

Background: Letm1 is a mitochondrial protein attributed disparate roles, including cation/proton antiport and translation.
Results: Letm1 RNAi silencing in Trypanosoma brucei triggers swelling mitochondria and translation arrest that is ameliorated by chemical potassium/proton exchangers.
Conclusion: The ancestral function of Letm1, to maintain mitochondrial potassium homeostasis, shows remarkable conservation.
Significance: Results from diverged T. brucei provide a better understanding of Letm1 function throughout eukaryotes.
Keywords: Bioenergetics, Mitochondria, Potassium Transport, Translation, Trypanosome, Letm1
Abstract
Letm1 is a conserved protein in eukaryotes bearing energized mitochondria. Hemizygous deletion of its gene has been implicated in symptoms of the human disease Wolf-Hirschhorn syndrome. Studies almost exclusively performed in opisthokonts have attributed several roles to Letm1, including maintaining mitochondrial morphology, mediating either calcium or potassium/proton antiport, and facilitating mitochondrial translation. We address the ancestral function of Letm1 in the highly diverged protist and significant pathogen, Trypanosoma brucei. We demonstrate that Letm1 is involved in maintaining mitochondrial volume via potassium/proton exchange across the inner membrane. This role is essential in the vector-dwelling procyclic and mammal-infecting bloodstream stages as well as in Trypanosoma brucei evansi, a form of the latter stage lacking an organellar genome. In the pathogenic bloodstream stage, the mitochondrion consumes ATP to maintain an energized state, whereas that of T. brucei evansi also lacks a conventional proton-driven membrane potential. Thus, Letm1 performs its function in different physiological states, suggesting that ion homeostasis is among the few characterized essential pathways of the mitochondrion at this T. brucei life stage. Interestingly, Letm1 depletion in the procyclic stage can be complemented by exogenous expression of its human counterpart, highlighting the conservation of protein function between highly divergent species. Furthermore, although mitochondrial translation is affected upon Letm1 ablation, it is an indirect consequence of K+ accumulation in the matrix.
Introduction
Letm1 (leucine zipper EF hand-containing transmembrane protein 1) is evolutionarily conserved in diverse eukaryotic lineages bearing energized mitochondria, ranging from opisthokonts, comprising metazoa and fungi, to plastid-containing plants and apicomplexans (1, 2). Letm1, a protein predicted to be embedded into the mitochondrial (mt)4 inner membrane (IM) via a predicted transmembrane domain (1, 3, 4), came into prominence because its gene locus is often within a deletion, occurring to different extents, on the short arm of human chromosome 4, causing Wolf-Hirschhorn syndrome (5). Symptoms of this disease, affecting 1 in 20,000–50,000 births, are multifarious but often include facial abnormalities, various degrees of mental retardation, and seizures (6). The loss of Letm1 has been implicated in the development of the final symptom because patients with deletions that exclude this locus do not exhibit seizures (7, 8).
The first hint of the role of Letm1 on the cellular level emerged from a deletion mutant screen for mt defects performed in Saccharomyces cerevisiae (9). The swollen appearance of the organelle in the Letm1 knock-out yeast strains prompted the authors to dub it MDM38, representing another alias for the protein, to reflect its effect on mitochondrial distribution and morphology. RNAi silencing of Letm1 in other opisthokont models like human cell cultures, Drosophila melanogaster, and Caenorhabditis elegans also resulted in swollen and fragmented mitochondria (3, 10–12), suggesting a conservation of function at least within this clade. This notion is further supported by the successful complementation of yeast Letm1 knockout by expression of the human ortholog (1).
However, how Letm1 operates on the cellular level remains debated. Given its dramatic effect on mt morphology, it has been proposed to play an undefined structural role in the human organelle, particularly in maintaining the cristae that form inner membrane invaginations into the matrix (12). This morphological function was determined to operate independently of the fission and fusion machineries that maintain the mt network in these cells (3, 12).
Letm1 has also been hypothesized to take part in maintaining matrix volume as a cation/proton (H+) antiporter. This function would also be consistent with the observed swollen mitochondria phenotype upon depletion of Letm1 because this treatment would negatively impact ion homeostasis and cause organellar osmotic stress. However, the identity of the cation that is translocated by Letm1 remains controversial. Several compelling studies in yeast, Drosophila, and human cell culture have shown that it has a central part in potassium/proton exchange (KHE), which maintains matrix volume by regulating potassium (K+) extrusion (1, 3, 10, 13). In all of these model systems, treatment with the chemical K+/H+ exchanger nigericin compensates for the loss of Letm1-mediated KHE. However, Letm1 was also identified as a calcium (Ca2+)/H+ antiporter in the genome-wide RNAi screen in Drosophila S2 cells (14), a finding corroborated in a later report (15).
Yet another role that has been attributed to Letm1 in S. cerevisiae is the anchoring of mt ribosomes to the inner membrane, into which it facilitates the incorporation of hydrophobic de novo translated subunits of the respiratory chain (4, 16, 17). This path of inquiry began with an observed reduction of the steady-state levels of a subset of mitochondrially encoded proteins in Letm1 knockouts (4). A similar phenomenon was also reported in Arabidopsis thaliana bearing simultaneous homozygous and hemizygous knockouts of its two Letm1 paralogs (18). Further support for this role, albeit indirect, was the report that Letm1 silencing in HeLa cells resulted in the disassembly of some respiratory chain complexes (12), which was nevertheless contradicted by another similar study on the same cell type (3).
To date, our understanding of Letm1 is rather convoluted. To shed light on this situation, we have undertaken functional analysis of Letm1 (TriTrypDB genome database accession number Tb927.3.4920 (19)) in the protozoan flagellate Trypanosoma brucei. As a member of the Kinetoplastea, it has a long and independent evolutionary history, perhaps due to its early branching from other eukaryotic lineages (20, 21). Kinetoplastids contain a single mitochondrion, with its organellar genome located in a discrete place as the giant kinetoplast DNA (kDNA) network (22). Most of the kDNA-encoded transcripts undergo elaborate post-transcriptional processing called RNA editing.
Research on kinetoplastids has proven to be invaluable to the field of mt comparative biochemistry. Notably, seminal work described Ca2+ influx into the trypanosome mitochondrion in a ruthenium red-sensitive fashion (23, 24). These data became critical for the algorithm to identify conserved proteins responsible for Ca2+ uptake, by testing a pool of mt proteins shared by kinetoplastids and vertebrates but absent in yeast, which lack this activity. Thus, after half a century of characterizing this activity, some of the responsible proteins have been identified (25–28).
T. brucei subspecies are the causative agents of a human disease with the familiar name sleeping sickness as well as the veterinarian disease nagana (29). These diseases are spread by the tsetse fly vector in sub-Saharan Africa. The parasite undergoes several morphological and physiological changes as it cycles between the mammalian host and insect vector (30), notably within its mitochondrion (22). In the procyclic stage (PS) that resides in the midgut of the vector, the organelle engages in oxidative phosphorylation to generate ATP, as in canonical mitochondria. The proliferative long slender bloodstream stage (BS) that is pathogenic for the mammalian host generates energy exclusively by glycolysis. In this milieu, the mitochondrion is not only reduced, as exemplified by its paucity of cristae and lack of cytochrome-containing respiratory complexes, but also becomes an energy consumer. Membrane potential is maintained by the remaining FOF1-ATP synthase, which hydrolyzes ATP to pump H+ out of the matrix (31, 32). However, the BS mitochondrion is not dormant because organellar gene expression is still needed for cell viability (33–36), and a handful of essential mt biochemical pathways have been revealed (31, 32, 37–39).
Interestingly, a subspecies called T. brucei evansi is a naturally occurring form of T. brucei without kDNA and the causative agent of the ungulate disease surra (31, 40). This parasite is the equivalent of petite mutant (ρ0) yeast, viable only in the fermentable medium of the bloodstream. As a consequence, it has lost the capacity to transform to the PS, which requires an energy-producing organelle and is instead spread mechanically by biting insects. T. brucei evansi bears compensatory mutations to the γ-subunit in the stalk of FoF1-ATP synthase, most likely facilitating the complex's competence for ATP hydrolysis in the absence of the mitochondrially encoded subunit A6 (31, 40). This activity in concert with that of the mt ATP/ADP carrier protein maintains the electrogenic component of the membrane potential by the antipodal exchange of ATP4− and ADP3−.
In this study, we take advantage of RNAi permissibility and ease of transgenesis of PS, BS, and T. brucei evansi in in vitro cultures to generate conditional knockdown cell lines to test the effect of Letm1 silencing in three different physiological states of the mitochondrion. We also compare our results in this highly diverged organism with the previously enumerated results from opisthokont model systems in order to elucidate the basal function of the evolutionarily conserved Letm1. Although this study does not represent the first one performed outside of the opisthokont clade, as the aforementioned report in Plantae can attest (18), this is for the first time when almost complete silencing of Letm1 has been achieved, yielding a clear and robust phenotype. This study also reveals yet another essential function of the T. brucei BS mitochondrion: the maintenance of ion homeostasis.
EXPERIMENTAL PROCEDURES
Cloning, Cultivation, Transfection, Growth Curves, and 5′-End Mapping of Letm1 mRNA
PS and BS T. brucei as well as T. brucei evansi were cultured, transfected, and selected for the relevant drug resistance for each of the given constructs and counted as described elsewhere (31, 34). A Letm1 gene fragment amplified using forward primer GGATCCGGTCAAGCCTACCCGATACA (introduced BamHI site underlined) and reverse primer AGGCCTTCGGTAATTGCCTTCACTCC (HindIII site underlined) was cloned into the p2T7-177 vector, bearing opposing T7 polymerase promoters/tetracycline operators and targeted to a transcriptionally silent part of the T. brucei genome (41), via the indicated restriction sites. For in situ C-terminal tagging of Letm1 with YFP, the full open reading frame (ORF) excluding the stop codon was PCR-amplified with the forward primer GGTACCATGTTGG CAGCAACGGGGTT (Acc65I restriction site underlined) and reverse primer GGATCCATTT TTTGCAATCACCTCTGAAGGCT (BamHI site underlined) and cloned into the p2937 vector, derived from the p2710 vector bearing the blasticidin resistance marker (42). The construct was linearized using the unique NcoI restriction site within the Letm1 ORF to yield homology flanks for integration into the endogenous locus. The full ORF of HsLetm1 was PCR-amplified from cDNA (clone FLJ81927AAAF) supplied by the National Institute of Technology and Evaluation Biological Resource Center (Japan) using the forward primer TCAGATCTGCTCTTCACCTCTGCGA and reverse primer TCAGATCTTTGCTTCATGGC GTTGA and cloned into the pABPURO vector (43) via the underlined BglII restriction sites. We took advantage of every mRNA bearing a spliced leader RNA sequence by amplifying the 5′-end of Letm1 with the canonical spliced leader RNA forward primer and the reverse primer AGACATTAAACGGCCCTTCC, as described previously (44).
Indirect Immunofluorescence and Confocal and Electron Microscopy
Indirect immunofluorescence was performed as described elsewhere (34) except that the fixed samples were permeabilized with 0.15% Triton X-100 in PBS (v/v). Samples were decorated with primary rabbit antibodies against either GFP or HA3, depending on the epitope and then subsequently with Alexa-488-conjugated anti-rabbit secondary antibody. Prior to this procedure, 2 × 106 live PS cells were incubated for 20 min at 27 °C with 100 nm MitoTracker Red CMXRos. All antibodies and dyes were from Molecular Probes. Transmission electron microscopy was performed as before (40).
Digitonin Fractionation, Western Blot Analysis, and Triton X-114 Separation of Membrane Proteins
Digitonin fractionation of cells into cytosolic and mitochondrial compartments and Western blots were performed as described previously (36). Antibodies against the T. brucei mitochondrial heat shock protein, cytochrome c, TrCOIV, and the β-subunit of F1-ATPase (31, 45) were used at 1:1000 dilutions, whereas enolase was used at 1:10,000 dilutions. The Triton X-114 isolation of membrane and soluble proteins was performed as described previously (46) on mitochondria hypotonically isolated from PS T. brucei by an established method (47). Acetone-precipitated proteins from the fractions were resuspended in equal volumes of ultrapure water.
Isolation of Submitochondrial Particles and Proteinase K Protection Assay
Hypotonically isolated mitochondria were further processed to generate submitochondrial particles (SMPs) by adapting procedures described previously (13, 45). Briefly, a mitochondria suspension corresponding to 2.4 mg/ml protein was sonicated with three 10-s pulses (50% amplitude, 1 Hz) followed by 1-min pauses in ice water using a UP200S Ultrasonic Processor (Hielscher Ultrasound Technology). The SMPs were sedimented at 31,000 × g for 5 h at 4 °C. For the proteinase K protection assay, the SMPs were resuspended at a concentration of 1 mg/ml protein and incubated with or without 200 μg/ml proteinase K for 15 min on ice and then treated with 1 mm PMSF for another 15 min on ice.
Treatment of PS T. brucei with Nigericin, Monensin, and Valinomycin
For the nigericin (all ionophores used were from Sigma-Aldrich) rescue experiment described in the legend of Fig. 4A, PS T. brucei were grown for 2 days in SDM-79 medium supplemented with tetracycline. Cells were subsequently diluted to 2 × 106 cells/ml into various media with a stepwise doubling of nigericin concentrations in the 0–100 nm range. The 0 nm medium was mock-treated with the nigericin solvent ethanol. This procedure was done in order to ensure that all cells had the same initial degree of Letm1 down-regulation before ionophore exposure. Cell density was then measured at each nigericin concentration every 24 h over a 3-day time course. The procedure for monensin treatment (Fig. 4B) was the same, except there was a stepwise 10-fold increase in this ionophore concentration at the 0–1000 ng/ml range. For the valinomycin treatment of PS cells described in the legend to Fig. 5, the PS cells were incubated with a 1 μm concentration of the compound for 2 h. A subset of the cells was pretreated with 2 μm of nigericin prior to valinomycin application.
FIGURE 4.

Treatment with the ionophore nigericin rescues the Letm1 knockdown in PS T. brucei. A, cell growth in a 0–100 nm range of nigericin concentrations (x axis), as measured by density (10x cells/ml, along the y axis) of cells grown in the presence (RNAi+) or absence of tetracycline (RNAi−). Each shaded line represents 1–3 days after treatment. n = 4; error bars indicate S.D. B, cell growth in a 0–100 ng/ml range of monensin concentrations (x axis); otherwise as in A. C, light micrographs showing cells after 3 days of RNAi induction either treated with 25 nm nigericin (+) or mock-treated with ethanol (−). Scale bar, 10 μm. D, flow cytometry histogram following membrane potential in MitoTracker-labeled PC T. brucei grown in the presence (RNAi+) or absence of tetracycline (RNAi−) and either mock-treated with ethanol (top plot; −) or treated with 25 nm nigericin (bottom plot; +). An increase in fluorescence is depicted on the x axis from left to right on a logarithmic scale plotted against a cell count on the y axis. Unfilled curves are measurements of cells also treated with 20 μm carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), an uncoupler of mitochondrial membrane potential.
FIGURE 5.
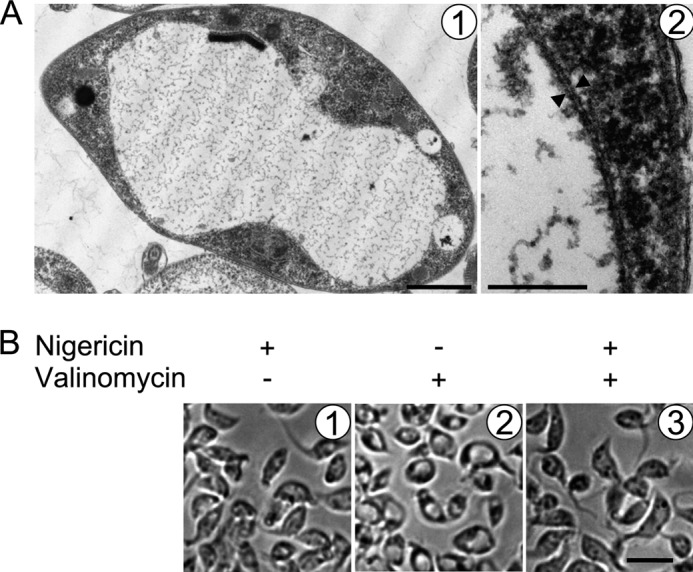
Treatment of PS T. brucei with the K+ ionophore valinomycin results in mitochondrial swelling. A, transmission electron micrographs of PS T. brucei treated with a 1 μm concentration of the K+ ionophore valinomycin. Double membranes are indicated by opposing arrowheads. Scale bars, 2 μm and 200 nm for pictures 1 and 2, respectively. Note the peripherally located kinetoplast DNA disk. B, light micrographs showing cells treated with either 2 μm nigericin or 1 μm valinomycin alone or pretreated with the former and then treated with the latter, as indicated at the top of the images from left to right. Scale bar, 10 μm.
Flow Cytometry for Membrane Potential
PS T. brucei incubated with MitoTracker Red CMXRos, as described above, were diluted 1:5 in PBS and placed into a FACSCanto II flow cytometer (BD Biosciences) for measurement of fluorescence. Twenty thousand cells were counted in each measurement. Controls in which membrane potential was collapsed by the simultaneous addition of 20 μm carbonyl cyanide p-trifluoromethoxyphenylhydrazone to cells were also measured. Data were analyzed using the Flowing Software program (Turku Centre for Biotechnology, Finland).
Quantitative Real-time PCR and Northern Blot Analysis
Quantitative real-time PCR was performed as described previously (34), using primers homologous to mt mRNAs as designed by Carnes et al. (48). Letm1 mRNA was measured using specific primers CGGAATACCTGTCGTCCACT and AGACATTAAACGGCCCTTCC. The relative abundance of the assayed mRNA in the induced RNAi knockdowns, as compared with the non-induced controls and normalized to either 18 S rRNA or β-tubulin levels, was determined by standard protocols (34, 48). Northern blots were done as described previously (44), using an antisense probe generated via the aforementioned Letm1 quantitative PCR primers. Both primers were used to amplify a product from T. brucei genomic DNA, which was subsequently used as a template for a 45-cycle PCR with the 32P-end-labeled reverse primer in the presence of [α-32P]dATP (6000 μCi/ml).
Mitochondrial Translation Assay
The mitochondrial translation assay is discussed in detail elsewhere (49). Briefly, 4 × 106 cells were incubated with [35S]methionine for 1 h (Easy Tag Express Protein Labeling Kit, PerkinElmer Life Sciences) in the presence of 10 mg/ml cyclohexamide to suppress cytoplasmic translation. The cells were lysed at 37 °C for 20 min in the loading buffer (2% SDS, 125 mm Tris-HCl, pH 6.8, 2% β-mercaptoethanol, 27% glycerol (v/v)) and then run on a 9% acrylamide SDS gel in the first dimension. Each lane was cut out and placed in a denaturing solution (1% SDS, 125 mm Tris-HCl, pH 6.8, 1% β-mercaptoethanol) at 37 °C for 1 h before being run in the second dimension on a 14% acrylamide-SDS gel. The gels were Coomassie-stained, incubated for 1 h in 1 m salicylate (Sigma-Aldrich), and then dried before exposure to BioMax Film (Eastman Kodak Co.).
RESULTS
Letm1 Is a Mitochondrial Inner Membrane Protein
In order to confirm that the annotated T. brucei Letm1 ortholog is targeted to the mitochondrion, cell lines were generated in which one of the gene loci was in situ tagged with sequence encoding a C-terminal YFP extension. Indirect immunofluorescence confocal microscopy revealed that Letm1 is indeed localized throughout the organelle because its signal overlaps with the specific marker MitoTracker Red CMXRos dye (Fig. 1A). This result was confirmed by digitonin permeabilization of cells into cytosolic and mitochondrial fractions, using antibodies immunodecorating mitochondrial heat shock protein 70 (mHSP70) and enolase as markers of these compartments, respectively (Fig. 1B). To verify that Letm1 is a membrane protein, isolated mitochondria from the YFP-tagged cell line were partitioned by Triton X-114 phase separation into membrane and soluble proteins, the former of which was retained in the detergent phase (Fig. 1C). The YFP antibody signal appeared in the detergent phase with the trypanosomatid-specific subunit of respiratory complex IV (trCOIV) (50), whereas the matrix marker mHSP70 remained in the aqueous phase (Fig. 1C). To determine the protein's IM localization, SMPs from the cell line where subjected to a proteinase K protection assay. In most SMPs, the inner leaflet of the IM faces outward, exposed to the protease. Indeed, trCOIV and matrix-facing F1-ATPase are susceptible to enzyme degradation, in contrast to cytochrome c, located in the intermembrane space, which is protected by the IM (Fig. 1D). The C-terminal YFP tag is also degraded by this treatment, indicating that this portion of the protein extrudes into the matrix from the IM (Fig. 1D), a topology kept by the human ortholog (3).
FIGURE 1.
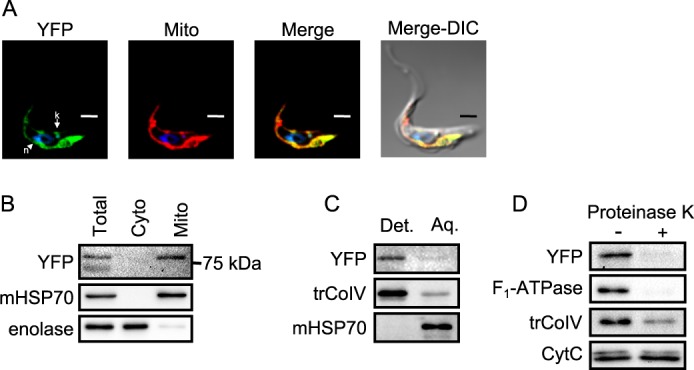
Localization of C-terminally YFP-tagged Letm1 to T. brucei mitochondrial inner membrane. A, indirect immunofluorescence of fixed PS T. brucei labeled with MitoTracker Red, which visualizes the single reticulated organelle. Labels above pictures indicate signal from YFP antibody (YFP), MitoTracker (Mito), and both (Merge) and signals overlaid with a differential contrast image (Merge-DIC). DAPI-stained nucleus (n) and kDNA (k) are indicated by an arrowhead and arrow, respectively. Scale bar, 2 μm. B, Western blot analysis of digitonin fractionation of cytoplasm (Cyto) and mitochondrial (Mito) compartments in comparison with an equivalent amount of lysate from whole cells (Total). The 75 kDa marker is indicated on the right. C, Western blot analysis of Triton X-114 fractionation of mitochondrial proteins into membrane and soluble fractions, which reside in the detergent (Det.) or aqueous (Aq.) phases, respectively. D, Western blot analysis of SMPs treated with proteinase K (+) or untreated controls (−). Antibodies used are indicated on the left.
Because the in situ tagged protein is bigger than the predicted size of the tagged Letm1 protein (Fig. 1A), about 80 versus 68 kDa, respectively, we decided to map the 5′-end of the mature Letm1 mRNA to define its ORF. The start codon is actually further upstream than predicted (supplemental Fig. 1A), encoding a protein with the observed size and of a length comparable with that of the ortholog from the related Trypanosoma cruzi (supplemental Fig. 1B). The revised ORF contains the transmembrane domain that defines all Letm1 orthologs and other well conserved features, such as a putative protein kinase C phosphorylation site and C-terminal coiled-coil regions, but lacks the two Ca2+-binding EF-hand domains present in orthologs of some other eukaryotes.
RNAi Silencing of Letm1 in Procyclic Stage Results in Mitochondrial Swelling and Inhibited Growth
To test whether Letm1 is an essential protein for the PS cells, a conditional RNAi cell line was generated using an established system in which dsRNA overexpression is induced by the addition of tetracycline, as described elsewhere (34). To test whether the dsRNA successfully targets Letm1 mRNA for degradation, RNA was harvested from cells grown in the presence and absence of the antibiotic for 48 h for subsequent Northern analysis using a radioactively labeled antisense probe that anneals to the Letm1 sequence. The transcript is undetectable in the RNAi-induced cells as compared with the non-induced controls (Fig. 2A). Ethidium bromide-stained rRNA was used as a control for equal loading. Next, we measured the growth of RNAi-induced and uninduced PS cells every 24 h over a 10-day course. Fig. 2B depicts a representative line graph showing absolute cell density at each time point, including the dilution of cultures every other day to 2 × 106 cells/ml. Reproducible growth inhibition is apparent 3 days after RNAi induction.
FIGURE 2.
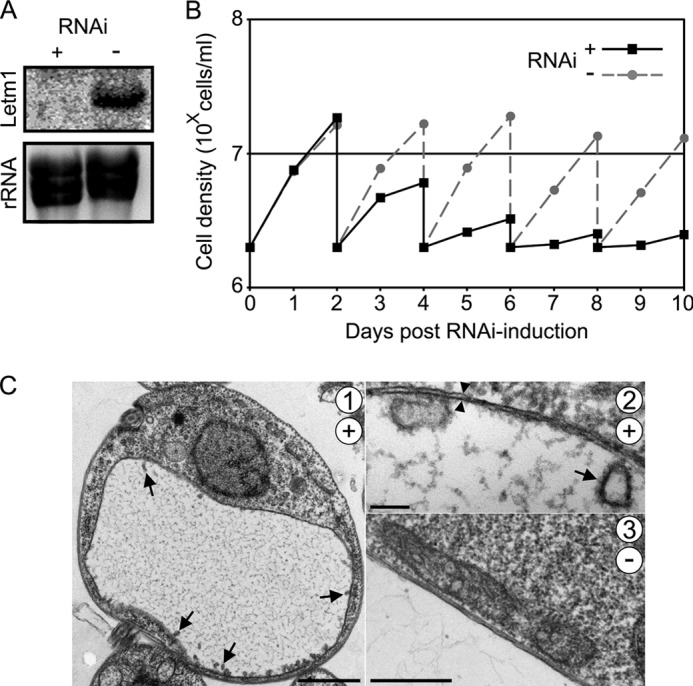
Knockdown of Letm1 in PS T. brucei results in growth inhibition and mitochondrial swelling. A, Northern blot analysis showing that Letm1 mRNA is depleted in cells grown with the RNAi-inducing tetracycline (+) as compared with the untreated controls (−) in the upper image (Letm1). The ethidium bromide-stained rRNA from the corresponding lanes is shown below as a loading control. B, PS T. brucei grown in the presence (RNAi+) or absence (RNAi−) of tetracycline. The x axis shows days post-RNAi induction; the y axis shows cell density in 10X cells/ml plotted on a logarithmic scale. C, transmission electron micrographs of cells grown in the presence (+; pictures 1 and 2) or absence (−; picture 3) of tetracycline for 3 days. Cristae and double membranes are indicated by arrows and double arrowheads, respectively. Scale bars, 1 μm, 100 nm, and 500 nm for pictures 1, 2, and 3, respectively.
The impairment of Letm1-depleted cells is most likely due to the appearance of a swollen mitochondrion, as revealed by transmission electron microscopy (Fig. 2C, 1). The identity of this massive, electron-lucent organelle, as compared with the unaltered mt electrodensity in the untreated controls (Fig. 2C, 3), is supported by the surrounding double membranes (Fig. 2C, 2, arrowheads). The discoidal cristae, which are characteristic for T. brucei and other members of the phylum Euglenozoa (21), remain upon Letm1 down-regulation in the periphery of the swollen organelle (Fig. 2C, 1 and 2, arrows).
Human Letm1 Complements the Endogenous Trypanosome Ortholog
To exclude the possibility that the Letm1-silencing phenotype is due to off target effects and also to determine the protein's functional homology across the enormous evolutionary distance separating kinetoplastids from opisthokonts, we examined whether a constitutively expressed human Letm1 (HsLetm1) could complement the depletion of its T. brucei ortholog (TbLetm1). First, we confirmed that the exogenous HsLetm1, bearing a 3× hemagglutinin (HA3) epitope tag on its C terminus, is targeted to the flagellate mitochondrion. Indirect immunofluorescence shows that HsLetm1 indeed co-localizes with the MitoTracker Red dye (Fig. 3A). This result was confirmed by digitonin subfractionation of cells into the cytosolic and mitochondrial compartments, in which the α-HA antibody signal associates with that of the mitochondrial marker mHSP70 (Fig. 3B). The abundant upper band migrated at the expected ∼90 kDa size with a less intense one just below, a pattern also observed when the protein was expressed in Escherichia coli (14).
FIGURE 3.
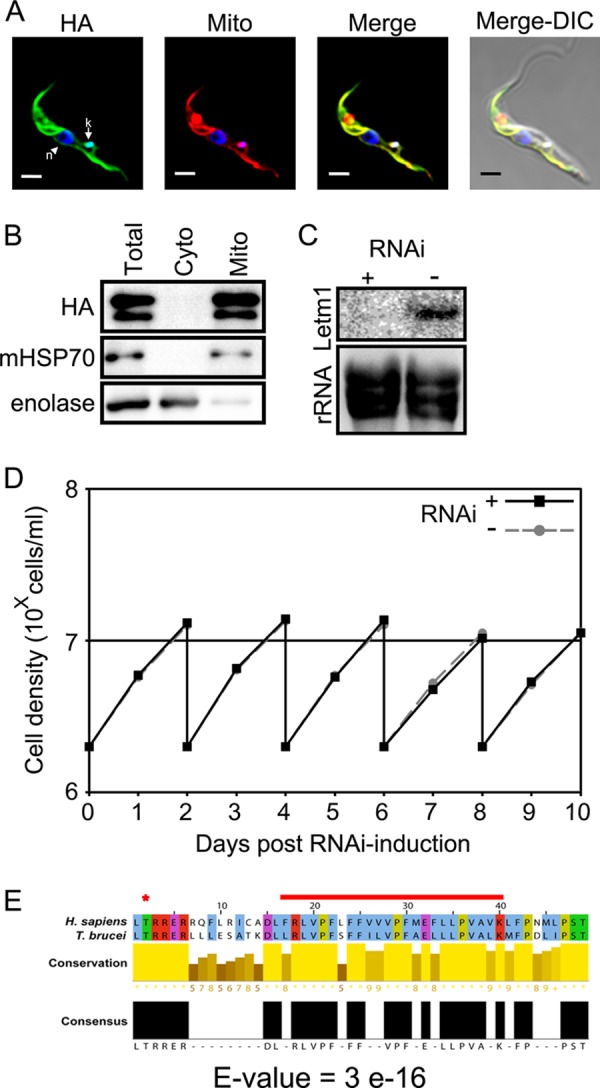
Expression of human Letm1 rescues knockdown of the T. brucei ortholog. A and B, indirect immunofluorescence and digitonin subfractionation of PS T. brucei confirm mitochondrial localization of the C-terminal HA3-tagged human Letm1 (HA). Other labels and the scale bar in A are as described for Fig. 1, A and B. C, Northern blot analysis confirms the ablation of the endogenous T. brucei Letm1 mRNA in cells constitutively overexpressing exogenous human Letm1. Labeling is as in Fig. 2A. D, growth of PS T. brucei constitutively overexpressing human Letm1 grown in the presence (RNAi+) or absence (RNAi−) of tetracycline. Labeling is as in Fig. 2B. E, alignment of the predicted transmembrane domain of the human and T. brucei orthologs. *, conserved predicted protein kinase C phosphorylation site; red line, transmembrane domain. The E-value of BLAST alignment of the two sequences is shown at the bottom.
After verifying that the endogenous TbLetm1 is still silenced in cells expressing HsLetm1 (Fig. 3C), the growth of cell lines with and without TbLetm1 down-regulation was compared. As shown in the line graph in Fig. 3D, both samples grew at the same rate. This result indicates that HsLetm1 can fully complement the ablation of TbLetm1 in T. brucei, suggesting functional homology of the two orthologs. Indeed, the two orthologs share sequence homology in the region of the predicted transmembrane domain (Fig. 3E).
The Nigericin Ionophore Restores Cell Viability and Mitochondrial Function in Letm1 Knockdowns
The manifested swollen mitochondria upon Letm1 ablation may be due to a consequent accumulation of ions. To test whether K+ is the cation in question, we attempted to treat RNAi-induced T. brucei with varying doses of nigericin, an ionophore that acts as an antiporter of K+ and H+ across membranes, as described under “Experimental Procedures.” Fig. 4A depicts a line graph in which average cell density among quadruplicates is plotted against nigericin concentration. Each line presents measurements made 1–3 days after ionophore treatment. Growth rates at each nigericin concentration are inferred from the difference between points relative to the y axis and also depicted in supplemental Fig. 2A.
In the cells depleted for Letm1 (RNAi+), there is a dose-dependent increase in growth from 0 to 50 nm nigericin, after which there is a decrease in growth. This latter trend is probably due to the intrinsic toxicity of the ionophore to T. brucei, as demonstrated by a line graph showing the dose-dependent decrease in growth of cells grown in the absence of tetracycline (RNAi−) throughout the whole concentration range. However, it should be mentioned that the Letm1-depleted cells grown in 100 nm nigericin still exhibit more rapid growth than those grown without the drug, indicating that this ionophore is able to partially restore cell viability upon loss of Letm1. The cells ablated for Letm1 exhibited comparable growth rates as compared with their RNAi-uninduced counterparts when treated with 25, 50, and 100 nm nigericin (supplemental Fig. 2A).
We next looked to see whether nigericin treatment restores mt morphology and physiology. Cells grown for 2 days in the presence of tetracycline to induce Letm1 silencing were subsequently diluted into media with or without 25 nm nigericin and grown for 24 h before being subjected to assays comparing RNAi-treated and untreated samples. Light microscopy reveals that about half of the cells grown in the absence of the ionophore (Fig. 4C, −) exhibit a rounded shape with lucent center, representing the swollen mitochondrion (arrows), whereas all of the treated cells exhibit normal gross morphology (Fig. 4C, +). The physiological state of the organelle in both samples was also determined using the MitoTracker Red dye, whose intercalation into the matrix correlates with membrane potential. According to the flow cytometry histogram (Fig. 4D, −), the membrane potential of Letm1-depleted cells (RNAi+) not treated with nigericin exhibits various degrees of membrane potential reduction compared with their non-induced counterparts (RNAi−), as represented by two broad fluorescence peaks. The broad range of membrane potential reduction also reflects the various morphological effects 3 days after Letm1 down-regulation. However, nigericin-treated cells with reduced or endogenous Letm1 levels exhibited the same membrane potential (Fig. 4D, +), suggesting that the ionophore mediates restoration of the physiological state of the organelle.
These results were further confirmed by treatment with monensin, a less specific ionophore that exchanges H+ and monovalent cations, such as K+. Using the same previously described scheme, 100 ng/ml was determined to restore growth in the Letm1-depleted cells compared with the uninduced cells treated with the same dose (Fig. 4B and supplemental Fig. 2B). Taken together, these results suggest that upon Letm1 silencing, K+ cations accumulate in the mt matrix, which cause subsequent osmotic stress and swelling mitochondria. The chemical action of the ionophores nigericin and monensin compensates for the ablation of Letm1 by mediating KHE across the inner membrane. Consequently, these cells exhibit normal mt morphology and physiology.
Depletion of Letm1 Phenocopies the K+ Ionophore Valinomycin
As further support for the notion that matrix K+ accumulation is behind the swelling of the PS T. brucei mitochondrion upon Letm1 silencing, the parental cell line used in the generation of the conditional RNAi knockdown was treated with the ionophore valinomycin. In contrast to nigericin, valinomycin acts in a Nerstian fashion by transporting K+ across lipid membranes down an electrochemical gradient. Thus, this compound is often used in other systems to dissipate mt membrane potential (3, 51). Valinomycin supplementation (1 μm) to SDM-79 medium was fatal to T. brucei after 2 h of exposure. As seen by subsequent observation by transmission electron microscopy, these cells exhibit a swollen mitochondrion in a manner reminiscent of the Letm1 knockdown (Fig. 5A).
We next asked whether nigericin pretreatment can prevent valinomycin-mediated swelling mitochondria. Cells were incubated with 2 μm nigericin for 15 min and then split into those exposed to or lacking valinomycin treatment as described above; cells without nigericin pretreatment prior to valinomycin exposure were included in this experiment. As visualized by light microscopy, T. brucei incubated only with this concentration of nigericin exhibit a shrunken appearance, possibly due to unspecified cell-wide effects (Fig. 5, B1). Nigericin pretreatment prevents the visible swelling of mitochondria caused by valinomycin (Fig. 5B, 2 and 3). It appears that Letm1 knockdown in PS phenocopies the swelling mitochondria effect of the K+ ionophore valinomycin in the parental cell line, including the susceptibility of this swelling to the K+/H+ antiporter nigericin.
Letm1 Is Essential for T. brucei evansi and the Bloodstream Stage of T. brucei
To gain insight into the role of Letm1 in mitochondria bearing different physiological states, we generated conditional RNAi knockdowns in T. brucei BS and T. brucei evansi, a petite mutant of T. brucei lacking any kDNA and hence a canonical proton gradient across the inner mt membrane (31, 52). Efficient degradation of the Letm1 transcript in either cell line was verified by Northern blot analysis (Fig. 6, A and C). Growth in the presence and absence of the RNAi induction agent was subsequently assayed as in PS, with a lower starting concentration of 5 × 105 cells/ml. As seen in the line graph in Fig. 6B, Letm1 depletion in BS resulted in growth inhibition already 2 days after RNAi induction, after the first daily dilution. A recovery of growth often occurring when essential proteins are down-regulated (34) is also observed to begin at day 4 of the time course. Obvious growth inhibition is also observed on day 3 after the first cell passage in T. brucei evansi (Fig. 6D), which is performed every other day due to the slower growth rate of these trypanosomes as compared with BS. Both cell types exhibited swelling mitochondria at these time points, as shown in a representative picture from T. brucei evansi (Fig. 6E, 1 and 2), a very dramatic contrast to the thin morphology of the organelle in the untreated controls (Fig. 6E, 3). Thus, Letm1 plays the same role in mediating KHE in the mitochondrion of trypanosome stages that lack a respiratory chain and cristae, as exemplified by the results in BS T. brucei. Furthermore, this ion exchange functions even in the absence of a canonical proton gradient across the inner membrane, which is not a component of the mitochondrial membrane potential in T. brucei evansi (31).
FIGURE 6.
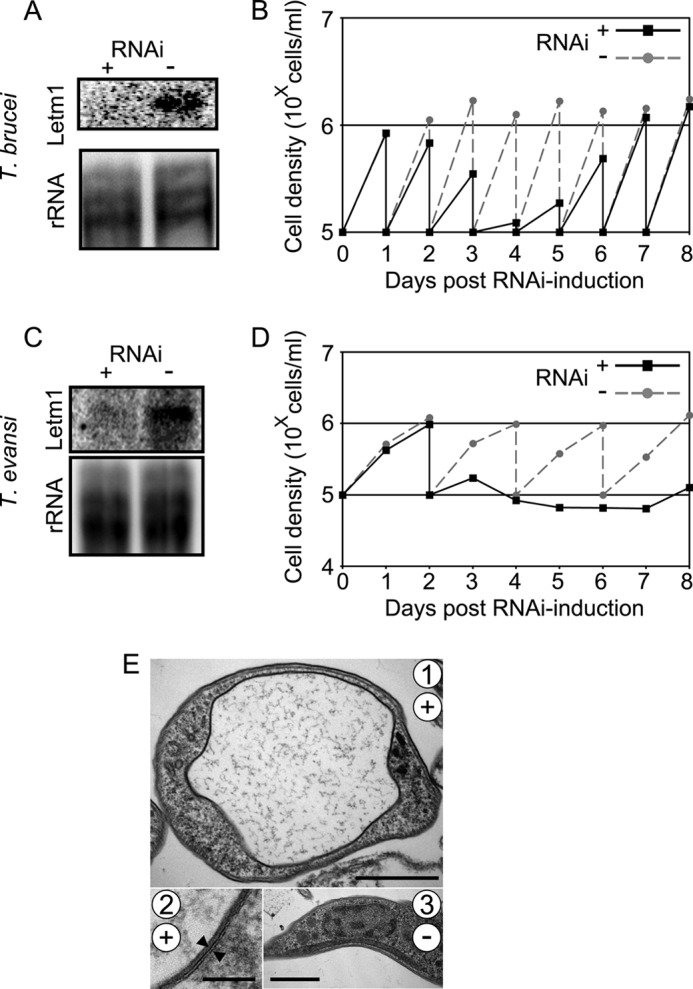
Knockdown of Letm1 in BS T. brucei and T. brucei evansi results in growth inhibition and mitochondrial swelling. A–D, Northern blot analysis confirming ablation of Letm1 mRNA in RNAi-induced cells versus non-induced controls plus growth analysis comparing these two samples in BS T. brucei (A and B) and T. brucei evansi (C and D). Labeling is as in Fig. 2, A and B. E, transmission electron micrographs of T. brucei evansi grown in the presence (+; pictures 1 and 2) or absence (−; picture 3) of tetracycline for 3 days. Double membranes are indicated by opposing arrowheads. Scale bars, 1 μm, 100 nm, and 1 μm for pictures 1, 2, and 3, respectively.
Letm1 Is Dispensable for Mitochondrial Translation
Because T. brucei evansi has lost its mt genome, components responsible for gene expression in the organelle have been rendered redundant (31, 36). Thus, the essential nature of Letm1 in these ρ0 trypanosomes argues ostensibly against a primary function in mt translation via its interaction with ribosomes. To further investigate, we decided to assay de novo translated apocytochrome b (CytB) and cytochrome c oxidase subunit 1 (Cox1) in PS (49), the T. brucei life cycle stage that assembles the respiratory complexes into which these proteins are incorporated (22).
Letm1 RNAi knockdowns grown in the presence of tetracycline for 2–4 days plus a non-induced control were subjected to the [35S]methionine labeling of de novo synthesized mt proteins, which were subsequently resolved on a 9%/14% acrylamide two-dimensional denaturing gel. A steady decrease in the labeled Cox1 and CytB, as well as still unidentified products, was observed over the time course (Fig. 7A). This decrease in 35S signal is compared with the Coomassie-stained cytosolic proteins (Fig. 7A, insets), which remain at a constant level in all samples. To ensure that this decrease was due to translational rather than transcriptional defects, steady-state levels of these transcripts were also determined by real-time quantitative PCR. Cox1 mRNA, which does not undergo RNA editing, and CytB mRNA, which is processed by moderate RNA editing, complemented by the massively edited Cox3 transcript, were virtually unaffected 3 days after RNAi induction (Fig. 7B). Therefore, it appears that mt translation is indeed compromised in Letm1-depleted T. brucei.
FIGURE 7.
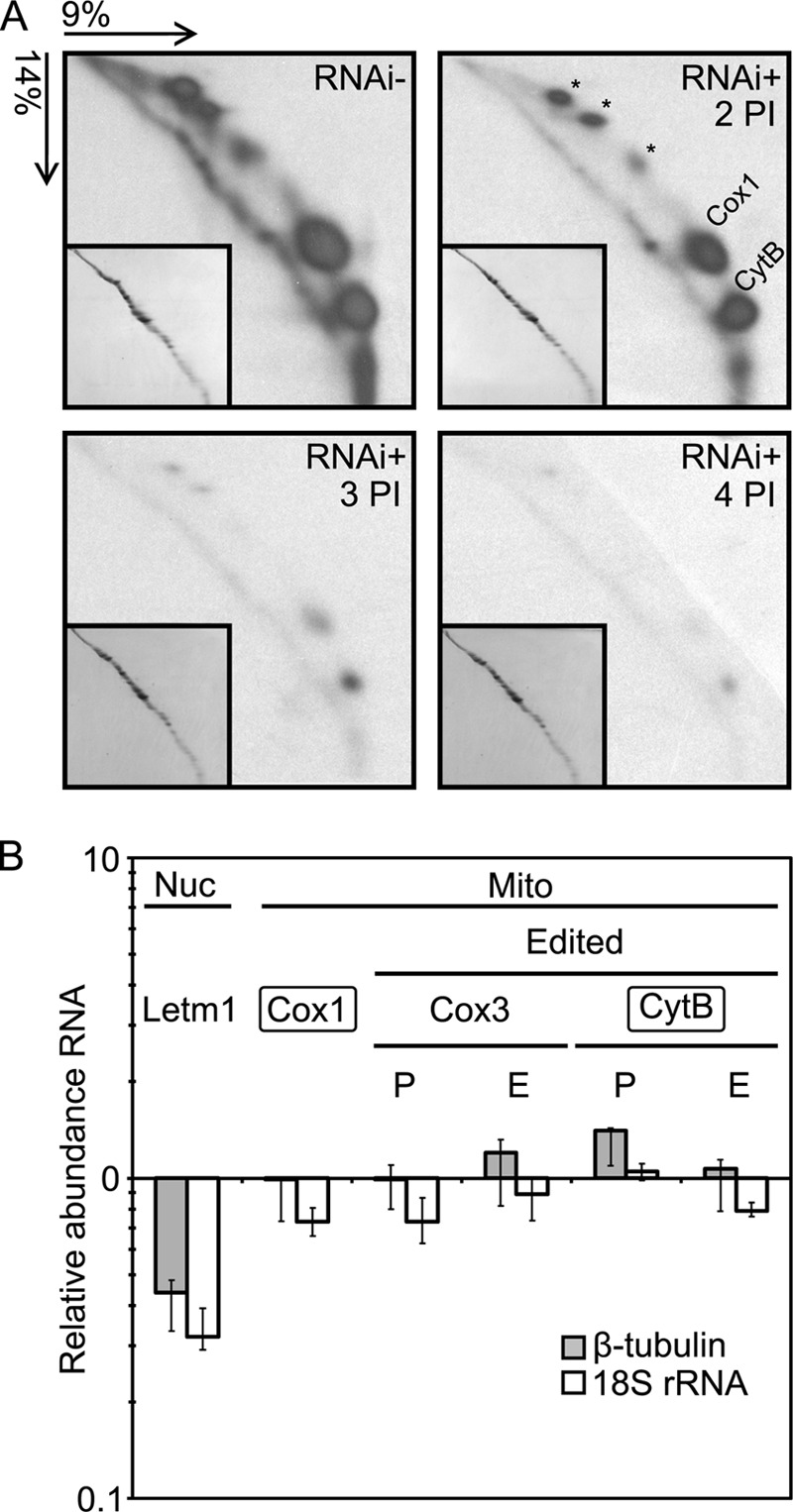
Knockdown of Letm1 has an effect on mitochondrial translation in PS T. brucei. A, resolution of de novo [35S]methionine-labeled mitochondrial proteins by two-dimensional SDS-PAGE. Acrylamide composition and the direction of each dimension are indicated at the top left. Insets show the same gels in which the cytoplasmic proteins are Coomassie-stained. Gels are RNAi− and RNA+ 2, 3, and 4 days postinduction (PI) by tetracycline. The identified spots corresponding to apocytochrome b (CytB) and cytochrome c oxidase subunit 1 (Cox1) are indicated along with hitherto unidentified mitochondrial proteins (*). B, quantitative real-time PCR assaying steady state levels of selected mitochondrial (Mito) and nucleus-encoded Letm1 mRNAs (Nuc). Transcripts undergoing RNA editing are indicated, and the pre-edited (P) and edited (E) forms were individually assayed. Boxed transcripts are those whose protein products were followed in A. The mean relative levels of the assayed amplicons in the RNAi-induced cells relative to the levels in the uninduced control are plotted logarithmically and normalized to the same calculation performed for the nucleus-encoded β-tubulin (gray bars) and 18 S rRNA cDNAs (white bars), which were unaffected by the Letm1 RNAi silencing. Error bars, range of obtained relative abundances; n = 3.
To resolve whether this phenomenon is directly due to the depletion of Letm1 or a downstream effect, mt translation was assayed in RNAi cell lines induced by tetracycline for 4 days with or without 25 nm nigericin treatment for the last 2 days plus the non-induced controls. As shown in Fig. 8A, translation proceeds in the Letm1-silenced cells, in which KHE is restored by nigericin. Letm1 mRNA was measured in the nigericin-treated cells to confirm that it was degraded upon RNAi induction (Fig. 8B). We conclude from these experiments that the observed hindering of mt translation in Letm1-depleted T. brucei is a secondary consequence of the disrupted ion homeostasis.
FIGURE 8.
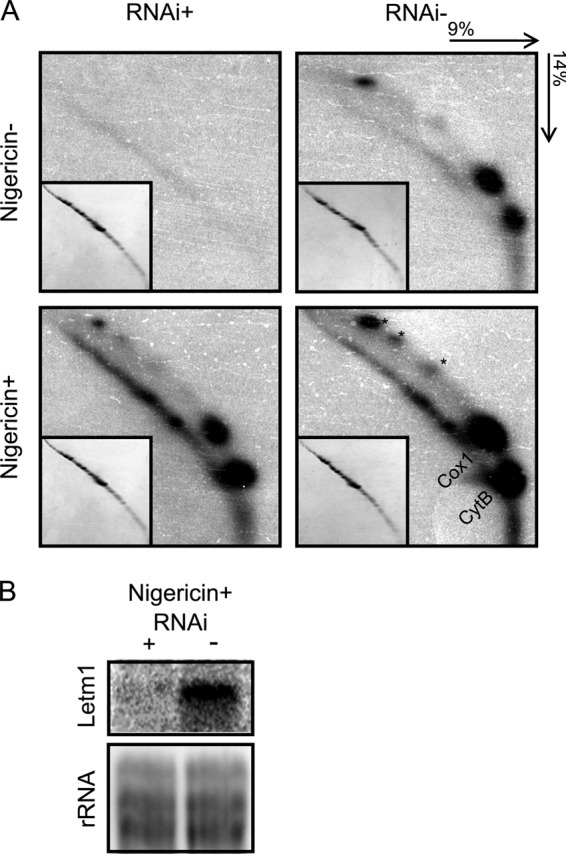
Mitochondrial translation persists in nigericin-treated PS T. brucei Letm1 knockdowns. A, resolution of de novo [35S]methionine-labeled mitochondrial proteins by two-dimensional SDS-PAGE. Acrylamide composition and the direction of each dimension are indicated at the top right. Other labels and features are as described for Fig. 7A. Gels corresponding to protein products from RNAi-induced cells (RNAi+) and uninduced controls (RNAi−) are indicated at the top, and those treated with 25 μm nigericin (Nigericin+) and untreated (Nigericin−) are designated on the left. B, Northern blot analysis showing that Letm1 mRNA is down-regulated in cells grown in the presence of tetracycline (+) as compared with the untreated controls (−), both grown in the presence of nigericin. Labeling is as in Fig. 2A.
DISCUSSION
Mitochondria are ancient organelles of an endosymbiotic origin that are, in one form or another, maintained in all extant eukaryotes living in very different ecological niches. As such, although mitochondria have undergone divergent evolution in these various organisms, they still retain basal characters that are common to them, such as being bound by double membranes (53). Using the highly diverged T. brucei as a study model, we have exploited its amenabilities and its evolutionary divergence to determine the ancestral function of the ubiquitous Letm1 protein, which exhibits remarkable conservation of function in different organellar contexts and across large evolutionary distances.
The T. brucei organelle is present in a single copy and carries discoidal cristae (21), and its energy metabolism undergoes dramatic alterations during the life cycle (22). However, despite such differences, our studies reproduced a phenotype seen in some of those previously carried out in other eukaryotes, namely that ablation of the Letm1 protein results in swelling mitochondria (1, 3, 10–12). This condition is alleviated by treatment with chemical monovalent cation/proton exchangers, such as nigericin or monensin. These results strongly suggest that Letm1 mediates KHE, the coupling of K+ extrusion from the mt matrix against its natural concentration gradient with the downhill movement of H+ into this space. In a Letm1-depleted background, K+ accumulates in the organelle, causing the osmotic stress that leads to the striking effect on organellar morphology. This RNAi phenotype is akin to the mt swelling observed when T. brucei is treated with a large dose of valinomycin, an ionophore that allows K+ to penetrate polarized membranes and accumulate in the enclosed space.
Peter Mitchell (54) originally recognized the hazard of cation sequestering within the negatively charged matrix of energized mitochondria as a consequence of membrane potential and predicted the existence of H+-driven mechanisms to mitigate this possibility as one of the postulates of his chemiosmotic hypothesis. The trypanosomatid organelle has a proven K+ uptake activity with pharmacological properties that are reminiscent of those possessed by an ATP-sensitive K+ channel embedded in the IM of other eukaryotes (55–57). Thus, the need for K+ efflux from the matrix via KHE is a logical requirement for the organelle in trypanosomes, and our results suggest that Letm1 has a role in this process.
Another study has implicated Letm1 in Ca2+ translocation across the IM (14, 15). Our results suggest otherwise, agreeing with those that have postulated a role in KHE (1, 3, 10). The reason for this disparity is unclear. Jiang and colleagues described Ca2+ translocation function in HeLa cell cultures and used liposome reconstitution of the recombinant protein (14), although it should be noted that this study described a significantly more limited effect on mt morphology than previous reports. However, in our study, the aforementioned ability of the K+/H+-exchanging ionophores nigericin and monensin to rescue the Letm1-depleted T. brucei presents convincing evidence that it is indeed K+ homeostasis that is disrupted. Also, in contrast to K+, which represents the most concentrated intracellular cation (58, 59), Ca2+ is a carefully regulated secondary messenger whose intracellular concentration is in the micromolar range (60), making its accumulation in the matrix a less likely cause of the swelling mitochondria phenotype. Finally, yeast still retain an ortholog of the Letm1 gene despite lacking an active Ca2+ uptake mechanism (25–27, 60), presumably removing the evolutionary pressure to maintain the protein in this organellar context.
In T. brucei, Letm1 does not appear to play a direct role in mitochondrial translation as has been suggested for the yeast ortholog (4, 16, 17). Although this process is indeed compromised in the Letm1 knockdowns, it persists when these cells are treated with nigericin. Because mitochondrial ribosomes disassociate in high K+ concentration environments (61, 62), we propose that the apparent effect on mitochondrial translation represents an epiphenomenon to the accumulation of the cation in the matrix upon Letm1 ablation. Furthermore, the similar growth rates of the Letm1-depleted and uninduced controls in the optimal doses of the ionophores nigericin and monensin suggest that additional processes are not affected besides mt K+/H+ antiport.
Taking all into account, we conclude that the ancestral function of Letm1 is KHE to maintain and/or regulate K+ homeostasis and consequently mt volume. This conservation of function throughout eukaryotes is underscored by the ability of the human Letm1 ortholog to complement the down-regulation of the T. brucei ortholog. This evidence of common function in trypanosomes and humans is also interesting, given the wide structural variation between the two orthologs; although they share sequence homology in the defining trans-membrane domain, only human Letm1 bears two EF hands, indicating that they are superfluous to Letm1 function in the parasite. This result suggests that the motif does not directly contribute to cation extrusion from the organelle but perhaps serves a regulatory role for the human protein.
Our model system provided additional insight into conserved Letm1 function in different mt physiological states, as represented by the BS T. brucei and the ρ0 T. brucei evansi, both of which we show require function of this protein. In the BS stage, mt membrane potential is exclusively generated by the FoF1-ATP synthase via ATP hydrolysis, whereas T. brucei evansi is quite extreme in that only the electrogenic component of mt membrane potential is responsible for maintaining the energized mitochondrion of the parasite. Interestingly, this component of the proton motive force appears to be sufficient to drive Letm1-mediated KHE.
The essentiality of Letm1 offers new insight into the functions of the BS mitochondrion, known to be massively reduced in function and morphology (22) yet still indispensable. Among the few active pathways in the BS organelle requiring the preservation of mt membrane potential are the maintenance of the glycosomal redox balance via the glycerol-3-phosphatase/dihydroxyacetone phosphate shuttle, fatty acid synthesis, and thymidylate production for DNA synthesis (31, 32, 37–39). Although Ca2+ uptake activity has previously been described in the BS mitochondrion, the essentiality of this pathway was not explored, and we are now therefore able to add ion homeostasis to the list of indispensable functions of the BS organelle. The requirement for Letm1 is perhaps more surprising still in the even further reduced mitochondrion of T. brucei evansi, because it suggests that one reason why this petite mutant undergoes extraordinary lengths to maintain an energized mitochondrion (31, 40) is to maintain mt matrix K+, an irony, considering that it is this very feature that predisposes the organelle to cation accumulation.
This study exploited many features of the T. brucei subspecies complex that makes it a suitable model for studying mitochondrial function that extends from its role in this neglected pathogen to other eukaryotes. Its high evolutionary divergence allows it to serve as a valuable outgroup for establishing whether pathways described in opisthokont models are well conserved and serve as a foundation for organelle function and biogenesis. The availability of genetically tractable in vitro cultures bearing very different mitochondrial states allows one to easily address the potential persistence or modulation of protein function in these milieus. Thus, we show that Letm1 serves an essential and extremely conserved function in T. brucei to maintain mitochondrial volume by KHE.
Acknowledgments
We thank Dmitri Maslov (University of California) for advice on the de novo mt protein labeling assay; Achim Schnaufer (University of Edinburgh) for providing the T. brucei evansi cell line used in the study and sequence information; Zhenqiu Huang (University of South Bohemia) for the Letm1-YFP construct made from a plasmid provided by Mark Carrington (Cambridge University); Julius Lukeš IV (Charles University) for help with initial experiments; Luděk Kořeny (Cambridge University) for assistance with the alignment of the human and T. brucei Letm1 orthologs; and Ken Stuart (Seattle Biomed), Paul Michels (Universidad de los Andes/University of Edinburgh), and Steve Hajduk (University of Georgia) for α-mHSP70, enolase, and cytochrome c antibodies, respectively.
This work was supported in part by Czech Grant Agency Grant P305/12/2261 and a Praemium Academiae award (to J. L.).

This article contains supplemental Figs. 1 and 2.
- mt
- mitochondrial
- IM
- inner membrane
- KHE
- potassium/proton exchange
- PS
- procyclic stage
- BS
- bloodstream stage
- SMP
- submitochondrial particle
- mHSP70
- mitochondrial heat shock protein 70
- HsLetm1 and TbLetm1
- human and T. brucei Letm1, respectively.
REFERENCES
- 1. Nowikovsky K., Froschauer E. M., Zsurka G., Samaj J., Reipert S., Kolisek M., Wiesenberger G., Schweyen R. J. (2004) The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf-Hirschhorn syndrome. J. Biol. Chem. 279, 30307–30315 [DOI] [PubMed] [Google Scholar]
- 2. Schlickum S., Moghekar A., Simpson J. C., Steglich C., O'Brien R. J., Winterpacht A., Endele S. U. (2004) LETM1, a gene deleted in Wolf-Hirschhorn syndrome, encodes an evolutionarily conserved mitochondrial protein. Genomics 83, 254–261 [DOI] [PubMed] [Google Scholar]
- 3. Dimmer K. S., Navoni F., Casarin A., Trevisson E., Endele S., Winterpacht A., Salviati L., Scorrano L. (2008) LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum. Mol. Genet. 17, 201–214 [DOI] [PubMed] [Google Scholar]
- 4. Frazier A. E., Taylor R. D., Mick D. U., Warscheid B., Stoepel N., Meyer H. E., Ryan M. T., Guiard B., Rehling P. (2006) Mdm38 interacts with ribosomes and is a component of the mitochondrial protein export machinery. J. Cell Biol. 172, 553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Endele S., Fuhry M., Pak S. J., Zabel B. U., Winterpacht A. (1999) LETM1, a novel gene encoding a putative EF-hand Ca2+-binding protein, flanks the Wolf-Hirschhorn syndrome (WHS) critical region and is deleted in most WHS patients. Genomics 60, 218–225 [DOI] [PubMed] [Google Scholar]
- 6. Battaglia A., Filippi T., Carey J. C. (2008) Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome. Experience with 87 patients and recommendations for routine health supervision. Am. J. Med. Genet. C Semin. Med. Genet. 148C, 246–251 [DOI] [PubMed] [Google Scholar]
- 7. South S. T., Bleyl S. B., Carey J. C. (2007) Two unique patients with novel microdeletions in 4p16.3 that exclude the WHS critical regions. Implications for critical region designation. Am. J. Med. Genet. A 143A, 2137–2142 [DOI] [PubMed] [Google Scholar]
- 8. Zollino M., Lecce R., Fischetto R., Murdolo M., Faravelli F., Selicorni A., Buttè C., Memo L., Capovilla G., Neri G. (2003) Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am. J. Hum. Genet. 72, 590–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dimmer K. S., Fritz S., Fuchs F., Messerschmitt M., Weinbach N., Neupert W., Westermann B. (2002) Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol. Biol. Cell 13, 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McQuibban A. G., Joza N., Megighian A., Scorzeto M., Zanini D., Reipert S., Richter C., Schweyen R. J., Nowikovsky K. (2010) A Drosophila mutant of LETM1, a candidate gene for seizures in Wolf-Hirschhorn syndrome. Hum. Mol. Genet. 19, 987–1000 [DOI] [PubMed] [Google Scholar]
- 11. Hasegawa A., van der Bliek A. M. (2007) Inverse correlation between expression of the Wolfs Hirschhorn candidate gene Letm1 and mitochondrial volume in C. elegans and in mammalian cells. Hum. Mol. Genet. 16, 2061–2071 [DOI] [PubMed] [Google Scholar]
- 12. Tamai S., Iida H., Yokota S., Sayano T., Kiguchiya S., Ishihara N., Hayashi J., Mihara K., Oka T. (2008) Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L. J. Cell Sci. 121, 2588–2600 [DOI] [PubMed] [Google Scholar]
- 13. Froschauer E., Nowikovsky K., Schweyen R. J. (2005) Electroneutral K+/H+ exchange in mitochondrial membrane vesicles involves Yol027/Letm1 proteins. Biochim. Biophys. Acta 1711, 41–48 [DOI] [PubMed] [Google Scholar]
- 14. Jiang D., Zhao L., Clapham D. E. (2009) Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326, 144–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Waldeck-Weiermair M., Jean-Quartier C., Rost R., Khan M. J., Vishnu N., Bondarenko A. I., Imamura H., Malli R., Graier W. F. (2011) Leucine zipper EF hand-containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J. Biol. Chem. 286, 28444–28455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bauerschmitt H., Mick D. U., Deckers M., Vollmer C., Funes S., Kehrein K., Ott M., Rehling P., Herrmann J. M. (2010) Ribosome-binding proteins Mdm38 and Mba1 display overlapping functions for regulation of mitochondrial translation. Mol. Biol. Cell 21, 1937–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lupo D., Vollmer C., Deckers M., Mick D. U., Tews I., Sinning I., Rehling P. (2011) Mdm38 is a 14-3-3-like receptor and associates with the protein synthesis machinery at the inner mitochondrial membrane. Traffic 12, 1457–1466 [DOI] [PubMed] [Google Scholar]
- 18. Zhang B., Carrie C., Ivanova A., Narsai R., Murcha M. W., Duncan O., Wang Y., Law S. R., Albrecht V., Pogson B., Giraud E., Van Aken O., Whelan J. (2012) LETM proteins play a role in the accumulation of mitochondrially encoded proteins in Arabidopsis thaliana and AtLETM2 displays parent of origin effects. J. Biol. Chem. 287, 41757–41773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aslett M., Aurrecoechea C., Berriman M., Brestelli J., Brunk B. P., Carrington M., Depledge D. P., Fischer S., Gajria B., Gao X., Gardner M. J., Gingle A., Grant G., Harb O. S., Heiges M., Hertz-Fowler C., Houston R., Innamorato F., Iodice J., Kissinger J. C., Kraemer E., Li W., Logan F. J., Miller J. A., Mitra S., Myler P. J., Nayak V., Pennington C., Phan I., Pinney D. F., Ramasamy G., Rogers M. B., Roos D. S., Ross C., Sivam D., Smith D. F., Srinivasamoorthy G., Stoeckert C. J., Jr., Subramanian S., Thibodeau R., Tivey A., Treatman C., Velarde G., Wang H. (2010) TriTrypDB. A functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 38, D457–D462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Philippe H., Lopez P., Brinkmann H., Budin K., Germot A., Laurent J., Moreira D., Müller M., Le Guyader H. (2000) Early-branching or fast-evolving eukaryotes? An answer based on slowly evolving positions. Proc. Biol. Sci. 267, 1213–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cavalier-Smith T. (2010) Kingdoms Protozoa and Chromista and the eozoan root of the eukaryotic tree. Biol. Lett. 6, 342–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lukeš J., Hashimi H., Verner Z., Čičová Z. (2010) The remarkable mitochondrion of trypanosomes and related flagellates. in Structures and Organelles in Pathogenic Protists (de Souza W., ed) pp. 227–252, Springer, Berlin [Google Scholar]
- 23. Xiong Z. H., Ridgley E. L., Enis D., Olness F., Ruben L. (1997) Selective transfer of calcium from an acidic compartment to the mitochondrion of Trypanosoma brucei. Measurements with targeted aequorins. J. Biol. Chem. 272, 31022–31028 [DOI] [PubMed] [Google Scholar]
- 24. Vercesi A. E., Docampo R., Moreno S. N. (1992) Energization-dependent Ca2+ accumulation in Trypanosoma brucei bloodstream and procyclic trypomastigotes mitochondria. Mol. Biochem. Parasitol. 56, 251–257 [DOI] [PubMed] [Google Scholar]
- 25. Perocchi F., Gohil V. M., Girgis H. S., Bao X. R., McCombs J. E., Palmer A. E., Mootha V. K. (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baughman J. M., Perocchi F., Girgis H. S., Plovanich M., Belcher-Timme C. A., Sancak Y., Bao X. R., Strittmatter L., Goldberger O., Bogorad R. L., Koteliansky V., Mootha V. K. (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Stefani D., Raffaello A., Teardo E., Szabò I., Rizzuto R. (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Docampo R., Lukeš J. (2012) Trypanosomes and the solution to a 50-year mitochondrial calcium mystery. Trends Parasitol. 28, 31–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barrett M. P., Burchmore R. J., Stich A., Lazzari J. O., Frasch A. C., Cazzulo J. J., Krishna S. (2003) The trypanosomiases. Lancet 362, 1469–1480 [DOI] [PubMed] [Google Scholar]
- 30. Matthews K. R. (2005) The developmental cell biology of Trypanosoma brucei. J. Cell Sci. 118, 283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schnaufer A., Clark-Walker G. D., Steinberg A. G., Stuart K. (2005) The F1-ATP synthase complex in bloodstream stage trypanosomes has an unusual and essential function. EMBO J. 24, 4029–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brown S. V., Hosking P., Li J., Williams N. (2006) ATP synthase is responsible for maintaining mitochondrial membrane potential in bloodstream form Trypanosoma brucei. Eukaryot. Cell 5, 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cristodero M., Seebeck T., Schneider A. (2010) Mitochondrial translation is essential in bloodstream forms of Trypanosoma brucei. Mol. Microbiol. 78, 757–769 [DOI] [PubMed] [Google Scholar]
- 34. Hashimi H., Cicová Z., Novotná L., Wen Y. Z., Lukes J. (2009) Kinetoplastid guide RNA biogenesis is dependent on subunits of the mitochondrial RNA binding complex 1 and mitochondrial RNA polymerase. RNA 15, 588–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schnaufer A., Panigrahi A. K., Panicucci B., Igo R. P., Jr., Wirtz E., Salavati R., Stuart K. (2001) An RNA ligase essential for RNA editing and survival of the bloodstream form of Trypanosoma brucei. Science 291, 2159–2162 [DOI] [PubMed] [Google Scholar]
- 36. Paris Z., Hashimi H., Lun S., Alfonzo J. D., Lukeš J. (2011) Futile import of tRNAs and proteins into the mitochondrion of Trypanosoma brucei evansi. Mol. Biochem. Parasitol. 176, 116–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clayton A. M., Guler J. L., Povelones M. L., Gluenz E., Gull K., Smith T. K., Jensen R. E., Englund P. T. (2011) Depletion of mitochondrial acyl carrier protein in bloodstream-form Trypanosoma brucei causes a kinetoplast segregation defect. Eukaryot. Cell 10, 286–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Helfert S., Estévez A. M., Bakker B., Michels P., Clayton C. (2001) Roles of triosephosphate isomerase and aerobic metabolism in Trypanosoma brucei. Biochem. J. 357, 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roldán A., Comini M. A., Crispo M., Krauth-Siegel R. L. (2011) Lipoamide dehydrogenase is essential for both bloodstream and procyclic Trypanosoma brucei. Mol. Microbiol. 81, 623–639 [DOI] [PubMed] [Google Scholar]
- 40. Lai D. H., Hashimi H., Lun Z. R., Ayala F. J., Lukes J. (2008) Adaptations of Trypanosoma brucei to gradual loss of kinetoplast DNA. Trypanosoma equiperdum and Trypanosoma evansi are petite mutants of T. brucei. Proc. Natl. Acad. Sci. U.S.A. 105, 1999–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wickstead B., Ersfeld K., Gull K. (2002) Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol. Biochem. Parasitol. 125, 211–216 [DOI] [PubMed] [Google Scholar]
- 42. Kelly S., Reed J., Kramer S., Ellis L., Webb H., Sunter J., Salje J., Marinsek N., Gull K., Wickstead B., Carrington M. (2007) Functional genomics in Trypanosoma brucei. A collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci. Mol. Biochem. Parasitol. 154, 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Long S., Jirků M., Mach J., Ginger M. L., Sutak R., Richardson D., Tachezy J., Lukes J. (2008) Ancestral roles of eukaryotic frataxin. Mitochondrial frataxin function and heterologous expression of hydrogenosomal Trichomonas homologues in trypanosomes. Mol. Microbiol. 69, 94–109 [DOI] [PubMed] [Google Scholar]
- 44. Kafková L., Ammerman M. L., Faktorová D., Fisk J. C., Zimmer S. L., Sobotka R., Read L. K., Lukes J., Hashimi H. (2012) Functional characterization of two paralogs that are novel RNA-binding proteins influencing mitochondrial transcripts of Trypanosoma brucei. RNA 18, 1846–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Speijer D., Breek C. K., Muijsers A. O., Hartog A. F., Berden J. A., Albracht S. P., Samyn B., Van Beeumen J., Benne R. (1997) Characterization of the respiratory chain from cultured Crithidia fasciculata. Mol. Biochem. Parasitol. 85, 171–186 [DOI] [PubMed] [Google Scholar]
- 46. Mathias R. A., Chen Y. S., Kapp E. A., Greening D. W., Mathivanan S., Simpson R. J. (2011) Triton X-114 phase separation in the isolation and purification of mouse liver microsomal membrane proteins. Methods 54, 396–406 [DOI] [PubMed] [Google Scholar]
- 47. Schneider A., Charrière F., Pusnik M., Horn E. K. (2007) Isolation of mitochondria from procyclic Trypanosoma brucei. Methods Mol. Biol. 372, 67–80 [DOI] [PubMed] [Google Scholar]
- 48. Carnes J., Trotter J. R., Ernst N. L., Steinberg A., Stuart K. (2005) An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 102, 16614–16619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nebohácová M., Maslov D. A., Falick A. M., Simpson L. (2004) The effect of RNA interference Down-regulation of RNA editing 3′-terminal uridylyl transferase (TUTase) 1 on mitochondrial de novo protein synthesis and stability of respiratory complexes in Trypanosoma brucei. J. Biol. Chem. 279, 7819–7825 [DOI] [PubMed] [Google Scholar]
- 50. Maslov D. A., Zíková A., Kyselová I., Lukes J. (2002) A putative novel nuclear-encoded subunit of the cytochrome c oxidase complex in trypanosomatids. Mol. Biochem. Parasitol. 125, 113–125 [DOI] [PubMed] [Google Scholar]
- 51. Malka F., Guillery O., Cifuentes-Diaz C., Guillou E., Belenguer P., Lombès A., Rojo M. (2005) Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 6, 853–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wirtz E., Leal S., Ochatt C., Cross G. A. (1999) A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99, 89–101 [DOI] [PubMed] [Google Scholar]
- 53. Vafai S. B., Mootha V. K. (2012) Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383 [DOI] [PubMed] [Google Scholar]
- 54. Mitchell P. (2011) Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. 1966. Biochim. Biophys. Acta 1807, 1507–1538 [DOI] [PubMed] [Google Scholar]
- 55. Paucek P., Mironova G., Mahdi F., Beavis A. D., Woldegiorgis G., Garlid K. D. (1992) Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. J. Biol. Chem. 267, 26062–26069 [PubMed] [Google Scholar]
- 56. Costa A. D., Krieger M. A. (2009) Evidence for an ATP-sensitive K+ channel in mitoplasts isolated from Trypanosoma cruzi and Crithidia fasciculata. Int. J. Parasitol. 39, 955–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Inoue I., Nagase H., Kishi K., Higuti T. (1991) ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 352, 244–247 [DOI] [PubMed] [Google Scholar]
- 58. Haddy F. J., Vanhoutte P. M., Feletou M. (2006) Role of potassium in regulating blood flow and blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R546–R552 [DOI] [PubMed] [Google Scholar]
- 59. Rodríguez-Navarro A. (2000) Potassium transport in fungi and plants. Biochim. Biophys. Acta 1469, 1–30 [DOI] [PubMed] [Google Scholar]
- 60. Rizzuto R., De Stefani D., Raffaello A., Mammucari C. (2012) Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578 [DOI] [PubMed] [Google Scholar]
- 61. Spremulli L., Kraus B. L. (1987) Bovine mitochondrial ribosomes. Effect of cations and heterologous dissociation factors on subunit interactions. Biochem. Biophys. Res. Commun. 147, 1077–1081 [DOI] [PubMed] [Google Scholar]
- 62. Maslov D., Agrawal R. (2012) Mitochondrial translation in trypanosomatids. in RNA Metabolism in Trypanosomes (Bindereif A., ed) pp. 215–236, Springer, Berlin [Google Scholar]