

Summary
The unfolded protein response (UPR) allows cells to cope with endoplasmic reticulum (ER) stress by adjusting the capacity of the ER to the load of ER-associated tasks. The UPR is important for maintaining ER homeostasis under extreme ER stress. UPR genes are important under normal growth conditions as well, but what they are required for under these conditions is less clear. Using C. elegans, we show that the ire-1/xbp-1 arm of the UPR plays a crucial role in maintaining ER plasticity and function also in the absence of external ER stress. We find that during unstressed growth conditions, loss of ire-1 or xbp-1 compromises basic ER functions required for the metabolism of secreted proteins, including translation, folding and secretion. Notably, by compromising ER-associated degradation (ERAD) and phagocytosis, loss of ire-1 hinders the clearance of misfolded proteins from the ER as well as the clearance of proteins that were secreted into the pseudocoleom. Whereas the basal activity of the UPR is beneficial under normal conditions, it accelerates the pathology caused by toxic Aβ protein in a C. elegans model of Alzheimer's disease. Taken together, our findings indicate that UPR genes are critical for maintaining secretory protein metabolism under normal growth conditions.
Key words: UPR, Caenorhabditis elegans, ER stress, ERAD, Coelomocytes, Alzheimer's disease
Introduction
The endoplasmic reticulum (ER) fulfills many roles in the cell, including the processing and trafficking of secreted proteins, lipid biosynthesis and metabolism, the production and storage of glycogen and other macromolecules and calcium sequestration. Accordingly, ER homeostasis is essential for proper cellular function; its disruption contributes to many diseases and impairs the development and function of dedicated secretory cells such as plasma cells, insulin secreting cells and liver cells (Lin et al., 2008; Yoshida, 2007).
The ER has a limited capacity and adjusts itself according to the load of its tasks. The accumulation of misfolded proteins in the ER lumen triggers the unfolded protein response (UPR), which initiates processes necessary for restoration of ER homeostasis. This is achieved by expanding the ER itself (Schuck et al., 2009) and by curtailing processes, such as translation, that further burden the ER, thus adjusting the capacity of the ER to the load of ER-associated tasks (Harding et al., 1999).
In Caenorhabditis elegans, as in humans, three proteins sense ER stress and activate the UPR: the ribonuclease inositol-requiring protein-1 (IRE-1), the PERK kinase homolog PEK-1 and activating transcription factor-6 (ATF-6). ire-1 is the only ER-stress sensor gene known in yeast. Upon ER stress, IRE-1 removes an intron from xbp-1 (X-box binding protein-1) mRNA through unconventional splicing. Spliced xbp-1 encodes a transcription factor that activates expression of UPR genes, such as chaperones and ER-associated degradation proteins (ERADs) (Calfon et al., 2002; Shen et al., 2001; Urano et al., 2002), which expand the folding capacity of the ER and increase degradation of misfolded proteins. Additional, xbp-1 independent functions of ire-1 also exist. These include activation of the cell death machinery (Urano et al., 2000; Yoneda et al., 2001), degradation of ER-localized mRNAs that encode secreted and membrane proteins through the RIDD (regulated Ire1-dependent decay) pathway (Hollien and Weissman, 2006) and induction of autophagosomes (Ogata et al., 2006).
The second branch of the UPR, mediated by PEK-1, a serine/threonine kinase, leads to the phosphorylation of the alpha subunit of the translation initiation factor eIF2. This phosphorylation inhibits the assembly of the 80S ribosome and thereby prevents protein translation initiation, thus reducing the load of proteins to be processed in the ER. In mammals, inhibition of eIF2α selectively increases the translation of ATF4, a transcription factor that regulates stress responses (Scheuner et al., 2001; Shen et al., 2005). ATF6, which mediates the third branch of the UPR, is a basic leucine-zipper transcription factor that is usually retained at the ER membrane. In response to ER stress, mammalian ATF6 transits to the Golgi complex, where it is processed by local proteases to yield an active transcription factor (Ye et al., 2000). At least in mammals, activation of ATF6 induces the expression of genes that increase the folding capacity of the ER.
A functional UPR is known to be required under conditions that abruptly disrupt the balance between ER demand and ER capacity. Most of these studies have been conducted in the presence of chemicals that disrupt protein folding, such as tunicamycin, thapsigargin and dithiothreitol (DTT), or upon expression of mutant proteins prone to misfolding, all of which abruptly disrupt ER homeostasis. Some evidence for a role of the UPR under physiological conditions has been inferred from studies of model animals lacking central UPR genes (Mori, 2009). However the role fulfilled by the UPR under these conditions has not been established yet.
In this study, we used C. elegans to investigate the consequences of UPR deficiency under physiological conditions, in the absence of any additional stress. The C. elegans genome encodes conserved homologues of all three known proximal stress-sensing components of the metazoan UPR, each represented by a single homologue, and mutant strains carrying null mutations in these genes are available and viable. By genetically manipulating each branch of the UPR and studying how these manipulations affect ER homeostasis and the life cycle of secreted proteins, we have uncovered a key role for the ire-1/xbp-1 arm of the UPR in maintaining secretory protein metabolism under basal physiological conditions.
Results
Activation of UPR pathways in ER stress-response mutants
To assess the need for a functional UPR under physiological conditions, we studied animals carrying loss-of-function mutations in central ER stress response genes in each of the UPR arms. We hypothesized that if basal ER homeostasis does not rely on the UPR, inactivation of one of the UPR arms would not disrupt ER homeostasis and hence would not activate the remaining arms of the UPR. In contrast, if a UPR arm is important for the maintenance of ER homeostasis under basal conditions, its inactivation would consequently activate the remaining intact arms of the UPR, in an attempt to restore ER homeostasis.
We assayed the activity of IRE-1 and PEK-1 in various UPR mutants. To assess the activity of IRE-1, we measured the ratio of spliced versus unspliced xbp-1 transcripts. [xbp-1 transcript is a direct substrate of the IRE-1 ribonuclease (Calfon et al., 2002; Shen et al., 2001).] This allowed us to compare the ribonuclease activity of IRE-1 in wild-type animals, to its activity in null mutants, in which the atf-6, pek-1 and ire-1/xbp-1 UPR genes are disrupted. To determine the activity of PEK-1, we measured the level of phosphorylated eIF2α. [eIF2α is a direct substrate of the PEK-1 kinase.] This allowed us to compare the kinase activity of PEK-1 in wild-type animals to its activity in mutants in which the ATF-6 and IRE-1/XBP-1 arms of the UPR are disrupted.
To analyze the importance of the IRE-1/XBP-1 branch of the UPR, we determined xbp-1 splicing in xbp-1(zc12) mutants, in which this branch has been silenced. The xbp-1(zc12) mutant contains a nonsense mutation at codon 34, terminating it before its functional domains, but leaving the xbp-1 splicing site intact (Richardson et al., 2011). Consistent with previous reports (Richardson et al., 2011), we found that the level of xbp-1 splicing in xbp-1(zc12) mutants was significantly higher than in wild-type animals (Fig. 1A). The level of xbp-1 splicing observed in xbp-1(zc12) mutants was significantly higher than in atf-6(ok551) or pek-1(ok275) null mutants as well. We did not detect a significant increase in xbp-1 splicing in pek-1 mutants compared with wild-type animals. A slightly increased level of xbp-1 splicing was observed in atf-6 mutants compared with wild-type animals (Fig. 1A). These results are consistent with previous reports that the levels of hsp-4 transcripts (hsp-4 encodes one of the major transcription targets of XBP-1 protein), as measured by qRT-PCR or using a transcriptional reporter, are altered similarly in atf-6- and pek-1-deficient animals (Shen et al., 2001).
Fig. 1.
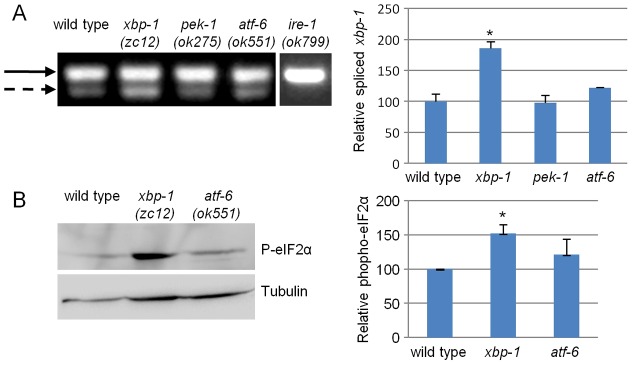
xbp-1 deficiency activates the ire-1 and pek-1 arms of the UPR. (A) Representative steady-state RT-PCR products of unspliced xbp-1 mRNA (solid arrow) and spliced xbp-1 mRNA (dashed arrow) of day-1 animals of the indicated genotypes. The xbp-1 segment was amplified by a single set of PCR primers encompassing the putative intron region. Note that no spliced xbp-1 mRNA was detected in ire-1 mutants and that a basal level of spliced xbp-1 was detected in wild-type animals. The bar graph shows the normalized mean ratio of spliced/unspliced xbp-1 transcripts in three independent biological experiments. (B) Representative western blot of phosphorylated eIF2α and tubulin of day-1 wild-type, xbp-1 and atf-6 animals. Bar graph presents the normalized mean ratio of phosphorylated eIF2α levels relative to tubulin in three independent experiments. *P<0.05 of the indicated genotype relative to wild type (Student's t-test).
Using the eIF2α phosphorylation assay, we found an increased level of phosphorylated eIF2α in xbp-1 and in ire-1(ok799) null animals relative to wild-type and atf-6 null animals (Fig. 1B; supplementary material Fig. S1). The phosphorylation of eIF2α in ire-1 mutants was very sensitive to pek-1 inactivation (supplementary material Fig. S1). In contrast, the phosphorylation of eIF2α in wild-type animals was most sensitive to RNAi inactivation of gcn-2, a cellular eIF2α-kinase that is activated in response to nutrient starvation rather than ER stress (supplementary material Fig. S1).
Taken together, we conclude that the contribution of each UPR arm to ER homeostasis under basal conditions is not equal. Assuming that the level of activation of the UPR arms directly correlates with the need to overcome an imbalance in the ER, then ER homeostasis appears to be most disrupted in ire-1 mutants and less disrupted in atf-6 mutants. No significant disruption of ER homeostasis was detected in pek-1 mutants.
Disruption of ER homeostasis and secretory protein expression in ire-1 mutants
Increased activation of the remaining arms of the UPR in the absence of one of the ER stress response genes may be sufficient to compensate and restore ER homeostasis and ER function. Alternatively, hyperactivation of the remaining arms of the UPR may be indicative of ER dysfunction as a result of chronic ER stress. To differentiate between these two scenarios, we examined the functionality of the ER in mutants defective in each of the three UPR arms. A main function of the ER is to serve as the entry point for proteins into the secretory pathway. Thus, we followed the expression of labeled secreted proteins in transgenic animals upon inactivation of ER stress-response genes. One set of transgenic lines expressed one of the worm insulin-like peptides, DAF-28, fused to GFP from the endogenous daf-28 promoter. As previously reported (Kao et al., 2007), in wild-type animals DAF-28::GFP was detected in the cells producing it (the ASI and ASJ neurons and the posterior intestine) as well as in the coelomocytes, which are macrophage-like scavenger cells that take up secreted material from the body cavity (Fig. 2A; supplementary material Fig. S2). A second set of transgenic lines expressed GFP fused to a secretion signal (referred to as ssGFP) from the muscle-specific myo-3 promoter. This strain has been used to follow GFP secretion by muscle cells, and its clearance from the body cavity by the coelomocytes (Fares and Greenwald, 2001). In wild-type animals, ssGFP was detected in the coelomocytes (supplementary material Fig. S3A), but was not clearly detected in the producing cells, probably due to its efficient secretion.
Fig. 2.

ire-1 deficiency alters DAF-28::GFP localization. (A–D) Representative fluorescence micrographs (original magnification, 100×) of day-3 wild-type, ire-1(ok799), atf-6(ok551) and pek-1(ok275) adults harboring an integrated DAF-28::GFP transgene. Blue arrows point to the ASI/ASJ neurons and red arrows indicate the hind gut. Wide white arrows point to coelomocytes, which are shown at high magnification in (A′–D′; fluorescence images) and (A′′–D′′; Nomarski images) (E) Quantification of fluorescence in ASI/ASJ neurons of the different strains, normalized to wild-type fluorescence levels. *P<0.0001 (Student's t-test). Similar results were obtained in two additional independent experiments. (F) Percentage of worms of the different genetic backgrounds in which fluorescent coelomocytes were detected. n, the number of animals analyzed. See supplementary material Fig. S2 for confocal images of DAF-28::GFP within the producing cells of wild-type animals and of ire-1, atf-6 or pek-1 mutants.
Analysis of both transgenes demonstrated that their overall expression patterns and levels in pek-1 and atf-6 mutants were similar to those seen in wild-type animals (Fig. 2; supplementary material Fig. S3). In contrast, mutating ire-1 appeared to impair protein secretion, as we observed ∼2.5-fold higher levels of the DAF-28::GFP reporter in producing ASI and ASJ neurons (Fig. 2E). DAF-28::GFP in the intestinal cells was also higher in ire-1 mutants compared with wild-type animals, but this was not quantified because of the autofluorescence of this tissue (Fig. 2). Likewise, accumulation of ssGFP in the muscle cells of ire-1 mutants could not be quantified because some of the muscle-produced ssGFP accumulated in the body cavity of ire-1(−) animals, making it difficult to distinguish between fluorescent protein in the body cavity of the animal and fluorescent protein within the adjacent muscle cells (supplementary material Fig. S3). We examined the effects of ire-1 mutation on a third secreted protein, NLP-21::YFP, a GGARAF neuropeptide expressed ectopically in the ventral cord neurons from the Punc-129 promoter. As previously reported, in wild-type animals, Punc-129::NLP-21::YFP was detected in the ventral cord neurons, and in the coelomocytes (Sieburth et al., 2007). In ire-1 mutants, the intensity of NLP-21::YFP in the producing cells was threefold higher than in wild-type animals (supplementary material Fig. S3).
We conclude that ER functionality, as judged by the ability to produce and secrete proteins passing through the secretory pathway, is maintained in pek-1 and atf-6 mutants, but not in ire-1 mutants. These data correlate with our findings that ER homeostasis is disrupted in ire-1 mutants to a greater extent than in atf-6 and pek-1 mutants.
IRE-1 is required cell-autonomously for coelomocyte function
Proteins harboring a secretion signal are translated and processed in the ER and secreted into the body cavity of the animal upon maturation and proper folding. Secreted proteins in the body cavity are eventually taken up and degraded by the coelomocyte cells. In transgenic animals expressing GFP fused to a secreted protein, the GFP is usually detected in the producing cells (before secretion) and in the coelomocytes (upon clearance from the body cavity). By following the expression pattern of DAF-28::GFP and ssGFP, we found that, unlike in wild-type animals, no GFP-labeled coelomocytes were detected in ire-1 mutants (Fig. 2B,F). Furthermore, in transgenic animals expressing secreted GFP from their muscle cells, GFP accumulated in the body cavity of the animals by day 1 of adulthood (Fig. 3E; supplementary material Fig. S3B).
Fig. 3.

ire-1 is required cell-autonomously for coelomocyte function. Representative fluorescence micrographs (original magnification, 100×) of day-3 adults harboring an integrated DAF-28::GFP transgene (A–C) and of day-1 adults harboring an integrated Pmyo-3::ssGFP transgene (D–F). In ire-1(ok799) mutants no GFP-labeled coelomocytes were detected (B,E). Rescue of ire-1 in the coelomocytes, achieved by the expression of Phat-1::ire-1 (see supplementary material Fig. S5 for rescue transgene expression) restored GFP fluorescence in coelomocytes (C,F). Wide white arrows point at coelomocytes shown at higher magnification in A′–F′′ (original magnification, 630×); fluorescence (A′–F′) and Nomarski (A″–F″) photographs are presented. (G) Percentage of animals in which fluorescent coelomocytes were detected in the different genetic backgrounds. n, the number of animals analyzed.
Why did we fail to observe fluorescent secreted proteins in the coelomocytes? First, we confirmed the presence of coelomocytes in animals lacking ire-1 both by identifying the cells using Nomarski optics (Fig. 2B′′; supplementary material Fig. S3E) and by labeling them with Pcup-4::GFP, which is expressed specifically in the coelomocytes (supplementary material Fig. S4). Next, we explored whether ire-1 may be a cup gene, required in coelomocytes for the uptake of secreted proteins from the body cavity. To this end, we tried to restore ire-1 expression specifically in the coelomocytes of ire-1(−) mutants by expressing ire-1 using the hat-1 promoter. This restored detection of the ire-1/xbp-1 pathway reporter Phsp-4::GFP only in coelomocyte cells of tunicamycin-treated ire-1(−) transgenic animals (supplementary material Fig. S5). This strongly suggested that the ire-1-rescuing construct was expressed in coelomocyte cells, but not in other cells in the animal. Expression of ire-1 using the hat-1 promoter in ire-1(−) mutants also restored detection of DAF-28::GFP and ssGFP in the coelomocytes (Fig. 3C,F). In addition, it reduced the accumulation of muscle-produced ssGFP in the body cavity of ire-1(−) mutants (Fig. 3F). Together, our data suggest that with ire-1 function intact, the coelomocytes were able to clear GFP from the body cavity of these animals.
IRE-1 is required cell-autonomously for protein secretion
The level of GFP-labeled proteins detected in the coelomocytes is usually used to estimate protein secretion rate. However, since the coelomocytes of ire-1 mutants are defective, they cannot be used for this purpose. Instead, we estimated protein secretion rate by the amount of labeled protein that accumulated in the body cavity of these animals. To this end, we deliberately prevented coelomocyte protein-uptake function in wild-type animals by introducing cup mutations. Notably, we never detected any secreted fluorescence signal taken up from the body cavity of animals by ire-1(−) or cup(−) coelomocytes. Even upon restoration of ire-1 expression in the producing cells of otherwise ire-1(−) mutants, where high levels of secreted fluorescent protein accumulated in the body cavity, no fluorescent protein was detected in the coelomocyte cells (Fig. 4). These findings suggest that the coelomocyte defect in ire-1 mutants is severe.
Fig. 4.
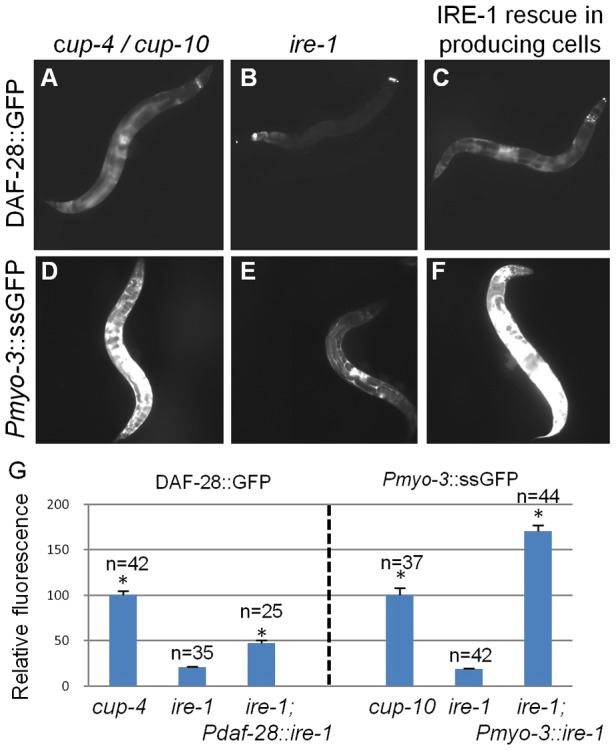
ire-1 deficiency reduces accumulation of secreted proteins in the body cavity of coelomocyte-defective animals. Representative fluorescence micrographs (100×) of (A–C) day-3 adults harboring an integrated DAF-28::GFP transgene, and (D–F) day-1 adults harboring a Pmyo-3::ssGFP transgene. Coelomocyte-defective cup-4(ok837) (A) and cup-10(ar479) (D) mutants accumulate secreted proteins in their body cavities. Coelomocyte-defective ire-1(ok799) mutants accumulate significantly less GFP in their body cavities (B,E). Expressing ire-1 specifically in the producing cells of ire-1(ok799) mutants using the daf-28 or the myo-3 promoters (see supplementary material Fig. S5 for rescue transgene expression) restored accumulation of secreted proteins in the body cavity of ire-1 mutants (C,F). (G) Bar graph showing the relative mean fluorescence measured in the whole body of the animals of the indicated genotypes. n, the number of animals analyzed. Similar results were obtained in two additional independent experiments. *P<0.0001compared with ire-1 mutants (Student's t-test).
We measured overall fluorescence to compare the amount of labeled protein that accumulated in the body of these animals. Using the Pmyo-3::ssGFP transgene, we observed accumulation of muscle-secreted GFP in the body cavity of ire-1(+);cup-10(−) and ire-1(−) mutants, both of which lack functional coelomocytes. Significantly higher levels of ssGFP were measured in ire-1(+); cup-10 mutants than in ire-1(−) mutants (Fig. 4D,E,G), suggesting less production of GFP from the muscle cells in ire-1 mutants. Similarly, using whole-body fluorescence measurements in animals expressing the DAF-28::GFP transgene, we detected relatively low fluorescence levels of DAF-28::GFP in ire-1(−) mutants compared with ire-1(+); cup-4 mutants (Fig. 4A,B,G). Curiously, this was in contrast to the fluorescence levels of DAF-28::GFP in the producing cells that were significantly higher in ire-1 mutants compared with wild-type animals (Fig. 2E). The accumulation of the secreted proteins in the producing cells, at the expense of their secretion into the body cavity of the animals, suggests that mutations in the ire-1 pathway interfere with protein secretion.
Next, we followed the expression pattern of DAF-28::GFP within the producing cells, focusing on the ASI/ASJ neurons. In wild-type animals DAF-28::GFP was detected surrounding the periphery of the nucleus as well as in the axons (Fig. 5A; supplementary material Fig. S2). In contrast, in ire-1 mutants DAF-28::GFP was exclusively detected surrounding the periphery of the nucleus, and was not detected in the axons (Fig. 5A; supplementary material Fig. S2). We suspected that DAF-28::GFP surrounding the periphery of the nucleus represents DAF-28::GFP within the ER. To confirm this, we generated a strain expressing an RFP reporter encompassing an N-terminal secretion signal and an ER retention KDEL signal at the C-terminus in the daf-28 producing cells (Fig. 5A). Upon co-expression with DAF-28::GFP, we observed that ∼85% of DAF-28::GFP colocalized with the ER marker in ire-1 mutants. In contrast, in wild-type animals, only ∼30% of DAF-28::GFP colocalized with the ER marker, as the rest of the DAF-28::GFP was detected outside of the ER (Fig. 5A,B). We conclude that in the absence of ire-1, DAF-28::GFP accumulates in the ER of the ASI/ASJ producing cells.
Fig. 5.
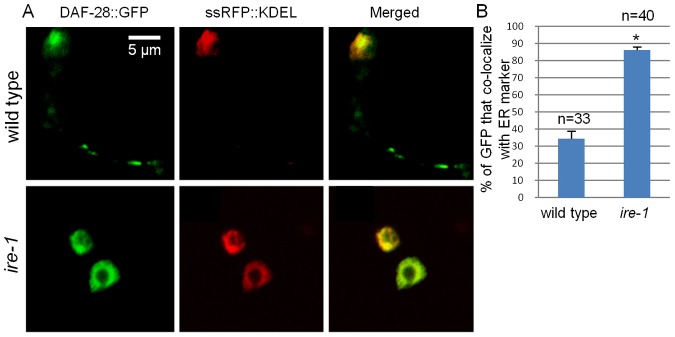
ire-1 deficiency traps DAF-28::GFP in the ER. (A) Representative fluorescence confocal micrographs (original magnification, 630×) of a 1.5-µm-thick section of ASI/ASJ neurons in day-3 wild type and ire-1 mutants co-expressing RFP (fused to a secretion signal and a KDEL-ER retention signal) and DAF-28::GFP. In ire-1 mutants most of the DAF-28::GFP colocalizes with the ER, whereas in wild-type animals the DAF-28::GFP is detected in the ER as well as in the axons. (B) Colocalization of DAF-28::GFP and the ER marker was quantified. n, the number of cells analyzed. *P<0.0001 (Student's t-test).
To explore the requirement for ire-1 in the cells producing the secreted proteins, we restored ire-1 expression using the daf-28 promoter or using the myo-3 promoter. When ire-1 expression was restored using the daf-28 promoter, the ire-1/xbp-1 pathway reporter Phsp-4::GFP was again detected in a few head neurons (most likely the ASI/ASJ neurons), in all animals examined. In some animals, Phsp-4::GFP expression was also detected in the hind gut (supplementary material Fig. S5C). Expression of ire-1 using the myo-3 promoter resulted in the Phsp-4::GFP reporter being expressed specifically in the muscle cells of tunicamycin-treated ire-1(−) transgenic animals (supplementary material Fig. S5B). These results strongly suggested that the ire-1-rescuing construct was specifically expressed in the producing cells, and not in other cells in the animal. In both cases, restoration of IRE-1 in the producing cells significantly increased the accumulation of labeled protein in the body cavity of the animals (Fig. 4C,F,G). Thus, we conclude that ire-1 functions autonomously to promote secretion by the producing cells.
These findings are consistent with concomitant defects in the cells producing the secreted proteins and in the coelomocytes of ire-1(−) mutants. Restoration of ire-1 in the producing cells restores production and secretion of secreted proteins. However, in the absence of functional coelomocytes, these proteins accumulate in the body cavity of the animal. Expression of ire-1 specifically in the coelomocytes restores uptake by the coelomocytes. However, with defective producing cells, only a small amount of the proteins intended for secretion reaches the body cavity of the animals, to be cleared by the rescued coelomocytes.
Misfolded secreted proteins accumulate in ire-1 mutants
Because the normal function of ire-1 is to decrease the load of misfolded secretory proteins, we sought a way to evaluate the level of misfolded proteins in the animal. One possibility was that misfolding could decrease the in vivo fluorescence of a GFP-tagged protein, whose absolute level could be detected by western blotting (Tanudji et al., 2002). If so, then the degree of mismatch between these two methods of protein quantification could reflect the extent of protein misfolding. To explore this, we compared DAF-28::GFP total protein levels and fluorescence levels before and after inactivation of edem-1(C47E12.3), an ERAD component, required for the degradation of misfolded proteins in the ER. Treatment with edem-1 RNAi increased total DAF-28::GFP levels in coelomocyte-defective cup-4 mutants by more than threefold, as determined by western blotting (Fig. 6A). However, treatment with edem-1 RNAi did not alter total DAF-28::GFP fluorescence in wild-type animals (Fig. 6B). This finding suggests that misfolded, non-fluorescent DAF-28::GFP is normally produced in wild-type animals, but is efficiently removed by the ERAD system. This finding provides a proof of principle that measurements of fluorescence levels versus total protein levels can be used to distinguish between properly folded and misfolded populations of this GFP-tagged protein.
Fig. 6.
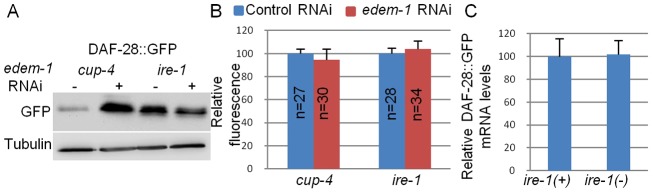
Misfolded DAF-28::GFP accumulates in ire-1 mutants. (A) Representative western blot of GFP and tubulin of coelomocyte-defective day-2 cup-4 and ire-1(ok799) mutants harboring an integrated DAF-28::GFP transgene. Animals were treated with control RNAi or edem-1 RNAi. edem-1 inactivation increased DAF-28::GFP levels in cup-4 mutants, but not in ire-1 mutants, whose basal DAF-28::GFP levels were high even before edem-1 inactivation. (B) Bar graph showing the relative mean whole-body fluorescence of DAF-28::GFP in day-2 cup-4 or ire-1 mutants treated with control RNAi (blue) or edem-1 RNAi (red). Fluorescence was calculated relative to control RNAi treatment of each strain. edem-1 inactivation did not increase DAF-28::GFP fluorescence. n, the number of animals analyzed. Similar results were obtained in two additional independent experiments. (C) Bar graph showing the average relative mRNA levels of daf-28::gfp in ire-1(+); cup-4(−) and ire-1(−) mutants, measured by qRT-PCR in three independent experiments. daf-28::gfp mRNA levels were not increased in ire-1 mutants (P>0.45, Student's t-test).
Next, we used this method to compare folded and misfolded populations of DAF-28::GFP in cup mutants and ire-1 mutants, both of which lack functional coelomocytes. We found higher levels of fluorescent DAF-28::GFP in cup mutants than in ire-1 mutants (Fig. 4G). In contrast, using the western blot measurements, we identified significantly higher levels of DAF-28::GFP in ire-1 mutants than in cup single mutants (Fig. 6A). This suggests that a significant portion of DAF-28::GFP protein in ire-1 mutants is not properly folded, and thus fails to contribute to the overall fluorescence.
Components of the ERAD machinery are well-established downstream targets of the ire-1/xbp-1 pathway. We hypothesized that the accumulation of misfolded proteins in ire-1 mutants may result from a compromised ERAD system. Thus, we asked whether edem-1 inactivation would further increase the level of misfolded DAF-28::GFP in ire-1 mutants. As predicted, treatment with edem-1 RNAi did not increase total DAF-28::GFP protein levels in ire-1 mutants, as determined by western blotting (Fig. 6A), and by fluorescence measurements (Fig. 6B). This suggests that, whereas misfolded DAF-28::GFP is normally produced in wild-type animals and efficiently removed by the ERAD system, in ire-1 mutants, the ERAD system does not remove misfolded proteins from the ER. These findings are consistent with the interpretation that the edem-1-related ERAD machinery is not functioning well in ire-1 mutants, thus leading to aberrant accumulation of misfolded proteins.
In principle, additional factors, such as increased transcript levels and/or increased translation rates, may contribute to the increased protein levels of DAF-28::GFP in ire-1 mutants. However, we did not detect significant differences in DAF-28::GFP mRNA levels between ire-1 and cup mutants using quantitative real-time PCR (Fig. 6C). Likewise, it is unlikely that translation rates are increased in ire-1 mutants, as the level of eIF2α phosphorylation, which limits translation initiation, was increased in ire-1 mutants (supplementary material Fig. S1). Furthermore, DAF-28::GFP steady-state levels, detected by western blotting, were lower in ire-1 mutants compared with cup mutants upon inactivation of ERAD (Fig. 6A). These conditions reflect DAF-28::GFP synthesis rate, lending further support to the notion that translation rate is slowed down in ire-1 mutants. We conclude that the increased protein levels of DAF-28::GFP in ire-1 mutants are mainly due to the increased stabilization of misfolded DAF-28::GFP.
Inactivation of xbp-1 is sufficient to disrupt ER homeostasis
The major mode of action of ire-1 upon ER stress is through xbp-1, a transcription factor that activates expression of downstream ER-stress-response genes. To assess whether ER homeostasis is disrupted in xbp-1 mutants, as in ire-1 mutants, we compared the levels of eIF2α phosphorylation, which reflects the activity of PEK-1. We found that eIF2α phosphorylation was significantly higher in xbp-1 mutants compared with wild-type animals (Fig. 1B).
Next, we found that the expression of labeled secreted proteins was disrupted in xbp-1 mutants, as in ire-1 mutants. As in ire-1 mutants, no labeled coelomocytes were detected in xbp-1 mutants expressing muscle-ssGFP or DAF-28::GFP (supplementary material Fig. S6), suggesting that xbp-1 is also required for coelomocytes to take up secreted proteins. Furthermore, the accumulation of GFP fluorescence in the body cavity of xbp-1 mutants was significantly lower compared with cup mutants. In the case of the DAF-28::GFP strain, an increased fluorescence of DAF-28::GFP in the producing cells was apparent (i.e. the ASI/ASJ neurons and the posterior intestinal cells) (Fig. 7A; supplementary material Fig. S6A). In contrast, total DAF-28::GFP levels in xbp-1 mutants, as detected by western blotting, were substantially higher compared with xbp-1(+) animals (Fig. 7B), indicative of the accumulation of misfolded proteins in these animals. Thus, we conclude that xbp-1 mutants display similar defects as ire-1 mutants, resulting in reduced protein secretion, protein folding and coelomocyte function.
Fig. 7.
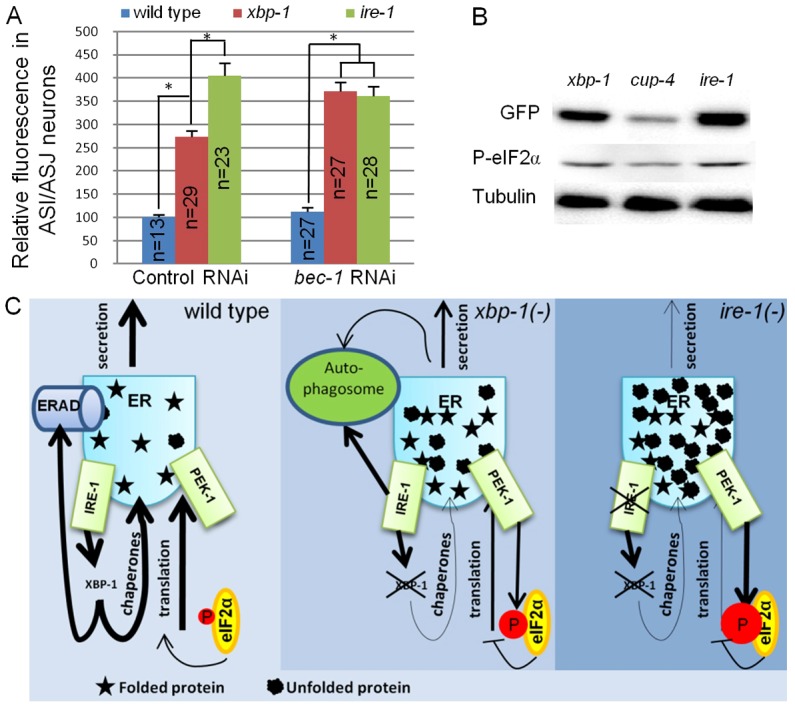
DAF-28::GFP accumulation in ASI/ASJ neurons is enhanced in ire-1 mutants and is bec-1 dependent. (A) Bar graph showing the relative mean fluorescence of DAF-28::GFP measured within the ASI/ASJ DAF-28::GFP-producing cells in wild type, xbp-1(tm2457) and ire-1(ok799) mutants treated with control or bec-1 RNAi. n, the number of animals analyzed. Similar results were obtained in two additional independent experiments. *P<0.0001 (Student's t-test). (B) Representative western blot of DAF-28::GFP, phosphorylated eIF2α and tubulin of day-1 xbp-1, cup-4 and ire-1 mutants expressing the daf-28::gfp transgene. (C) Model for disruption of ER homeostasis in ire-1 and xbp-1 mutants. Normally, xbp-1 transcribes target genes that promote ER homeostasis by preventing the accumulation of misfolded proteins in the ER. In xbp-1-deficient animals, misfolded proteins are not cleared by ERAD. These accumulate and activate IRE-1. ire-1 cannot activate xbp-1. Instead, it induces autophagosomes that clear some of the misfolded proteins from the ER. In ire-1-deficient animals, misfolded proteins accumulate in the ER but the ERAD and ire-1-induced autophagosomes do not clear them. High levels of misfolded proteins burden the ER, activate the remaining UPR arms and interfere with protein secretion.
ER homeostasis is not equally disrupted in ire-1 and xbp-1 mutants, due to autophagy
Even though ire-1 and xbp-1 mutants display similar secretion defects, direct comparison of ER homeostasis measures between these two mutants suggested that it was not disrupted equally. Specifically, we found that ire-1 mutants had higher levels of phosphorylated eIF2α than did xbp-1 mutants (Fig. 7B). Likewise, ire-1 mutants accumulated more DAF-28::GFP in the ASI and ASJ neurons than did xbp-1 mutants (Fig. 7A). These findings suggest that ER homeostasis is disrupted to a greater extent in ire-1 mutants than in xbp-1 mutants.
Previous studies in mammalian cell culture demonstrated that IRE1 promotes autophagosome formation in response to tunicamycin-induced ER stress in an xbp-1-independent manner (Ogata et al., 2006). We hypothesized that in the absence of xbp-1, the imbalanced ER activates IRE-1, which in turn promotes autophagy and possibly ER-phagy, resulting in lysosome/phagosome-mediated degradation of the cargo that accumulated in the stressed ER. This mechanism, which may partially relieve ER stress, is xbp-1 independent, but cannot be activated in the absence of ire-1. To test this hypothesis, we used RNAi to inactivate the Beclin homologue bec-1, which is needed for autophagosome formation, and compared DAF-28::GFP levels in the producing cells of wild type, ire-1 and xbp-1 mutants. bec-1 RNAi did not affect DAF-28::GFP fluorescence levels in the ASI/ASJ neurons of wild type or ire-1 mutants. In contrast, bec-1 RNAi treatment increased DAF-28::GFP fluorescence levels in the producing cells of xbp-1 mutants, to levels comparable with those of ire-1 mutants (Fig. 7A). Thus, it seems that in xbp-1 mutants, some of the DAF-28::GFP is cleared from the stressed ER through autophagosomes. We propose that autophagosomes formation is initiated by ire-1 when ER homeostasis is disrupted sufficiently, resulting in the partial clearance of protein that accumulated in the ER. In the absence of ire-1, although ER homeostasis is disrupted severely, autophagosome formation cannot be induced, resulting in even higher levels of DAF-28::GFP that cannot be cleared from the ER (see summary model in Fig. 7C).
xbp-1 and bec-1 depletion delay Aβ-associated toxicity
Our findings suggest that inhibition of the ire-1/xbp-1 pathway interferes with the translation and secretion of proteins in the secretory pathway even in the absence of exogenous ER stressors. We hypothesized that this activity may be beneficial in diseases associated with the production of toxic secreted proteins, such as Alzheimer's disease. To test this hypothesis, we used a C. elegans Alzheimer's disease model in which the expression of the Aβ peptide can be induced in its muscle cells upon temperature shift (Link et al., 2003). This Aβ peptide is equivalent to the 43 amino acid tail cleaved off the terminus of the amyloid precursor protein (APP), which is thought to be toxic to cells. Consistent with this, otherwise wild-type worms expressing this Aβ peptide are paralyzed ∼24 hours after Aβ induction (Link et al., 2003). We found that xbp-1 RNAi treatment delayed paralysis in this inducible Aβ C. elegans strain by ∼3 hours (Fig. 8A). This delay correlated with reduced Aβ protein levels (Fig. 8B,C), but did not correlate with any change in Aβ mRNA levels (Fig. 8C). Thus, this reduced Aβ toxicity may derive from the inhibition of translation in the ire-1/xbp-1 mutants.
Fig. 8.

xbp-1 inactivation reduces Aβ levels and delays paralysis. Eggs from animals expressing inducible Aβ in their muscles were grown at the permissive temperature and treated with control RNAi, xbp-1 RNAi or bec-1 RNAi for 48 hours. Animals were then shifted to the non-permissive temperature and Aβ expression was induced. (A) Paralyzed animals were scored each hour between 24 to 31 hours after temperature shift. xbp-1 RNAi significantly delayed paralysis of Aβ-expressing animals (control RNAi: mean = 25.9 hours; n = 124; xbp-1 RNAi: mean = 28.7 hours, n = 134; P<0.0001) (B) Representative western blots of Aβ (top panel) and tubulin (bottom panel) of Aβ-expressing animals treated with control RNAi and xbp-1 RNAi. Most of the Aβ protein was detected in high-molecular-mass aggregates. A very weak Aβ band was in a monomeric form. (C) Bar graph showing the average relative protein and mRNA levels of Aβ in control and xbp-1-RNAi-treated animals measured by qRT-PCR (blue bars) and western blot (red bars). Measurements were normalized to actin mRNA and tubulin protein levels, respectively. Values are the average of three independent experiments. *P>0.05, Student's t-test. (D–F) Paralyzed animals were scored every 2 hours between 20 and 32 hours after temperature shift. (D) bec-1 RNAi did not delay paralysis of wild-type Aβ-expressing animals (control RNAi: mean = 24.1 hours; n = 600; bec-1 RNAi: mean = 24.2 hours, n = 317; P = 0.27). (E) bec-1 RNAi significantly delayed paralysis of Aβ-expressing xbp-1 mutants (control RNAi: mean = 27.4 hours, n = 577; bec-1 RNAi: mean = 29.6 hours, n = 488; P<0.0001). (F) bec-1 RNAi did not delay paralysis of Aβ-expressing ire-1 mutants (control RNAi: mean = 30.4 hours, n = 380; bec-1 RNAi: mean = 28.8 hours, n = 250; P<0.0001).
Based on our finding that RNAi against bec-1 further enhanced the secretory defects in xbp-1 mutants, we hypothesized that reduction of autophagosome formation in an xbp-1-mutant background would further delay the toxicity of Aβ as well. To test this, Aβ-expressing xbp-1 mutants were treated with control or bec-1 RNAi. We found that RNAi against bec-1 further delayed the paralysis of xbp-1 mutants by ∼2 hours (Fig. 8E). Importantly, bec-1 RNAi did not delay the paralysis of ire-1 animals or wild-type animals expressing the Aβ peptide (Fig. 8D,F). Thus, counterintuitively, inhibition of the UPR through xbp-1 inactivation, in conjunction with inhibition of autophagosome formation, might be beneficial in diseases associated with toxic secreted proteins. We speculate that this is due to enhanced interference with the production of the toxic proteins in the ER.
Discussion
The ire-1/xbp-1 UPR pathway fulfills crucial functions required for embryonic development and viability in Drosophila, Xenopus and mice (Reimold et al., 2000; Souid et al., 2007; Zhao et al., 2003). A functional UPR is also required for the development of hepatic, bone marrow and B cells, as well as for yeast cytokinesis (Bicknell et al., 2007; Hu et al., 2009; Lee et al., 2008; Reimold et al., 2001; Todd et al., 2008). In C. elegans, ire-1/xbp-1-depleted animals are hypersensitive to ER stress (Shen et al., 2001) and pathogens (Richardson et al., 2010) and have a shortened lifespan (Henis-Korenblit et al., 2010). However, these mutants are viable under physiological conditions, generating a unique opportunity to study the cellular requirements of this pathway at the organism level.
One of the main functions of the ER is to serve as an entry point into the secretory pathway of the cell. In this study, we used the model organism C. elegans to investigate the physiological requirement of UPR signaling for ER homeostasis and for the life-cycle of proteins passing through the secretory pathway under standard growth conditions and in an Alzheimer's disease model. Using loss-of-function mutations disrupting the ire-1, pek-1 and atf-6 arms of the UPR, we found that elimination of one UPR arm leads to increased activation of the remaining UPR arms in adult animals. This is consistent with a comprehensive microarray analysis of UPR mutants that demonstrated that some genes associated with ER-stress conditions were upregulated in some UPR mutants (Shen et al., 2005).
Elimination of the ire-1, pek-1 and atf-6 arms of the UPR leads to increased activation of the remaining UPR arms in adult animals, albeit to different extents. Consistent with a previous report (Richardson et al., 2011), we found that inactivation of the ire-1/xbp-1 pathway significantly increased the UPR in adult animals. This was reflected in increased splicing of the xbp-1 transcript by IRE-1 and increased level of phosphorylation of eIF2α by PEK-1. Nevertheless, ER homeostasis and function were not restored in ire-1 and xbp-1 mutants, in spite of strong activation of the remaining UPR arms. In our hands, no significant disruption in ER function or activation of the UPR was detected in pek-1 mutants. In contrast, although atf-6 mutants maintained ER function, weak activation of the UPR was observed in atf-6 mutants. We propose that activation of the UPR arms in atf-6 mutants, whose ER homeostasis was mildly disturbed, served as a compensatory response that successfully restored ER homeostasis and maintained ER function. Our findings indicate that animals are constantly subjected to a basal level of ER stress and that UPR genes, and especially the ire-1/xbp-1 arm of the UPR, are required to maintain ER homeostasis under these physiological conditions.
In ire-1 and xbp-1 mutants, high levels of labeled secreted proteins were detected in the producing cells, suggesting a secretion defect. Previously, the glutamate receptor was shown to require xbp-1 to exit the ER and take its place in the plasma membrane in C. elegans (Shim et al., 2004). However, the same study demonstrated that this phenomenon was not general, as other transmembrane proteins reached their destination in the outer membrane in the absence of xbp-1. This unique feature of the glutamate receptor could be attributed to its tendency to fold improperly and its requirement for additional cofactors as it exits the ER (Vandenberghe and Bredt, 2004). However, xbp-1 appears to be more generally required for protein secretion, as we found that the ire-1/xbp-1 pathway was required for efficient protein secretion of three independent proteins expressed in the muscle, the intestine and neurons.
Our findings suggest that misfolded proteins accumulate in ire-1/xbp-1 mutants. We were able to draw this conclusion by comparing DAF-28::GFP levels of wild type and ire-1/xbp-1 mutants expressing the DAF-28::GFP transgene in two different ways: by fluorescence intensity in the animal, which reflects the level of properly folded protein, and by western blotting, which detects DAF-28::GFP irrespective of its folding state. In this way, we found that ire-1/xbp-1 mutants accumulate more DAF-28::GFP than control animals, however, most of this protein is in a misfolded state in which the GFP region cannot contribute to the fluorescence measurements.
Incorrect folding of proteins in the ER is a normal phenomenon. However, typically, misfolded proteins are cleared by the ERAD machinery and thus do not accumulate. Interestingly, inactivation of ERAD did not further increase the level of DAF-28::GFP protein in ire-1/xbp-1 mutants. This may indicate that the majority of DAF-28::GFP in these animals is properly folded and thus not affected by ERAD. However, given the lack of correlation between the fluorescence of DAF-28::GFP and DAF-28::GFP increased protein levels, this is unlikely. Instead, we favor the interpretation that the ERAD machinery in these animals is inactive to begin with. As a result, DAF-28::GFP levels are not further increased in these mutants upon ERAD inactivation. This interpretation is consistent with the fact that genes encoding ERAD components, such as hrd-1 and edem-1, are XBP-1 target genes (Yamamoto et al., 2007). Curiously, microarray studies comparing constitutive and inducible transcriptional changes in ire-1 and xbp-1 mutants demonstrated that ire-1 and xbp-1 were not required for their constitutive transcription under physiological conditions (Shen et al., 2005). Nevertheless, our findings suggest that even if ERAD components are actively transcribed in ire-1 and xbp-1 mutants, the ERAD machinery in these mutants does not function properly, leading to the accumulation of misfolded proteins instead of their degradation.
In addition to the reduced ability of cells to fold and secrete proteins into the pseudocoelom, we find that the ability of ire-1 and xbp-1 mutants to clear proteins that have been deposited in the pseudocoelom is also compromised. Using cell-specific rescue of ire-1 expression, we found that ire-1 is required cell-autonomously in specialized scavenger cells called coelomocytes to enable their efficient uptake of secreted material from the body cavity. This further supports a previous report that identified a C. elegans Derlin homolog, associated with ERAD function, as being required for coelomocyte function (Schaheen et al., 2009). It is not clear why coelomocyte scavenger function would be disrupted in the absence of ire-1 and xbp-1. Given that coelomocytes endocytose a wide variety of secreted proteins in the animal, it is possible that their level of membrane-protein trafficking and recycling is higher than that of many other cell types. Therefore their scavenger function may be particularly sensitive to reduced ER efficiency.
This coelomocyte defect explains our counterintuitive finding that although protein secretion is severely disrupted in ire-1 and xbp-1 mutants, some secreted proteins can be detected in high amounts in the body cavity of ire-1 and xbp-1 animals compared with control animals (this is the case for ssGFP expressed from the muscle cells). Thus, if proteins leak from the producing cells into the pseudoceolom, even at a very low rate, these proteins will not be cleared from the body cavity because of the coelomocyte defect, and will accumulate there.
In ire-1 and xbp-1 mutants, the pek-1 UPR pathway is activated and protein secretion and coelomocyte function are disrupted. Thus, it could be that these defects of ire-1 and xbp-1 mutants are mediated by the compensatory activation of the remaining UPR pathways. However, we were unable to support or disprove this hypothesis. This was because treatment of ire-1 mutants, expressing the DAF-28::GFP transgene, with atf-6 or pek-1 RNAi from the L3 stage did not alter protein secretion or coelomocyte function (data not shown). More robust treatment with atf-6 and pek-1 RNAi was incompatible with the animals' development.
Interestingly, ire-1 mutants accumulate higher levels of DAF-28::GFP in their producing cells than do xbp-1 mutants, suggesting that the secretory defect of ire-1 is more severe than that of xbp-1. Furthermore, ire-1 mutants exhibit higher levels of phosphorylated eIF2α than do xbp-1 mutants, reflecting increased activity of the PEK-1 UPR pathway. These finding suggest that ire-1 mutants experience more ER stress than do xbp-1 mutants, and thus that xbp-1-independent functions of ire-1 also contribute to the maintenance of physiological ER homeostasis.
What xbp-1-independent functions of ire-1 might contribute to the maintenance of ER homeostasis? One plausible hypothesis is that autophagosome formation, mediated by ire-1 independently of xbp-1, might counteract and partially relieve the accumulation of secreted proteins in the ER. In support of this hypothesis, we find that inactivation of the C. elegans Beclin homologue, which is needed for phagosome formation, further disrupted ER homeostasis in xbp-1 mutants. Furthermore, since bec-1 inactivation increased DAF-28::GFP levels in the producing cells, matching them to those detected in ire-1 mutants, autophagy may account for most of the difference in ER homeostasis between ire-1 and xbp-1 mutants.
In summary, our data support the idea that UPR signaling, and especially ire-1/xbp-1 signaling, are crucial for ER homeostasis and its ability to metabolize secretory proteins not only under conditions of environmental stress, but also under normal growth conditions. Under these conditions, ire-1 and xbp-1 deficiency reduces basic ER functions, including translation, folding, secretion and degradation of proteins passing through the secretory pathway. These findings may explain a wide range of physiological defects associated with UPR deficiencies (Bicknell et al., 2007; Fonseca et al., 2012; Henis-Korenblit et al., 2010; Hu et al., 2009; Lee et al., 2008; Reimold et al., 2000; Reimold et al., 2001; Richardson et al., 2010; Souid et al., 2007; Todd et al., 2008; Zhao et al., 2003).
Finally, although disruption of the UPR impairs many aspects of normal cellular function, it may be beneficial under extreme circumstances, such as disease onset associated with toxic protein production. Specifically, we find that reducing xbp-1 levels in a C. elegans Alzheimer's disease model limited the levels of the toxic peptide Aβ and delayed its toxicity. Inhibition of the UPR through xbp-1 inactivation in conjunction with inhibition of autophagosome formation further delayed Aβ toxicity. We speculate that this delay is due to enhanced interference with the production and processing of the toxic proteins in the ER. These findings may have implications for higher organisms, because targeting of xbp-1 in a Huntington's disease model in mice proved to be protective as well (Vidal et al., 2012). Thus, better understanding of the consequences of UPR inactivation in the context of normal growth and disease, in C. elegans and in higher organisms, may lead to new therapeutic strategies to combat diseases that are associated with the production of toxic proteins in the ER.
Materials and Methods
Strains
Strains were cultured as described previously (Brenner, 1974) other than strain CL4176, which was maintained at 15°C and shifted to 25°C for Aβ induction. A list of strains used in this study is provided in supplementary material Table S1.
xbp-1 splicing
On day 1 of adulthood, animals were collected for RNA extraction, purification and reverse transcription, using random 9-mers and standard protocol. A primers set encompassing the noncanonical intron of the xbp-1 transcript was used, giving rise to two PCR products of amplified spliced and unspliced xbp-1 transcript (primers: 5′-TCCGCTTGGGCTCTTGAGATGTTC-3′ and 5′-TGTCGTCGTCGGAGGAGAGGATCG-3′). PCR products were visualized on a 2% agarose gel stained with ethidium bromide. Gels were scanned and analyzed using ImageJ software.
Fluorescence microscopy and quantification
Animals were anaesthetized on 2% agarose pads containing 2 mM levamisol.
Images were taken with a CCD digital camera using a Nikon 90i fluorescence microscope. For each trial, exposure time was calibrated to minimize the number of saturated pixels and was kept constant through the experiment. The NIS element software was used to quantify mean fluorescence intensity as measured by intensity of each pixel in the selected area.
Confocal microscopy
Confocal images were taken using an LSM 510 confocal scanning microscope (Carl Zeiss, Jena, Germany) with a 63 NA objective lens. Worms were mounted on 2% agarose pads containing 2 mM levamisol. Sections of 1.5 µm were taken. Colocalization was measured using ImageJ software.
Quantitative RT-PCR analysis
Animals were raised at 20°C until day 1 of adulthood, unless indicated otherwise. On day 1 animals were collected for RNA extraction. RNA extraction, purification and reverse transcription were carried out using standard protocols. Real-time PCR was performed using Maxima SYBR (Fermentas) in a StepOnePlus instrument. Transcript levels of act-1 were used for normalization.
Primers used for qPCR:
act-1: 5′-CCAATCCAAGAGAGGTATCCTTAC-3′ and 5′-CATTGTAGAAGGTGTGATGCCAG-3′; gfp: 5′-CTGTTCCATGGCCAACACTTG-3′ and 5′-GTCATGCTGTTTCATATGATCTGG-3′; Aβ: 5′-CCGACATGACTCAGGATATGAAGT-3′ and 5′-CACCATGAGTCCAATGATTGCA-3′.
Plasmids and transgenic animals
The ire-1 coding sequence was amplified from cDNA and cloned into the NheI and KpnI sites in the L3691/promoterless plasmid. Genomic DNA-amplified promoters were inserted upstream of the ire-1 coding sequence (CDS) as follows: Pdaf-28 (∼3.3 kb), Phat-1 (∼2.9 kb), Pmyo-3 (∼2.6 kb).
To generate Pdaf-28::RFP::KDEL, the ire-1 CDS of L3691/Pdaf-28::ire-1 was replaced with a PCR product of RFP fused to KDEL followed by a stop codon. The ire-1 CDS was excised such that the first 118 amino acids of ire-1, including the signal peptide, was fused in frame to the RFP-KDEL. Germline transformations were performed by injection of 25 ng/µl plasmids and 100 ng/µl of rol-6(su1006) as a co-transformation marker.
Western blot
A similar number of animals were boiled in protein sample buffer containing 2% SDS. Proteins were separated using standard PAGE separation, transferred to a nitrocellulose membrane and detected by western blotting using the following antibodies: anti-GFP (Roche, 1∶1000), anti-tubulin (DHSB, 1∶5000), anti-phospho-eIF2α (Cell signaling, 1∶500) and anti-Aβ (6E10, Covans, 1∶1000).
Paralysis assay
Synchronized worms of strain CL4176 laid eggs on plates RNAi, which were grown at 15°C. After ∼48 hours the worms were shifted to 25°C for induction of Aβ expression. After 24 hours animals were scored hourly for paralysis or harvested for western blot and RT-PCR analysis.
Statistical analysis
Error bars represent the standard error of the mean (s.e.m.). P-values were calculated using the unpaired Student's t-test. Statistical analysis of paralyzed worms was done using Statview 5.0.1 software (SAS) and P-values were calculated using the log-rank Mantel–Cox method.
Supplementary Material
Acknowledgments
Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR) and by Dr Shohei Mitani, National Bioresource Project for the nematode, Tokyo Women's Medical University School of Medicine, Japan. We thank Dr Hanna Fares (University of Arizona, Tucson, AZ) for coelomocyte marker strains, Prof. Peter Naredi (Umea University, Sweden) for the DAF-28::GFP-expressing strain and Dr QueeLim Ch'ng (King's College, London, UK) for the NLP-21::YFP.
Footnotes
Author contributions
M.S., C.K. and S.H.K. conceived and designed the experiments. M.S. and S.B.H. performed the experiments. M.S., C.K. and S.H.K. analyzed the data and wrote the manuscript.
Funding
This study was supported by the National Institutes of Health [grant number R37 AG011816 to C.K.]; the United States–Israel Binational Science Foundation [grant number 2009356 to S.H.K. and C.K.]; the Israel Science Foundation [grant number 1749/11 to S.H.K.]; and a Marie Curie International Reintegration Grant [grant number 256551 to S.H.K.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.123000/-/DC1
References
- Bicknell A. A., Babour A., Federovitch C. M., Niwa M. (2007). A novel role in cytokinesis reveals a housekeeping function for the unfolded protein response. J. Cell Biol. 177, 1017–1027 10.1083/jcb.200702101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., Ron D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- Fares H., Greenwald I. (2001). Genetic analysis of endocytosis in Caenorhabditis elegans: coelomocyte uptake defective mutants. Genetics 159, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca S. G., Urano F., Weir G. C., Gromada J., Burcin M. (2012). Wolfram syndrome 1 and adenylyl cyclase 8 interact at the plasma membrane to regulate insulin production and secretion. Nat. Cell Biol. 14, 1105–1112 10.1038/ncb2578 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Harding H. P., Zhang Y., Ron D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- Henis-Korenblit S., Zhang P., Hansen M., McCormick M., Lee S. J., Cary M., Kenyon C. (2010). Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc. Natl. Acad. Sci. USA 107, 9730–9735 10.1073/pnas.1002575107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J., Weissman J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107 10.1126/science.1129631 [DOI] [PubMed] [Google Scholar]
- Hu C. C., Dougan S. K., McGehee A. M., Love J. C., Ploegh H. L. (2009). XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. 28, 1624–1636 10.1038/emboj.2009.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao G., Nordenson C., Still M., Rönnlund A., Tuck S., Naredi P. (2007). ASNA-1 positively regulates insulin secretion in C. elegans and mammalian cells. Cell 128, 577–587 10.1016/j.cell.2006.12.031 [DOI] [PubMed] [Google Scholar]
- Lee A. H., Scapa E. F., Cohen D. E., Glimcher L. H. (2008). Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496 10.1126/science.1158042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. H., Walter P., Yen T. S. (2008). Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 3, 399–425 10.1146/annurev.pathmechdis.3.121806.151434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link C. D., Taft A., Kapulkin V., Duke K., Kim S., Fei Q., Wood D. E., Sahagan B. G. (2003). Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer's disease model. Neurobiol. Aging 24, 397–413 10.1016/S0197-4580(02)00224-5 [DOI] [PubMed] [Google Scholar]
- Mori K. (2009). Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem. 146, 743–750 10.1093/jb/mvp166 [DOI] [PubMed] [Google Scholar]
- Ogata M., Hino S. i., Saito A., Morikawa K., Kondo S., Kanemoto S., Murakami T., Taniguchi M., Tanii I., Yoshinaga K. et al. (2006). Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231 10.1128/MCB.01453-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold A. M., Etkin A., Clauss I., Perkins A., Friend D. S., Zhang J., Horton H. F., Scott A., Orkin S. H., Byrne M. C. et al. (2000). An essential role in liver development for transcription factor XBP-1. Genes Dev. 14, 152–157 [PMC free article] [PubMed] [Google Scholar]
- Reimold A. M., Iwakoshi N. N., Manis J., Vallabhajosyula P., Szomolanyi-Tsuda E., Gravallese E. M., Friend D., Grusby M. J., Alt F., Glimcher L. H. (2001). Plasma cell differentiation requires the transcription factor XBP-1. Nature 412, 300–307 10.1038/35085509 [DOI] [PubMed] [Google Scholar]
- Richardson C. E., Kooistra T., Kim D. H. (2010). An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature 463, 1092–1095 10.1038/nature08762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C. E., Kinkel S., Kim D. H. (2011). Physiological IRE-1-XBP-1 and PEK-1 signaling in Caenorhabditis elegans larval development and immunity. PLoS Genet. 7, e1002391 10.1371/journal.pgen.1002391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaheen B., Dang H., Fares H. (2009). Derlin-dependent accumulation of integral membrane proteins at cell surfaces. J. Cell Sci. 122, 2228–2239 10.1242/jcs.048892 [DOI] [PubMed] [Google Scholar]
- Scheuner D., Song B., McEwen E., Liu C., Laybutt R., Gillespie P., Saunders T., Bonner-Weir S., Kaufman R. J. (2001). Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7, 1165–1176 10.1016/S1097-2765(01)00265-9 [DOI] [PubMed] [Google Scholar]
- Schuck S., Prinz W. A., Thorn K. S., Voss C., Walter P. (2009). Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 187, 525–536 10.1083/jcb.200907074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X., Ellis R. E., Lee K., Liu C. Y., Yang K., Solomon A., Yoshida H., Morimoto R., Kurnit D. M., Mori K. et al. (2001). Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107, 893–903 10.1016/S0092-8674(01)00612-2 [DOI] [PubMed] [Google Scholar]
- Shen X., Ellis R. E., Sakaki K., Kaufman R. J. (2005). Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet. 1, e37 10.1371/journal.pgen.0010037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J., Umemura T., Nothstein E., Rongo C. (2004). The unfolded protein response regulates glutamate receptor export from the endoplasmic reticulum. Mol. Biol. Cell 15, 4818–4828 10.1091/mbc.E04-02-0108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburth D., Madison J. M., Kaplan J. M. (2007). PKC-1 regulates secretion of neuropeptides. Nat. Neurosci. 10, 49–57 10.1038/nn1810 [DOI] [PubMed] [Google Scholar]
- Souid S., Lepesant J. A., Yanicostas C. (2007). The xbp-1 gene is essential for development in Drosophila. Dev. Genes Evol. 217, 159–167 10.1007/s00427-006-0124-1 [DOI] [PubMed] [Google Scholar]
- Tanudji M., Hevi S., Chuck S. L. (2002). Improperly folded green fluorescent protein is secreted via a non-classical pathway. J. Cell Sci. 115, 3849–3857 10.1242/jcs.00047 [DOI] [PubMed] [Google Scholar]
- Todd D. J., Lee A. H., Glimcher L. H. (2008). The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 8, 663–674 10.1038/nri2359 [DOI] [PubMed] [Google Scholar]
- Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., Ron D. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 10.1126/science.287.5453.664 [DOI] [PubMed] [Google Scholar]
- Urano F., Calfon M., Yoneda T., Yun C., Kiraly M., Clark S. G., Ron D. (2002). A survival pathway for Caenorhabditis elegans with a blocked unfolded protein response. J. Cell Biol. 158, 639–646 10.1083/jcb.200203086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe W., Bredt D. S. (2004). Early events in glutamate receptor trafficking. Curr. Opin. Cell Biol. 16, 134–139 10.1016/j.ceb.2004.01.003 [DOI] [PubMed] [Google Scholar]
- Vidal R. L., Figueroa A., Court F. A., Thielen P., Molina C., Wirth C., Caballero B., Kiffin R., Segura-Aguilar J., Cuervo A. M. et al. (2012). Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 21, 2245–2262 10.1093/hmg/dds040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K., Sato T., Matsui T., Sato M., Okada T., Yoshida H., Harada A., Mori K. (2007). Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 13, 365–376 10.1016/j.devcel.2007.07.018 [DOI] [PubMed] [Google Scholar]
- Ye J., Rawson R. B., Komuro R., Chen X., Davé U. P., Prywes R., Brown M. S., Goldstein J. L. (2000). ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364 10.1016/S1097-2765(00)00133-7 [DOI] [PubMed] [Google Scholar]
- Yoneda T., Imaizumi K., Oono K., Yui D., Gomi F., Katayama T., Tohyama M. (2001). Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276, 13935–13940 [DOI] [PubMed] [Google Scholar]
- Yoshida H. (2007). ER stress and diseases. FEBS J. 274, 630–658 10.1111/j.1742-4658.2007.05639.x [DOI] [PubMed] [Google Scholar]
- Zhao H., Cao Y., Grunz H. (2003). Xenopus X-box binding protein 1, a leucine zipper transcription factor, is involved in the BMP signaling pathway. Dev. Biol. 257, 278–291 10.1016/S0012-1606(03)00069-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.