oxygen species. BMB Rep. 41, 555–559 [DOI] [PubMed] [Google Scholar]
- 13. Rocconi R. P., Kirby T. O., Seitz R. S., Beck R., Straughn J. M., Jr., Alvarez R. D., Huh W. K. (2008) Lipoxygenase pathway receptor expression in ovarian cancer. Reprod. Sci. 15, 321–326 [DOI] [PubMed] [Google Scholar]
- 14. Sveinbjörnsson B., Rasmuson A., Baryawno N., Wan M., Pettersen I., Ponthan F., Orrego A., Haeggström J. Z., Johnsen J. I., Kogner P. (2008) Expression of enzymes and receptors of the leukotriene pathway in human neuroblastoma promotes tumor survival and provides a target for therapy. FASEB J. 22, 3525–3536 [DOI] [PubMed] [Google Scholar]
- 15. Hennig R., Osman T., Esposito I., Giese N., Rao S. M., Ding X. Z., Tong W. G., Büchler M. W., Yokomizo T., Friess H., Adrian T. E. (2008) BLT2 is expressed in PanINs, IPMNs, pancreatic cancer and stimulates tumour cell proliferation. Br. J. Cancer 99, 1064–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choi J. A., Lee J. W., Kim H., Kim E. Y., Seo J. M., Ko J., Kim J. H. (2010) Pro-survival of estrogen receptor-negative breast cancer cells is regulated by a BLT2-reactive oxygen species-linked signaling pathway. Carcinogenesis 31, 543–551 [DOI] [PubMed] [Google Scholar]
- 17. Cork D. J., Lembark S., Tovanabutra S., Robb M. L., Kim J. H. (2010) W-curve alignments for HIV-1 genomic comparisons. PLoS One 5, e10829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tong W. G., Ding X. Z., Talamonti M. S., Bell R. H., Adrian T. E. (2005) LTB4 stimulates growth of human pancreatic cancer cells via MAPK and PI-3 kinase pathways. Biochem. Biophys. Res. Commun. 335, 949–956 [DOI] [PubMed] [Google Scholar]
- 19. Ihara A., Wada K., Yoneda M., Fujisawa N., Takahashi H., Nakajima A. (2007) Blockade of leukotriene B4 signaling pathway induces apoptosis and suppresses cell proliferation in colon cancer. J. Pharmacol. Sci. 103, 24–32 [DOI] [PubMed] [Google Scholar]
- 20. Yoo M. H., Song H., Woo C. H., Kim H., Kim J. H. (2004) Role of the BLT2, a leukotriene B4 receptor, in Ras transformation. Oncogene 23, 9259–9268 [DOI] [PubMed] [Google Scholar]
- 21. Haribabu B., Zhelev D. V., Pridgen B. C., Richardson R. M., Ali H., Snyderman R. (1999) Chemoattractant receptors activate distinct pathways for chemotaxis and secretion: role of G-protein usage. J. Biol. Chem. 274, 37087–37092 [DOI] [PubMed] [Google Scholar]
- 22. Gaudreau R., Le Gouill C., Venne M. H., Stankova J., Rola-Pleszczynski M. (2002) Threonine 308 within a putative casein kinase 2 site of the cytoplasmic tail of leukotriene B4 receptor (BLT1) is crucial for ligand-induced, G-protein-coupled receptor-specific kinase 6-mediated desensitization. J. Biol. Chem. 277, 31567–31576 [DOI] [PubMed] [Google Scholar]
- 23. Woo C. H., You H. J., Cho S. H., Eom Y. W., Chun J. S., Yoo Y. J., Kim J. H. (2002) Leukotriene B4 stimulates Rac-ERK cascade to generate reactive oxygen species that mediates chemotaxis. J. Biol. Chem. 277, 8572–8578 [DOI] [PubMed] [Google Scholar]
- 24. Wang D., Li Z., Messing E. M., Wu G. (2002) Activation of Ras/Erk pathway by a novel MET-interacting protein RanBPM. J. Biol. Chem. 277, 36216–36222 [DOI] [PubMed] [Google Scholar]
- 25. Leff A. R. (2000) Role of leukotrienes in bronchial hyperresponsiveness and cellular responses in airways. Am. J. Respir. Crit. Care Med. 161, S125–S132 [DOI] [PubMed] [Google Scholar]
- 26. Funk C. D. (2001) Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875 [DOI] [PubMed] [Google Scholar]
- 27. Tager A. M., Luster A. D. (2003) BLT1 and BLT2: the leukotriene B4 receptors. Prostaglandins Leukot. Essent. Fatty Acids 69, 123–134 [DOI] [PubMed] [Google Scholar]
- 28. Peres C. M., Aronoff D. M., Serezani C. H., Flamand N., Faccioli L. H., Peters-Golden M. (2007) Specific leukotriene receptors couple to distinct G proteins to effect stimulation of alveolar macrophage host defense functions. J. Immunol. 179, 5454–5461 [DOI] [PubMed] [Google Scholar]
- 29. Chen Z., Gaudreau R., Le Gouill C., Rola-Pleszczynski M., Stanková J. (2004) Agonist-induced internalization of leukotriene B4 receptor 1 requires G-protein-coupled receptor kinase 2 but not arrestins. Mol. Pharmacol. 66, 377–386 [DOI] [PubMed] [Google Scholar]
- 30. Nakamura M., Masuda H., Horii J., Kuma K. i., Yokoyama N., Ohba T., Nishitani H., Miyata T., Tanaka M., Nishimoto T. (1998) When overexpressed, a novel centrosomal protein, RanBPM, causes ectopic microtubule nucleation similar to γ-tubulin. J. Cell Biol. 143, 1041–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nishitani H., Hirose E., Uchimura Y., Nakamura M., Umeda M., Nishii K., Mori N., Nishimoto T. (2001) Full-sized RanBPM cDNA encodes a protein possessing a long stretch of proline and glutamine within the N-terminal region, comprising a large protein complex. Gene 272, 25–33 [DOI] [PubMed] [Google Scholar]
- 32. Rao M. A., Cheng H., Quayle A. N., Nishitani H., Nelson C. C., Rennie P. S. (2002) RanBPM, a nuclear protein that interacts with and regulates transcriptional activity of androgen receptor and glucocorticoid receptor. J. Biol. Chem. 277, 48020–48027 [DOI] [PubMed] [Google Scholar]
- 33. Cheng L., Lemmon S., Lemmon V. (2005) RanBPM is an L1-interacting protein that regulates L1-mediated mitogen-activated protein kinase activation. J. Neurochem. 94, 1102–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bai D., Chen H., Huang B. R. (2003) RanBPM is a novel binding protein for p75NTR. Biochem. Biophys. Res. Commun. 309, 552–557 [DOI] [PubMed] [Google Scholar]
- 35. Togashi H., Schmidt E. F., Strittmatter S. M. (2006) RanBPM contributes to Semaphorin3A signaling through plexin-A receptors. J. Neurosci. 26, 4961–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rhodes D. A., de Bono B., Trowsdale J. (2005) Relationship between SPRY and B30.2 protein domains. Evolution of a component of immune defence? Immunology 116, 411–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lakshmana M. K., Yoon I. S., Chen E., Bianchi E., Koo E. H., Kang D. E. (2009) Novel role of RanBP9 in BACE1 processing of amyloid precursor protein and amyloid-β peptide generation. J. Biol. Chem. 284, 11863–11872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murrin L. C., Talbot J. N. (2007) RanBPM, a scaffolding protein in the immune and nervous systems. J. Neuroimmune Pharmacol. 2, 290–295 [DOI] [PubMed] [Google Scholar]
- 39. Zou Y., Lim S., Lee K., Deng X., Friedman E. (2003) Serine/threonine kinase Mirk/Dyrk1B is an inhibitor of epithelial cell migration and is negatively regulated by the Met adaptor Ran-binding protein M. J. Biol. Chem. 278, 49573–49581 [DOI] [PubMed] [Google Scholar]
- 40. Suresh B., Ramakrishna S., Kim Y. S., Kim S. M., Kim M. S., Baek K. H. (2010) Stability and function of mammalian lethal giant larvae-1 oncoprotein are regulated by the scaffolding protein RanBPM. J. Biol. Chem. 285, 35340–35349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Denti S., Sirri A., Cheli A., Rogge L., Innamorati G., Putignano S., Fabbri M., Pardi R., Bianchi E. (2004) RanBPM is a phosphoprotein that associates with the plasma membrane and interacts with the integrin LFA-1. J. Biol. Chem. 279, 13027–13034 [DOI] [PubMed] [Google Scholar]
- 42. Sugimoto N., Takuwa N., Yoshioka K., Takuwa Y. (2006) Rho-dependent, Rho kinase-independent inhibitory regulation of Rac and cell migration by LPA1 receptor in Gi-inactivated CHO cells. Exp. Cell Res. 312, 1899–1908 [DOI] [PubMed] [Google Scholar]
- 43. Talbot J. N., Skifter D. A., Bianchi E., Monaghan D. T., Toews M. L., Murrin L. C. (2009) Regulation of mu opioid receptor internalization by the scaffold protein RanBPM. Neurosci Lett 466, 154–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seebahn A., Rose M., Enz R. (2008) RanBPM is expressed in synaptic layers of the mammalian retina and binds to metabotropic glutamate receptors. FEBS Lett. 582, 2453–2457 [DOI] [PubMed] [Google Scholar]
- 45. Kim E. Y., Seo J. M., Kim C., Lee J. E., Lee K. M., Kim J. H. (2010) BLT2 promotes the invasion and metastasis of aggressive bladder cancer cells through a reactive oxygen species-linked pathway. Free Radic. Biol. Med. 49, 1072–1081 [DOI] [PubMed] [Google Scholar]
- 46. Emberley E. D., Gietz R. D., Campbell J. D., HayGlass K. T., Murphy L. C., Watson P. H. (2002) RanBPM interacts with psoriasin in vitro, and their expression correlates with specific clinical features in vivo in breast cancer. BMC Cancer 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Donepudi M., Resh M. D. (2008) c-Src trafficking and co-localization with the EGF receptor promotes EGF ligand-independent EGF receptor activation and signaling. Cell. Signal. 20, 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]