

Background: An eight-amino acid segment lying within the αC-β4 loop region of many protein kinases determines sensitivity to Hsp90 inhibitors.
Results: A peptide comprised of this segment of the EGFR inhibits both Hsp90 binding and EGF-dependent EGFR dimerization.
Conclusion: The peptide selectively degrades EGFR versus other Hsp90 clients.
Significance: This peptide represents a unique approach to the therapy of EGFR-driven tumors.
Keywords: Cancer, Epidermal Growth Factor Receptor (EGFR), Hsp90, Protein Degradation, Receptor Tyrosine Kinase, EGFR Degradation
Abstract
An eight-amino acid segment is known to be responsible for the marked difference in the rates of degradation of the EGF receptor (ErbB1) and ErbB2 upon treatment of cells with the Hsp90 inhibitor geldanamycin. We have scrambled the first six amino acids of this segment of the EGF receptor (EGFR), which lies in close association with the ATP binding cleft and the dimerization face. Scrambling these six amino acids markedly reduces EGFR stability, EGF-stimulated receptor dimerization, and autophosphorylation activity. Two peptides were synthesized as follows: one containing the wild-type sequence of the eight-amino acid segment, which we call Disruptin; and one with the scrambled sequence. Disruptin inhibits Hsp90 binding to the EGFR and causes slow degradation of the EGFR in two EGFR-dependent cancer cell lines, whereas the scrambled peptide is inactive. This effect is specific for EGFR versus other Hsp90 client proteins. In the presence of EGF, Disruptin, but not the scrambled peptide, inhibits EGFR dimerization and causes rapid degradation of the EGFR. In contrast to the Hsp90 inhibitor geldanamycin, Disruptin inhibits cancer cell growth by a nonapoptotic mechanism. Disruptin provides proof of concept for the development of a new class of anti-tumor drugs that specifically cause EGFR degradation.
Introduction
The epidermal growth factor receptor (EGFR)2 is a member of the ErbB family of transmembrane tyrosine kinases (EGFR/ErbB1/HER1, ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4), which play critical roles in regulating cell proliferation, differentiation, and migration (1). The EGFR is activated by ligand-induced dimerization (2), and abnormal activation is associated with a variety of human cancers (1), making the EGFR a target of considerable interest for anti-cancer drug development. The major clinically approved approaches have been to develop EGFR kinase inhibitors, such as gefitinib or erlotinib, and the production of monoclonal anti-EGFR antibodies like cetuximab. An experimental approach has been to target the EGFR for degradation by using inhibitors of Hsp90, which cycles with the EGFR monomer to stabilize it (3).
The EGFR is activated by forming an asymmetric homodimer with itself (4, 5) or by forming a heterodimer with another member of the ErbB family. ErbB2 (HER2) functions as a ligandless coreceptor that heterodimerizes with other members of the ErbB family to amplify signaling. The kinase domain of ErbB2 forms a stable complex with Hsp90, and Hsp90 regulates ErbB2 function by limiting heterodimer formation (6). In contrast, the EGFR undergoes dynamic cycling with Hsp90 and is less stringently regulated by the chaperone machinery (7). Upon Hsp90 inhibition by geldanamycin, ErbB2 is polyubiquitinated and rapidly degraded, whereas the EGFR is modestly ubiquitinated and more slowly degraded (8, 9). This difference in geldanamycin sensitivity is accounted for by a short eight-amino acid segment within the highly homologous kinase domains (6, 10). Swapping the eight-amino acid segments between the EGFR and ErbB2 yields the appropriate exchange of dynamic versus stable cycling with Hsp90 and the corresponding change in geldanamycin sensitivity (6).
This eight-amino acid segment lies within the αC-β4 loop region of many protein kinases, and it is proposed to define a common surface with which Hsp90 interacts (11). The αC helix is a region that regulates kinase activity (12), and it forms part of the dimerization interface that interacts with activator kinase in the asymmetric EGFR dimer (4, 5). Although the function and turnover of a wide variety of signaling proteins are regulated by Hsp90 (13), there is no specific motif that determines interaction with the chaperone, and the eight-amino acid segments of the EGFR and ErbB2 are unrelated to a seven-amino acid region of the glucocorticoid receptor that similarly determines its stable versus dynamic cycling with Hsp90 (14). There is considerable evidence supporting the proposal that Hsp90 interacts with proteins in the region where their ligand binding clefts open onto the protein surface (reviewed in Ref. 7).
Inasmuch as the eight-amino acid segment lies in close association with the EGFR ATP binding cleft (12) and the receiver dimerization face (4, 5) as well as controlling Hsp90 binding (6), we explore here the role of the segment in determining EGFR stability. We show first that scrambling the first six amino acids of the wild-type segment markedly reduces EGFR stability and function, which was assessed by EGF-dependent dimerization and phosphorylation. We then synthesized two peptides, one with the wild-type sequence, which we call Disruptin, and one with the scrambled sequence. Treatment of EGFR-dependent cancer cell lines with Disruptin inhibits EGFR binding to Hsp90 and destabilizes the receptor. The effect is specific for the EGFR and does not pertain to other Hsp90 client proteins. Disruptin, but not the scrambled peptide, also inhibits EGF-dependent dimerization of the EGFR and cell growth. These observations suggest a model in which Disruptin interacts directly with the EGFR to inhibit an intermolecular or intramolecular protein interaction to inhibit both Hsp90 binding and dimerization. They also provide a basis for the development of unique drugs that will specifically target EGFR-driven tumors.
EXPERIMENTAL PROCEDURES
Materials
Geldanamycin was acquired from Enzo Life Sciences, Farmingdale, NY. EGFR (sc-03) antibody was acquired from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Hsp70, cleaved PARP, Src, and Akt were purchased from Cell Signaling Technology (Danvers, MA), and antibodies to detect ErbB2 and Hsp90 were purchased from Neomarkers (Kalamazoo, MI), and Pharmingen, respectively. Cycloheximide and the cross-linking agent disuccinimidyl suberate were obtained from Sigma. Peptides were synthesized by Peptide 2.0 (Chantilly, VA) and American Peptide Co. (Sunnyvale, CA). The peptide transfection reagent Chariot was purchased from Active Motif (Carlsbad, CA).
Methods
Cell Culture
EGFR-null CHO cells were purchased from the American Type Culture Collection. The human head and neck squamous cell carcinoma cell line UMSCC1 was kindly provided by Dr. Thomas Carey (University of Michigan, Ann Arbor, MI). The lung cancer cell line NCI-H1975 was provided by Dr. J. A. Engelman (Massachusetts General Hospital, Boston). All cell lines were grown in RPMI 1640 medium supplemented with 10% cosmic calf serum. For all in vitro experiments, cells were released from flasks using PBS containing 0.25% trypsin and 0.2 mm EDTA, and cells were plated onto culture dishes 2 days prior to any treatment.
Immunoblotting
Cells were scraped into PBS containing a sodium orthovanadate and protease inhibitor mixture (Roche Diagnostics). Cells were incubated for 15 min on ice in Laemmli buffer (63 mm Tris-HCl, 2% (w/v) SDS, 10% (v/v) glycerol, and 0.005% (w/v) bromphenol blue) containing 100 mm NaF, 1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, and 1 μg/ml aprotinin. After sonication, particulate material was removed by centrifugation at 13,000 rpm for 15 min at 4 °C. The soluble protein fraction was heated to 95 °C for 5 min, then applied to a 4–12% BisTris precast gel (Invitrogen), and transferred onto a PVDF membrane. Membranes were incubated for 1 h at room temperature in blocking buffer consisting of 3% BSA and 1% normal goat serum in Tris-buffered saline (137 mm NaCl, 20 mm Tris-HCl (pH 7.6), 0.1% (v/v) Tween 20). Membranes were subsequently incubated overnight at 4 °C with 1 μg/ml primary antibody in blocking buffer, washed, and incubated for 1 h with horseradish peroxidase-conjugated secondary antibody (Cell Signaling). After three additional washes in Tris-buffered saline, bound antibody was detected by enhanced chemiluminescence plus reagent (GE Healthcare). For quantification of relative protein levels, immunoblot films were scanned and analyzed using ImageJ 1.32j software (National Institutes of Health, Bethesda). The relative protein levels shown represent a comparison with untreated controls.
Immunoprecipitation
Cells were trypsinized and washed twice with 1× PBS, and cell lysates were prepared by incubation for 30 min on ice in fresh lysis buffer (1% Triton X-100, 0.1% SDS, 150 mm sodium chloride, 10 mm sodium phosphate (pH 7.2), 1 mm phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin, 0.2 mm sodium orthovanadate, 50 mm sodium fluoride, 2 mm EDTA) containing 20 mm ammonium molybdate. Immunoprecipitation of EGFR and Hsp90 was performed as described previously (3).
Site-directed Mutagenesis of EGFR Constructs and Transfection
A modified site-directed mutagenesis protocol was used to create the desired mutations in EGFR. The protocol includes 5′ end phosphorylation of the primer using T4 polynucleotide kinase enzyme followed by PCR with single primer and DpnI enzyme treatment. Primers for site-directed mutagenesis were designed by introducing minimal nucleotide changes in the DNA sequence of EGFR cloned into the N1-EYFP vector (Clontech). Mutations in EGFR were confirmed by the University of Michigan DNA sequencing core facility. CHO cells were transiently transfected with the constructs using Lipofectamine (Invitrogen) according to the instructions of the manufacturer.
Clonogenic Cell Survival Assay
Clonogenic assays were performed using a standard technique described previously (15). Briefly, 500 cells were plated in 60-mm dishes in triplicate, and the next day, cells were treated with Disruptin or the scrambled peptide. Eight to 10 days later, cells were fixed with acetic acid/methanol (1:7, v/v), stained with crystal violet (0.5%, w/v), and counted using a stereomicroscope. The fraction surviving each treatment was normalized to the survival of the control cells. Cell survival curves were fitted using the following equation: SF = (C50)m/((C50)mcm), where SF is the surviving fraction; C is the peptide concentration; C50 is the concentration of peptide that produces a 50% cell survival, and m is the slope of the sigmoid curve.
Half-life Study
CHO cells were transfected with an equal amount of DNA template (1 μg) of WT or 768–773 EGFR (scrambled mutant) constructs. Twelve h post-transfection, cycloheximide (50 μg/ml) was added to cells expressing each of these constructs. Cells were harvested at the indicated times post-treatment, and immunoblotting was carried out for EGFR and GAPDH to analyze the protein half-life of WT and 768–773 scrambled EGFR.
ATP Binding Assay
Cell lysates were prepared in RIPA buffer. About 500 μg of protein was incubated overnight at 4 °C with 25 μl of γ-linked ATP-agarose beads (Innova Biosciences, Cambridge, UK). After centrifugation, beads were washed six times in PBS, and ATP bound proteins were extracted in Laemmli buffer, resolved on a SDS-polyacrylamide gel, and immunoblotted with anti-Hsp90 antibody to detect changes in ATP-bound Hsp90 levels.
RESULTS
Mutation of the Eight-amino Acid Segment and EGFR Stability
The eight-amino acid segment conferring dynamic cycling with Hsp90 consists of residues 768–775 (SVDNPHVC) of the human EGFR. To determine whether this segment plays a major role in receptor stability, we switched amino acids 768–773 (SVDNPH) to a scrambled sequence (NHVPSD), which we call 768–773 ScramEGFR. As shown in Fig. 1A, the level of EGFR was markedly reduced by scrambling the 768–773 amino acids from the wild-type sequence. The reduction is independent of transcription as determined by RT-PCR (data not shown), but as shown in Fig. 1B, the degradation rate is much faster for the scrambled mutant than for the wild-type EGFR. Several single amino acid mutants and one double amino acid mutant within the 768–773 segment were prepared and expressed in CHO cells. The steady state levels of mutant S768A and the double mutant D770A/N771A were reduced relative to wild-type EGFR, but the reduction was minimal compared with the scrambled mutant (Fig. 1C). The amount of mutant EGFR immunoprecipitated with Hsp90 is decreased roughly in proportion to the decreased level of EGFR protein (Input) in Fig. 1C. Even when the 768–773 amino acids are scrambled, the mutant EGFR is bound to Hsp90 in rough proportion to its level in the cell (Fig. 1, A and C), consistent with the notion that Hsp90 associates with a topological feature of the protein rather than a specific motif comprised by this segment.
FIGURE 1.
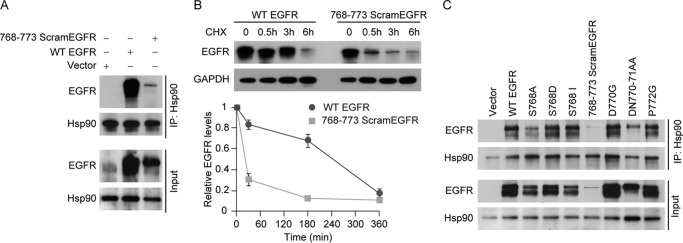
Effect of scrambling amino acids 768–773 on EGFR stability. A, EGFR-null CHO cells transiently expressing wild-type (WT) EGFR or 768–773 ScramEGFR for 24 h were lysed and immunoprecipitated (IP) for Hsp90. Samples of immunoprecipitate or cytosol (Input) were immunoblotted for EGFR and Hsp90. B, to assess receptor stability, cells transfected with WT EGFR or 768–773 ScramEGFR for 12 h were treated with 50 μg/ml of cycloheximide (CHX) for the indicated times, at which cell lysates were immunoblotted for EGFR and GAPDH. The graph shows the relative densities of bands for three experiments expressed as a percent of the time 0 value. Mean ± S.E. C, indicated mutant EGFRs were expressed in CHO cells (24 h); cells were lysed and immunoprecipitated for Hsp90. Samples of immunoprecipitate or cytosol were immunoblotted for EGFR or Hsp90.
Effect of Scrambled Mutation on EGFR Dimerization and Phosphorylation
To determine the effect of scrambling the amino acids in the 768–773 segment on EGFR function, cells expressing full-length wild-type EGFR or the scrambled mutant were treated with EGF, and the dimer was trapped by exposing the cells to the cross-linker disuccinimidyl suberate. As shown in Fig. 2A (top panel), EGF promoted dimerization of the wild-type EGFR, but only a trace amount of dimer was produced by the scrambled mutant. Consistent with the lack of dimerization, the scrambled mutant does not undergo phosphorylation (Fig. 2A, bottom panel). When the V5-tagged kinase domain of wild-type EGFR is expressed in CHO cells, a kinase domain dimer is detected in cells treated with cross-linker, but dimerization is not detected in cells expressing the scrambled mutant kinase domain (Fig. 2B). As shown in Fig. 2C, Hsp90 binds preferentially, and perhaps solely, to the EGFR monomer. One possibility is that the segment determining dynamic versus stable cycling with Hsp90 either overlaps with part of the dimerization interface or that it somehow affects the conformation of the dimerization interface for asymmetric dimerization. In either case, the segment may participate in an intermolecular or intramolecular interaction that could be competed with a peptide containing the eight-amino acid segment.
FIGURE 2.
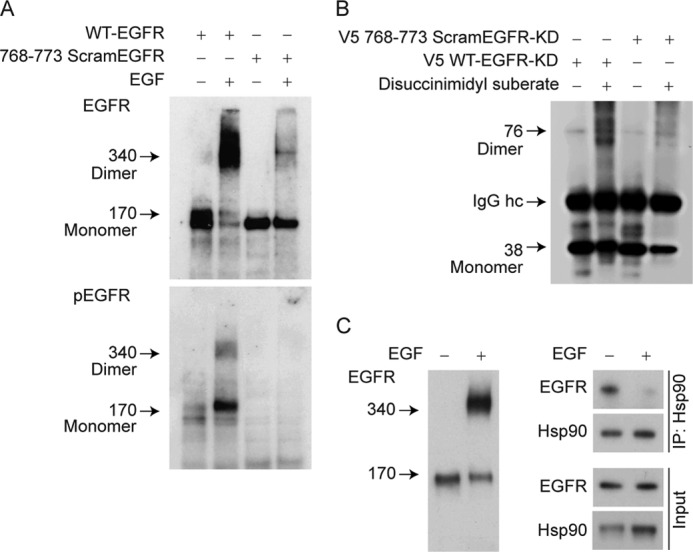
Effect of scrambling amino acids 768–773 on EGF-mediated EGFR dimerization and autophosphorylation. A, effect of scrambling on the full-length EGFR. CHO cells transfected for 24 h with either full-length wild-type EGFR or 768–773 ScramEGFR were treated with EGF (30 ng/ml for 30 min) followed by disuccinimidyl suberate (150 μm for 30 min). Monomers were separated from dimers using reducing SDS-PAGE, and EGFR was immunoblotted for EGFR protein and with anti-EGFR-P845 antiserum to detect tyrosine phosphorylation. B, effect of scrambling on dimer formation by the expressed EGFR kinase domain (EGFR-KD). CHO cells transfected with V5-WT-EGFR- KD or V5–768-773 ScramEGFR-KD were treated with disuccinimidyl suberate. The EGFR-KD was immunoadsorbed with anti-V5 antibody and immunoblotted with the same antibody. IgG hc refers to the immunoglobulin heavy chain. C, Hsp90 binds to the EGFR monomer. UMSCC1 cells were treated with EGF and disuccinimidyl suberate, and a portion of the cell lysate was immunoblotted for EGFR (left panel). A replicate sample of cells treated with EGF in the absence of cross-linker was lysed and immunoadsorbed for Hsp90 (right panel).
Disruptin Peptide Inhibits Hsp90 Binding to EGFR
To determine whether a peptide consisting of the 8-amino acid segment of the EGFR was able to affect cell growth, UMSCC1 cells were treated with the peptide in the presence or absence of the peptide delivery reagent Chariot. As shown in Fig. 3A, the eight-amino acid segment peptide reduced the survival fraction only when Chariot was present, indicating that peptide internalization is required. A peptide consisting of the scrambled sequence had no activity with or without Chariot. Thus, we synthesized two peptides representing the eight-amino acid segment of EGFR, one with the wild-type sequence, which we will call Disruptin, and a second eight-amino acid peptide with the sequence of the first six amino acids scrambled, which we will call Scram-peptide (Fig. 3B). Both peptides were synthesized along with 11 amino acids selected from the HIV-TAT gene to enable cellular uptake (16), and a biotin moiety was attached for molecular studies. To determine the effect of the peptides on EGFR levels, we selected two EGFR-driven cancer cell lines. UMSCC1 is derived from a head and neck tumor, and NCI-H1975 is derived from a non-small cell adenocarcinoma of the lung, which contains the gefitinib/erlotinib-resistant T790M-EGFR mutation. Both cell lines were treated for 24 h with peptide, and treatment with the Hsp90 inhibitor geldanamycin was used as a positive control. In both cell lines, the level of EGFR was reduced by treatment with geldanamycin (Fig. 3C). The levels of Hsp90 and Hsp70 also increase in geldanamycin-treated cells because cycling of heat shock factor 1 (HSF1) with Hsp90 maintains it in an inactive state, and treatment of cells with geldanamycin activates HSF1, which induces the stress proteins (17, 18). Treatment with Disruptin also reduced the level of EGFR bound to Hsp90, but there is little decrease in EGFR protein at a treatment time of 24 h (Fig. 3C). After a longer treatment period, Disruptin causes a marked decrease in EGFR protein levels (Fig. 3D). Because the levels of EGFR are markedly decreased after longer times of Disruptin treatment, the effect of Disruptin treatment on EGFR binding to Hsp90 was quantitated at 24 h, at which time binding to Hsp90 was reduced by 30–40% (Fig. 3E).
FIGURE 3.

Disruptin inhibits Hsp90 binding to the EGFR and promotes EGFR degradation. A, peptide SVDNPHVC must be internalized to be active at growth inhibition. UMSCC1 cells were grown in 6-well plates to clonal density and treated with 30 μg/ml peptide SVDNPHVC or scrambled peptide with or without mixing with the peptide delivery reagent Chariot. Colonies were counted 8 days after treatment. * denotes significant difference from control at p < 0.05. B, structures of Disruptin and the scrambled peptide. C, effect of Disruptin on Hsp90 interaction with the full-length wild-type EGFR. UMSCC1 or NCI-H1975 cells were treated for 24 h with 50 nm geldanamycin (GA), 30 μg/ml Disruptin, or 30 μg/ml scrambled peptide. Hsp90 was immunoprecipitated (IP), and Hsp90 and EGFR in the immunoprecipitate were detected by immunoblotting. D, time course of Disruptin effect on EGFR levels. UMSCC1 and NCI-H1975 cells treated for 24, 48, or 72 h with 30 μg/ml Disruptin or scrambled peptide were lysed and immunoblotted for EGFR. E, quantitation of Disruptin effect on Hsp90 binding to the EGFR. Hsp90 was immunoprecipitated from UMSCC1 cells treated for 24 h with Disruptin or scrambled peptide as in B. Immunoblots of Hsp90-bound EGFR and total EGFR (Input) were scanned, and the Hsp90-bound EGFR is expressed as % of total receptor. Mean ± S.E. from four experiments. F, disruptin does not inhibit Hsp90 binding to the GR. Aliquots of immunoadsorbed GR were stripped of chaperones with NaCl and incubated with rabbit reticulocyte lysate as described previously (19). After washing, the immune pellets were immunoblotted for GR and associated Hsp90 and Hsp70 or bound with [3H]dexamethasone for assay of steroid binding activity. Lane 1, stripped GR; lanes 2–4, stripped GR incubated with 50 μl of reticulocyte lysate alone (lane 2) or reticulocyte lysate plus 300 μg/ml Disruptin (lane 3) or reticulocyte lysate plus 10 μm geldanamycin (lane 4). G, disruptin does not affect ATP binding activity of Hsp90. UMSCC1 cells were treated for 3 h with 50 nm geldanamycin or 30 μg/ml of Disruptin or scrambled peptide. Cell lysates were prepared and incubated overnight with ATP-agarose beads, and bound Hsp90 was immunoblotted.
Disruptin does not increase stress protein levels (Fig. 3C), suggesting that the peptide is not an inhibitor of Hsp90 cycling in general. To test this notion further, we asked whether Disruptin would affect assembly of the glucocorticoid receptor (GR)·Hsp90 heterocomplex by the Hsp90/Hsp70-based chaperone machinery in reticulocyte lysate. The GR is the most studied of the Hsp90 client proteins, and it must be in heterocomplex with Hsp90 for it to have high affinity steroid binding activity (7, 13). When GR that is stripped of Hsp90 is incubated with reticulocyte lysate, GR·Hsp90 complexes are formed, and steroid binding activity is activated (19). As shown in Fig. 3F, even when it is present at 10 times the concentration used elsewhere in this work, Disruptin does not affect GR·Hsp90 heterocomplex assembly or activation of steroid binding activity. The failure of Disruptin to inhibit functional Hsp90 heterocomplex assembly with this classic Hsp90 client protein argues strongly that Disruptin does not function by interacting directly with Hsp90. Also, in contrast to geldanamycin, Disruptin clearly does not affect ATP binding by Hsp90 (Fig. 3G).
Selectivity of Disruptin for EGFR in UMSCC1 Cells
The selectivity of the Disruptin effect is highlighted by the experiments of Fig. 4. UMSCC1 cells grown for 72 h in the presence of geldanamycin or Disruptin were lysed and immunoblotted for ErbB2, Src, and Akt, three signaling protein kinases that are well established Hsp90 client proteins (13). As shown in Fig. 4A, geldanamycin decreases the levels of the three established Hsp90 clients as well as EGFR. In contrast, Disruptin decreases the level of EGFR but not the levels of ErbB2, Src, or Akt. The effect of Disruptin is dependent upon peptide concentration, and the established Hsp90 client proteins are not affected even at very high levels of peptide (Fig. 4B).
FIGURE 4.
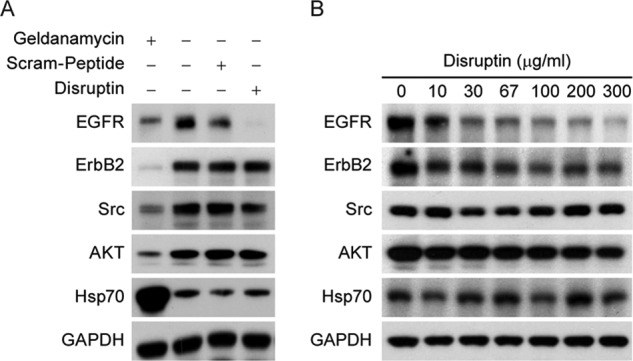
Selectivity of Disruptin for EGFR. A, selectivity of 72 h of treatment with Disruptin. UMSCC1 cells were treated for 72 h with 50 nm geldanamycin, 30 μg/ml Disruptin, or 30 μg/ml scrambled peptide, and cells were lysed and immunoblotted for the indicated proteins. B, concentration dependence of Disruptin effect at 24 h. UMSCC1 cells were treated with the indicated concentrations of Disruptin for 24 h, lysed, and immunoblotted for the indicated proteins.
Disruptin Inhibits EGFR Dimerization
The specificity of Disruptin for EGFR may reflect direct interaction of the peptide with the receptor to inhibit dimerization. To test this possibility, UMSCC1 cells were first treated with Disruptin and then with EGF and with disuccinimidyl suberate to trap the dimer. We have found that EGFR degradation is especially rapid in cells treated with Disruptin and EGF together. Although cells treated in this manner have less overall (monomer plus dimer) EGFR, receptor dimerization is inhibited by Disruptin but not by the scrambled peptide (Fig. 5A). The fraction of EGFR in dimer form was determined by scanning the bands in three experiments to demonstrate the inhibition of EGFR dimerization (Fig. 5B). The time courses in Fig. 5, C and D, illustrate the rapidity of EGFR degradation in the presence of EGF and Disruptin. To test for a direct interaction, we have incubated cell lysate with streptavidin-immobilized Disruptin and immunoblotted the washed pellet proteins for EGFR and Hsp90. Unfortunately, we have not been able to detect a clearly specific binding to either protein by this approach.
FIGURE 5.

Disruptin inhibits EGFR dimerization. A, inhibition of dimerization. UMSCC1 cells were treated for 1 h with 30 μg/ml Disruptin or scrambled peptide and an additional 30 min with EGF (30 ng/ml) and disuccinimidyl suberate (150 μm). Lysates were prepared and immunoblotted for EGFR. B, quantitation of inhibition. The monomer and dimer bands from three experiments performed as in A were scanned to determine total EGFR and the fraction of receptor in dimer form. C, rate of EGFR degradation. UMSCC1 cells were treated simultaneously with vehicle (control), 30 ng/ml EGF, 30 μg/ml Disruptin, or both EGF and Disruptin, and at the indicated times, lysates were prepared and immunoblotted for EGFR. D, blots were scanned, and data are mean ± S.E. for three experiments. ** denotes significant difference from EGF alone at p < 0.01.
Disruptin Inhibits Cell Growth
To determine whether Disruptin inhibits the growth of the two EGFR-driven cancer cell lines, UMSCC1 and NCI-H1975 cells were treated with Disruptin for 72 h, and cell survival was determined with a clonogenic assay. Again, the effect of Disruptin was compared with that of the Hsp90 inhibitor geldanamycin, which is known to kill cancer cells through an apoptotic mechanism (20, 21). Using PARP cleavage as an indicator of apoptosis, we found that geldanamycin induced PARP cleavage but Disruptin did not (Fig. 6A). Disruptin decreased the survival fraction about as much as geldanamycin, but the scrambled peptide did not (Fig. 6B). This suggests that Disruptin is inhibiting cell replication or may be killing cells through a nonapoptotic mechanism.
FIGURE 6.
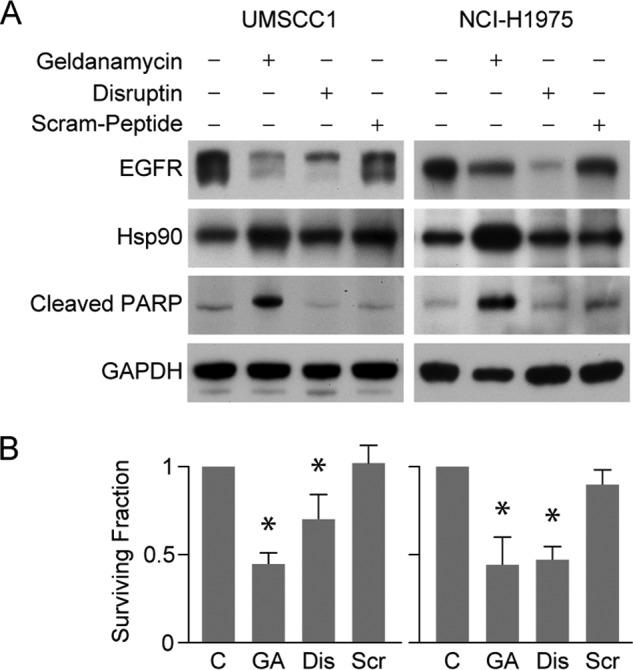
Disruptin inhibits cell growth. A, in contrast to geldanamycin, Disruptin does not induce PARP cleavage. UMSCC1 or NCI-H1975 cells were treated for 72 h with 50 nm geldanamycin, 30 μg/ml Disruptin, or 30 μg/ml scrambled peptide. Cell lysates were then immunoblotted for the indicated proteins. B, cell survival was assessed using a colony formation assay. C, control; GA, geldanamycin; Dis, Disruptin; Scr, Scram-peptide. Mean ± S.E. from three experiments. * denotes significant difference from control at p < 0.05.
DISCUSSION
Several mutational studies of EGFR (ErbB1) and ErbB2 have established the importance of the eight-amino acid segment within the αC-β4 loop region of the kinase for Hsp90 binding and regulation (6, 10, 11, 22). In a broad examination of this segment in multiple human kinases, Citri et al. (11) established that of 105 kinases, 80 were Hsp90 clients and 25 were non-clients. They concluded that although the αC-β4 loop is critical for defining interaction with Hsp90, recognition of kinases in general by Hsp90 does not depend on sequence motifs within this region but is most likely based on surface characteristics of the kinase within this region (11). Thus, within the kinases in general, it would seem that surface charge and hydrophobicity are critical properties determining Hsp90 interaction as they are for ErbB2 (22). In reviewing Hsp90 interaction with a variety of Hsp90-regulated proteins, we have proposed that the general feature on protein surfaces with which Hsp90 interacts is the region where hydrophobic ligand binding clefts merge with the charged surface of the protein (7, 13). In the case of the kinases, it would be the opening of the ATP binding cleft that would provide the interaction site for the chaperone.
In their original paper showing that the kinase domain of ErbB2 is the site of Hsp90 binding, Xu et al. (8) found that only the nascent EGFR was sensitive to geldanamycin and that, in contrast to ErbB2, Hsp90 was not coimmunoprecipitated with mature EGFR. Thus, until we recently reported that mature, wild-type EGFR is stabilized by direct interaction with Hsp90 in cancer cells (3), the EGFR was not considered to be an Hsp90-regulated protein. Over the course of the past decade, it has been established that the turnover of proteins that cycle more dynamically with Hsp90 than the classic “client” proteins, such as ErbB2, are regulated in a less stringent manner (7). Some of these more dynamically cycling proteins, such as nitric-oxide synthases, EGFR, and perhaps some of the 25 kinases designated as non-clients by Citri et al. (11), may prove to be useful therapeutic targets for chaperone manipulation. The close proximity of the EGFR dimerization interface, the Hsp90 binding region, and the ATP-binding site to the eight-amino acid segment determining geldanamycin sensitivity makes this segment an attractive target for developing drugs that are different from the kinase inhibitors gefitinib and erlotinib but specifically target the EGFR for degradation.
Here, we have shown that scrambling the sequence of six amino acids within the segment markedly destabilizes the EGFR with respect to the wild-type receptor (Fig. 1) and impairs EGF-dependent receptor dimerization and phosphorylation (Fig. 2). The peptide Disruptin containing the wild-type sequence selectively (with respect to the scrambled peptide) disrupts EGFR interaction with Hsp90 and destabilizes both wild-type and gefitinib/erlotinib-resistant EGFR (T790M mutant in NCI-H1975 cells). Both disruption of the interaction with Hsp90 and destabilization are specific for EGFR as opposed to classic Hsp90 clients, such as the GR, ErbB2, Src, and Akt (Figs. 3 and 4).
In a recent crystallographic study of the kinase domain of EGFR in complex with a dual EGFR/HER2 inhibitor, TAK-285, the eight-amino acid segment, was shown to be located on the surface of EGFR at the heterodimerization interface (23). The initial SVDN in the eight-amino acid segment is thought to play a critical role in kinase activation (24). It is not unlikely that Disruptin specifically affects the EGFR by competing with the eight-amino acid segment for binding at the dimerization interface, which is also where Hsp90 binds. This would account for both Disruptin inhibition of Hsp90 binding to the EGFR monomer and inhibition of EGF-stimulated EGFR dimerization. It is also consistent with the original proposal of Citri et al. (6) that Hsp90 binds to the eight-amino acid segment in ErbB2/HER2 to restrain heterodimer formation and catalytic function.
Both inhibition of stabilization by Hsp90 and inhibition of EGF-stimulated dimerization appear to be involved in EGFR degradation upon Disruptin treatment. In the absence of EGF, Disruptin-induced EGFR degradation occurs over several days (Fig. 3D), whereas in the presence of EGF, there is marked Disruptin-induced EGFR degradation within 2 h (Fig. 5C). It seems reasonable to propose that in the absence of EGF, cycling with Hsp90 keeps a monomer/dimer equilibrium strongly in favor of the monomer. In this case, Disruptin is competing with the stabilizing effect of the Hsp90/Hsp70-based chaperone machinery in cycling Hsp90 with the otherwise unstable monomer. Upon EGF binding, the EGFR undergoes a conformational change in the region of the αC helix (4) such that the receptor can no longer cycle with Hsp90. Disruptin inhibits EGFR dimerization, but the EGF-bound EGFR cannot be stabilized by Hsp90. Thus, in the presence of EGF, the EGFR is degraded very rapidly when Disruptin is also present. It would seem that Disruptin is more effective at inhibiting EGFR dimerization than cycling of the ligand-free EGFR with Hsp90.
We and others have found that EGFR degradation increases tumor cell-specific cytotoxicity of chemotherapy and radiotherapy beyond that of EGFR inhibition alone (25–27). These studies suggest that not just inhibition of EGFR tyrosine kinase activity but down-regulation of EGFR is an important target in cancer therapy (28, 29). EGFR degradation can be achieved with ansamycin analogues, such as geldanamycin or allylamino-17-demethoxygeldanamycin, which significantly enhance both chemosensitivity and radiosensitivity (30, 31). However, as illustrated in Fig. 4A, an inhibitor of Hsp90 causes the degradation of the many proteins that are Hsp90 clients, and such inhibitors have proven to be very toxic in clinical trials (32, 33). In contrast, an approach that promotes the selective degradation of EGFR might be considerably less toxic in the treatment of EGFR-driven tumors.
We have developed Disruptin for use in mechanistic studies as reported here and as proof of concept for an agent that is specific for promoting EGFR degradation. However, it should be noted that the perception that peptides make poor drugs is rapidly changing as an increasing number of peptides have entered the therapeutic pipeline (34–37). Although most therapeutic peptides are directed toward extracellular targets (38), the increasing use of cell-penetrating peptide sequences to transport “cargo,” including peptide therapeutics, across the cell membrane has opened the door to many intracellular targets, including many proteins implicated in cancer (39, 40). Recently, small peptides have shown efficacy in blocking the interaction of Bcl-XL with Bax (34) and of p53 with MDM2 (36) in preclinical cancer therapy models. At present, we have no indication that the use of Disruptin would be therapeutically viable, either alone or by potentiating the effects of radiation or chemotherapy. It is possible that the transfer of key structural features from the peptide to a peptidomimetic scaffold of smaller size will yield molecules with similar activity to Disruptin but with improved pharmacokinetic properties.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA131290 (to M. K. N.), R01CA160981 (to D. R.), GM077430 (to Y. O.), P50CA097248 (project IV to M. K. N.), P50CA097248 (Career Development award to A. A.), and Cancer Center Support Grant P30CA46592. This work was also supported by the Michigan Institute for Clinical and Health Research (to M. K. N.), the James Stuart and Barbara Padnos Funds for Cancer Research (to M. K. N.), and an Alfred Taubman Scholarship (to T. S. L.). This project is a component of the University of Michigan Medical School's Fast Forward Strategic Research Initiative.
This article was selected as a Paper of the Week.
- EGFR
- EGF receptor
- Hsp
- heat shock protein
- PARP
- poly(ADP ribose) polymerase
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- GR
- glucocorticoid receptor.
REFERENCES
- 1. Yarden Y., Sliwkowski M. X. (2001) Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137 [DOI] [PubMed] [Google Scholar]
- 2. Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 [DOI] [PubMed] [Google Scholar]
- 3. Ahsan A., Ramanand S. G., Whitehead C., Hiniker S. M., Rehemtulla A., Pratt W. B., Jolly S., Gouveia C., Truong K., Van Waes C., Ray D., Lawrence T. S., Nyati M. K. (2012) Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors. Neoplasia 14, 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang X., Gureasko J., Shen K., Cole P. A., Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 5. Shan Y., Eastwood M. P., Zhang X., Kim E. T., Arkhipov A., Dror R. O., Jumper J., Kuriyan J., Shaw D. E. (2012) Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 149, 860–870 [DOI] [PubMed] [Google Scholar]
- 6. Citri A., Gan J., Mosesson Y., Vereb G., Szollosi J., Yarden Y. (2004) Hsp90 restrains ErbB-2/HER2 signalling by limiting heterodimer formation. EMBO Rep. 5, 1165–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pratt W. B., Morishima Y., Osawa Y. (2008) The Hsp90 chaperone machinery regulates signaling by modulating ligand binding clefts. J. Biol. Chem. 283, 22885–22889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu W., Mimnaugh E., Rosser M. F., Nicchitta C., Marcu M., Yarden Y., Neckers L. (2001) Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J. Biol. Chem. 276, 3702–3708 [DOI] [PubMed] [Google Scholar]
- 9. Citri A., Alroy I., Lavi S., Rubin C., Xu W., Grammatikakis N., Patterson C., Neckers L., Fry D. W., Yarden Y. (2002) Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J. 21, 2407–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tikhomirov O., Carpenter G. (2003) Identification of ErbB-2 kinase domain motifs required for geldanamycin-induced degradation. Cancer Res. 63, 39–43 [PubMed] [Google Scholar]
- 11. Citri A., Harari D., Shohat G., Ramakrishnan P., Gan J., Lavi S., Eisenstein M., Kimchi A., Wallach D., Pietrokovski S., Yarden Y. (2006) Hsp90 recognizes a common surface on client kinases. J. Biol. Chem. 281, 14361–14369 [DOI] [PubMed] [Google Scholar]
- 12. Huse M., Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282 [DOI] [PubMed] [Google Scholar]
- 13. Pratt W. B., Toft D. O. (2003) Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. (Maywood) 228, 111–333 [DOI] [PubMed] [Google Scholar]
- 14. Kaul S., Murphy P. J., Chen J., Brown L., Pratt W. B., Simons S. S., Jr. (2002) Mutations at positions 547–553 of rat glucocorticoid receptors reveal that hsp90 binding requires the presence, but not defined composition, of a seven-amino acid sequence at the amino terminus of the ligand binding domain. J. Biol. Chem. 277, 36223–36232 [DOI] [PubMed] [Google Scholar]
- 15. Lawrence T. S. (1988) Reduction of doxorubicin cytotoxicity by ouabain: correlation with topoisomerase-induced DNA strand breakage in human and hamster cells. Cancer Res. 48, 725–730 [PubMed] [Google Scholar]
- 16. Futaki S., Suzuki T., Ohashi W., Yagami T., Tanaka S., Ueda K., Sugiura Y. (2001) Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 276, 5836–5840 [DOI] [PubMed] [Google Scholar]
- 17. Zou J., Guo Y., Guettouche T., Smith D. F., Voellmy R. (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94, 471–480 [DOI] [PubMed] [Google Scholar]
- 18. Bagatell R., Paine-Murrieta G. D., Taylor C. W., Pulcini E. J., Akinaga S., Benjamin I. J., Whitesell L. (2000) Induction of heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin. Cancer Res. 6, 3312–3318 [PubMed] [Google Scholar]
- 19. Morishima Y., Lau M., Peng H. M., Miyata Y., Gestwicki J. E., Pratt W. B., Osawa Y. (2011) Heme-dependent activation of neuronal nitric-oxide synthase by cytosol is due to an Hsp70-dependent, thioredoxin-mediated thiol-disulfide interchange in the heme/substrate binding cleft. Biochemistry 50, 7146–7156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nimmanapalli R., O'Bryan E., Bhalla K. (2001) Geldanamycin and its analogue 17-allylamino-17-demethoxygeldanamycin lowers Bcr-Abl levels and induces apoptosis and differentiation of Bcr-Abl-positive human leukemic blasts. Cancer Res. 61, 1799–1804 [PubMed] [Google Scholar]
- 21. Hostein I., Robertson D., DiStefano F., Workman P., Clarke P. A. (2001) Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 61, 4003–4009 [PubMed] [Google Scholar]
- 22. Xu W., Yuan X., Xiang Z., Mimnaugh E., Marcu M., Neckers L. (2005) Surface change and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat. Struct. Mol. Biol. 12, 120–126 [DOI] [PubMed] [Google Scholar]
- 23. Aertgeerts K., Skene R., Yano J., Sang B. C., Zou H., Snell G., Jennings A., Iwamoto K., Habuka N., Hirokawa A., Ishikawa T., Tanaka T., Miki H., Ohta Y., Sogabe S. (2011) Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 286, 18756–18765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Monsey J., Shen W., Schlesinger P., Bose R. (2010) Her4 and Her2/neu tyrosine kinase domains dimerize and activate in a reconstituted in vitro system. J. Biol. Chem. 285, 7035–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Feng F. Y., Lopez C. A., Normolle D. P., Varambally S., Li X., Chun P. Y., Davis M. A., Lawrence T. S., Nyati M. K. (2007) Effect of epidermal growth factor receptor inhibitor class in the treatment of head and neck cancer with concurrent radiotherapy in vivo. Clin. Cancer Res. 13, 2512–2518 [DOI] [PubMed] [Google Scholar]
- 26. Ahsan A., Hiniker S. M., Davis M. A., Lawrence T. S., Nyati M. (2009) Role of cell cycle in epidermal growth factor receptor inhibitor-mediated radiosensitization. Cancer Res. 69, 5108–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ahsan A., Hiniker S. M., Ramanand S. G., Nyati S., Hegde A., Helman A., Menawat R., Bhojani M. S., Lawrence T. S., Nyati M. K. (2010) Role of epidermal growth factor receptor degradation in cisplatin-induced cytotoxicity in head and neck cancer. Cancer Res. 70, 2862–2869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burtness B. (2005) The role of cetuximab in the treatment of squamous cell cancer of the head and neck. Expert Opin. Biol. Ther. 5, 1085–1093 [DOI] [PubMed] [Google Scholar]
- 29. Kirisits A., Pils D., Krainer M. (2007) Epidermal growth factor receptor degradation: an alternative view of oncogenic pathways. Int. J. Biochem. Cell Biol. 39, 2173–2182 [DOI] [PubMed] [Google Scholar]
- 30. Kim W. Y., Oh S. H., Woo J. K., Hong W. K., Lee H. Y. (2009) Targeting heat shock protein 90 overrides the resistance of lung cancer cells by blocking radiation-induced stabilization of hypoxia-inducible factor-1α. Cancer Res. 69, 1624–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bisht K. S., Bradbury C. M., Mattson D., Kaushal A., Sowers A., Markovina S., Ortiz K. L., Sieck L. K., Isaacs J. S., Brechbiel M. W., Mitchell J. B., Neckers L. M., Gius D. (2003) Geldanamycin and 17-allylamino-17-demethoxygeldanamycin potentiate the in vitro and in vivo radiation response of cervical tumor cells via the heat shock protein 90-mediated intracellular signaling and cytotoxicity. Cancer Res. 63, 8984–8995 [PubMed] [Google Scholar]
- 32. Sharp S., Workman P. (2006) Inhibitors of the HSP90 molecular chaperone: current status. Adv. Cancer Res. 95, 323–348 [DOI] [PubMed] [Google Scholar]
- 33. Pacey S., Wilson R. H., Walton M., Eatock M. M., Hardcastle A., Zetterlund A., Arkenau H. T., Moreno-Farre J., Banerji U., Roels B., Peachey H., Aherne W., de Bono J. S., Raynaud F., Workman P., Judson I. (2011) A phase 1 study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin. Cancer Res. 17, 1561–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. LaBelle J. L., Katz S. G., Bird G. H., Gavathiotis E., Stewart M. L., Lawrence C., Fisher J. K., Godes M., Pitter K., Kung A. L., Walensky L. D. (2012) A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J. Clin. Invest. 122, 2018–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rérole A. L., Gobbo J., De Thonel A., Schmitt E., Pais de Barros J. P., Hammann A., Lanneau D., Fourmaux E., Deminov O., Micheau O., Lagrost L., Colas P., Kroemer G., Garrido C. (2011) Peptides and aptamers targeting HSP70: a novel approach for anticancer chemotherapy. Cancer Res. 71, 484–495 [DOI] [PubMed] [Google Scholar]
- 36. Liu M., Li C., Pazgier M., Li C., Mao Y., Lv Y., Gu B., Wei G., Yuan W., Zhan C., Lu W. Y., Lu W. (2010) D-peptide inhibitors of the p53-MDM2 interaction for targeted therapy of malignant neoplasms. Proc. Natl. Acad. Sci. U.S.A. 107, 14321–14326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen S., Zhang P., Lovchik M. A., Li Y., Tang L., Chen Z., Zeng R., Ma D., Yuan J., Yu Q. (2009) Cyclodepsipeptides toxin promotes the degradation of Hsp90 client proteins through chaperone-mediated autophagy. J. Cell Biol. 185, 629–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shrivastava A., Nunn A. D., Tweedle M. F. (2009) Designer peptides: learning from nature. Curr. Pharm. Des. 15, 675–681 [DOI] [PubMed] [Google Scholar]
- 39. Heitz F., Morris M. C., Divita G. (2009) Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br. J. Pharmacol. 157, 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sugahara K. N., Teesalu T., Karmali P. P., Kotamraju V. R., Agemy L., Greenwald D. R., Ruoslahti E. (2010) Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 328, 1031–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]