

Background: Dysregulation of Ca2+ homeostasis has been implicated in Alzheimer disease pathogenesis, but the effects of Ca2+ on amyloid precursor protein processing are not well understood.
Results: Constitutive activation of the store-operated calcium entry pathway reduces β-amyloid generation.
Conclusion: Elevation of Ca2+ influx affects amyloid precursor protein processing.
Significance: Alteration of Ca2+ homeostasis in Alzheimer disease may influence pathogenesis directly through modulation of β-amyloid production.
Keywords: Alzheimer Disease, Amyloid, Amyloid Precursor Protein, Calcium Imaging, Calcium Signaling, Orai1, STIM1, β-Amyloid, Store-operated Calcium Entry
Abstract
Alzheimer disease (AD), the leading cause of dementia, is characterized by the accumulation of β-amyloid peptides (Aβ) in senile plaques in the brains of affected patients. Many cellular mechanisms are thought to play important roles in the development and progression of AD. Several lines of evidence point to the dysregulation of Ca2+ homeostasis as underlying aspects of AD pathogenesis. Moreover, direct roles in the regulation of Ca2+ homeostasis have been demonstrated for proteins encoded by familial AD-linked genes such as PSEN1, PSEN2, and APP, as well as Aβ peptides. Whereas these studies support the hypothesis that disruption of Ca2+ homeostasis contributes to AD, it is difficult to disentangle the effects of familial AD-linked genes on Aβ production from their effects on Ca2+ homeostasis. Here, we developed a system in which cellular Ca2+ homeostasis could be directly manipulated to study the effects on amyloid precursor protein metabolism and Aβ production. We overexpressed stromal interaction molecule 1 (STIM1) and Orai1, the components of the store-operated Ca2+ entry pathway, to generate cells with constitutive and store depletion-induced Ca2+ entry. We found striking effects of Ca2+ entry induced by overexpression of the constitutively active STIM1D76A mutant on amyloid precursor protein metabolism. Specifically, constitutive activation of Ca2+ entry by expression of STIM1D76A significantly reduced Aβ secretion. Our results suggest that disruptions in Ca2+ homeostasis may influence AD pathogenesis directly through the modulation of Aβ production.
Introduction
Alzheimer disease (AD)3 is a progressive neurodegenerative disorder, the number one cause of dementia in the elderly and the sixth leading cause of death in the United States (1). Pathologically, AD is characterized by the accumulation of 38–43-amino acid-long amyloid β peptides (Aβ) in senile plaques and the presence of tangles composed of hyperphosphorylated tau in the brains of affected individuals (2). Clinically, >90% of cases of AD are classified as non-familial or sporadic disease, with aging as the main risk factor. However, causative mutations leading to early-onset familial AD have been identified in three genes: APP, PSEN1, and PSEN2 (3). These mutations all appear to lead to AD by increasing overall levels of Aβ or by promoting production of Aβ peptides (Aβ42) that are more prone to oligomerization and deposition. As such, the “amyloid cascade” hypothesis of AD was developed, and Aβ is still considered to be one of, if not the most, important factors in the pathogenesis of AD (4).
Aβ peptides are generated through sequential proteolytic cleavage of amyloid precursor protein (APP), which is a type I transmembrane protein (5). In the Aβ-producing amyloidogenic pathway, APP is first cleaved by the aspartyl protease β-secretase (β-site APP-cleaving enzyme (BACE1)) within its extracellular domain, liberating the soluble ectodomain sAPPβ and generating a membrane-tethered β-C-terminal fragment (β-CTF) (6). The β-CTF is then cleaved within its transmembrane domain by γ-secretase, releasing Aβ and the cytoplasmic APP intracellular domain (AICD). γ-Secretase is an unusual aspartyl protease made up of four transmembrane subunits: nicastrin, APH-1, PEN-2, and presenilin (PS) 1 or PS2 (7). The β-CTF is cleaved serially by γ-secretase at multiple sites producing Aβ fragments of varying size, with Aβ40 and Aβ42 being the most abundant (5). Alternatively, APP can be processed in a non-amyloidogenic manner. Cleavage by α-secretase generates sAPPα and an α-CTF, which is further cleaved by γ-secretase to produce the small peptide p3 and AICD (5). Because α-secretase processing precludes formation of Aβ, the non-amyloidogenic processing of APP is thought to be potentially beneficial.
Multiple lines of evidence suggest that Ca2+ homeostasis is deregulated in AD (8, 9). For example, alterations in the levels of Ca2+ channels, exchangers, and Ca2+-dependent enzymes have been demonstrated in the brains of affected patients (10–12). Several studies have also found altered Ca2+ homeostasis in fibroblasts isolated from patients with AD compared with controls (13–15). In fact, both PS1 and APP have been shown to mediate changes in Ca2+ homeostasis. Recent studies have proposed a variety of functions for PS1 in Ca2+ homeostasis, including modulation of store-operated Ca2+ entry (SOCE), formation of ER Ca2+ leak channels, and regulation of sarcoendoplasmic reticulum calcium transport ATPase, inositol trisphosphate receptors, and ryanodine receptors (16–21). APP appears to have numerous effects on Ca2+ homeostasis as well. Expression of full length APP, for instance, affects spontaneous Ca2+ oscillations in cultured neurons (22, 23). Effects on intracellular Ca2+ stores, on the other hand, have been attributed to the APP cleavage product AICD (24, 25). Perhaps most intriguingly of all, however, are the effects mediated directly by the interaction of Aβ with Ca2+-permeable channels. These include functional alterations of plasma membrane ion channels such as voltage-gated Ca2+ channels, nicotinic acetylcholine channels, and glutamate, serotonin, and dopamine receptors, alterations of intracellular Ca2+ channels such as ryanodine receptors and inositol trisphosphate receptors, and even the direct formation of Ca2+-permeable ion channels (26).
Although a role for disruptions in Ca2+ homeostasis in the pathogenesis of AD has been studied in the past using pharmacological manipulations, the effects of these changes on the processing of APP to generate Aβ are not well understood due to conflicting results. Therefore, we devised a genetic approach to alter [Ca2+]i levels that would allow us to more precisely investigate the effects of Ca2+ influx on Aβ generation. Recently, stromal interaction molecule 1 (STIM1) and Orai1 have been identified as the molecular components of the SOCE machinery. STIM1 is a type I transmembrane protein that resides within the ER membrane as dimers under basal conditions. Upon ER Ca2+ store depletion, STIM1 rapidly oligomerizes and translocates within the ER membrane to plasma-membrane adjacent regions where it binds, clusters, and activates the store-operated Ca2+ channel Orai1 (27, 28). Coexpression of these components is sufficient to reconstitute and potentiate SOCE (29–31). Additionally, expression of a well characterized mutant of the luminal EF-hand domain of STIM1, STIM1D76A, leads to constitutive activation of Ca2+ influx even under store-replete conditions (32). Therefore, we utilized these components of the SOCE pathway to specifically modulate Ca2+ influx and isolate the effects of these manipulations on Aβ generation in the absence of confounding mutations in PSEN genes or the use of non-physiologic pharmacologic agents. In particular, we found that increased Ca2+ influx resulting from overexpression of the constitutively active STIM1D76A mutant led to dramatic reductions in the secretion of Aβ. Our results indicate that Ca2+ influx pathways have multiple effects on APP maturation and processing and provide insights into the importance of Ca2+ homeostasis to neuronal pathophysiology and AD.
EXPERIMENTAL PROCEDURES
Cells, Plasmids, and Antibodies
Cells were cultured in DMEM supplemented with 10% bovine growth serum (Hyclone). Human embryonic kidney HEK293 (HEK) cells stably expressing c-Myc epitope-tagged wild-type APP695 (HEK-APP) and APP695 harboring the “Swedish” double mutation (HEK-APPswe) have been previously described (33). The YFP-STIM1 and YFP-STIM1D76A expression vectors were used for transient expression of STIM1 (32). For stable expression of Orai1, the Orai1myc cDNA was subcloned into the pMXs-puro retroviral expression vector (provided by Dr. Toshio Kitamura (University of Tokyo)). Phoenix packaging cells (ATCC) were used to generate retroviruses, and stably transduced pools of HEK-Orai, HEK-APP-Orai, and HEK-APPswe-Orai cells were selected in the presence of 1 μg/ml puromycin. The pMX-C99–6myc plasmid has been previously described (34). Rabbit polyclonal antisera against STIM1, APP, PS1, and Flotillin-2 were previously generated in our laboratory and have been described (35–38). The following antibodies were purchased: anti-Nicastrin (Santa Cruz), mAb 9E10 (ATCC), anti-protein disulfide isomerase (Stressgen), and mAb 4G8 (Covance).
Protein Analysis
For immunoblotting, cells were lysed in cold lysis buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 0.25% sodium dodecyl sulfate, 5 mm EDTA, and protease inhibitor mixture (1:200; Sigma)) and briefly sonicated. Aliquots of lysates were fractionated by SDS-PAGE on 4–20% Tris-glycine gradient gels (Invitrogen) or 16.5% Tris-Tricine gels for APP CTF and Aβ analysis and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were sequentially incubated with primary antibodies and horseradish peroxidase-conjugated protein A (Sigma) or goat anti-mouse IgG (Jackson ImmunoResearch Laboratories), and signals were visualized by enhanced chemiluminescence detection (PerkinElmer Life Sciences). Alternatively, the blots were incubated with IR-dye-conjugated secondary antibodies and visualized by the Odyssey infrared imaging system (LI-COR Biosciences) as described (39).
Analysis of APP processing by metabolic labeling with [35S]Met/Cys was performed as previously described (40). CTM1 antiserum was used to immunoprecipitate full-length APP and APP CTFs from cell lysates, and mAb 4G8 was used to immunoprecipitate Aβ and p3 from conditioned media (40).
Immunofluorescence Microscopy
HEK cells were grown on poly-l-lysine-coated coverslips and transfected with YFP-STIM1 or YFP-STIM1D76A. After ∼36 h cells were either washed in Hanks' balanced salt solution (HBSS) and fixed in 4% paraformaldehyde or treated with 1 μm thapsigargin in 0 Ca2+ HBSS for 10 min before fixation. Cells were permeabilized with 0.2% Triton X-100, and immunofluorescence staining was performed using mAb 9E10 (0.2 μg/ml) and anti-protein disulfide isomerase (1:5000) as described (35). HEK-Orai1 cells were also double-stained with 7 μg/ml wheat germ agglutinin-rhodamine (Vector Laboratories). Alexa Fluor-555 or 647-conjugated secondary antibodies (Molecular Probes) were used at 1:500 and 1:250 dilutions, respectively. Images were acquired on Leica SP5II STED-CW Superresolution laser confocal microscope using100× objective (NA 1.4; zoom 2.5) and processed using ImageJ software.
For total internal reflection microscopy (TIRF), HEK-APP cells plated on poly-l-lysine-coated 35-mm glass-bottom dishes were transfected with YFP-STIM1 or YFP-STIM1D76A. After ∼24 h cells were placed in warm HBSS before mounting on the microscope stage maintained at 37 °C using a custom-designed environment chamber. TIRF images were acquired every 15 s using a 100× TIRF objective (1.45 NA) and an EMCCD camera (Photometrics Cascade II). After acquiring base-line TIRF images, cells were briefly washed in Ca2+-free (0 Ca2+) HBSS, and ER Ca2+ stores were depleted by the addition of 1 μm thapsigargin (Tg). Images were analyzed using Metamorph imaging software.
Ca2+ Imaging
Intracellular Ca2+ concentration ([Ca2+]i) was measured in cells loaded with 5 μm Fura-2 AM using a Nikon Diaphot inverted epifluorescence microscope and InCyt IM2TM fluorescence imaging system as previously described (38, 41). A three-part protocol was utilized to measure Ca2+ entry under basal conditions (Ca2+ stores full) and Ca2+ entry after Ca2+ store depletion. Specifically, after incubation for ∼2–3 min in 0 Ca2+ HBSS, cells were perfused in HBSS ([Ca2+] = ∼1.3 mm) to measure basal Ca2+ entry. Then cells were perfused in 0 Ca2+ HBSS for 2 min before the addition of 1 μm thapsigargin to deplete ER Ca2+ stores. Finally, cell perfusion was switched back to HBSS to trigger SOCE. Individual responses from ∼50 cells per coverslip were monitored and averaged. Each experiment was repeated on 2–4 independent coverslips. In some experiments cells were pretreated with 50 μm 2-aminoethyldiphenyl borate (2-APB) during the Fura-2 AM loading and unloading periods (∼2 h), and the cells were perfused in HBSS + 50 μm 2-APB for initial Ca2+ measurements (7 min). Then cells were perfused with HBSS for 7 min before switching back to HBSS + 50 μm 2-APB.
Aβ ELISA
Conditioned media from transfected HEK cells were collected after overnight incubation, and sAPPα, Aβ40, and Aβ42 levels were quantified by ELISA using specific monoclonal antibodies for capture (B113 for Aβ40, A387 for Aβ42, and 5228 for sAPPα) followed by alkaline phosphatase detection with monoclonal antibody B436 and CSPD Sapphire II Luminescence Substrate (Applied Biosystems) (42).
Statistics
All statistical analyses were calculated using Prism software (GraphPad Software, Inc.). Statistical tests used are indicated in the corresponding figure legends. All data are represented as the mean ± S.E.
RESULTS
Generation of a Genetic Model to Modulate Ca2+ Influx
To establish a cell culture model for studying the effects of Ca2+ influx on APP processing and Aβ production, we generated cell lines overexpressing the components of the SOCE pathway. Specifically, we transduced HEK cells stably overexpressing human wild-type APP with retroviruses carrying empty vector DNA (HEK-APP vector) or a cDNA encoding the store-operated Ca2+ channel Orai1 (HEK-APP-Orai1). For individual experiments, we then transiently transfected these cells with empty vector DNA (Control), the store-operated channel activator YFP-STIM1, or the constitutively active YFP-STIM1D76A mutant. Western blot analysis confirmed expression of Orai1 and YFP-STIM1 or YFP-STIM1D76A in these cells (Fig. 1A). Importantly, steady state levels of transgene-derived full-length APP were similar between vector transduced cells and cells overexpressing Orai1 and STIM1 (Fig. 1A). Additionally, levels of nicastrin and PS1 N-terminal fragment were similar among all groups of cells, indicating that endogenous γ-secretase subunit expression was unaffected by overexpression of the SOCE machinery (Fig. 1A). Similar results were obtained for HEK cells stably expressing APPswe (data not shown). Because expression of full-length APP and endogenous γ-secretase components are unaffected by manipulation of the SOCE machinery, we reasoned that these cells would provide a good model for investigating the effects of Ca2+ influx on APP processing.
FIGURE 1.
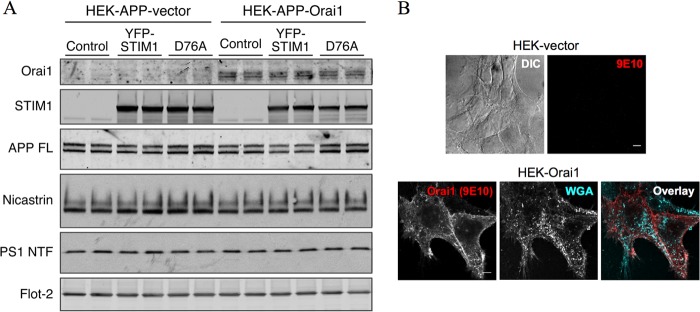
Overexpression of the SOCE machinery in HEK-APP cells. A, HEK-APP cells were stably transduced with recombinant retrovirus containing empty vector or Orai1myc cDNA followed by transient transfection with YFP-STIM1 or YFP-STIM1D76A expression plasmids. Expression of Orai1myc, YFP-STIM1, and YFP-STIM1D76A was analyzed by Western blotting. In addition, the levels of full-length APP (APP FL), nicastrin, PS1 N-terminal fragment, and flotillin-2 (Flot-2) were analyzed. B, stable HEK-vector and HEK-Orai1 cells were immunostained with mAb 9E10 to detect Myc-tagged Orai1. Cells were incubated with fluorescent wheat germ agglutinin (WGA) before permeabilization to stain surface glycoproteins and glycolipids. DIC, differential interference contrast. Scale bars, 5 μm.
Having established expression of the components of the SOCE pathway, we utilized immunofluorescence staining to characterize their subcellular localization. In accordance with previously reported studies, Orai1 appeared to localize on the surface with little intracellular accumulation (Fig. 1B). On the other hand, YFP-STIM1-transfected cells exhibited a diffuse reticular pattern of fluorescence that overlapped with the ER marker, protein disulfide isomerase (Fig. 2A). As expected, YFP-STIM1 fluorescence appeared as multiple discrete puncta after ER Ca2+ store depletion with Tg that was distinct from the ER (Fig. 2A). In accordance with previous findings (32), STIM1D76A formed puncta even under store-replete conditions (Fig. 2A). YFP-STIM1 fluorescence showed some overlap with surface localization of Orai under basal conditions (Fig. 2B). After store depletion, obvious overlap between YFP-STIM1 and Orai1 could be observed in discrete puncta. Consistent with previous studies, puncta containing STIM1D76A were also positive for Orai 1 under basal conditions (Fig. 2B).
FIGURE 2.

Subcellular localization of YFP-STIM1 and Orai1. HEK-Orai1 cells transfected with YFP-STIM1 or YFP-STIM1D76A (D76A) were fixed and imaged under basal, ER Ca2+ store-replete conditions or after a 10-min treatment with 1 μm Tg in 0 Ca2+ HBSS. A, cells were immunostained with an antibody against the ER marker, protein disulfide isomerase (PDI). Scale bars, 5 μm. B, cells were immunostained with mAb 9E10 to detect Orai1. Insets show enlarged regions indicated by boxes. Scale bars, 5 μm. C, HEK-APP cells transfected with YFP-STIM1 or YFP-STIM1D76A were imaged using TIRF microscopy. Cells were maintained in HBSS in a humidified environment at 37 °C, and images were acquired every 15 s. After 2 min, 1 μm Tg was added in 0 Ca2+ HBSS to deplete Ca2+ stores. Representative frames from the TIRF image sequence taken before (0 min) and 10 min after the addition of Tg (12 min) are shown.
Next, we used TIRF microscopy to observe the dynamic behavior of YFP-STIM1 at sites adjacent to the plasma membrane. Using this approach, we confirmed the dynamic translocation of YFP-STIM1 to plasma-membrane adjacent sites in HEK-APP cells after store depletion with Tg (Fig. 2C; supplemental movie, left panel). In contrast, after transfection with YFP-STIM1D76A, we observed numerous puncta near the plasma membrane under store-replete conditions, and the sizes of these puncta were not significantly affected by store depletion (Fig. 2C; supplemental movie, right panel). These results confirm that STIM1 and STIM1D76A are not only expressed but also dynamically localize as expected in our cell culture model.
STIM1D76A Alters Ca2+ Homeostasis in HEK-APP Cells
Next, we loaded cells with Fura-2 AM and directly measured [Ca2+]i levels. Because the EF-hand mutation of STIM1, STIM1D76A, activates SOCE independently of ER store depletion, we used a three-part protocol to measure both store-dependent (SOCE) and store-independent Ca2+ influx (Fig. 3, A and D). First, cells were switched from perfusion in 0 Ca2+ HBSS to HBSS to measure store-independent, basal Ca2+ entry. After [Ca2+]i levels plateaued, cells were perfused in 0 Ca2+ HBSS, and 1 μm Tg was added to deplete ER Ca2+ stores followed by Ca2+ add-back in HBSS to trigger SOCE. The plateau for basal Ca2+ entry (Fig. 3, B and E) and the peak for SOCE (Fig. 3, C and F) were then quantified for each stable cell pool. In vector-transduced HEK-APP cells, overexpression of YFP-STIM1 produced a small, but non-significant potentiation of basal Ca2+ entry, whereas overexpression of YFP-STIM1D76A dramatically increased basal Ca2+ entry (Fig. 3B). Upon Ca2+ add-back, robust SOCE was induced in all cases without any discernible difference between the three groups (Fig. 3C).
FIGURE 3.

Characterization of calcium entry in HEK-APP and HEK-APP-Orai1 cells. A, Fura-2 AM-loaded HEK-APP cells were imaged to quantify Ca2+ entry phenotypes induced by YFP-STIM1 or YFP-STIM1D76A expression. First, cells were switched from 0 Ca2+HBSS to HBSS to assess basal Ca2+ levels. Subsequently 1 μm Tg was added in 0 Ca2+HBSS to deplete Ca2+ stores followed by calcium add-back to trigger SOCE. B and C, shown is quantification of the plateau of basal Ca2+ entry (B) and peak SOCE (C) in HEK-APP cells. D, Fura-2 AM loaded HEK-APP-Orai cells were imaged as in A. E and F, shown is quantification of basal Ca2+ entry (E) and peak SOCE (F) in HEK-APP-Orai1 cells. Data represent 3–4 experiments, with ∼50 cells per experiment; one-way analysis of variance; **, p < 0.01; ***, p < 0.001. G, HEK-APP-Orai1 cells transiently expressing STIM1D76A were pretreated with 50 μm 2-APB for 2 h during the Fura-2 loading and unloading period, the cells were transferred to the imaging chamber containing HBSS + 2-APB, and the basal Ca2+ levels recorded. The effect of 2-APB addition on the basal Ca2+ levels is plotted; unpaired t test with Welch's correction; ****, p < 0.0001. H, HEK-APP-Orai1 cells transiently expressing STIM1D76A were pretreated with 50 μm 2-APB as above, and the basal Ca2+ levels recorded. The cells were then perfused with HBSS for 7 min, and then 2-APB was added back to the chamber. The trace represents the mean of six experiments, with ∼50 cells per experiment.
Consistent with previous reports showing a dominant negative effect of expression of Orai1 alone (29, 31), HEK-APP-Orai1 cells, which stably overexpress Orai1, had reduced SOCE (Fig. 3F) as compared with parental HEK-APP cells (Fig. 3C; Table 1). The transient expression of YFP-STIM1 in HEK-APP-Orai1 cells produced a modest increase in basal Ca2+ entry and a large increase in SOCE compared with the transient transfection control (HEK-APP-Orai1 cells transfected with an empty vector), as expected (Fig. 3, E and F). The transient expression of YFP-STIM1D76A in HEK-APP-Orai1 cells, in contrast, led to dramatic increases in basal Ca2+ entry along with significantly elevated basal [Ca2+]i levels even in 0 Ca2+ HBSS (Fig. 3, D–F). Similar alterations in Ca2+ homeostasis were observed in HEK-APPswe cells (Fig. 4). Thus, our data demonstrate significant modulation of Ca2+ homeostasis in these cells (summarized in Table 1), with the most dramatic changes observed in cells expressing STIM1D76A.
TABLE 1.
Summary of changes in Ca2+ homeostasis in HEK-APP cells
| Protein expression | Basal [Ca2+]i | Basal Ca2+ entry (plateau) | SOCE peak |
|---|---|---|---|
| nm | nm | nm | |
| Vector_Control | 30.4 | 28.0 | 565.5 |
| Vector_YFP-STIM1 | 42.7 | 65.2 | 649.2 |
| Vector_YFP-STIM1D76A | 54.5 | 209.3 | 513.0 |
| Orai_Control | 29.4 | 27.4 | 187.0 |
| Orai _YFP-STIM1 | 60.9 | 138.1 | 548.6 |
| Orai _YFP-STIM1D76A | 191.7 | 259.0 | 426.9 |
FIGURE 4.

Generation and characterization of Ca2+ homeostasis in HEK-APPswe cells. A, HEK cells stably expressing APPswe were transduced with empty vector or Orai1myc followed by transient transfection with YFP-STIM1 or YFP-STIM1D76A. Western blot analysis confirmed successful expression of Orai1myc, YFP-STIM1, or YFP-STIM1D76A, and APPswe. B and C, Fura-2 AM-loaded HEK-APPswe (B) and HEK-APPswe-Orai (C) cells were imaged to quantify Ca2+ entry phenotypes induced by YFP-STIM1 or YFP-STIM1D76A expression as described in Fig. 2. D, stably-transduced HEK-Orai cells (without APP overexpression) were transfected with empty vector control or YFP-STIM1 and subsequently loaded with Fura-4F for Ca2+ imaging. After perfusion in HBSS, 1 μm Tg was added in 0 Ca2+ HBSS to deplete Ca2+ stores followed by perfusion in HBSS to trigger SOCE. Data represent four experiments with ∼50 cells per experiment.
To confirm that the elevated basal Ca2+ levels in the HEK-APP-Orai1 cells transiently expressing YFP-STIM1D76A were indeed the result of elevated SOCE, the effect of the SOCE inhibitor 2-APB was investigated. Cells were pretreated with 50 μm 2-APB during the Fura-2 loading and unloading periods, and the cells were placed in HBSS + 50 μm 2-APB for the initial Ca2+ measurements. The high basal levels are clearly inhibited by 2-APB as seen in the statistical comparison of Ca2+ basal levels in HEK-APP-Orai1-YFP-STIM1D76A cells in the presence and absence of 2-APB (Fig. 3G). Furthermore, the Ca2+ levels in these cells dynamically changed when 2-APB was removed for a brief period and then added back (Fig. 3H).
It is interesting to note that although the transient expression of STIM1 significantly potentiated SOCE in HEK-APP-Orai1 cells, the overall magnitude of SOCE was not increased compared with HEK-APP cells transiently expressing STIM1 (Fig. 3, F versus C; Table 1). Previously published data (29–31) in HEK cells overexpressing both Orai1 and STIM1 show much higher levels of SOCE than seen in HEK-APP cells overexpressing Orai1 and STIM1. Our own experiments overexpressing Orai1 and STIM1 in HEK cells (without overexpression of APP) also showed a much higher level of SOCE (Fig. 4D), suggesting that the apparent difference in the magnitude of SOCE potentiation by overexpressing Orai1 and STIM1 may result from the overexpression of APP. In support of this notion, we observed that the magnitude of SOCE in HEK-APPswe cells overexpressing Orai1 and STIM1 was also different from that seen in HEK cells overexpressing Orai1 and STIM1 (Fig. 4).
STIM1D76A Expression Leads to Accumulation of APP CTFs and Reduced Aβ Secretion
Having confirmed significant alterations in Ca2+ homeostasis in our cell culture system, we began to investigate the effects of these changes on APP processing. First, we performed Western blot analyses to assess the steady-state levels of full-length APP and APP CTFs derived from α-secretase and BACE1 processing (α- and β-CTFs, respectively) (Fig. 5A). We found that overexpression of YFP-STIM1D76A significantly increased β-CTF levels in both HEK-APP vector and HEK-APP-Orai1 cells compared with control transfection (Fig. 5B). Similarly, significant accumulation of β-CTFs also occurred in HEK-APPswe-Orai cells after the expression of YFP-STIM1D76A (Fig. 5, C and D). Although we also observed a trend in the accumulation of α-CTFs after the overexpression of STIM1D76A, the difference did not reach statistical significance (Fig. 5). These results raised the possibility that STIM1-mediated alterations in Ca2+ homeostasis may have effects on APP processing and/or the fate of APP CTFs.
FIGURE 5.

Constitutive activation of SOCE leads to accumulation of APP β-CTFs. A, Western blot analysis of full-length APP and APP CTFs in HEK-APP vector and HEK-APP-Orai1 cells transfected with control, YFP-STIM1, or YFP-STIM1D76A plasmids is shown. B, quantification of α- and β-CTF levels, normalized to loading control (Flot-2) HEK-APP-vector cells transfected with control plasmid is shown. C, Western blot analysis of transfected HEK-APPswe and HEK-APPswe-Orai1 cells is as described in A. D, quantification of α- and β-CTF levels is as described in B. Data represent three experiments performed in duplicate; one-way analysis of variance; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To further characterize the effects of Orai1 and STIM1 expression on APP processing, we performed metabolic labeling in the HEK-APP vector and HEK-APP-Orai1 cells transfected with YFP-STIM1D76A (Fig. 6, left panels). In parallel, we also transfected HEK-APPswe vector and HEK-APPswe-Orai1 cells with YFP-STIM1D76A (Fig. 6, right panels). As described above, expression of YFP-STIM1D76A alone or coexpression with Orai1 results in significantly elevated levels of Ca2+ entry, even in the absence of store depletion (Figs. 3 and 4, Table 1). We first measured APP synthesis by pulse-labeling cells for 15 min with [35S]Met/Cys and found similar levels of immature full-length APP in control and STIM1D76A-transfected cells (Fig. 6). After 3 h of continuous labeling [35S]Met/Cys, we observed similar overall levels of full-length APP among groups but found that compared with control cells, STIM1D76A-transfected cells had a shift in the ratio of mature to immature APP, favoring immature APP (Fig. 6). In contrast, steady-state levels of mature and immature APP were similar across cells lines (Figs. 1A and 4A), suggesting a delay rather than an absolute blockade in APP maturation induced by elevation of [Ca2+]i levels.
FIGURE 6.

Analysis of APP processing in HEK-APP and HEK-APPswe cells by metabolic labeling. The indicated HEK cells were transiently transfected with YFP-STIM1D76A and pulse-labeled for 15 min or continuously labeled for 3 h with [35S]Met/Cys. Full-length (FL) APP and APP CTFs were immunoprecipitated from cell lysates with CTM1 antibody and analyzed by phosphorimaging. Secreted Aβ was analyzed by immunoprecipitation of conditioned media collected after 3 h labeling and using the monoclonal antibody 4G8. The bands corresponding to Aβ and +11 Aβ, which are generated by BACE1 cleavage at alternate sites followed by γ-secretase cleavage, are indicated. The peptide p3 is released by sequential cleavage of APP by α- and γ-secretases. Imm, immature APP; Mat, mature APP.
Next, we immunoprecipitated APP CTFs from lysates of cells after 3 h of continuous labeling using the APP C-terminal-specific antibody CTM1. We observed a small but reproducible increase in both α- and β-CTFs in STIM1D76A-transfected cells (Fig. 6), consistent with the analysis of steady-state APP CTFs in these cells (Fig. 5). To examine the levels of secreted Aβ-related peptides, we then subjected conditioned media from these experiments to immunoprecipitation using monoclonal antibody 4G8. Notably, in cells transfected with STIM1D76A, we observed decreased levels of secreted Aβ (Fig. 6) both from cells stably expressing wild-type APP and from cells expressing APPswe. Furthermore, in STIM1D76A-transfected cells expressing APPswe we were also able to observe decreases in secretion of the alternate β-site cleavage-derived product Aβ11–40 and a corresponding increase in the levels of α-secretase cleavage-derived p3 peptide (Fig. 6, long exposure (long exp.)). Unfortunately, the levels of Aβ11–40 and p3 were too low to detect in cells expressing wild-type APP. Importantly, overall protein secretion was not reduced in STIM1D76A-transfected cells (data not shown), suggesting that the observed effects on Aβ secretion are not due to a generalized impairment in secretory protein trafficking or secretion of lumenal cargo.
To confirm the results observed in our metabolic labeling experiments, we collected media conditioned by HEK-APP and HEK-APP-Orai1 cells after transfection with YFP-STIM1D76A and quantified levels of Aβ40, Aβ42, and sAPPα by ELISA. In both wild-type APP- and APPswe-expressing cells, elevation of [Ca2+]i levels by transfection of STIM1D76A produced significant decreases in the amount of secreted Aβ40 and Aβ42 (Fig. 7, A and C). Transfection of STIM1D76A also resulted in increased secretion of sAPPα in cells expressing APPswe, although no increase in sAPPα levels was detected in cells expressing wild-type APP (Fig. 7, B and D).
FIGURE 7.

Modulation of SOCE alters levels of secreted Aβ in HEK-APP and HEK-APPswe cells. A and B, HEK-APP-vector and HEK-APP-Orai cells were transiently transfected with control plasmid, YFP-STIM1, or YFP-STIM1D76A. The levels of Aβ40, Aβ42 (A), and sAPPα (B) in conditioned media were quantified by ELISA. C and D, HEK-APPswe-vector and HEK-APPswe-Orai1 cells were transfected as above, and the levels of secreted Aβ40, Aβ42, and sAPPα were analyzed by ELISA. Data represent three experiments performed in duplicate; one-way analysis of variance; *, p < 0.05; **, p < 0.01; ***, p < 0.001. E, HEK cells stably expressing Orai1myc were co-transfected with C99–6myc and either empty vector or YFP-STIM1D76A. Western blot analysis of C99 and AICD was then performed using an anti-myc antibody (9E10), and the ratio of AICD:C99 was quantified; n = 8 transfections; Student's t test; **, p < 0.01.
Based on the observations from metabolic labeling experiments, we reasoned that accumulation of APP CTFs and diminution of Aβ secretion by elevation of [Ca2+]i levels in cells expressing STIM1D76A might be due to reduced γ-secretase processing of APP β-CTFs. However, it was also possible that elevated [Ca2+]i levels independently influenced BACE1 and γ-secretase processing. To directly test the effects of [Ca2+]i levels on γ-secretase processing, we co-transfected HEK cells stably expressing Orai1 with YFP-STIM1D76A and a plasmid encoding C99–6myc, the APP β-CTF. This construct is often used to examine γ-secretase cleavage of the APP β-CTF in the absence of any confounding effects of α- and β-secretase processing of full-length APP. In agreement with our prediction, we found significantly reduced cleavage of C99 to AICD in cells expressing STIM1D76A compared with controls (Fig. 7E). Together, these findings suggest that elevation of [Ca2+]i levels through the SOCE pathway leads to multiple effects on APP metabolism, including reduced amyloidogenic processing of APP β-CTF by γ-secretase.
DISCUSSION
In recent years several alternative hypotheses to the amyloid cascade have been advanced to explain the synaptic dysfunction and neuronal death that occurs in AD. Dysregulation of Ca2+ homeostasis is one such example, and numerous studies have lent support to this hypothesis, from data demonstrating alterations in Ca2+ handling in cells from affected patients to the discoveries of molecular roles for presenilins, APP, and Aβ peptides in the regulation of [Ca2+]i levels (26). Although Ca2+ has well established roles in neurotransmission and synaptic plasticity, its effects on the processing of APP and Aβ generation are less well known.
Studies to date have demonstrated conflicting results. In non-excitable cells, pharmacological elevation of [Ca+2]i has been shown to both increase and decrease Aβ levels (43–45). In neurons, the data on the relationship between Ca2+ influx and Aβ production is equally unclear. For example, Tg- and depolarization-induced elevations of [Ca+2]i have been reported to selectively increase intraneuronal Aβ42 (46). Likewise, ionomycin treatment of primary cortical neurons overexpressing human APPswe resulted in an increase in Aβ production through an increase in BACE1 expression (47). In contradiction, familial AD-linked mutations in PS1 that increase Aβ42 production were found to decrease SOCE (16). Additionally, Ca2+ influx through NMDA receptors has been shown to stimulate non-amyloidogenic α-secretase processing and inhibit Aβ production (48). In general, these studies have been hampered by one of two limitations: modulation of [Ca+2]i by the expression of PS1 or PS2 mutants, which lends itself to ambiguity as PS1 or PS2 functions as the catalytic subunit of γ-secretase, and the use of non-physiologic pharmacologic agents (Ca2+ ionophore, thapsigargin) that often have pleiotropic effects on cellular function.
To avoid these limitations we utilized the molecular components of the SOCE system, STIM1 and Orai1, to genetically alter Ca2+ levels in HEK cells. In this way we hypothesized that we would be able to observe the effects of a primary alteration in Ca2+ homeostasis on APP processing in the absence of confounding effects due to presenilin mutations or pharmacologic manipulations. Based on data from previous studies (29–31), we initially expected to observe a potentiation of SOCE in HEK-APP-Orai cells overexpressing STIM1. In contrast to this well characterized effect, we found that the magnitude of SOCE was similar in HEK-APP and HEK-293-APP-Orai1 cells transfected with STIM1. Because we were able to observe STIM1-mediated potentiation of SOCE in HEK-Orai1 cells, but not HEK-APP-Orai1 cells (Figs. 3 and 4), we conclude that this discrepancy is likely attributable to the overexpression of APP. Although numerous papers have demonstrated effects of APP expression on Ca2+ homeostasis, the data are often conflicting (24, 25, 49–51). Additionally, no studies to date have examined the effects of APP on SOCE in cells overexpressing STIM1 and Orai1, and thus the exact mechanism by which APP overexpression is affecting this process remains unclear. It will be interesting to further investigate this effect in future studies.
Fortunately, we also utilized the constitutively active STIM1D76A mutant in our study. It is known that expression of this well characterized mutant of the luminal EF-hand domain of STIM1 leads to constitutive activation of Ca2+ influx even under store-replete conditions (29, 32). As expected, in co-transfected cells, Orai1 with STIM1D76A oligomerized and formed numerous puncta near the plasma membrane even under store-replete conditions (Fig. 2), in agreement with constitutive Ca2+ entry. Indeed, this reconstituted SOCE channel function was inhibited by preincubation with the SOCE blocker, 2-APB (Fig. 3) (31). Although in early studies 2-APB was thought to inhibit inositol trisphosphate receptors in addition to store-operated Ca2+ channels, as a result of intense research conducted since the discovery of Orai and STIM family proteins, the complex actions of 2-APB effects on SOCE have been attributed to a direct block of Orai subunits at the channel level as well as an additional uncoupling of STIM1 and Orai subunits (52–57). Thus, the most dramatic derangements in Ca2+ homeostasis in HEK-APP-Orai1 cells transfected with STIM1D76A is due to activation of constitutive SOCE. Interestingly, HEK-APP-Orai1 cells transfected with STIM1D76A had significant accumulation of β-CTFs, suggesting potential alterations in APP processing and/or metabolism. Therefore, we chose to focus further investigations of APP processing on these cells with constitutive Ca2+ entry. Using metabolic labeling and ELISA we found striking reductions in the secretion of Aβ with concomitant increases in the levels of both α- and β-CTFs. Moreover, we observed reduced APP maturation, a small increase in p3 secretion, and reduced γ-secretase cleavage of APP C99. These results suggest that elevations in [Ca2+]i levels resulting from constitutive activation of Ca2+ entry affects APP metabolism at multiple levels.
Notably, the magnitude of the effect on APP processing we observed appears to be proportional to the level of derangement in cellular Ca2+ homeostasis. HEK-APP-Orai1 cells transfected with STIM1D76A exhibited the most prominent elevations in [Ca2+]i levels to such a degree that resting cytosolic Ca2+ levels were significantly elevated even in nominally Ca2+-free buffer. This elevation of basal Ca2+ levels suggests significant alterations in the homeostatic mechanisms controlling resting [Ca2+]i levels. Although this effect has been observed previously, the mechanisms mediating it have not been well characterized (29). However, the alterations in Ca2+ homeostatic mechanisms are clearly dependent on the elevated SOCE, as the elevated basal Ca2+ levels in HEK-APP-Orai1-YFP-STIM1D76A cells were returned to almost normal levels by preincubating cells for 2 h with 2-APB, a widely used pharmacological agent that (at the concentration used in our study) inhibits SOCE and calcium release-activated Ca2+ currents (Fig. 3, G and H). For the purposes of our study, these dramatic alterations in Ca2+ handling were correlated with both greater accumulation of APP CTFs and reduced Aβ generation compared with STIM1D76A-transfected cells that do not coexpress Orai1. Therefore, there may be a dose-response relationship between the magnitude of elevation in [Ca2+]i levels and the impairment in Aβ generation, strengthening the correlation between dysregulation of Ca2+ homeostasis and reduced amyloidogenic APP processing.
Throughout our investigations we utilized cells overexpressing wild-type APP and the familial AD-linked Swedish APP mutation. In almost all experiments we found a similar effect of alterations in Ca2+ homeostasis on APP metabolism. These include a delay in maturation of APP, accumulation of APP CTFs, reduced secretion of Aβ, and a small increase in p3 levels. The one exception was the effect of STIM1D76A transfection on secretion of sAPPα. In cells expressing APPswe we found that elevation of [Ca2+]i levels resulted in increased sAPPα secretion, and again greater alterations in Ca2+ homeostasis were correlated with larger increases in sAPPα secretion (Fig. 7D). However, in cells expressing wild-type APP, STIM1D76A transfection produced no change in secretion of sAPPα (Fig. 7B). We suggest that the reason for this difference is likely due to the extent to which full-length APP molecules are subject to amyloidogenic versus non-amyloidogenic processing in these cells. Whereas most wild-type APP undergoes non-amyloidogenic processing, the presence of Swedish mutations in APP leads to preferential BACE1 cleavage and consequently a greater proportion of APP undergoing amyloidogenic processing (58). Notably, BACE1 processing of APPswe can occur as early as during transit of nascent APPswe polypeptides through the cis-Golgi (58). Thus, in HEK-APPswe cells even a small reduction in amyloidogenic processing would allow more APP to reach the cell surface and be subject to non-amyloidogenic processing, resulting in a readily observable increase in sAPPα. On the other hand, in HEK-APP-overexpressing cells where most APP is already undergoing non-amyloidogenic processing, further increases in APP available for non-amyloidogenic processing result in a proportionally smaller effect on the total levels of sAPPα produced.
Taken together, our results demonstrate that elevation of [Ca2+]i levels by Ca2+ influx through store-operated channels leads to reduced amyloidogenic processing of APP and a dramatic decrease in the generation of Aβ40 and Aβ42. Although Aβ has been implicated in the disruption of intracellular Ca2+ homeostasis through a variety of mechanisms, including membrane disruption, Ca2+ pore formation, and ion channel modulation, our data suggest that the relationship between Ca2+ and Aβ may be reciprocal. Specifically, it appears that Aβ species (peptides, oligomers, and/or fibrils) may lead to elevations in [Ca2+]i levels that then negatively regulate amyloidogenic APP processing, reducing further production of Aβ. This reciprocity could serve as a protective cellular mechanism, which limits production of Aβ when extracellular concentrations are high, preventing pathologic accumulation of potentially toxic Aβ peptides. Alterations in the mechanisms regulating Ca2+ accumulated during aging or through the acquisition of a mutation in PSEN1 or other AD-associated genes could then potentially disrupt this homeostatic balance, favoring AD pathogenesis. Alternatively, Ca2+ induced accumulation of APP CTFs may result in alterations in intracellular signaling, as has been recently demonstrated (59).
In neurons, the principal cell type affected in AD, the relationship between [Ca2+]i levels and Aβ generation and secretion is likely to be more complex than observed in our experiments in HEK cells. Calcium-regulating systems are markedly more complex in neurons, and Ca2+ signals are involved in diverse processes such as protein and secretory vesicle trafficking for neurotransmission, endocytosis, gene transcription, and synaptic plasticity. Neurons are also polarized cells, and many Ca2+ signaling events are restricted to specific microdomains. Overall, this results in a system in which the effects of Ca2+ signaling on APP processing will depend on the localization, magnitude, and mode of Ca2+ entry. For example, in presynaptic nerve terminals Aβ secretion and intraneuronal accumulation of Aβ have been linked to Ca2+-dependent neuronal activity (46, 60). On the other hand, at post-synaptic sites Ca2+ influx through NMDA receptors has been reported to reduce Aβ release (48).
We chose to utilize simplified non-neuronal cells for this work precisely because we wanted to avoid the complexity in neuronal Ca2+-regulating systems. Thus, we believe our work presents strong evidence of a direct role for elevated [Ca2+]i levels in the negative regulation of amyloidogenic APP processing. However, because we utilized the components of the SOCE pathway (STIM1 and Orai1) to manipulate [Ca2+]i levels, we cannot rule out the possibility that the effects we have observed are specific to STIM1-mediated Ca2+ influx through store-operated Ca2+ channels. The implications of this possibility on disease pathogenesis are difficult to predict because, although some studies in neurons have demonstrated functional SOCE, the precise roles of STIM1 and Orai1 in the central nervous system remain unknown (61, 62). In fact, STIM1 likely has functions independent of SOCE in neurons as it has been shown to be a negative regulator of voltage-gated Ca2+ channels (63, 64). Further studies of the specific role of STIM1 on neuronal Ca2+ regulation and APP processing are warranted in the future.
Acknowledgments
We thank Dr. S. Sisodia (University of Chicago) for the HEK-APP and HEK-APPswe cells. The YFP-STIM1 and YFP-STIM1-D76A expression vectors were a generous gift of T. Meyer (Stanford University). The Orai1myc cDNA was a generous gift of M. Prakriya (Northwestern University). Confocal imaging was performed at the Integrated Microscopy Core Facility at the University of Chicago (supported by Grant S10OD010649).
This work was supported, in whole or in part, by National Institutes of Health Grants AG021495 and AG019070 (to G. T.) and National Research Service Award NS065660 (to W. Z.). This work was also supported by grants from the Alzheimer's Association (to G. T. and K. S. V.).

This article contains a supplemental movie.
- AD
- Alzheimer disease
- Aβ
- amyloid β peptide
- APP
- amyloid precursor protein
- AICD
- APP intracellular domain
- BACE1
- β-site APP-cleaving enzyme
- CTF
- C-terminal fragment
- PS
- presenilin
- SOCE
- store-operated Ca2+ entry
- STIM1
- stromal interaction molecule 1
- Tg
- thapsigargin
- TIRF
- total internal reflection microscopy
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- 2-APB
- 2-aminoethyldiphenyl borate
- ER
- endoplasmic reticulum
- HBSS
- Hanks' balanced salt solution.
REFERENCES
- 1. Alzheimer's Association (2012) 2012 Alzheimer's disease facts and figures. Alzheimers Dement. 8, 131–168 [DOI] [PubMed] [Google Scholar]
- 2. Selkoe D. J., Mandelkow E., Holtzman D. M. (2011) The Biology of Alzheimer Disease, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 3. Tanzi R. E., Bertram L. (2001) New frontiers in Alzheimer's disease genetics. Neuron 32, 181–184 [DOI] [PubMed] [Google Scholar]
- 4. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease. Progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 5. Haass C., Kaether C., Thinakaran G., Sisodia S. (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2, a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vassar R., Kovacs D. M., Yan R., Wong P. C. (2009) The β-secretase enzyme BACE in health and Alzheimer's disease. Regulation, cell biology, function, and therapeutic potential. J. Neurosci. 29, 12787–12794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Strooper B., Iwatsubo T., Wolfe M. S. (2012) Presenilins and γ-secretase. Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2, a006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindholm D., Wootz H., Korhonen L. (2006) ER stress and neurodegenerative diseases. Cell Death Differ 13, 385–392 [DOI] [PubMed] [Google Scholar]
- 9. Green K. N., Smith I. F., Laferla F. M. (2007) Role of calcium in the pathogenesis of Alzheimer's disease and transgenic models. Subcell. Biochem. 45, 507–521 [DOI] [PubMed] [Google Scholar]
- 10. Colvin R. A., Bennett J. W., Colvin S. L. (1991) Na+-Ca2+ exchange activity is increased in Alzheimer's disease brain tissues. Ann. N.Y. Acad. Sci. 639, 325–327 [DOI] [PubMed] [Google Scholar]
- 11. Grynspan F., Griffin W. R., Cataldo A., Katayama S., Nixon R. A. (1997) Active site-directed antibodies identify calpain II as an early-appearing and pervasive component of neurofibrillary pathology in Alzheimer's disease. Brain Res 763, 145–158 [DOI] [PubMed] [Google Scholar]
- 12. Coon A. L., Wallace D. R., Mactutus C. F., Booze R. M. (1999) L-type calcium channels in the hippocampus and cerebellum of Alzheimer's disease brain tissue. Neurobiol. Aging 20, 597–603 [DOI] [PubMed] [Google Scholar]
- 13. Peterson C., Ratan R. R., Shelanski M. L., Goldman J. E. (1988) Altered response of fibroblasts from aged and Alzheimer donors to drugs that elevate cytosolic free calcium. Neurobiol. Aging 9, 261–266 [DOI] [PubMed] [Google Scholar]
- 14. Peterson C., Ratan R. R., Shelanski M. L., Goldman J. E. (1986) Cytosolic free calcium and cell spreading decrease in fibroblasts from aged and Alzheimer donors. Proc. Natl. Acad. Sci. U.S.A. 83, 7999–8001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirashima N., Etcheberrigaray R., Bergamaschi S., Racchi M., Battaini F., Binetti G., Govoni S., Alkon D. L. (1996) Calcium responses in human fibroblasts. A diagnostic molecular profile for Alzheimer's disease. Neurobiol. Aging 17, 549–555 [DOI] [PubMed] [Google Scholar]
- 16. Yoo A. S., Cheng I., Chung S., Grenfell T. Z., Lee H., Pack-Chung E., Handler M., Shen J., Xia W., Tesco G., Saunders A. J., Ding K., Frosch M. P., Tanzi R. E., Kim T. W. (2000) Presenilin-mediated modulation of capacitative calcium entry. Neuron 27, 561–572 [DOI] [PubMed] [Google Scholar]
- 17. Tu H., Nelson O., Bezprozvanny A., Wang Z., Lee S. F., Hao Y. H., Serneels L., De Strooper B., Yu G., Bezprozvanny I. (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell 126, 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stutzmann G. E., Smith I., Caccamo A., Oddo S., Parker I., Laferla F. (2007) Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer's mouse models. Ann. N.Y. Acad. Sci. 1097, 265–277 [DOI] [PubMed] [Google Scholar]
- 19. Cheung K. H., Shineman D., Müller M., Cárdenas C., Mei L., Yang J., Tomita T., Iwatsubo T., Lee V. M., Foskett J. K. (2008) Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58, 871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Green K. N., Demuro A., Akbari Y., Hitt B. D., Smith I. F., Parker I., LaFerla F. M. (2008) SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J. Cell Biol. 181, 1107–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang C., Wu B., Beglopoulos V., Wines-Samuelson M., Zhang D., Dragatsis I., Südhof T. C., Shen J. (2009) Presenilins are essential for regulating neurotransmitter release. Nature 460, 632–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kloskowska E., Malkiewicz K., Winblad B., Benedikz E., Bruton J. D. (2008) APPswe mutation increases the frequency of spontaneous Ca2+-oscillations in rat hippocampal neurons. Neurosci. Lett. 436, 250–254 [DOI] [PubMed] [Google Scholar]
- 23. Santos S. F., Tasiaux B., Sindic C., Octave J. N. (2011) Inhibition of neuronal calcium oscillations by cell surface APP phosphorylated on T668. Neurobiol. Aging 32, 2308–2313 [DOI] [PubMed] [Google Scholar]
- 24. Hamid R., Kilger E., Willem M., Vassallo N., Kostka M., Bornhövd C., Reichert A. S., Kretzschmar H. A., Haass C., Herms J. (2007) Amyloid precursor protein intracellular domain modulates cellular calcium homeostasis and ATP content. J. Neurochem. 102, 1264–1275 [DOI] [PubMed] [Google Scholar]
- 25. Leissring M. A., Murphy M. P., Mead T. R., Akbari Y., Sugarman M. C., Jannatipour M., Anliker B., Müller U., Saftig P., De Strooper B., Wolfe M. S., Golde T. E., LaFerla F. M. (2002) A physiologic signaling role for the γ-secretase-derived intracellular fragment of APP. Proc. Natl. Acad. Sci. U.S.A. 99, 4697–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Demuro A., Parker I., Stutzmann G. E. (2010) Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem. 285, 12463–12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liou J., Fivaz M., Inoue T., Meyer T. (2007) Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. U.S.A. 104, 9301–9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luik R. M., Wang B., Prakriya M., Wu M. M., Lewis R. S. (2008) Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 454, 538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mercer J. C., Dehaven W. I., Smyth J. T., Wedel B., Boyles R. R., Bird G. S., Putney J. W., Jr. (2006) Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J. Biol. Chem. 281, 24979–24990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peinelt C., Vig M., Koomoa D. L., Beck A., Nadler M. J., Koblan-Huberson M., Lis A., Fleig A., Penner R., Kinet J. P. (2006) Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat. Cell Biol. 8, 771–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soboloff J., Spassova M. A., Tang X. D., Hewavitharana T., Xu W., Gill D. L. (2006) Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 281, 20661–20665 [DOI] [PubMed] [Google Scholar]
- 32. Liou J., Kim M. L., Heo W. D., Jones J. T., Myers J. W., Ferrell J. E., Jr., Meyer T. (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim S. H., Ikeuchi T., Yu C., Sisodia S. S. (2003) Regulated hyperaccumulation of presenilin-1 and the “γ-secretase” complex. Evidence for differential intramembranous processing of transmembrane substrates. J. Biol. Chem. 278, 33992–34002 [DOI] [PubMed] [Google Scholar]
- 34. Gong P., Vetrivel K. S., Nguyen P. D., Meckler X., Cheng H., Kounnas M. Z., Wagner S. L., Parent A. T., Thinakaran G. (2010) Mutation analysis of the presenilin 1 N-terminal domain reveals a broad spectrum of γ-secretase activity toward amyloid precursor protein and other substrates. J. Biol. Chem. 285, 38042–38052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vetrivel K. S., Meckler X., Chen Y., Nguyen P. D., Seidah N. G., Vassar R., Wong P. C., Fukata M., Kounnas M. Z., Thinakaran G. (2009) Alzheimer disease Aβ production in the absence of S-palmitoylation-dependent targeting of BACE1 to lipid rafts. J. Biol. Chem. 284, 3793–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thinakaran G., Regard J. B., Bouton C. M., Harris C. L., Price D. L., Borchelt D. R., Sisodia S. S. (1998) Stable association of presenilin derivatives and absence of presenilin interactions with APP. Neurobiol. Dis. 4, 438–453 [DOI] [PubMed] [Google Scholar]
- 37. Meckler X., Roseman J., Das P., Cheng H., Pei S., Keat M., Kassarjian B., Golde T. E., Parent A. T., Thinakaran G. (2010) Reduced Alzheimer's disease β-amyloid deposition in transgenic mice expressing S-palmitoylation-deficient APH1aL and nicastrin. J. Neurosci. 30, 16160–16169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zeiger W., Ito D., Swetlik C., Oh-hora M., Villereal M. L., Thinakaran G. (2011) Stanniocalcin 2 is a negative modulator of store-operated calcium entry. Mol. Cell. Biol. 31, 3710–3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gong P., Roseman J., Fernandez C. G., Vetrivel K. S., Bindokas V. P., Zitzow L. A., Kar S., Parent A. T., Thinakaran G. (2011) Transgenic neuronal overexpression reveals that stringently regulated p23 expression is critical for coordinated movement in mice. Mol. Neurodegener. 6, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vetrivel K. S., Barman A., Chen Y., Nguyen P. D., Wagner S. L., Prabhakar R., Thinakaran G. (2011) Loss of cleavage at β′-site contributes to apparent increase in β-amyloid peptide (Aβ) secretion by β-secretase (BACE1)-glycosylphosphatidylinositol (GPI) processing of amyloid precursor protein. J. Biol. Chem. 286, 26166–26177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu X., Zagranichnaya T. K., Gurda G. T., Eves E. M., Villereal M. L. (2004) A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19-7 hippocampal neuronal cells. J. Biol. Chem. 279, 43392–43402 [DOI] [PubMed] [Google Scholar]
- 42. Vetrivel K. S., Gong P., Bowen J. W., Cheng H., Chen Y., Carter M., Nguyen P. D., Placanica L., Wieland F. T., Li Y. M., Kounnas M. Z., Thinakaran G. (2007) Dual roles of the transmembrane protein p23/TMP21 in the modulation of amyloid precursor protein metabolism. Mol. Neurodegener. 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buxbaum J. D., Ruefli A. A., Parker C. A., Cypess A. M., Greengard P. (1994) Calcium regulates processing of the Alzheimer amyloid protein precursor in a protein kinase C-independent manner. Proc. Natl. Acad. Sci. U.S.A. 91, 4489–4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Querfurth H. W., Jiang J., Geiger J. D., Selkoe D. J. (1997) Caffeine stimulates amyloid β-peptide release from β-amyloid precursor protein-transfected HEK293 cells. J. Neurochem. 69, 1580–1591 [DOI] [PubMed] [Google Scholar]
- 45. Querfurth H. W., Selkoe D. J. (1994) Calcium ionophore increases amyloid β peptide production by cultured cells. Biochemistry 33, 4550–4561 [DOI] [PubMed] [Google Scholar]
- 46. Pierrot N., Ghisdal P., Caumont A. S., Octave J. N. (2004) Intraneuronal amyloid-β1–42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J. Neurochem. 88, 1140–1150 [DOI] [PubMed] [Google Scholar]
- 47. Cho H. J., Jin S. M., Youn H. D., Huh K., Mook-Jung I. (2008) Disrupted intracellular calcium regulates BACE1 gene expression via nuclear factor of activated T cells 1 (NFAT 1) signaling. Aging Cell 7, 137–147 [DOI] [PubMed] [Google Scholar]
- 48. Hoey S. E., Williams R. J., Perkinton M. S. (2009) Synaptic NMDA receptor activation stimulates α-secretase amyloid precursor protein processing and inhibits amyloid-β production. J. Neurosci. 29, 4442–4460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chatzistavraki M., Kyratzi E., Fotinopoulou A., Papazafiri P., Efthimiopoulos S. (2013) Down-regulation of AβPP enhances both calcium content of endoplasmic reticulum and acidic stores and the dynamics of store operated calcium channel activity. J. Alzheimers Dis. 34, 407–415 [DOI] [PubMed] [Google Scholar]
- 50. Stieren E., Werchan W. P., El Ayadi A., Li F., Boehning D. (2010) FAD mutations in amyloid precursor protein do not directly perturb intracellular calcium homeostasis. PLoS ONE 5, e11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Linde C. I., Baryshnikov S. G., Mazzocco-Spezzia A., Golovina V. A. (2011) Dysregulation of Ca2+ signaling in astrocytes from mice lacking amyloid precursor protein. Am. J. Physiol. Cell Physiol. 300, C1502–C1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bootman M. D., Collins T. J., Mackenzie L., Roderick H. L., Berridge M. J., Peppiatt C. M. (2002) 2-Aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 16, 1145–1150 [DOI] [PubMed] [Google Scholar]
- 53. Peppiatt C. M., Collins T. J., Mackenzie L., Conway S. J., Holmes A. B., Bootman M. D., Berridge M. J., Seo J. T., Roderick H. L. (2003) 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps, and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium 34, 97–108 [DOI] [PubMed] [Google Scholar]
- 54. DeHaven W. I., Smyth J. T., Boyles R. R., Bird G. S., Putney J. W., Jr. (2008) Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 283, 19265–19273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peinelt C., Lis A., Beck A., Fleig A., Penner R. (2008) 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J. Physiol. 586, 3061–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schindl R., Bergsmann J., Frischauf I., Derler I., Fahrner M., Muik M., Fritsch R., Groschner K., Romanin C. (2008) 2-Aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J. Biol. Chem. 283, 20261–20267 [DOI] [PubMed] [Google Scholar]
- 57. Zhang S. L., Kozak J. A., Jiang W., Yeromin A. V., Chen J., Yu Y., Penna A., Shen W., Chi V., Cahalan M. D. (2008) Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J. Biol. Chem. 283, 17662–17671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thinakaran G., Teplow D. B., Siman R., Greenberg B., Sisodia S. S. (1996) Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the “β-secretase” site occurs in the Golgi apparatus. J. Biol. Chem. 271, 9390–9397 [DOI] [PubMed] [Google Scholar]
- 59. Deyts C., Vetrivel K. S., Das S., Shepherd Y. M., Dupré D. J., Thinakaran G., Parent A. T. (2012) Novel GαS-protein signaling associated with membrane-tethered amyloid precursor protein intracellular domain. J. Neurosci. 32, 1714–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kamenetz F., Tomita T., Hsieh H., Seabrook G., Borchelt D., Iwatsubo T., Sisodia S., Malinow R. (2003) APP processing and synaptic function. Neuron 37, 925–937 [DOI] [PubMed] [Google Scholar]
- 61. Baba A., Yasui T., Fujisawa S., Yamada R. X., Yamada M. K., Nishiyama N., Matsuki N., Ikegaya Y. (2003) Activity-evoked capacitative Ca2+ entry. Implications in synaptic plasticity. J. Neurosci. 23, 7737–7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bouron A., Altafaj X., Boisseau S., De Waard M. (2005) A store-operated Ca2+ influx activated in response to the depletion of thapsigargin-sensitive Ca2+ stores is developmentally regulated in embryonic cortical neurons from mice. Brain Res. Dev. Brain Res. 159, 64–71 [DOI] [PubMed] [Google Scholar]
- 63. Park C. Y., Shcheglovitov A., Dolmetsch R. (2010) The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330, 101–105 [DOI] [PubMed] [Google Scholar]
- 64. Wang Y., Deng X., Mancarella S., Hendron E., Eguchi S., Soboloff J., Tang X. D., Gill D. L. (2010) The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330, 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]