

Background: FcγR are critical for antibody therapy.
Results: Monocyte FcγR activation leads to production of sFlt-1 that inhibits angiogenesis in vitro and tumor growth in vivo. This production is negatively regulated by miR-181a.
Conclusion: FcγR lead to production of biologically active sFlt-1, which has antitumor functions.
Significance: This finding represents a novel antitumor mechanism of antibodies.
Keywords: Angiogenesis, FCγ Receptors, MicroRNA, Monocytes, Signal Transduction
Abstract
Monocytes/macrophages are potent mediators of antitumor antibody therapy, where they engage target cells via Fcγ receptors (FcγR). Binding of these cells to opsonized tumor targets elicits cytokine production, phagocytosis, and antibody-mediated cellular cytotoxicity. Here we show for the first time that activation of monocyte FcγR results in the secretion of soluble vascular endothelial growth factor receptor-1 (VEGFR-1/sFlt-1), which serves to antagonize VEGF-mediated angiogenesis and tumor growth. Consistent with this, using a murine solid tumor model of antibody therapy, we show that sFlt-1 is involved in restricting tumor growth. Analyzing the mechanism of induction of sFlt-1, we found that the Erk and PI3K pathways were required for transcription, and NF-κB was required for translation. Upon closer examination of the role of NF-κB, we found that a microRNA, miR181a, negatively regulates FcγR-mediated sFlt-1 production and that NF-κB serves to antagonize this microRNA. Taken together, these results demonstrate a novel and biologically important function of monocytes and macrophages during antibody therapy.
Introduction
Monocytes are important for proper immune function and are key mediators of antibody therapy. Binding of immunoglobulins by monocyte FcγR2 triggers important effector functions such as phagocytosis (1), release of reactive oxygen species (2), and cytokine production (3). Although some success of antibody therapy in the treatment of malignancies can be attributed to the direct activity of antibodies on tumor cells, the bulk of antibody function is mediated through Fc receptor-bearing cells, of which monocytes/macrophages have been shown to be critical. Multiple strategies are currently being pursued to both enhance the binding of cells to antibody-immune complexes as well as to enhance the effector mechanisms used to mediate tumor cell destruction.
Tumor angiogenesis involves the recruitment of normal vasculature to tumor sites, and this is required for tumor growth (4). Although many factors and pathways are known to mediate blood vessel recruitment to the tumor site, activation of vascular endothelial growth factor-2 (VEGFR-2) by VEGF-A is the most important (5, 6). It has also been found that VEGFR-driven expression of anti-apoptotic molecules (survivin and XIAP) in endothelial cells reduces the effectiveness of chemotherapeutics (7). Therefore, sequestering VEGF may be an effective antitumoral strategy because it can deprive these tumors of blood supply as well as of an essential growth/survival factor (6).
Alternative splicing of the VEGFR1/Flt-1 (Fms-like tyrosine kinase 1) receptor results in a truncated form of the receptor, which does not include the transmembrane nor intracellular domain and is unable to initiate intracellular signaling (8, 9). The resulting truncated protein is believed to be a decoy used by the organism to deplete excess VEGF under certain situations (10, 11) and is commonly referred to as the soluble form of the receptor or sVEGFR1/sFlt-1. It has previously been shown that granulocyte macrophage colony-stimulating factor and hypoxia potently stimulate sFlt-1 production from monocytes/macrophages. Additionally, sFlt-1 expressed from these cells effectively blocks VEGF activity in vitro and in vivo (12, 13).
Here, we show that monocyte FcγR clustering leads to sFlt-1 production and consequent VEGF blockade. Such monocyte-derived sFlt-1 can inhibit VEGF-dependent tube formation in an angiogenic assay, demonstrating its ability to block VEGF signaling in vitro. We also found that inhibition of sFlt-1 with a neutralizing antibody abrogated the antibody-mediated reduction of tumor burden in a mouse model. Finally, we identified NF-κB-mediated reduction of microRNA-181a (miR-181a) as a critical step in permitting FcγR-mediated up-regulation of sFlt-1 transcription and translation. To our knowledge this work represents the first description of angiogenic regulation mediated by monocytes as a consequence of interaction with antibodies.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
To prepare IgG-coated plates human or mouse immunoglobulin (Jackson ImmunoResearch) was added to wells (10 μg/ml) and incubated (sterile) for 24 h at 4 °C to allow IgG to attach to well surface. Primers to detect Flt-1 were: human membrane mFlt1 (mFlt-1) primer F5′-GTT CAA AGG GAA CCT CGG ACA A, human mFlt1 primer R5′-GCT CAC ACT GCT CAT CCA A, human soluble Flt-1 (sFlt-1) primer F5′-ATG GCC ATC ACT AAG GAG CA, and human sFlt-1 primer R5′-TTT GTT GCA GTG CTC ACC TC. Antibodies for flow cytometry were obtained from BD Biosciences Pharmingen: CD14-PECy7 (catalogue #557742), mPE-Cy7-IgG2a (catalogue #557907), CD56-PE (catalogue #555516), and m-PE-IgG1 (catalogue #340761). Protein kinase inhibitor BAY 11-7085 (5 μm) IKK (IκB kinase) inhibitor was a gift from Dr. Denis Guttridge (The Ohio State University). LY294002 (20 μm) PI3K inhibitor, U0126 (2.5 μm) MEK 1/2 inhibitor, SB203580 (5 μm) SAPK/p38 inhibitor, and SP600125 (5 μm) JNK inhibitor were obtained from Calbiochem. DMSO vehicle control was obtained from Sigma.
Microarrays
RNA was processed and hybridized to Affymetrix hgu133plus2 chips at The Ohio State University Medical Center Genetics Core Facility. Pre-processing and analysis were performed using R and Bioconductor (14, 15), testing for differentially expressed genes using the “limma” package (16).
qRT-PCR
RNA was extracted from peripheral blood monocytes (PBMs) using TRIzol. Reverse-transcribed cDNA was then run on the Applied Biosystems Step-One Plus System. Relative expression was calculated as 2−ΔCt, with Ct calculated by subtracting the average cycle threshold of two housekeeping controls (TRAF3 and GAPDH) from the experimental sample (17, 18).
Antibody-coated Tumor Targets
Raji and MDA-MB-468 cells were obtained from the American Type Culture Collection (Manassas, VA). SKBR3 cells were a kind gift from Dr. William Carson (The Ohio State University). All cells were cultured in RPMI 1640 supplemented with 10% FBS and 50 units/ml penicillin-streptomycin and l-glutamate. Rituximab (10 μg/ml) was added to Raji cells, cetuximab (10 μg/ml) was added to MDA-MB-468 cells, and trastuzumab (10 μg/ml) was added to SKBR3 cells. Cells were incubated on ice for 1 h. After 3 washes in cold RPMI 1640, Raji cells were fixed using 1% paraformaldehyde at room temperature for 20 min. After 3 washes in PBS, antibody-coated cells were resuspended in RPMI1640 supplemented with 10% FBS and penicillin-streptomycin and l-glutamate. Target cells were added to the PBMs at an effector cell-to-target cell ratio of 1:1 and incubated at 37 °C for 24 h. Cell-free supernatants were collected and kept sterile for subsequent experiments and analyses.
ELISAs
Cell supernatants were collected, centrifuged at full speed to clear cellular debris, then assayed for cytokine through sandwich ELISA (R&D Systems) according to the manufacturer's protocol. Human IFNγ (hIFNγ) DuoSet (catalogue #DY285), hTNFα DuoSet (catalogue #DY210), hVEGF-A DuoSet (catalogue #DY293B), hVEGFR1/Flt1 Quantikine (catalogue #SVR100B), mVEGF-A DuoSet (catalogue #DY493), and mVEGFR1 Quantikine (catalogue # MVR100) were used.
Peripheral Blood Monocyte/Natural Killer (NK) Cell Isolation
Cells were isolated from American Red Cross Leukopaks through Ficoll centrifugation (Mediatech) followed by CD14-positive selection or CD56-positive selection as relevant. Cells or peripheral blood mononuclear cells (PBMCs) were resuspended in RPMI 1640 containing 10% heat inactivated FBS (Hyclone), penicillin-streptomycin, and l-glutamate (Invitrogen).
HUVEC Tube Assay
Isolated monocytes were left unstimulated or were incubated with antibody-coated tumor cells and incubated at 37 °C for 24 h. Cell-free supernatants were harvested and stored at −80 °C until use for this assay. On 96-well plates, growth factor depleted Matrigel (60 μl/well) and was allowed to solidify at 37 °C. Cell-free supernatants were rotated in Matrigel-coated wells for 1 h at 4 °C. HUVECs were serum-starved in Vascular Cell Basal Medium (American Type Culture Collection) for 2 h. All control and experimental cells were resuspended at 5 × 106 cells/ml. 20,000 HUVEC cells were added to wells containing Matrigel and cell-free supernatants. Plates were incubated at 37 °C for 8 h. Digital photography was used to document the results in the well using an Olympus IX50 inverted fluorescence microscope with a 10× objective. Nine fields of view were photographed per well, and each treatment group was done in duplicate wells for each of three donors. Examination of these photographs was used to determine the number of branch points as a quantitative measurement of the response of the HUVEC cells to PBM-derived sFlt-1. Anti-angiogenic activity was assessed by the inhibition of branch points from capillary-like tube structures formed between the endothelial cells. The number of branch points from HUVEC cells incubated with cell-free supernatant from unstimulated PBMs was set as the base line (12).
Western Blotting
Cells were lysed in TN1 buffer (50 nmol/liter Tris (pH 8.0), 10 nmol/liter EDTA, 10 nmol/liter Na4P2O7, 10 nmol/liter NaF, 1% Triton X-100, 125 mmol/liter NaCl, 10 mmol/liter Na3VO4, and 10 μg/ml each aprotinin and leupeptin). Postnuclear protein-matched lysates were boiled in Laemmli sample buffer and separated by SDS-PAGE, transferred to nitrocellulose membranes, probed with the antibody of interest, and then developed by enhanced chemiluminescence (GE Healthcare).
miR-181a Mimic and PBM Transfection
PBMs were transfected with 500 nm miR-181a mimic (Dharmacon RNAi Technologies, Lafayette CO, item #C-300552-05-0005) using the Amaxa Nucleofector apparatus (Amaxa Biosystems, Cologne, Germany). Briefly, 1 × 107 cells were resuspended in 100 μl of Cell Line Nucleofector Solution T (Amaxa Biosystems). Immediately after transfection the cells were transferred to 10 ml of prewarmed media (RPMI 1640 supplemented with 10% FBS) and incubated on IgG-coated plates for 8 h. Cell-free supernatants were collected and examined by ELISA. Cells were lysed in TRIzol reagent, and RNA was extracted and examined by qRT-PCR using Applied Biosystems Taqman miRNA assays for miR-181a expression (assay #000480) and RNU44 (assay #001094).
Transfection of RAW 264 Murine Macrophage Cell Line
RAW 264 cells were obtained from American Type Culture Collection and cultured in RPMI 1640 (Invitrogen) supplemented with 5% heat-inactivated fetal bovine serum (FBS), l-glutamine, and penicillin-streptomycin. These cells were transfected with the appropriate plasmid DNA using the Amaxa Nucleofector (Amaxa Biosystems, Gaithersburg, MD) as previously described (19). Briefly, 5 × 106 cells were resuspended in 100 μl of Nucleofector Solution, then 5 μg of empty vector (pcDNA3.1) or plasmid containing IκBαSR (20, 21) (a kind gift from Dr. Denis Guttridge, The Ohio State University, Columbus, OH) was nucleofected according to the manufacturer's instructions. Cells were then incubated for 12 h in a CO2 incubator at 37 °C, verified for transfection efficiency by Western blotting, and used in experiments.
Bone Marrow Macrophage (BMM) Isolation and Culture
BMM were cultured from femurs of wild-type mice by flushing the marrow from femurs and planting the cells on plastic dishes in RPMI 1640 supplemented with 5% FBS (heat inactivated), 20 ng/ml mouse recombinant macrophage colony-stimulating factor, and 10 μg/ml polymyxin B. Four days after extraction, media were replaced with fresh RPMI 1640, supplemented with 5% FBS, 20 ng/ml recombinant MCSF, and 10 μg/ml polymyxin B. Six days after extraction media were supplemented with mouse recombinant MCSF (20 ng/ml) and polymyxin B (10 μg/ml). After 6–7 days, non-adherent cells were washed away using PBS, and the remaining BMMs were used for experiments.
Murine Solid Tumor Model
CT26-Her2/neu colon carcinoma cells (22) were grown in 1640 RPMI supplemented with 10% FBS, penicillin/streptomycin, 200 μg/ml Geneticin and l-glutamate, washed to remove non-adherent cells, then resuspended using enzyme-free cell dissociation buffer (Invitrogen). Cells were centrifuged and resuspended at 10 × 106 cells/ml in RPMI 1640. This tumor model has been described before (22–24); therefore, a brief description follows. Five-week-old female Balb/cJ mice (The Jackson Laboratory) were injected subcutaneously with 1 × 106 cells of syngeneic CT26-Her2/neu cells. Mice were left for 7 days, and tumors were allowed to develop. Intraperitoneal injections with treatments were done three times per week, and tumor measurements were performed on each treatment day. Tumor volumes were calculated as (0.5 × (length measurement) × (height measurement)2), in which length was the longest diameter of the tumor. Treatments consisted of vehicle control (PBS), 1 mg/kg 4D5 (anti-Her2 antibody; National Cell Culture Center, Minneapolis, MN), 4 μg/injection anti-Flt1 (13), 4D5, and anti-Flt1. All in vivo experiments were done in strict accordance with guidelines established by the Institutional Animal Care and Use Committee.
Statistical Analyses
Student's t tests were used to test for statistical significance. Statistics for the murine solid tumor model were performed by the Center for Biostatistics at The Ohio State University. For this tumor, volume data were normalized by cubic-root transformation, and mixed-effect modeling (incorporating dependences across observations) was used to analyze the transformed data. Hypothesis testing focused on the rate of tumor growth, and the mixed model we chose purposely allowed most liberally for mouse variation in the growth rate (25). An interaction contrast was used to test the antagonist effect of anti-sFlt-1 on 4D5. In brief, comparing the effect on growth rate of combining 4D5 and anti-sFlt1 with that of 4D5 and anti-sFlt1 when each was given alone provides an estimate of antagonist interaction. Holm's method was used to adjust for multiple testing (26).
RESULTS
Monocytes Express sFlt-1 upon FcγR Clustering
To identify novel transcriptional targets of FcγR signaling, we performed a microarray experiment after stimulation of PBM FcγR for 24 h. Interestingly, VEGFR-1/Flt-1 was found to be transcriptionally up-regulated (Fig. 1A). Flt-1 is a receptor for VEGF-A, a potent and principal mediator of angiogenesis. Flt-1 expression is expected on monocytes and functions as a chemotactic receptor for VEGF-A yet has not been previously reported to be up-regulated as a result of FcγR activation. To verify the expression and to determine the kinetics of the observed transcriptional up-regulation of Flt-1 after FcγR clustering, we performed qRT-PCR. However, we found no detectable increases of Flt-1 transcript at any of the time points examined (data not shown). Because our microarray data did not distinguish between membrane-bound and soluble Flt-1 and because other stimuli such as granulocyte macrophage colony-stimulating factor have been shown to elicit the soluble form (12, 13), we performed qRT-PCR using primers specific to each mRNA splice variant. Results showed significant increases in mRNA for the soluble form of Flt-1 (sFlt-1) at 5 and 8 h after PBM stimulation (Fig. 1B).
FIGURE 1.
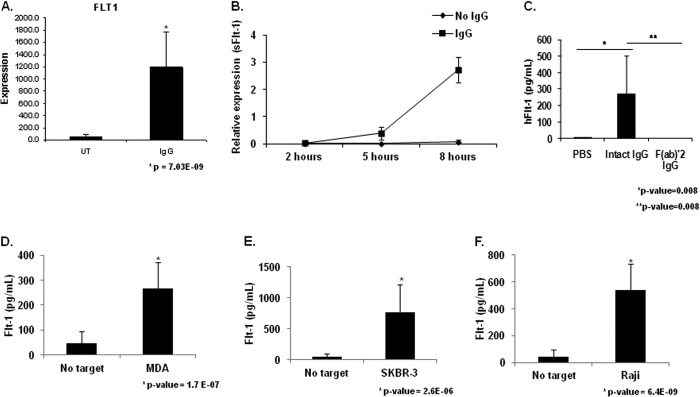
FcγR clustering by IgG leads to sFlt-1 expression by human peripheral blood monocytes. A, human PBMs were stimulated by incubation with heat-aggregated human IgG. Microarray analysis revealed increased Flt-1 expression in IgG-stimulated PBMs compared with unstimulated control (UT). B, human PBMs were incubated for the indicated time points on IgG coated wells. Quantitative real-time RT-PCR was performed with primers specific to the membrane-bound and soluble forms of Flt-1 to determine the kinetics of FcγR induction of Flt-1 expression. Significant differences in mRNA levels of sFlt-1 were observed at 5 h and 8 h relative to unstimulated controls, whereas no detectable levels of membrane bound Flt-1 were observed under any treatment condition or time point (representative donor; n ≥ 3). C, human PBMs were incubated on IgG coated plates for 24 h. Cell-free supernatants were obtained and examined for sFlt-1. No detectable levels of sFlt-1 were found in wells containing unstimulated PBMs. Whole molecule IgG is capable of inducing detectable levels of sFlt-1, whereas the F(ab)′2 portion of the IgG molecule is not (representative donor; n ≥ 3). D and F, to determine if tumor cell-bound IgG was capable of inducing sFlt-1 production after FcγR clustering, human PBMs were incubated with MDA-MB-468 (D), SKBR3 (E), or Raji (F) tumor cell lines opsonized with 10 μg/ml cetuximab, trastuzumab, or rituximab, respectively. Induction of sFlt-1 was observed for all three cell lines tested (representative donor; n ≥ 3).
Monocyte FcγR Clustering Induces sFlt-1 Protein
To verify that the sFlt-1 message was effectively translated into protein by stimulated monocytes, we incubated PBMs on IgG-coated plates for 24 h and examined cell-free supernatants by sFlt-1 ELISA. Unstimulated PBMs produced no detectable sFlt-1, but PBMs incubated with immobilized IgG produced significant levels of sFlt-1 (Fig. 1C). To verify that this was an FcγR-mediated event, we incubated PBMs on plates with immobilized F(ab)′2 IgG fragments and found no detectable induction of sFlt-1 (Fig. 1C).
We then sought to determine whether sFlt-1 expression occurred within the context of antitumor antibody therapy. For this we coated 3 tumor cell lines (MDA-MB-468, SKBR3, and Raji) with cetuximab, trastuzumab, and rituximab, respectively. These were then fixed and added to monocyte cultures for 24 h. Cell-free supernatants were examined by ELISA for sFlt-1. All three therapeutic antibodies were able to induce robust levels of sFlt-1 (Fig. 1, D–F).
Monocytes Are the Primary Source of FcγR-induced sFlt-1
Monocytes and NK cells are both FcγR-bearing cells known to actively target antibody-coated tumor cells. This raised the possibility that NK cells might also produce sFlt-1 upon FcγR clustering. To test this, we examined the ability of intact, NK cell-depleted, and monocyte-depleted PBMCs to produce sFlt-1 when incubated on IgG-coated plates. We found that both intact and NK cell-depleted PBMCs were capable of producing sFlt-1 after stimulation (Fig. 2A). However, monocyte-depleted PBMCs yielded no detectable levels of sFlt-1 (Fig. 2A), demonstrating that monocytes are the primary cell responsible for sFlt-1 production via FcγR. We verified depletion of each of the cells from PBMCs by use of specific markers for each leukocyte (monocytes (CD14) and NK cells (CD56)) on whole PBMCs, monocyte-depleted PBMCs, or in NK-cell-depleted PBMCs (data not shown).
FIGURE 2.

Peripheral blood monocytes are responsible for biologically active sFlt-1 production following FcγR stimulation. To determine which FcγR bearing leukocyte(s) is capable of mediating sFlt-1 synthesis through activation of FcγR, we examined whole PBMCs, monocyte depleted PBMCs, and NK-cell depleted PBMCs by incubation on IgG-coated plates. A, ELISA shows sFlt-1 production by intact PBMCs, NK cell-depleted PBMCs, or monocyte-depleted PBMCs after FcγR clustering (representative donor; n ≥ 3). B, isolated monocytes and NK cells were incubated on IgG-coated plates, and secreted sFlt-1 was measured by ELISA. C and D, human PBMs were incubated with antibody-coated tumor cells, and cell-free supernatants were collected. These supernatants were then used to determine the ability of sFlt-1 to inhibit VEGF-A-induced tube formation by HUVEC cells. C, supernatants from unstimulated PBMs or PBMs stimulated with antibody-coated tumor cells were tested in a tube-forming assay using HUVEC cells. These assays were also done with or without a neutralizing antibody to sFlt-1. Graphed are the percentages of branch points compared with those treated with supernatants from unstimulated PBMs (representative donor; n = 3). D, shown are representative photographs of HUVEC cell branch points quantified in C.
We then examined the depleted cells themselves for their ability to produce sFlt-1 independently from the remaining cells in the PBMC mix. We found that the NK cells separated from PBMCs were not capable of producing FcγR-induced sFlt-1 (Fig. 2B) but were capable of producing IFN-γ (data not shown). In contrast, monocytes separated from the PBMCs showed detectable levels of sFlt-1 after clustering of FcγR (Fig. 2B). Hence, because PBMCs depleted of monocytes show no IgG-mediated sFlt-1 production and the isolated monocytes produce measurable sFlt-1 upon incubation with immobilized IgG, we conclude that monocytes are the major cell type contributing to FcγR-mediated sFlt-1 production.
sFlt-1 Produced by Monocytes after FcγR Clustering Is Biologically Active
To determine whether the sFlt-1 produced by PBMs as a result of FcγR clustering could elicit biological responses, we treated HUVEC cells with conditioned media from FcγR-clustered or non-stimulated PBMs. The tube formation assay is an established assay for determining the ability of factors to regulate VEGF-A dependent angiogenesis in vitro (27). As shown in Fig. 2, C and D, supernatants from unstimulated PBMs (Fig. 2C) permitted significantly greater tube formation than supernatant from FcγR-clustered PBMs (Fig. 2D). Inhibition of sFlt-1 by use of a neutralizing antibody resulted in a return to the base-line number of branch points (Fig. 2, C and D), confirming that sFlt-1 produced by monocytes as a consequence of FcγR clustering was biologically active.
sFlt-1 Contributes to Antibody-mediated Tumor Inhibition in Vivo
To determine whether FcγR clustering in primary murine macrophages also elicited sFlt-1 production, we prepared murine BMM and incubated them on IgG-coated plates. ELISAs with cell supernatants showed a significant increase in sFlt-1 production by BMM incubated in IgG-coated wells, whereas there was no detectable sFlt-1 from BMM in control wells (Fig. 3A).
FIGURE 3.

FcγR-induced sFlt-1 can influence tumor growth. A, bone marrow macrophages were incubated on plates coated with mouse IgG for 24 h. Supernatants were collected and murine, soluble Flt-1(sFlt-1) was measured using ELISA. N.D., not determined. B, mice were injected in the right flanks with CT26-Her2/neu cells, then treated 3 times per week for 2 weeks with vehicle, 4D5 (anti-Her2/neu), anti-Flt-1, or 4D5 plus anti-Flt-1. Tumors growth was measured on each treatment day. Each circle represents an observation of a mouse under each treatment. Crosses represent the mean volume for each treatment.
It had been previously reported that granulocyte macrophage colony-stimulating factor-induced sFlt-1 could be neutralized in a mouse tumor model and that this resulted in a reversal of granulocyte macrophage colony-stimulating factor-mediated reduction of tumor growth (13). Hence, we wondered whether FcγR-induced sFlt-1 was similarly involved with antagonizing tumor growth within the context of antibody therapy. To test this we used the CT-26-Her2/neu model. In this model mice are injected with syngeneic adenocarcinoma cells (CT-26) that express human Her2/neu. These cells form tumors within ∼7 days. The antibody 4D5 targets Her2/neu, providing a murine model of antibody therapy (22). Here, we used this model to test the effect of neutralizing sFlt-1 on antibody-mediated tumor inhibition. The therapeutic antibody alone (4D5) reduced tumor growth by 69.2 mm3/day compared with PBS control (Fig. 3B). When 4D5 was combined with an antibody to neutralize sFlt-1, the combination decreased tumor growth rate by only 0.2 mm3/day (Fig. 3B), which is 105.3 mm3/day greater than the effect of each drug alone (4D5, −69.2 mm3/day; anti-sFlt, −36.3 mm3/day). An interaction contrast showed that this increase was significant (p value = 0.016). Notably, a control antibody combined with 4D5 did not enhance tumor reduction (data not shown). These results suggest that FcγR-induced sFlt-1 contributes to the antitumor effects of antibody therapy.
FcγR Clustering Leads to Up-regulation of sFlt-1 Protein via NF-κB Activation
Because FcγR-mediated production of sFlt-1 is a novel form of angiogenic regulation, we sought to determine the signaling pathways responsible. Clustering of FcγR results in the activation of PI3K and ERK signaling pathways, both of which are involved in phagocytosis of the immune complex and/or induction of inflammation (28). To determine the signaling pathway(s) responsible for sFlt-1 production via FcγR clustering, we incubated PBMs on IgG-coated plates with or without pharmacologic inhibitors and measured sFlt-1 production by ELISA. Results showed that inhibition of Akt (PI3K pathway) led to significant reduction of sFlt-1 (Fig. 4A). Additionally we found that inhibition of ERK signaling significantly decreased the levels of detectable sFlt-1 (Fig. 4B). We verified the inhibition of these pathways by determining the phosphorylation status of the targeted molecules and found that, as expected, Akt was not phosphorylated after LY-294002 treatment and Erk was not phosphorylated after U0126 treatment (Fig. 4, A and B, respectively, lower panels). In contrast, inhibition of p38 or JNK did not result in significant down-regulation of sFlt-1, suggesting that these pathways do not contribute to FcγR-mediated production of sFlt-1 (data not shown).
FIGURE 4.

FcγR clustering regulates of sFlt-1 synthesis through NF-κB activation. A–C, PBMs were pretreated with inhibitors of the PI3K (LY294002) (A), MAPK (U0126) (B), or NF-κB pathways (C), then FcγRs were clustered using immobilized IgG for 24 h. Supernatants were collected, and sFlt-1 was measured by ELISA (top panels) and Western blots with accompanying densitometry done with the protein lysates (middle and bottom panels, respectively) to verify inhibitor efficacy. Representative graphs and blots are shown (n = 4 separate donors). No-IgG controls were done for each donor and resulted in undetectable levels of sFlt-1 (data not shown). D, RAW 264.7 cells were nucleofected to express the IκBαSR. Expression of the transfected molecule was confirmed by Western blotting with IκBα antibody (lower panel). Transfected cells were incubated in IgG-coated plates and analyzed by ELISA for the production of sFlt-1 (upper panel).
Because NF-κB activation is a downstream target of FcγR clustering in monocytes, we examined the consequences of NF-κB inhibition on sFlt-1 production. Treatment with BAY 11-7085 inhibits IκBα phosphorylation and degradation, resulting in a block of NF-κB phosphorylation and nuclear translocation. In the presence of BAY 11-7085, FcγR clustering led to a significant decrease in sFlt-1 production (Fig. 4C). Inhibition of NF-κB phosphorylation was verified by Western blot (Fig. 4C, lower panels). To additionally confirm the involvement of NF-κB, we transfected RAW 264 murine macrophage cell line with a IκBα super-repressor (SR) and examined FcγR-induced sFlt-1 production. Consistent with the inhibitor data, the expression of the IκBαSR significantly suppressed sFlt-1 production (Fig. 4D). RAW 264 cells were used in these experiments as sufficient levels of the IκBαSR could not be achieved in primary human monocytes. These results indicate that NF-κB mediates the production of sFlt-1 downstream of FcγR.
De Novo Protein Synthesis Is Not Required for sFlt-1 Transcriptional Up-regulation
Next, we sought to determine whether the increase in sFlt-1 via FcγR clustering was a direct effect or required synthesis of additional proteins. To test this we treated PBMs with cycloheximide, a translation inhibitor. Treatment of PBMs with cycloheximide and concomitant stimulation with immobilized IgG led to increased expression of the sFlt-1 transcript (Fig. 5A). As expected, no sFlt-1 protein was detected (Fig. 5B). These results indicate that sFlt-1 transcription via FcγR does not require de novo protein synthesis and argues for a direct pathway from FcγR clustering to sFlt-1 mRNA production.
FIGURE 5.

Protein synthesis is not required for sFlt-1 transcription, but NF-κB is necessary for translation. A, PBMs were pretreated with cycloheximide, activated as above, then transcripts of sFlt-1 were measured using real-time RT-PCR (representative donor; n ≥ 3). B, PBMs were pretreated with cycloheximide and activated as above, then ELISAs were performed to measure sFlt-1 in the supernatants (representative donor; n ≥ 3). C, PBMs were pretreated with or without BAY 11-7085, then FcγR clustered as described above. ELISAs were done to measure sFlt-1 in supernatants (representative donor; n ≥ 3). D, PBMs were pretreated and activated as in A, then the transcript of sFlt-1 was measured using real-time RT-PCR. (representative donor; n ≥ 3).
sFlt-1 Protein Synthesis but Not Transcription Requires NF-κB Activation
We then decided to examine the effect of FcγR-mediated NF-κB activation on sFlt-1 transcription and translation. After pretreating PBMs with or without BAY 11-7085, they were incubated with immobilized IgG for 24 h. After this, lysates and supernatants were collected, and sFlt-1 was measured by ELISA. Results showed that inhibition of NF-κB led to decreased sFlt-1 protein in PBM lysates and supernatants (Fig. 5C). RNA was also extracted from these cells, and qRT-PCR was performed to measure sFlt-1 transcript. Strikingly, results showed that up-regulation of sFlt-1 mRNA was not affected (Fig. 5D). These results indicate that NF-κB is indeed required for sFlt-1 production but only at the post-transcriptional level.
NF-κB Promotes sFlt-1 Translation by Targeting miR181a
Our data suggest that FcγR clustering leads directly to sFlt-1 transcription and that FcγR-mediated activation of NF-κB is required for sFlt-1 translation. Given that the cytosolic translational machinery itself was presumably intact, it seemed likely that NF-κB served to neutralize a negative regulator of sFlt-1 translation. Therefore, we chose to examine the effect of FcγR clustering (and subsequent NF-κB activation) on microRNAs (miRNAs), which are powerful regulators of translation. miRNA-mediated regulation of gene expression results when a miRNA interacts with a specific recognition element located within the target mRNA and subsequently suppresses its translation or initiates its degradation (29). We analyzed the sFlt-1 transcript (NM_001159920) using TargetScan (Release 6.2, June 2012), which predicted several putative miRNA target sites, including one for miR-181abcd, in the 3′-untranslated region. We focused on miR-181a, as miR-181 has been shown to be basally expressed in human monocytes from healthy donors (30) (which would putatively keep the basal expression of sFlt-1 low) and as miR-181a has been studied extensively within the context of tumorigenesis (31–35).
To test whether miR-181a targets sFlt-1, we transfected PBMs with a miR-181a mimic. The transfection resulted in a greater than 10-fold increase in miR-181a in PBMs (Fig. 6A) and a subsequent reduced FcγR-mediated sFlt-1 protein production by ∼30% (Fig. 6B).
FIGURE 6.
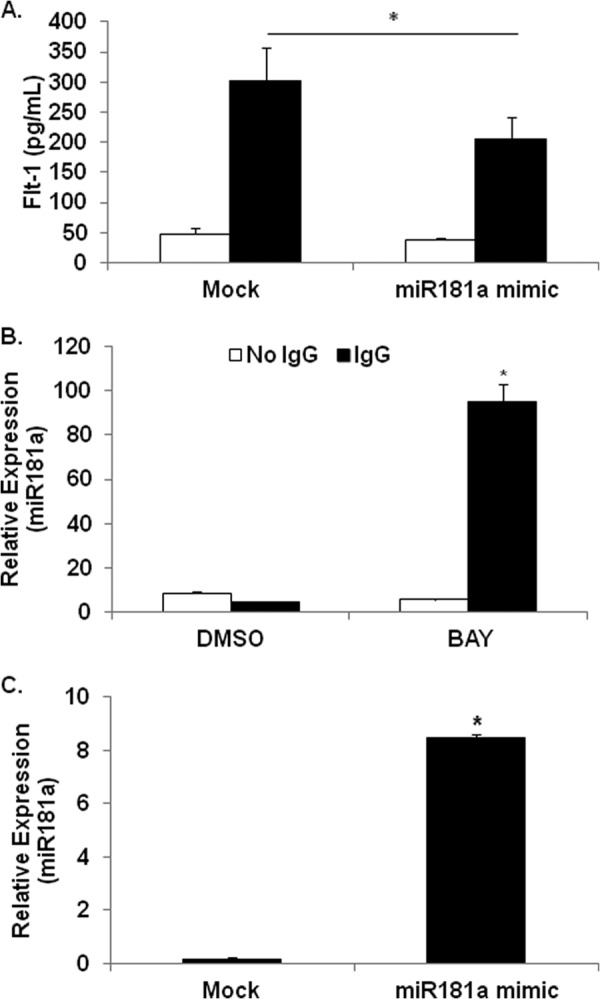
FcγR clustering down-regulates miR181a while up-regulating sFlt-1 transcription and protein synthesis. A, PBMs were transfected with miR181a mimic, then FcγR clustered as above. ELISAs were done to measure the levels of sFlt-1 in the supernatants (representative donor; n ≥ 3). B, PBMs were pretreated with or without BAY 11-7085 and incubated on IgG-coated plates for 24 h. RNA was collected and assayed for miR181a expression by real-time RT-PCR (representative donor; n ≥ 3). C, PBMs were transfected with a miR181a mimic, then real-time RT-PCR was performed after 8 h to verify increased miR181a levels (representative donor; n ≥ 3).
Because FcγR clustering induces sFlt-1 and because miR-181a was shown to negatively regulate sFlt-1, we asked whether miR-181a was basally expressed in monocytes but down-regulated upon FcγR activation. In addition, because NF-κB activity is required for sFlt-1 production, we sought to determine whether it was also involved in regulating miR-181a. To test this hypothesis, PBMs were pretreated with either vehicle or Bay-11-7085 and then subsequently incubated with or without immobilized IgG to cluster the FcγR. RNA was extracted after 24 h, and qRT-PCR was performed to measure miR-181a levels. Results showed that there was a detectable basal level of miR-181a but that FcγR clustering significantly reduced this (Fig. 6C). Strikingly, in the presence of the NF-κB inhibitor, FcγR clustering led to roughly a 10-fold increase in miR-181a (Fig. 6C). These results indicate that FcγR activation promotes the induction of miR-181a, but the corresponding FcγR-mediated NF-κB activation serves to repress this.
DISCUSSION
sFlt-1 is an important regulator of angiogenesis (36), and it has been previously reported that monocyte production of sFlt-1 can reduce tumor burden by inhibiting angiogenesis at tumor sites (13). However, results from this study suggest that sFlt-1 is produced in response to antibody therapy and that it represents a significant component that contributes to the observed antitumor effects. To our knowledge this is the first report of a link between FcγR and expression of this angiogenic regulator.
Our finding that Flt-1 was up-regulated as a consequence of FcγR signaling led us to examine the transcriptional and protein expression of Flt-1 by monocytes. Previous reports have shown that monocytes produce the soluble form of Flt-1, sFlt-1, as a consequence of cytokine stimulus and by hypoxia (12, 37). Studies are ongoing to delineate the similarities and differences between cytokine- and FcγR-mediated sFlt-1 production.
Monocytes are important effectors of antibody therapy and are commonly found at tumor sites mediating tumor clearance. Previous reports have shown that through interactions with FcγR, these cells can be recruited and initiate anti-tumor responses. Known effector mechanisms mediated by monocytes that contribute to tumor elimination include antibody-dependent cell cytotoxicity (38), phagocytosis (39–41), and cytokine production (42, 43). Our current findings are the first to demonstrate the induction of sFlt-1 through FcγR signaling.
Monocyte participation in regulation of angiogenesis has been previously reported (13). Through its ability to block VEGF-A, sFlt-1 is able to suppress signaling downstream of VEGFR-1, therefore, restricting monocyte recruitment to the tumor by blocking the chemotactic function mediated by this receptor on monocytes. Additionally, sFlt-1 blockade of VEGFR-2 leads to inhibition of endothelial cell proliferation and migration toward the tumor site, thereby restricting tumor vascularization.
Activation of the PI3K and MAPK are common after FcγR clustering. Consequences of signaling downstream of FcγR are the ingestion and destruction of immune complexes or direct cytotoxicity. We found that FcγR clustering and subsequent activation of PI3K and MAPK are important mediators of sFlt-1 production. Additionally, these pathways appear to mediate activation of the NF-κB pathway. Our results indicate that NF-κB activation is not required for the transcription of sFlt-1 but instead down-regulates a negative regulator of sFlt-1 transcription, miR181a. There is evidence for regulation of angiogenesis by miRNAs (44). HIF1α, an important regulator of VEGF transcription and consequently angiogenesis, is in turn subject to regulation by von Hippel-Lindau factor. von Hippel-Lindau was shown to be subject to regulation by miR92-1 (45). It has been previously suggested that miRNAs may play a role in Flt-1 regulation in monocytes (46). Targeting these miRNA regulators of sFlt-1 may lead to innovative strategies and/or agents that may enhance monocyte sFlt-1 production.
It has been proposed that tumor angiogenesis results in a “self-reinforcing vicious cycle” in which highly metabolic cancer cells, spurred by oncogene activation and exposure to a highly abnormal microenvironment, lead tumor cells to synthesize proangiogenic factors. These excessive proangiogenic signals result in abnormal vasculature incapable of proper provision of nutrients and metabolic waste removal leading to hypoxia and increased acidity, which are both strong stimuli for angiogenesis (47–49). VEGF blockade has been shown to reduce interstitial pressure and edema (50–52). Because monocytes/macrophages are known to infiltrate tumors, our results suggest that antibody-mediated FcγR activation may aid in blocking this angiogenic cycle. Importantly, antibody-mediated sFlt-1 production would be more localized at the tumor sites where the antibody binds, thereby potentially sidestepping deleterious effects of global VEGF inhibition. A greater understanding and perhaps enhancement of this novel effector function of monocytes may lead to new avenues of improving the outcome of antibody therapy.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA162411, P01 CA095426, and K12CA133250 (to J. P. B., X. M., N. M., J. C. B., and S. T.) and Postdoctoral Fellowship T32 60013191 (to S. E. J.). This work was also supported by American Cancer Society Grant IRG-67-003-47 (to J. P. B.) and The Leukemia and Lymphoma Society Grant P50-CA140158, The Harry Mangurian Foundation, and The D. Warren Brown Foundation (to J. P. B. and N. M.).
- FcγR
- Fcγ receptor
- sFlt-1
- soluble Flt-1
- VEGFR1
- VEGF receptor 1
- NF-κB
- nuclear factor κ-light-chain-enhancer of activated B cells
- miR
- microRNA
- PBM
- peripheral blood monocyte
- PBMC
- peripheral blood mononuclear cell
- BMM
- bone marrow macrophage
- NK
- natural killer
- SR
- super-repressor
- qRT
- quantitative real-time.
REFERENCES
- 1. Anderson C. L., Shen L., Eicher D. M., Wewers M. D., Gill J. K. (1990) Phagocytosis mediated by three distinct Fcγ receptor classes on human leukocytes. J. Exp. Med. 171, 1333–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Casado J. A., Merino J., Cid J., Subira M. L., Sanchez-Ibarrola A. (1994) The type of interaction with FcγR in human monocytes determines the efficiency of the generation of oxidative burst. Immunology 83, 148–154 [PMC free article] [PubMed] [Google Scholar]
- 3. Kindt G. C., Moore S. A., She Z. W., Wewers M. D. (1993) Endotoxin priming of monocytes augments Fcγ receptor cross-linking-induced TNF-α and IL-1β release. Am. J. Physiol. 265, L178–L185 [DOI] [PubMed] [Google Scholar]
- 4. Folkman J. (1971) Tumor angiogenesis. Therapeutic implications. N. Engl. J. Med. 285, 1182–1186 [DOI] [PubMed] [Google Scholar]
- 5. Dvorak H. F. (2002) Vascular permeability factor/vascular endothelial growth factor. A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 20, 4368–4380 [DOI] [PubMed] [Google Scholar]
- 6. Hicklin D. J., Ellis L. M. (2005) Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 23, 1011–1027 [DOI] [PubMed] [Google Scholar]
- 7. Tran J., Master Z., Yu J. L., Rak J., Dumont D. J., Kerbel R. S. (2002) A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc. Natl. Acad. Sci. U.S.A. 99, 4349–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kendall R. L., Thomas K. A. (1993) Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. U.S.A. 90, 10705–10709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kondo K., Hiratsuka S., Subbalakshmi E., Matsushime H., Shibuya M. (1998) Genomic organization of the flt-1 gene encoding for vascular endothelial growth factor (VEGF) receptor-1 suggests an intimate evolutionary relationship between the 7-Ig and the 5-Ig tyrosine kinase receptors. Gene 208, 297–305 [DOI] [PubMed] [Google Scholar]
- 10. Banks R. E., Forbes M. A., Searles J., Pappin D., Canas B., Rahman D., Kaufmann S., Walters C. E., Jackson A., Eves P., Linton G., Keen J., Walker J. J., Selby P. J. (1998) Evidence for the existence of a novel pregnancy-associated soluble variant of the vascular endothelial growth factor receptor, Flt-1. Mol. Hum. Reprod. 4, 377–386 [DOI] [PubMed] [Google Scholar]
- 11. Krüssel J. S., Casañ E. M., Raga F., Hirchenhain J., Wen Y., Huang H. Y., Bielfeld P., Polan M. L. (1999) Expression of mRNA for vascular endothelial growth factor transmembraneous receptors Flt1 and KDR, and the soluble receptor sFlt in cycling human endometrium. Mol. Hum. Reprod. 5, 452–458 [DOI] [PubMed] [Google Scholar]
- 12. Eubank T. D., Roberts R., Galloway M., Wang Y., Cohn D. E., Marsh C. B. (2004) GM-CSF induces expression of soluble VEGF receptor-1 from human monocytes and inhibits angiogenesis in mice. Immunity 21, 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eubank T. D., Roberts R. D., Khan M., Curry J. M., Nuovo G. J., Kuppusamy P., Marsh C. B. (2009) Granulocyte macrophage colony-stimulating factor inhibits breast cancer growth and metastasis by invoking an anti-angiogenic program in tumor-educated macrophages. Cancer Res. 69, 2133–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dudoit S., Gentleman R. C., Quackenbush J. (2003) Open source software for the analysis of microarray data. BioTechniques Suppl 45–51 [PubMed] [Google Scholar]
- 15. Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A. J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J. Y., Zhang J. (2004) Bioconductor. Open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smyth G. K. (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 [DOI] [PubMed] [Google Scholar]
- 17. Gavrilin M. A., Bouakl I. J., Knatz N. L., Duncan M. D., Hall M. W., Gunn J. S., Wewers M. D. (2006) Internalization and phagosome escape required for Francisella to induce human monocyte IL-1β processing and release. Proc. Natl. Acad. Sci. U.S.A. 103, 141–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Butchar J. P., Cremer T. J., Clay C. D., Gavrilin M. A., Wewers M. D., Marsh C. B., Schlesinger L. S., Tridandapani S. (2008) Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS ONE 3, e2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rajaram M. V., Butchar J. P., Parsa K. V., Cremer T. J., Amer A., Schlesinger L. S., Tridandapani S. (2009) Akt and SHIP modulate Francisella escape from the phagosome and induction of the Fas-mediated death pathway. PLoS ONE 4, e7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brockman J. A., Scherer D. C., McKinsey T. A., Hall S. M., Qi X., Lee W. Y., Ballard D. W. (1995) Coupling of a signal response domain in IκBα to multiple pathways for NF-κB activation. Mol. Cell. Biol. 15, 2809–2818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cogswell P. C., Kashatus D. F., Keifer J. A., Guttridge D. C., Reuther J. Y., Bristow C., Roy S., Nicholson D. W., Baldwin A. S., Jr. (2003) NF-κ B and I κ B α are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-κB. J. Biol. Chem. 278, 2963–2968 [DOI] [PubMed] [Google Scholar]
- 22. Penichet M. L., Challita P. M., Shin S. U., Sampogna S. L., Rosenblatt J. D., Morrison S. L. (1999) In vivo properties of three human HER2/neu-expressing murine cell lines in immunocompetent mice. Lab. Anim. Sci. 49, 179–188 [PubMed] [Google Scholar]
- 23. Roda J. M., Parihar R., Lehman A., Mani A., Tridandapani S., Carson W. E., 3rd (2006) Interleukin-21 enhances NK cell activation in response to antibody-coated targets. J. Immunol. 177, 120–129 [DOI] [PubMed] [Google Scholar]
- 24. Butchar J. P., Mehta P., Justiniano S. E., Guenterberg K. D., Kondadasula S. V., Mo X., Chemudupati M., Kanneganti T. D., Amer A., Muthusamy N., Jarjoura D., Marsh C. B., Carson W. E., 3rd, Byrd J. C., Tridandapani S. (2010) Reciprocal regulation of activating and inhibitory Fcγ receptors by TLR7/8 activation. Implications for tumor immunotherapy. Clin. Cancer Res. 16, 2065–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Verbeke G., Molenberghs G. (2000) Linear Mixed Models for Longitudinal Data, pp. 23–26, Springer Verlag, New York [Google Scholar]
- 26. Holm S. (1979) A simple sequentially rejective multiple test procedure. Scand. J. Statistics 6, 65–70 [Google Scholar]
- 27. Madri J. A., Pratt B. M., Tucker A. M. (1988) Phenotypic modulation of endothelial cells by transforming growth factor-β depends upon the composition and organization of the extracellular matrix. J. Cell Biol. 106, 1375–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fanger M. W., Erbe D. V. (1992) Fcγ receptors in cancer and infectious disease. Immunol. Res. 11, 203–216 [DOI] [PubMed] [Google Scholar]
- 29. Bartel D. P. (2009) MicroRNAs. Target recognition and regulatory functions. Cell 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramkissoon S. H., Mainwaring L. A., Ogasawara Y., Keyvanfar K., McCoy J. P., Jr., Sloand E. M., Kajigaya S., Young N. S. (2006) Hematopoietic-specific microRNA expression in human cells. Leuk. Res. 30, 643–647 [DOI] [PubMed] [Google Scholar]
- 31. Debernardi S., Skoulakis S., Molloy G., Chaplin T., Dixon-McIver A., Young B. D. (2007) MicroRNA miR-181a correlates with morphological sub-class of acute myeloid leukaemia and the expression of its target genes in global genome-wide analysis. Leukemia 21, 912–916 [DOI] [PubMed] [Google Scholar]
- 32. Cuesta R., Martínez-Sánchez A., Gebauer F. (2009) miR-181a regulates cap-dependent translation of p27(kip1) mRNA in myeloid cells. Mol. Cell. Biol. 29, 2841–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maillot G., Lacroix-Triki M., Pierredon S., Gratadou L., Schmidt S., Bénès V., Roché H., Dalenc F., Auboeuf D., Millevoi S., Vagner S. (2009) Widespread estrogen-dependent repression of microRNAs involved in breast tumor cell growth. Cancer Res. 69, 8332–8340 [DOI] [PubMed] [Google Scholar]
- 34. Schwind S., Maharry K., Radmacher M. D., Mrózek K., Holland K. B., Margeson D., Whitman S. P., Hickey C., Becker H., Metzeler K. H., Paschka P., Baldus C. D., Liu S., Garzon R., Powell B. L., Kolitz J. E., Carroll A. J., Caligiuri M. A., Larson R. A., Marcucci G., Bloomfield C. D. (2010) Prognostic significance of expression of a single microRNA, miR-181a, in cytogenetically normal acute myeloid leukemia. A cancer and leukemia group B study. J. Clin. Oncol. 28, 5257–5264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seoudi A. M., Lashine Y. A., Abdelaziz A. I. (2012) MicroRNA-181a. A tale of discrepancies. Expert. Rev. Mol. Med. 14, e5. [DOI] [PubMed] [Google Scholar]
- 36. Olsson A. K., Dimberg A., Kreuger J., Claesson-Welsh L. (2006) VEGF receptor signaling. In control of vascular function. Nat. Rev. Mol. Cell. Biol. 7, 359–371 [DOI] [PubMed] [Google Scholar]
- 37. Roda J. M., Sumner L. A., Evans R., Phillips G. S., Marsh C. B., Eubank T. D. (2011) Hypoxia-inducible factor-2α regulates GM-CSF-derived soluble vascular endothelial growth factor receptor 1 production from macrophages and inhibits tumor growth and angiogenesis. J. Immunol. 187, 1970–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shaw G. M., Levy P. C., LoBuglio A. F. (1978) Human monocyte antibody-dependent cell-mediated cytotoxicity to tumor cells. J. Clin. Invest. 62, 1172–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Watanabe M., Wallace P. K., Keler T., Deo Y. M., Akewanlop C., Hayes D. F. (1999) Antibody-dependent cellular phagocytosis (ADCP) and antibody-dependent cellular cytotoxicity (ADCC) of breast cancer cells mediated by bispecific antibody, MDX-210. Breast Cancer Res. Treat. 53, 199–207 [DOI] [PubMed] [Google Scholar]
- 40. Akewanlop C., Watanabe M., Singh B., Walker M., Kufe D. W., Hayes D. F. (2001) Phagocytosis of breast cancer cells mediated by anti-MUC-1 monoclonal antibody, DF3, and its bispecific antibody. Cancer Res. 61, 4061–4065 [PubMed] [Google Scholar]
- 41. Oflazoglu E., Stone I. J., Brown L., Gordon K. A., van Rooijen N., Jonas M., Law C. L., Grewal I. S., Gerber H. P. (2009) Macrophages and Fc-receptor interactions contribute to the anti-tumour activities of the anti-CD40 antibody SGN-40. Br. J. Cancer 100, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ansell S. M., Witzig T. E., Kurtin P. J., Sloan J. A., Jelinek D. F., Howell K. G., Markovic S. N., Habermann T. M., Klee G. G., Atherton P. J., Erlichman C. (2002) Phase 1 study of interleukin-12 in combination with rituximab in patients with B-cell non-Hodgkin lymphoma. Blood 99, 67–74 [DOI] [PubMed] [Google Scholar]
- 43. Parihar R., Nadella P., Lewis A., Jensen R., De Hoff C., Dierksheide J. E., VanBuskirk A. M., Magro C. M., Young D. C., Shapiro C. L., Carson W. E., 3rd (2004) A phase I study of interleukin 12 with trastuzumab in patients with human epidermal growth factor receptor-2-overexpressing malignancies. Analysis of sustained interferon γ production in a subset of patients. Clin. Cancer Res. 10, 5027–5037 [DOI] [PubMed] [Google Scholar]
- 44. Wang S., Olson E. N. (2009) AngiomiRs. Key regulators of angiogenesis. Curr. Opin. Genet. Dev. 19, 205–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghosh A. K., Shanafelt T. D., Cimmino A., Taccioli C., Volinia S., Liu C. G., Calin G. A., Croce C. M., Chan D. A., Giaccia A. J., Secreto C., Wellik L. E., Lee Y. K., Mukhopadhyay D., Kay N. E. (2009) Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in CLL B cells. Blood 113, 5568–5574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eubank T. D., Roda J. M., Liu H., O'Neil T., Marsh C. B. (2011) Opposing roles for HIF-1α and HIF-2α in the regulation of angiogenesis by mononuclear phagocytes. Blood 117, 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Carmeliet P., Jain R. K. (2011) Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 10, 417–427 [DOI] [PubMed] [Google Scholar]
- 48. Fukumura D., Duda D. G., Munn L. L., Jain R. K. (2010) Tumor microvasculature and microenvironment. Novel insights through intravital imaging in pre-clinical models. Microcirculation 17, 206–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hunt T. K., Aslam R., Hussain Z., Beckert S. (2008) Lactate, with oxygen, incites angiogenesis. Adv. Exp. Med. Biol. 614, 73–80 [DOI] [PubMed] [Google Scholar]
- 50. Tong R. T., Boucher Y., Kozin S. V., Winkler F., Hicklin D. J., Jain R. K. (2004) Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 64, 3731–3736 [DOI] [PubMed] [Google Scholar]
- 51. Kamoun W. S., Ley C. D., Farrar C. T., Duyverman A. M., Lahdenranta J., Lacorre D. A., Batchelor T. T., di Tomaso E., Duda D. G., Munn L. L., Fukumura D., Sorensen A. G., Jain R. K. (2009) Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J. Clin. Oncol. 27, 2542–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tailor T. D., Hanna G., Yarmolenko P. S., Dreher M. R., Betof A. S., Nixon A. B., Spasojevic I., Dewhirst M. W. (2010) Effect of pazopanib on tumor microenvironment and liposome delivery. Mol. Cancer Ther. 9, 1798–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]