

Background: Vps4 functions in eukaryotic cells in membrane fission reactions.
Results: Mutational analysis suggests that the long linker region of Vps4 is not essential but regulates this ATPase.
Conclusion: The binding to the substrate ESCRT-III drives formation of the active Vps4 complex.
Significance: Understanding the mechanism of Vps4 function gives insights into protein trafficking, cytokinesis, and virus formation.
Keywords: ATPases, Endosomes, Enzyme Mechanisms, Membrane, Yeast
Abstract
The AAA-type ATPase Vps4 functions with components of the ESCRT (endosomal sorting complex required for transport) machinery in membrane fission events that are essential for endosomal maturation, cytokinesis, and the formation of retroviruses. A key step in these events is the assembly of monomeric Vps4 into the active ATPase complex, which is aided in part by binding of Vps4 via its N-terminal MIT (microtubule interacting and trafficking) domain to its substrate ESCRT-III. We found that the 40-amino acid linker region between the MIT and the ATPase domain of Vps4 is not required for proper function but plays a role in regulating Vps4 assembly and ATPase activity. Deletion of the linker is expected to bring the MIT domains into close proximity to the central pore of the Vps4 complex. We propose that this localization of the MIT domain in linker-deleted Vps4 mimics a repositioning of the MIT domain normally caused by binding of Vps4 to ESCRT-III. This structure would allow the Vps4 complex to engage ESCRT-III subunits with both the pore and the MIT domain simultaneously, which might be essential for the ATP-driven disassembly of ESCRT-III.
Introduction
In eukaryotes, the Vps4 ATPase functions as part of the ESCRT2 (endosomal sorting complex required for transport) machinery in the formation of multivesicular bodies. Furthermore, in mammalian cells, Vps4 is involved in the execution of cytokinesis and the liberation of newly formed retroviral particles (for reviews, see Refs. 1 and 2). In all these cases, Vps4 functions together with ESCRT-III, a heterologous, membrane-associated polymer required for membrane remodeling. ATP hydrolysis by Vps4 results in the disassembly of the bound ESCRT-III complex, which has been proposed to drive the membrane fission reactions that represent the final steps in MVB vesicle formation, cytokinesis, and viral budding.
Vps4 belongs to the AAA protein family, ATPases that use hydrolysis energy to conduct mechanical work, including inducing conformational changes in proteins and DNA and the movement of cargo along microtubules (reviewed in Refs. 3 and 4). AAA members form oligomeric, ring-shaped complexes, often containing 6 or 12 subunits. These rings form a central pore that engages the substrate and performs the ATP-driven work. In the case of the type-I AAA protein Vps4 (which contains one AAA domain in contrast to type-II proteins, which contain two), structural studies have been complicated by the fact that in the cytoplasm, this protein is present as a monomer or dimer, which when recruited to ESCRT-III assembles into the active oligomer (5). Therefore, the quaternary structure of Vps4 is dynamic and in part determined by the presence of the substrate ESCRT-III and other co-factors, making the analysis of the Vps4 oligomer difficult. Thus, it is not surprising that the structure of the active Vps4 oligomer remains a subject of debate. X-ray crystallography and NMR analysis were able to resolve the Vps4 AAA domain and the N-terminal ESCRT-III-binding region, called the MIT domain (6–8). The problem in resolving the complete Vps4 structure is in part caused by the highly flexible linker between the AAA and MIT domain. Three independent single-particle EM studies attempted to solve the oligomeric structure of Vps4 (9–11). These studies came to different conclusions with regard to the number of subunits present and the arrangement of these subunits within the complex. However, two of these studies concluded that the Vps4 oligomer is composed of two hexameric rings stacked tail-to-tail on top of each other (9, 10). Furthermore, both studies observed that the two rings differed in their conformation, suggesting different functionalities for the two rings.
The oligomerization of Vps4 is regulated and aided by the four proteins Vta1, Ist1, Did2, and Vps60 (see review in Ref. 12). Based on structural analyses, the latter three of these proteins belong to the family of ESCRT-III-related proteins. Ist1 binds to monomeric Vps4 in the cytoplasm and together with Did2 seems to regulate the recruitment of Vps4 to endosome-associated ESCRT-III (13–15). Vta1 and Vps60 are involved in the stabilization and activation of the Vps4 complex (16–20). An additional layer of regulation has recently been proposed by studies of human Vps4. The deletion of the N-terminal MIT domain and adjacent linker resulted in increased ATPase activity of Vps4 in vitro, suggesting that these regions of the proteins are part of an auto-inhibitory mechanism that ensures Vps4 activation only when associated with the substrate ESCRT-III (21). In our study, we demonstrate that deletion of just the Vps4 linker region results in an active enzyme that is functional in vivo. Interestingly, the deletion of the linker restricts the position of the MIT domain in close proximity to the pore region, consistent with collaboration between the MIT domain and pore in the disassembly of ESCRT-III.
EXPERIMENTAL PROCEDURES
Strains, Media, and Antibodies
The Saccharomyces cerevisiae strains used for this study (MBY2 and MBY3 (22)) were grown for microscopy and cell extract preparation in synthetic minimal medium at 30 °C and harvested at exponential growth phase. For protein purification, Escherichia coli strains were grown in auto-induction medium (50 mm Na2HPO4, 22 mm KH2PO4, 20 g/liter Tryptone, 5 g/liter NaCl, 5 g/liter yeast extract, 0.2% lactose, 0.6% glycerol, 0.05% glucose, pH 7.2). The anti-Vps4 antibody was described previously (22).
DNA Manipulations
Plasmids used in this study are listed in Table 1. Plasmids obtained by PCR-based cloning techniques were confirmed by DNA sequencing. Point mutations were introduced using the Stratagene QuikChange site-directed mutagenesis kit (Agilent Technologies, La Jolla, CA). The pRS4XX shuttle vectors used in this study have been described previously (23). The plasmids used to express Vps4 protein in E. coli were constructed using the GST fusion vector pGEX-2T (GE Healthcare).
TABLE 1.
Plasmids used in this study
| Plasmid | Descriptive name | Vector | Reference or source |
|---|---|---|---|
| pMB28 | VPS4 | pRS416 | This study |
| pMB419 | vps4 (Δ89–95) | pRS416 | This study |
| pAS86 | vps4 (Δ85–95GS) | pRS416 | This study |
| pAS87 | vps4 (Δ85–106GS) | pRS416 | This study |
| pAS90 | vps4 (Δ85–112GS) | pRS416 | This study |
| pAS91 | vps4 (Δ85–115GS) | pRS416 | This study |
| pAS109 | vps4 (Δ85–118GS) | pRS416 | This study |
| pAS92 | vps4 (Δ85–120GS) | pRS416 | This study |
| pAS118 | vps4 (Δ84–118GS) | pRS416 | This study |
| pAS115 | vps4 (Δ83–118GS) | pRS416 | This study |
| pAS117 | vps4 (Δ81–118GS) | pRS416 | This study |
| pAS116 | vps4 (Δ79–118GS) | pRS416 | This study |
| pAS114 | vps4 (Δ78–118GS) | pRS416 | This study |
| pAS125 | vps4 (Δ79–118) | pRS416 | This study |
| pAS126 | vps4 (Δ82–118) | pRS416 | This study |
| pMB480 | vps4 (Δ1–79) | pRS416 | This study |
| pMB481 | vps4 (Δ1–116) | pRS416 | This study |
| pAS111 | vps4 (L119A) | pRS416 | This study |
| pAS88 | vps4 (+12aa) | pRS416 | This study |
| pMB54 | GST-VPS4 | pGEX-KG | 29 |
| pMB63 | GST-vps4 (E233Q) | pGEX-KG | 5 |
| pAS136 | GST-vps4 (Δ79–118) | pGEX-KG | This study |
| pAS142 | GST-vps4 (Δ79–118, E233Q) | pGEX-KG | This study |
| pAS137 | GST-vps4 (Δ82–118) | pGEX-KG | This study |
| pAS143 | GST-vps4 (Δ82–118, E233Q) | pGEX-KG | This study |
| pAS131 | GST-vps4 (Δ85–118GS) | pGEX-KG | This study |
| pAS132 | GST-vps4 (Δ85–118GS, E233Q) | pGEX-KG | This study |
| pMB468 | GST-vps4 (Δ1–79) | pGEX-KG | This study |
| pMB479 | GST-vps4 (Δ1–79, E233Q) | pGEX-KG | This study |
| pAS106 | GST-vps4 (Δ1–116) | pGEX-KG | This study |
| pAS108 | GST-vps4 (Δ1–116, E233Q) | pGEX-KG | This study |
| pMB343 | vps4 (E233Q)-HA-GFP | pRS416 | 15 |
| pAS123 | vps4 (Δ85–118GS, E233Q)-HA-GFP | pRS416 | This study |
| pAS124 | vps4 (Δ78–118GS, E233Q)-HA-GFP | pRS416 | This study |
| pMB118 | GFP-Cps1 | pRS425 | 30 |
Biochemical Procedures
Vps4 and Did2 proteins were purified as described previously (5). In brief, E. coli expressing the GST fusion proteins were grown in auto-induction medium at 18 °C for 24 h. The cells were harvested, lysed, and centrifuged at 100,000 × g for 20 min. The resulting supernatant was separated using a GST-Sepharose column (GE Healthcare). The resulting GST fusion proteins were incubated with thrombin (Sigma-Aldrich) at 25 °C for 1 h. The Vps4 and Did2 proteins were separated from GST and thrombin by ion-exchange chromatography using a ResourceQ column (GE Healthcare). The buffer for the in vitro ATPase activity assays was composed of 100 mm KAc, 5 mm MgAc2, 20 mm HEPES, pH 7.4, 1 mm ATP. At different time points, 10-μl samples were taken from the assay and added to 10 μl of methanol. The mixture was centrifuged for 10 min at 20,000 × g, and the resulting supernatant was separated by HPLC on a C18 reverse-phase column (buffer: 30 mm KH2PO4, 15 mm tetrabutylammonium bisulfate, 19% acetonitrile, pH 6.7). The amount of ADP and ATP was determined spectroscopically by measuring the absorption at 260 nm. Superose 6 (3.2/30 column, GE Healthcare) gel filtration analysis of purified proteins was performed in the presence of 1 mm ATP, 150 mm KAc, 20 mm HEPES, pH 7.4, and 5 mm MgAc2 at a flow rate of 40 μl/min. Chemical cross-linking of Vps4 was performed at 25 °C in the ATPase assay buffer in the presence of 0.02% glutaraldehyde. After 5 min of incubation, the reaction was terminated by the addition of equal volume 1 m glycine. The cross-linked proteins were precipitated by adding 10% trichloroacetic acid and centrifuged at 20,000 × g for 10 min. The resulting pellet was washed with acetone, dried, resuspended in SDS sample buffer (2% SDS, 0.1 m Tris, pH 6.8, 10% glycerol, 0.01% bromphenol blue, 5% β-mercaptoethanol), and separated by SDS-PAGE. CPY-Invertase assays were performed as described previously (24). Sedimentation equilibrium experiments were performed in an XL-I analytical ultracentrifuge (Beckman Coulter) with two-channel external loading cells. The cells were filled with water and aged as described previously (25). Blank scans were taken at all speeds used for the experiment with 150 μl of water in each sector. Protein samples were prepared by gel filtration into 25 mm Tris/HCl, pH 7.4, 100 mm NaCl, 2 mm magnesium chloride, 1 mm ATP, and 1 mm DTT. 120 μl of protein at different concentrations was loaded in the sample sector with 125 μl of gel filtration buffer in the reference sector. Interference data were collected at equilibrium at 4 °C and rotor speeds of 3000 and 5000 rpm. The Heteroanalysis software (version 1.1.56) (26) was used to analyze the data sets.
RESULTS
An ∼40-amino acid linker region connects the N-terminal MIT domain of Vps4 with the AAA-type ATPase domain (Fig. 1A). Comparison of yeast Vps4 with the two human homologues, hVps4A and hVps4B, showed low evolutionary conservation of the linker region both in amino acid sequence as well as in length. In addition, x-ray crystallography studies have suggested that the linker is unstructured (6), allowing for great flexibility between the MIT and AAA domain. To test whether this structural flexibility is important for function, we deleted different parts of the Vps4 linker region (Fig. 1A). For the construction of most of these VPS4 mutants, a BamHI site was introduced, resulting in 2 additional amino acids at the deletion site (Fig. 1A, Gly and Ser labeled in blue). The resulting vps4 mutant genes were expressed in a VPS4 deletion strain (vps4Δ) that contained a GFP-tagged cargo of the MVB pathway, GFP-Cps1, and the trafficking phenotype was determined by fluorescence microscopy. The newly synthesized transmembrane protein GFP-Cps1 traffics via endoplasmic reticulum and Golgi to the endosome where it is packaged into MVB vesicles and delivered to the vacuolar lumen (27). As a consequence, the majority of GFP-Cps1 localizes to the lumen of the vacuole in wild-type cells (Fig. 1B). In contrast, cells deleted for ESCRT components (e.g. vps4Δ) exhibit a block in MVB vesicle formation, resulting initially in the accumulation of GFP-Cps1 in aberrant endosomes and ultimately the delivery of GFP-Cps1 to the limiting membrane of the vacuole (see control in Fig. 1B).
FIGURE 1.

Trafficking phenotypes of vps4 linker mutants. A, amino acid sequence alignment of the linker regions of yeast and human (h) Vps4 proteins. The linker mutations analyzed in this study are listed. Dotted regions indicate deletions, whereas amino acid exchanges are labeled in blue. The mutants important for this study are highlighted in green. The +, +/−, and − labels indicate the MVB sorting efficiency of GFP-Cps1 in the corresponding vps4 mutant background. B, fluorescence microscopy of wild-type and vps4 mutants expressing GFP-Cps1. The images are inverted for better visualization of the GFP localization. The +, +/−, and − labels refer to the GFP-Cps1 sorting efficiency. con., control. C, CPY-Invertase secretion assays. The invertase activity is shown relative to the negative control vps4Δ (100%). Error bars indicate S.D.
To our surprise, most of the vps4 mutants were able to complement vps4Δ and restore normal protein sorting (summarized in Fig. 1A, + indicates wild-type GFP-Cps1 sorting; Fig. 1B shows GFP-Cps1 sorting in a subset of mutants). However, vps4 mutant Δ85–120GS, which removed the first 2 predicted amino acids of the AAA domain, exhibited severe MVB trafficking defects (the deletion did not affect protein stability, data not shown). The importance of the conserved leucine at position 119 for Vps4 function was further demonstrated by the fact that mutating this amino acid to an alanine caused partial loss of GFP-Cps1 sorting (L119A, Fig. 1, A and B), consistent with structural studies of Vps4 in which Leu-119 was found to be the first structured amino acid of the AAA domain (6). Thus, defects associated with Vps4(Δ85–120GS) are attributed to a defective AAA domain.
The MIT domain has been predicted to end at amino acid 77 (7, 8). Removing the complete linker region, from amino acid 78 to 118, resulted in loss of Vps4 activity (Δ78–118GS, Fig. 1A). However, the presence of 3 amino acids between the MIT domain and the AAA domain restored some Vps4 functionality. The amino acid sequence of this minimal linker seemed to be less important for Vps4 activity than the number of amino acids present, as demonstrated by the comparable activity of Vps4(Δ82–118) and Vps4(Δ79–118GS) (Fig. 1, A and B). Adding 4 more amino acids to the linker, to a total of 7 amino acids, completely restored Vps4 function in GFP-Cps1 sorting (Vps4(Δ83–118GS), Fig. 1A). These results suggested that a minimal linker between the MIT and AAA domains is important for normal Vps4 function in the MVB pathway, although most of the linker region is dispensable. In addition, we found that extending the linker by 12 amino acids did not interfere with Vps4 activity (Vps4(+12aa), Fig. 1, A and B), further supporting the idea that the linker region is not highly structured but functions mainly in providing flexibility between the MIT and the AAA domain. In contrast, the deletion of the MIT domain alone or in combination with the linker resulted in complete loss of Vps4 activity (Vps4(Δ1–79) and Vps4(Δ1–116), Fig. 1A). These results are consistent with the essential role of the MIT domain in Vps4 recruitment to MVBs (5).
Based on the observed MVB sorting efficiency, a subset of vps4 mutants was chosen for a detailed phenotypic analysis. These include the functional Δ85–118GS deletion, the partially functional vps4(Δ82–118) mutant, and the nonfunctional vps4(Δ79–118) (highlighted in Fig. 1A). To quantify the trafficking phenotype of these mutants, we performed CPY-Invertase secretion assays (Fig. 1C). In wild-type cells, the fusion protein CPY-Invertase traffics from the endoplasmic reticulum to the trans-Golgi, where it is packed into vesicles en route to the vacuole. However, ESCRT mutants are defective in the transport of cargo from the Golgi to the vacuole and thus secrete CPY-Invertase, a phenotype that can be quantified by an invertase activity assay (22). The CPY-Invertase secretion analysis supported the findings of the GFP-Cps1 sorting assay. Cells expressing Vps4(Δ85–118GS) exhibited a very mild secretion phenotype, indicating functionality comparable with wild-type Vps4 (Fig. 1C). In contrast, the mutant Vps4(Δ82–118) was only partially functional, whereas cells expressing Vps4(Δ79–118) showed a secretion phenotype similar to that of the vps4Δ strain (Fig. 1C, con.).
One possible explanation for the impaired function of Vps4(Δ82–118) and Vps4(Δ79–118) is that these mutant proteins are defective in the recruitment of Vps4 to the MVB. Therefore, we performed subcellular fractionation experiments. Cell extracts were separated by centrifugation into soluble (S) and membrane-bound pellet (P) fractions, and the resulting samples were analyzed by Western blot for the presence of Vps4. Consistent with previously published data (5), wild-type Vps4 was found mainly in the soluble fraction, whereas Vps4(E233Q) accumulated in the membrane-containing pellet fraction (Fig. 2A). Vps4(E233Q) contains a mutation in the Walker B motif of the AAA domain that blocks hydrolysis activity and thus results in accumulation of the ATP-bound form of Vps4 on MVBs (5, 28). The Vps4 linker mutants tested, Vps4(Δ85–118GS), Vps4(Δ82–118), and Vps4(Δ79–118), were present in the pellet fraction at levels comparable with wild-type Vps4, suggesting that these proteins are recruited to MVBs (Fig. 2A). This notion was further supported by fluorescence microscopy, which demonstrated that the ATP-locked versions of the Vps4 linker mutants co-localized with the MIT-RFP (Fig. 2B), a fusion protein that binds to ESCRT-III (15). Together, both subcellular fractionation and microscopy indicated that the impaired function of Vps4(Δ82–118) and Vps4(Δ79–118) was not due to a problem of recruitment of these mutant proteins to the substrate ESCRT-III.
FIGURE 2.
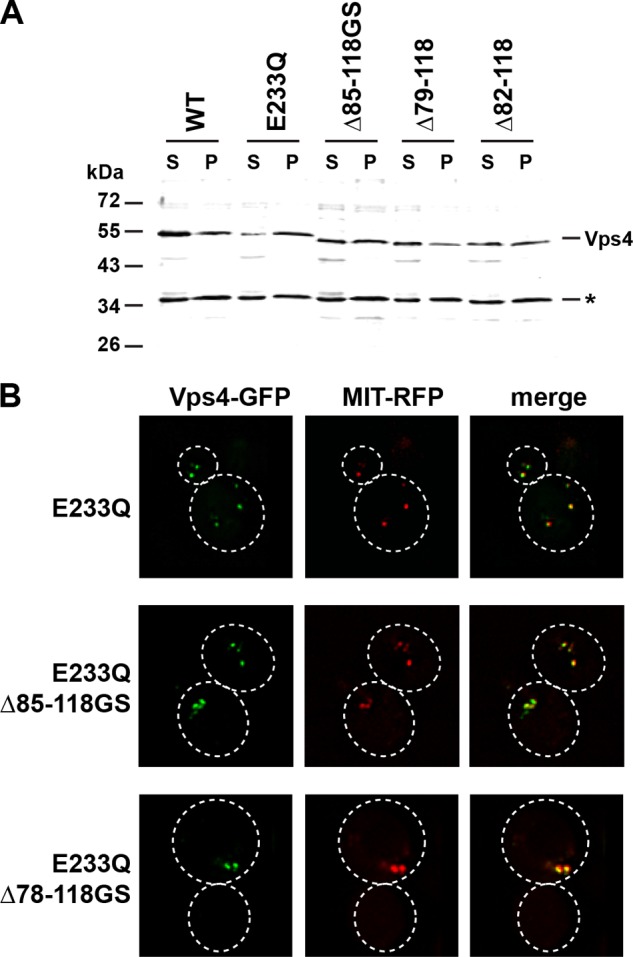
Subcellular localization of mutant Vps4 proteins. A, Western blot analysis of soluble (S) and membrane-bound pellet (P) cell fractions using anti-Vps4 antiserum. The cross-reacting protein at 35 kDa (*) serves as a loading control. B, fluorescence microscopy of cells expressing GFP-tagged versions of Vps4 mutants and the MVB marker MIT-RFP.
Linker Deletions Alter Vps4 ATPase Activity in Vitro
The fact that most of the linker region was dispensable for Vps4 function in MVB sorting was surprising. To further understand the function of this linker region, in vitro characterizations of recombinant Vps4 linker region mutants were performed. For these experiments, we purified recombinant wild-type and mutant Vps4 (Fig. 3A). Although ESCRT-III and other factors facilitate Vps4 oligomerization to disassemble ESCRT-III in vivo, high concentrations of soluble Vps4 in vitro can assemble into the oligomeric form in the absence of any other ESCRT proteins. Because only the oligomeric form of Vps4 is able to hydrolyze ATP, ATPase activity is indicative of Vps4 assembly.
FIGURE 3.

In vitro analysis of linker-deleted Vps4 proteins. A, SDS-PAGE analysis of the recombinant Vps4 proteins used in the in vitro assays. Proteins are visualized by Coomassie Blue staining. B–D, Vps4 ATPase activity in the absence (B) or presence of the substrate Did2 (C) or the co-factor Vta1 (D). E, in vitro ESCRT-III disassembly by wild-type and mutant Vps4. Error bars in panels B–E indicate S.D.
Consistent with previous publications, we observed that increasing the concentration of wild-type Vps4 from 0.5 to 2 μm resulted in an increase of ATPase activity from ∼18 to ∼35 ATP/min/Vps4 (5) (Fig. 3B). Similarly, all linker deletion mutants tested showed concentration-dependent ATPase activity. However, the ATPase activity was very low at 0.5 μm protein concentration but increased to higher than wild-type activity at 2 μm (Fig. 3B). These data suggested that the deletion of the linker impaired assembly of Vps4, therefore requiring higher protein concentration to initiate oligomerization. However, when assembled, the mutant Vps4 proteins showed higher ATPase activity than wild-type protein. The ATPase activity of the mutant proteins did not correlate with the observed in vivo activity in MVB cargo sorting. Even Vps4(Δ79–118), which was found to be a loss-of-function mutant in vivo, demonstrated in vitro the ability to hydrolyze ATP (Fig. 3B). Similarly, the nonfunctional Vps4 mutant lacking the MIT domain was found to have ATPase activity at high concentrations (Vps4(Δ1–79), Fig. 3B). However, deleting both the MIT domain and the linker abolished Vps4 hydrolysis activity at the concentrations examined (Vps4(Δ1–116), Fig. 3B), which is in contrast to human VPS4A, where deletion of the corresponding residues (1–100) results in higher ATPase activity when compared with the wild-type protein (21). Our data suggested that the MIT domain and the linker affect both oligomerization and ATPase activity of the assembled Vps4 complex.
To further explore the activity of the wild-type and mutant Vps4 oligomers, ATPase activity was examined in the presence of Vta1. Vta1 is an important co-factor of Vps4 that has been shown to play a role in both assembly of Vps4 and its ATPase activity (29). As expected, the addition of Vta1 to the in vitro assay increased the ATPase activity of wild-type Vps4 ∼6-fold (Fig. 3D). Similarly, the activity of the mutant Vps4 proteins was strongly enhanced by the presence of Vta1. In particular, Vta1 dramatically enhanced the ATPase activity of MIT-deleted Vps4, reaching levels of ∼450 ATP/min/Vps4 (Fig. 3D). Even Vps4(Δ1–116), which by itself did not exhibit appreciable activity, showed in the presence of Vta1 near wild-type ATP hydrolysis (Fig. 3D). Together, these data indicated that Vta1 was able to bind to the different mutant versions of Vps4 and positively affect their ATPase activity. Thus, the in vivo defect of these mutants was not due to a lack of ATP hydrolysis or a defect in binding the co-factor Vta1.
Vps4 Linker Mutants Exhibit Reduced Stimulation by ESCRT-III and in Vitro Disassembly of ESCRT-III
When assembled, the Vps4 linker mutants exhibited higher ATPase activity than the wild-type protein, suggesting that these mutants are in a more active state. Substrate binding has been shown to increase the hydrolysis activity of Vps4 (16, 21). Therefore, one possible model is that the Vps4 mutants assemble into a conformation that resembles the ESCRT-III bound state of Vps4. This model predicts that these Vps4 linker mutants would be resistant to further stimulation by ESCRT-III subunits, such as Did2. To test this idea, we examined Did2-His6 stimulation of wild-type Vps4 and Vps4(Δ82–118), the linker deletion mutant with the highest ATPase activity. Did2-His6 increased the ATPase activity of wild-type Vps4 ∼1.5-fold (Fig. 3C). In contrast, the ATPase activity of the linker mutant Vps4(Δ82–118) was not affected by the presence of the substrate Did2-His6. This result was consistent with the model that deletion of the linker region resulted in a Vps4 structure that mimicked that of substrate-bound Vps4.
Vps4 functions in the disassembly and recycling of ESCRT-III subunits from MVBs. This activity of Vps4 can be tested in vitro using recombinant protein (30). Increasing concentrations of purified wild-type or Vps4 linker mutants were added to ESCRT-III-containing endosome fractions, and the resulting solubilization of the ESCRT-III subunit Snf7 was determined by centrifugation and Western blot analysis of the resulting pellet and supernatant fractions. Vps4(Δ85–118GS) and Vps4(Δ82–118), which were at least partially functional in vivo, also showed disassembly activity in vitro (Fig. 3E). However, the activity of both of these linker mutants was lower than that observed for the wild-type protein. The mutant Vps4(Δ79–118) lacked in vitro disassembly activity, consistent with the inability of Vps4(Δ79–118) to function in vivo (Figs. 1, A and B, and 3E). Although the linker mutants exhibited enhanced in vitro ATPase activity, the mutants exhibited defects in ESCRT-III stimulation and ESCRT-III disassembly. These observations suggest that the hyperactive Vps4 oligomers formed by the Vps4 linker region mutants are less efficient at ESCRT-III disassembly than the wild-type oligomers.
Deletion of the Linker Region Enhanced Vps4 Oligomer Assembly
The Vps4 mutants tested exhibited ATPase activity, suggesting that they were able to assemble into a higher-ordered complex. To further characterize the oligomeric state of the different Vps4 mutant proteins, we performed gel filtration analysis. For these studies, we purified the E233Q mutant versions of full-length Vps4 and the linker deletions of Vps4. Introducing the E233Q mutation blocks ATP hydrolysis by Vps4 and thus stabilizes Vps4 in the ATP-bound, oligomeric state (5). The purified proteins were loaded onto a Superose S6 column and separated by size in the presence of ATP.
As previously shown, ATP-bound Vps4(E233Q) eluted from the column in the size range of ∼450 kDa (5) (Fig. 4A, oligomer #1). In comparison, deletions in the linker of Vps4 caused the appearance of a larger Vps4 species in a size range of ∼600 kDa (oligomer #2). Interestingly, we observed a correlation between the in vivo observed phenotype of the linker mutants and the amount of mutant protein present in the higher molecular weight form. The E233Q form of the functional Vps4(Δ85–118GS) was mainly found in the higher oligomeric form, whereas E233Q forms of Vps4(Δ82–118) and Vps4(Δ79–118), which exhibited progressively increased trafficking defects, showed correspondingly increased amounts of the protein in the lower molecular weight form (Fig. 4A, oligomer #1). The MIT-deleted Vps4 mutant, Vps4(Δ1–79, E233Q), eluted from the gel filtration column with an apparent molecular weight similar to that of the full-length protein. In contrast, deletion of both the MIT domain and the linker region shifted the peak fraction to a smaller size oligomer, which likely represented dimeric Vps4 (Vps4(Δ1–116, E233Q), Fig. 4A). This result was consistent with the lack of ATPase activity exhibited by this N-terminal-deleted Vps4 mutant (Fig. 3B).
FIGURE 4.

Gel filtration analysis of wild-type or mutant Vps4 in the presence (B) or absence (A) of Vta1. All experiments were performed in the presence of 1 mm ATP.
Vta1 has been shown to bind to the oligomeric form of Vps4 and stabilize the complex (29). Consistent with these published results, we observed by gel filtration analysis the formation of an ∼1-MDa protein complex composed of Vps4(E233Q) and Vta1 (Fig. 4B). All Vps4 mutant proteins tested assembled with Vta1 into a large protein complex, consistent with Vta1 stimulation of the Vps4 linker mutants (Fig. 3D). Even Vps4(Δ1–116, E233Q), a protein that by itself was found to form only dimers, assembled with Vta1 into a complex that was similar in size to the oligomer formed by the full-length protein. However, the apparent molecular weight of these protein complexes varied according to the variation in size we observed for the gel filtration analysis shown in Fig. 4A. The Vps4(Δ85–118GS,E233Q) mutant, which most efficiently formed the ∼600-kDa homo-oligomer (Fig. 4A, oligomer #2), also showed the largest increase in size in the presence of Vta1 (Fig. 4B).
The gel filtration analysis indicated that deleting the linker region resulted in the formation of a homo-oligomeric complex that is larger than that observed with full-length Vps4(E233Q) (Fig. 4A, oligomers #1 and #2). To further analyze this change in oligomeric structure, we performed glutaraldehyde cross-linking experiments. In the presence of ADP or ATP, purified Vps4(E233Q) and Vps4(Δ85–118GS, E233Q) were exposed to 0.02% glutaraldehyde for 5 min, and the resulting cross-linked products were analyzed using a 4–15% SDS-PAGE gel. The analysis of the ADP-bound full-length protein showed a ladder of bands indicating a mixture of cross-linked oligomers containing different numbers of subunits (Fig. 5A). Because chemical cross-linking changes the SDS-PAGE running behavior of a protein, determining the molecular weight of such a protein by comparison with a protein standard is not possible. We observed three distinct Vps4(E233Q) bands in the size range of 50–70 kDa (based on the protein standard), all of which are likely representing differently cross-linked Vps4(E233Q) monomers (Fig. 5A). However, the spacing of the band pattern, the number of bands observed (six major bands) and the fact that Vps4 is predicted to form hexameric rings (9, 10) fit best to the interpretation that the protein bands represented the cross-linking of 1–6 Vps4 monomers. In the presence of ATP, glutaraldehyde cross-linking resulted mainly in the formation of the predicted hexameric Vps4(E233Q) (Fig. 5A), consistent with the observation that ATP stabilizes the oligomeric form of Vps4. The same result was observed by cross-linking Vps4(E233Q) from the peak fraction of the gel filtration analysis shown in Fig. 4A (Fig. 5A, GF).
FIGURE 5.
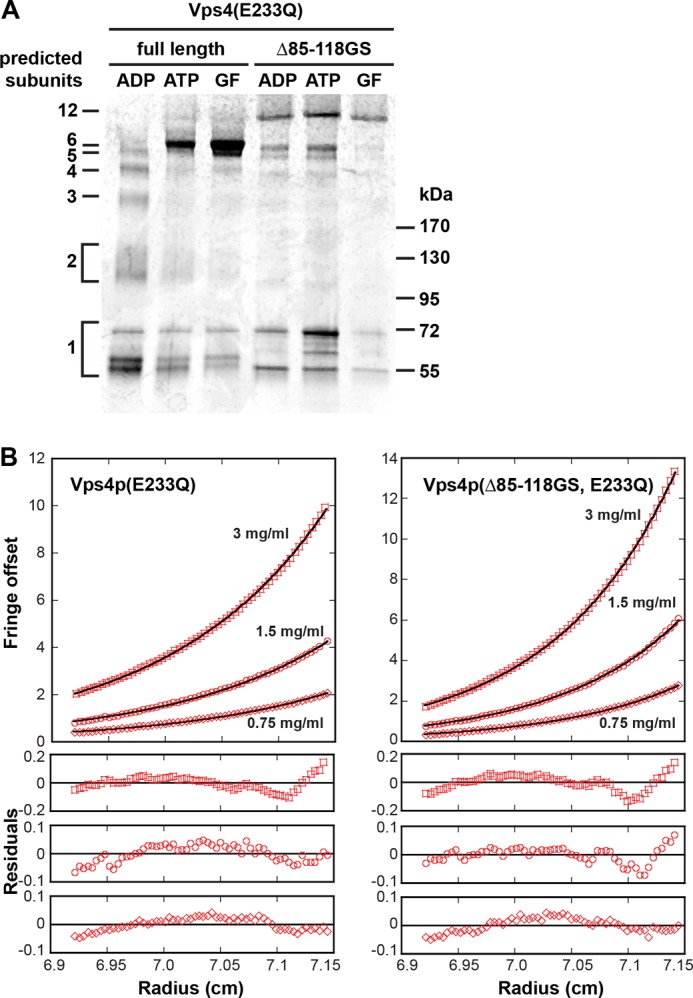
Analysis of the quaternary structure of Vps4. A, recombinant Vps4 protein was cross-linked by glutaraldehyde in the presence of ADP or ATP, and the products were analyzed by SDS-PAGE (4–15% gradient gel) followed by Coomassie Blue staining. The GF lanes contain the cross-linking products of the Vps4 peak fractions from the gel filtration analyses shown in Fig. 4A. B, analytical ultracentrifugation of Vps4(Δ85–118GS, E233Q) and Vps4(E233Q) in the presence of ATP. The interference signal from scans taken at equilibrium at 5000 rpm is plotted versus the distance from the axis of rotation (radius) as red, with open symbols representing different protein concentrations (squares, 3 mg/ml; circles, 1.5 mg/ml; diamonds, 0.75 mg/ml). The black solid line represents a global fit over all concentrations and including data taken at 3000 rpm (not shown). Residuals are shown below for all concentrations.
Interestingly, when the same cross-linking experiments were performed with Vps4(Δ85–118GS, E233Q) a higher molecular band appeared under all conditions tested, consistent with the higher molecular weight oligomer observed by gel filtration analysis (Fig. 4A, oligomer #2). The size of this protein band was difficult to estimate because it was much larger then the predicted hexameric complex observed with full-length protein. However, the cross-linking data indicated that the size increase observed by gel filtration analysis of Vps4(Δ85–118GS, E233Q) was due to an increased number of subunits and was not caused simply by a change in shape.
We used sedimentation equilibrium analytical ultracentrifugation to further investigate how the linker deletion affects the oligomeric state of the protein. To this end, we analyzed the radial distribution of Vps4(Δ85–118GS, E233Q) and Vps4p(E233Q) in the presence of ATP. In both cases, the molecular weight average increased with increasing concentrations when the data for each concentration were fit separately, indicating that there is a mixture of oligomeric states. We were unable to determine the size of these oligomers as all attempts to fit the data resulted in biased residuals with both ideal species and self-association models. Fig. 5B shows the result of a global fit using an ideal species model. The S-shaped residuals suggest that there are both smaller and larger species present than the molecular mass average suggests. The molecular size average is larger for Vps4(Δ85–118GS, E233Q) (408 kDa, 8–9 subunits) than for Vps4(E233Q) (314 kDa, 6–7 subunits). This is consistent with the gel filtration and cross-linking experiments described above, suggesting that the equilibrium is shifted toward a larger species for Vps4(Δ85–118GS, E233Q).
In summary, none of the three methods used to analyze the Vps4 structure were able to unequivocally determine the subunit composition of the formed protein complexes. However, taking previous structural studies of both Vps4 and other AAA proteins into account, the most likely explanation for the data is that in vitro, full-length Vps4(E233Q) assembles into a hexameric ring, whereas linker-deleted Vps4(E233Q) prefers to form a dodecameric, two-ring structure. Because of its shape (flat ring), the hexamer elutes from gel filtration columns in the size range of 450 kDa instead of the theoretical ∼300 kDa. Deletion of the linker stabilizes the assembly of two hexameric rings with a more globular shape, resulting in the elution of the complex from the gel filtration column close to the theoretical size of ∼600 kDa. Unlike the complex formed by full-length protein, the formation of the dodecamer is less dependent on the nucleotide-bound state and thus can occur in the presence of ADP (Fig. 5A). As the Vps4(Δ85–118GS) oligomer exhibited increased ATPase activity reminiscent of the ESCRT-III-stimulated Vps4 oligomer (Fig. 3, B and C), we propose that the ESCRT-III-bound Vps4 oligomer may adopt a dodecameric structure to disassemble ESCRT-III as observed for Vps4(Δ85–118GS, E233Q).
DISCUSSION
The assembly of Vps4 into the active ATPase complex is a key step that spatially and temporally regulates its function. The inactive Vps4 monomers or dimers are recruited from the cytoplasm to ESCRT-III where they assemble into a higher oligomeric complex that functions in the ATP-driven disassembly of the bound ESCRT-III complex. This ESCRT-III disassembly reaction occurs on endosomes as part of the ESCRT-mediated formation of MVBs. Furthermore, in mammalian cells, ESCRT-III disassembly by Vps4 is observed at the midbody during cytokinesis and at the plasma membrane during the release of newly formed retroviral particles. Both recruitment and assembly of Vps4 are aided by the substrate ESCRT-III and the co-factors/regulators Did2, Vta1, Ist1, and Vps60, which work together to ensure that the Vps4 ATPase is only active when assembled at the proper site.
Vps4 is composed of the enzymatic AAA domain, the substrate binding MIT domain, and a linker region connecting the MIT domain to the AAA domain. To our surprise, we found that removing 33 of the 40 amino acids in the linker region did not interfere with Vps4 function in the MVB pathway. Even Vps4 mutants that contained only 3 amino acids of the linker showed partial in vivo activity. The exact amino acid sequence of the minimal linker region did not seem to matter as much as the number of amino acids present, suggesting that a minimal distance between MIT and AAA domain was important for normal Vps4 function.
Although the linker region exhibits little sequence conservation between yeast to human, the continued presence of the region suggested that the linker contributes to Vps4 function in ways unappreciated by the in vivo phenotypic analysis. To better understand the contributions of the linker region, biochemical characterization was undertaken. The analysis of recombinant Vps4 mutant proteins demonstrated that loss of the linker region caused an increase in maximal ATPase activity, a result that is consistent with the idea that the linker is involved in an auto-inhibition mechanism (21). However, the protein concentration necessary to induce oligomerization was higher for linker-deleted Vps4 than for the wild-type protein. The additional deletion of the MIT domain (Vps4(Δ1–116)) resulted in a Vps4 mutant that oligomerized only in the presence of the co-factor Vta1, a result that differs from data obtained with human VPS4A (21). Together, our data indicated that the AAA domain alone is not sufficient for efficient oligomerization and that the N-terminal MIT domain and adjacent linker play a role in stabilizing the Vsp4 oligomer.
Deletions of the linker region affected not only efficiency of Vps4 oligomerization but also the size of the oligomer formed. Data from gel filtration analysis, chemical cross-linking, and analytical ultracentrifugation fit best to a model in which deletion of the linker region stabilized a two-ring, dodecameric structure of Vps4. In contrast, full-length Vps4(E233Q) seemed to prefer the formation of a hexameric complex. Even in the presence of Vta1, a protein that supports oligomerization of Vps4, the linker-deleted mutant protein formed a Vta1-Vps4 protein complex that was larger then the complex formed with full-length Vps4. Because the linker-deleted versions of Vps4 are active in vivo, we predict that the larger, dodecameric form of Vps4 represents an assembly that plays an important role in the function of Vps4.
Although Vps4 is recruited to ESCRT-III via the MIT domain, the pore region of the Vps4 ring structure is thought to perform the ATP-driven disassembly of the ESCRT-III complex. Therefore, an explanation for the presence of a long linker between the MIT domain and the AAA domain could be that the MIT-ESCRT-III interaction has to occur far away from the pore region to allow the pore to engage the substrate without steric hindrance by the MIT domain. In contrast, the linker mutant Vps4(Δ82–118) showed activity in vivo with only 4 amino acids between the MIT domain and the ATPase domain. Interestingly, in the postulated hexameric structure of the Vps4 AAA domain, the first amino acids of this domain are localized close to the pore region of the complex (Fig. 6A), indicating that in the case of the linker deletions, the MIT domains surround the Vps4 pore, thereby blocking direct access of the pore to the substrate. This observation suggested a coordinated activity of the MIT domain and pore during ESCRT-III disassembly, a model that has been previously proposed based on studies on human VPS4A (21).
FIGURE 6.

Model of the MIT-regulated assembly of Vps4. A, model for the hexameric structure of the Vps4 AAA domain based on the p97 D1 structure (6). The first amino acid of the AAA domain (Leu-119) is labeled in red. B, model for the oligomerization of Vps4 into a double-ring structure triggered by either linker deletion (in vitro) or substrate binding (in vivo).
In summary, our data fit well to the published single-particle EM analyses of Vps4 that predicted the formation of a dodecameric complex containing two rings with different conformations and different localizations of the MIT domains (9, 10) (Fig. 6B). We predict that the positioning of the MIT determines the conformation of the two distinct Vps4 rings. The Vps4 hexamer with the MIT domains distant from the pore (hexamer #1) exhibits low ATPase activity but assembles efficiently at lower Vps4 concentration. This type of hexamer is preferentially formed by recombinant, ATP-locked Vps4. In contrast, the repositioning of the MIT domains close to the pore causes the formation of a hexameric Vps4 ring with high ATPase activity but lower affinity between the subunits (hexamer #2). Furthermore, this second type of hexamer prefers the assembly into the dodecameric double-ring structure. In vivo the repositioning of the MIT domain is likely caused by the simultaneous engagement of the substrate ESCRT-III with the MIT domains as well as the pore loops, thereby bringing the MIT domains into proximity of the pore, a model that has been previously proposed for the activation of human Vps4 (21). It is likely that the MIT domains and pore are functioning together in the disassembly of the bound ESCRT-III complex. In the case of the Vps4 mutants tested in vitro, the deletion of the linker repositions the MIT domains close to the pore of the Vps4 complex, mimicking the substrate-bound state of Vps4. Consistent with this idea, we observed that the in vitro ATPase activity of linker-deleted Vps4 was similar to that of the wild-type protein in the presence of substrate (Fig. 3C). However, Vps4 mutants with linkers shorter than 7 amino acids (Δ79–118, Δ82–118) exhibited problems both in the formation of the dodecamer (Fig. 4A) as well as in the in vivo function (Fig. 1C). A likely explanation for these observations is that these Vps4 mutants are impaired in the formation of hexamer #1 and thus are not able to efficiently form a double-ring structure (Fig. 6B) that is essential for normal Vps4 activity.
The proposed arrangement of two asymmetrical hexameric Vps4 rings fits well to the known structures of type-II AAA ATPases, such as p97 and N-ethylmaleimide-sensitive factor (NSF). These ATPases contain two AAA domains, which after assembly into a hexamer form two rings with discrete structure and function. One ring acts as the ATP-driven disassembly machine, whereas the second ring functions mainly as a structural component that stabilizes the hexameric complex. Similarly, the Vps4 dodecamer seems to be composed of two discrete hexameric rings, the one with high ATPase activity engaging the substrate and the other providing stability to the complex.
Acknowledgments
We thank Nicole Monroe for analysis of Vps4 by analytical ultracentrifugation. We thank Wes Sundquist and Chris Hill for insightful discussions and help with the three-dimensional modeling of Vps4.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM074171 (to M. B.) and R01GM73024 (to D. J. K.).
- ESCRT
- endosomal sorting complex required for transport
- MIT
- microtubule interacting and trafficking
- MVB
- multivesicular body
- CPY
- carboxypeptidase Y.
REFERENCES
- 1. Hill C. P., Babst M. (2012) Structure and function of the membrane deformation AAA ATPase Vps4. Biochim. Biophys. Acta 1823, 172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McCullough J., Colf L. A., Sundquist W. I. (2013) Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 82, 663–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. White S. R., Lauring B. (2007) AAA+ ATPases: achieving diversity of function with conserved machinery. Traffic 8, 1657–1667 [DOI] [PubMed] [Google Scholar]
- 4. Wendler P., Ciniawsky S., Kock M., Kube S. (2012) Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta 1823, 2–14 [DOI] [PubMed] [Google Scholar]
- 5. Babst M., Wendland B., Estepa E. J., Emr S. D. (1998) The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17, 2982–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scott A., Chung H. Y., Gonciarz-Swiatek M., Hill G. C., Whitby F. G., Gaspar J., Holton J. M., Viswanathan R., Ghaffarian S., Hill C. P., Sundquist W. I. (2005) Structural and mechanistic studies of VPS4 proteins. EMBO J. 24, 3658–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scott A., Gaspar J., Stuchell-Brereton M. D., Alam S. L., Skalicky J. J., Sundquist W. I. (2005) Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc. Natl. Acad. Sci. U.S.A. 102, 13813–13818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takasu H., Jee J. G., Ohno A., Goda N., Fujiwara K., Tochio H., Shirakawa M., Hiroaki H. (2005) Structural characterization of the MIT domain from human Vps4b. Biochem. Biophys. Res. Commun. 334, 460–465 [DOI] [PubMed] [Google Scholar]
- 9. Yu Z., Gonciarz M. D., Sundquist W. I., Hill C. P., Jensen G. J. (2008) Cryo-EM structure of dodecameric Vps4p and its 2:1 complex with Vta1p. J. Mol. Biol. 377, 364–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Landsberg M. J., Vajjhala P. R., Rothnagel R., Munn A. L., Hankamer B. (2009) Three-dimensional structure of AAA ATPase Vps4: advancing structural insights into the mechanisms of endosomal sorting and enveloped virus budding. Structure 17, 427–437 [DOI] [PubMed] [Google Scholar]
- 11. Hartmann C., Chami M., Zachariae U., de Groot B. L., Engel A., Grütter M. G. (2008) Vacuolar protein sorting: two different functional states of the AAA-ATPase Vps4p. J. Mol. Biol. 377, 352–363 [DOI] [PubMed] [Google Scholar]
- 12. Babst M., Davies B. A., Katzmann D. J. (2011) Regulation of Vps4 during MVB sorting and cytokinesis. Traffic 12, 1298–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bajorek M., Schubert H. L., McCullough J., Langelier C., Eckert D. M., Stubblefield W. M., Uter N. T., Myszka D. G., Hill C. P., Sundquist W. I. (2009) Structural basis for ESCRT-III protein autoinhibition. Nat. Struct. Mol. Biol. 16, 754–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiao J., Chen X. W., Davies B. A., Saltiel A. R., Katzmann D. J, Xu Z. (2009) Structural basis of Ist1 function and Ist1-Did2 interaction in the multivesicular body pathway and cytokinesis. Mol. Biol. Cell 20, 3514–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shestakova A., Hanono A., Drosner S., Curtiss M., Davies B. A., Katzmann D. J, Babst M. (2010) Assembly of the AAA ATPase Vps4 on ESCRT-III. Mol. Biol. Cell 21, 1059–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Azmi I. F., Davies B. A., Xiao J., Babst M., Xu Z., Katzmann D. J. (2008) ESCRT-III family members stimulate Vps4 ATPase activity directly or via Vta1. Dev. Cell 14, 50–61 [DOI] [PubMed] [Google Scholar]
- 17. Yang Z., Vild C., Ju J., Zhang X., Liu J., Shen J., Zhao B., Lan W., Gong F., Liu M., Cao C., Xu Z. (2012) Structural basis of molecular recognition between ESCRT-III-like protein Vps60 and AAA-ATPase regulator Vta1 in the multivesicular body pathway. J. Biol. Chem. 287, 43899–43908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ward D. M., Vaughn M. B., Shiflett S. L., White P. L., Pollock A. L., Hill J., Schnegelberger R., Sundquist W. I., Kaplan J. (2005) The role of LIP5 and CHMP5 in multivesicular body formation and HIV-1 budding in mammalian cells. J. Biol. Chem. 280, 10548–10555 [DOI] [PubMed] [Google Scholar]
- 19. Shim S., Merrill S. A., Hanson P. I. (2008) Novel interactions of ESCRT-III with LIP5 and VPS4 and their implications for ESCRT-III disassembly. Mol. Biol. Cell 19, 2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Skalicky J. J., Arii J., Wenzel D. M., Stubblefield W. M., Katsuyama A., Uter N. T., Bajorek M., Myszka D. G., Sundquist W. I. (2012) Interactions of the human LIP5 regulatory protein with endosomal sorting complexes required for transport. J. Biol. Chem. 287, 43910–43926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Merrill S. A., Hanson P. I. (2010) Activation of human VPS4A by ESCRT-III proteins reveals ability of substrates to relieve enzyme autoinhibition. J. Biol. Chem. 285, 35428–35438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Babst M., Sato T. K., Banta L. M., Emr S. D. (1997) Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO J. 16, 1820–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Christianson T. W., Sikorski R. S., Dante M., Shero J. H., Hieter P. (1992) Multifunctional yeast high-copy-number shuttle vectors. Gene 110, 119–122 [DOI] [PubMed] [Google Scholar]
- 24. Darsow T., Odorizzi G., Emr S. D. (2000) Invertase fusion proteins for analysis of protein trafficking in yeast. Methods Enzymol. 327, 95–106 [DOI] [PubMed] [Google Scholar]
- 25. Cole J. L., Lary J. W., Moody T. P., Laue T. M. (2008) Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol. 84, 143–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cole J. L. (2004) Analysis of heterogeneous interactions. Methods Enzymol. 384, 212–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Odorizzi G., Babst M., Emr S. D. (1998) Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell 95, 847–858 [DOI] [PubMed] [Google Scholar]
- 28. Bishop N., Woodman P. (2000) ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol. Biol. Cell 11, 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Azmi I., Davies B., Dimaano C., Payne J., Eckert D., Babst M., Katzmann D. J. (2006) Recycling of ESCRTs by the AAA-ATPase Vps4 is regulated by a conserved VSL region in Vta1. J. Cell Biol. 172, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davies B. A., Azmi I. F., Payne J., Shestakova A., Horazdovsky B. F., Babst M., Katzmann D. J. (2010) Coordination of substrate binding and ATP hydrolysis in Vps4-mediated ESCRT-III disassembly. Mol. Biol. Cell 21, 3396–3408 [DOI] [PMC free article] [PubMed] [Google Scholar]