

Summary
The aggresome is a key cytoplasmic organelle for sequestration and clearance of toxic protein aggregates. Although loading misfolded proteins cargos to dynein motors has been recognized as an important step in the aggresome formation process, the molecular machinery that mediates the association of cargos with the dynein motor is poorly understood. Here, we report a new aggresome-targeting pathway that involves isoforms of 14-3-3, a family of conserved regulatory proteins. 14-3-3 interacts with both the dynein-intermediate chain (DIC) and an Hsp70 co-chaperone Bcl-2-associated athanogene 3 (BAG3), thereby recruiting chaperone-associated protein cargos to dynein motors for their transport to aggresomes. This molecular cascade entails functional dimerization of 14-3-3, which we show to be crucial for the formation of aggresomes in both yeast and mammalian cells. These results suggest that 14-3-3 functions as a molecular adaptor to promote aggresomal targeting of misfolded protein aggregates and may link such complexes to inclusion bodies observed in various neurodegenerative diseases.
Key words: Aggresome, 14-3-3, Dynein, BAG3, Adaptor
Introduction
Misfolded proteins are prone to forming aggregates that perturb normal cellular functions and lead to cytotoxicity (Ross and Poirier, 2005). In addition to chaperone-assisted refolding and ubiquitin-proteasome mediated degradation, the aggresome-autophagy dependent clearance has recently been identified as an important cellular response to toxic misfolded proteins (Tyedmers et al., 2010). When proteasome-dependent degradation fails, misfolded proteins can be sequestered into large juxtanuclear inclusion bodies known as aggresomes, which not only help alleviate the toxicity of small aggregates, but also facilitate their clearance by macroautophagy (Garcia-Mata et al., 2002; Kopito, 2000). Aggresome formation is an active process in which misfolded proteins are loaded onto the dynein–dynactin motor complex by protein adaptors for transport along microtubules to the microtubule organizing center (MTOC) (Chin et al., 2010; Ravikumar et al., 2005). While the underlying molecular details are not fully understood, previous studies have identified the dynein motor and molecular adaptors [e.g. histone deacetylase 6 (HDAC6)] as critical components in the aggresome formation process (Kawaguchi et al., 2003; Ravikumar et al., 2005). More recently, heat-shock protein 70 (Hsp70) and its co-chaperone Bcl-2-associated athanogene 3 (BAG3) were shown to be critical for targeting misfolded proteins to aggresomes (Gamerdinger et al., 2011; Zhang and Qian, 2011).
We report here that aggresome formation is profoundly regulated by 14-3-3, a family of ubiquitous proteins that are abundantly expressed in the brain (Boston et al., 1982; Moore, 1967). 14-3-3 proteins are highly conserved from yeast to human and consist of seven mammalian isoforms (β, ε, η, γ, τ/θ, ζ and σ) (Rosenquist et al., 2000; Wang and Shakes, 1996). The crystal structures of human 14-3-3 proteins reveal that they exist as homo- and heterodimers, with each monomer comprising nine α-helices organized in an anti-parallel array (Liu et al., 1995; Xiao et al., 1995). Co-crystallization of several ligand-bound 14-3-3 complexes further demonstrate that the 14-3-3 dimer is arranged in such a way that the ligand binding groove runs in opposite directions for each monomer, thereby allowing simultaneous binding of two ligands for one 14-3-3 dimer (Rittinger et al., 1999).
14-3-3 proteins are known to interact with over 200 proteins that contain specific phosphoserine/phosphothreonine motifs (Furukawa et al., 1993; Muslin et al., 1996). Through binding to their target proteins, 14-3-3 participates in a wide variety of biological processes, ranging from transcription to neuronal development (Datta et al., 2000; Fantl et al., 1994; Fu et al., 2000; Peng et al., 1997; Skoulakis and Davis, 1998; Tzivion and Avruch, 2002; Zha et al., 1996). In addition, 14-3-3 proteins have been implicated in a number of neurodegenerative disorders, largely based on the observation that 14-3-3 proteins co-localize with the pathological inclusion bodies associated with these diseases, including Lewy bodies in Parkinson's disease, neurofibrillary tangles in Alzheimer's disease, mutant huntingtin aggregates in Huntington's disease, etc. (Chen et al., 2003; Kawamoto et al., 2002; Umahara et al., 2004). However, it is not known how and why 14-3-3 assembles in these inclusion bodies (Foote and Zhou, 2012).
It has been hypothesized that the presence of 14-3-3 proteins in various inclusion bodies may be a consequence of their associations with disease-related proteins such as α-synuclein (α-Syn), parkin and tau, which are major components of inclusions in certain neurodegenerative diseases (Hashiguchi et al., 2000; Ostrerova et al., 1999; Sato et al., 2006). On the other hand, 14-3-3 has been proposed to actively promote the formation of aggresome-like inclusions, thus acting as a sweeper to facilitate the sequestration and deposition of disease-associated toxic proteins (Kaneko and Hachiya, 2006). In support of this hypothesis, previous studies have shown that 14-3-3 plays an important role in aggresome formation. For example, 14-3-3ζ is required for aggresome formation induced by the expression of a polyglutamine-expanded huntingtin protein (Htt86Q) in mammalian cells (Omi et al., 2008). Likewise, deletion of bmh1, which encodes one of two yeast 14-3-3 homologs Bmh1, inhibits aggresomal targeting of another disease-related huntingtin protein (Htt103QP) ectopically expressed in yeast cells (Wang et al., 2009). Current studies, however, have not yet addressed whether the effect of 14-3-3 on aggresome formation is specific for the huntingtin proteins, and more importantly, which molecular step 14-3-3 might participate in the aggresome formation pathway.
In this study, we have determined that 14-3-3 functions as a molecular adaptor to couple chaperone-associated misfolded proteins with dynein motors through its dimeric binding to both BAG3 and the dynein intermediate chain (DIC). 14-3-3 thus plays a critical role in recruiting misfolded protein cargos onto the cytoplasmic motor complex, thereby promoting aggresome formation in response to accumulation of various misfolded proteins. Our findings set a stage for further determination of the molecular detail and functional significance of the 14-3-3-mediated aggresomal targeting complex and provide a foundation to investigate the protective role of 14-3-3 in neurodegenerative diseases.
Results
14-3-3 facilitates the formation of α-Syn aggresomes
14-3-3 proteins have previously been reported to interact with α-Syn, one of the major components of Lewy bodies (Ostrerova et al., 1999; Sato et al., 2006). To examine the potential role of 14-3-3 in α-Syn aggregation, we adopted an established method to induce α-Syn aggregation by inhibition of proteasome functions in cells expressing α-Syn-EGFP fusion proteins (McLean et al., 2001). Consistent with the previous report, α-Syn-EGFP fusion protein underwent C-terminal cleavage, and treatment of cells with the proteasome inhibitor ALLN resulted in a marked accumulation of C-terminal truncated fragments in the Triton-X-100-insoluble fraction (pellet) (Fig. 1A). Strikingly, the ALLN-induced α-Syn-EGFP fragments were markedly reduced in the pellet from cells co-transfected with pSCM138, which encodes the YFP fused difopein (dimeric fourteen-three-three peptide inhibitor) that antagonizes the interaction of 14-3-3 proteins with their endogenous partners (Masters and Fu, 2001) (Fig. 1A, bottom panel). Conversely, co-expression of exogenous 14-3-3 led to a moderate increase in the accumulation of insoluble α-Syn-EGFP fragments after ALLN treatment (Fig. 1A, bottom panel). Since the cleavage of α-Syn-EGFP proteins was only moderately affected by 14-3-3 (Fig. 1A, top panel), a major effect of 14-3-3 appears to be enhancing α-Syn aggregation.
Fig. 1.

14-3-3 promotes α-Syn aggresome formation. (A) α-Syn-EGFP was transfected into tsA201 cells together with 14-3-3γHA, empty vector or pSCM138. Followed by a treatment of either ALLN (proteasome inhibitor, 50 µM) (A) or DMSO (D) as control for 24 h, whole-cell lysates were separated into Triton X-100 soluble (supernatant) and insoluble (pellet) fractions, and samples from both fractions were analyzed by western blotting using antibody against α-Syn. Arrows and arrowheads denote full-length and truncated α-Syn-EGFP, respectively. (B) The percentage of cells bearing large α-Syn-containing inclusion bodies are shown in the bar graph (*P<0.05, n = 5). (C) Representative confocal images of CHO cells into which α-Syn-EGFPΔ155 was co-transfected with 14-3-3γHA, pSCM138 or a control vector. After treatment with either DMSO or ALLN for 24 h, cells were immunostained using specific antibodies against α-Syn (panels a, e, i, m, q and u; green) or vimentin (panels b, f, j, n, r and v; red), and nuclei were visualized by DAPI (panels c, g, k, o, s and w; blue). Superimposed images (panels d, h, l, p, t and x; merge) show colocalization of α-Syn-EGFPΔ155 and vimentin in aggresomes (indicated by arrows). Scale bar: 10 µm.
Next, we generated a construct encoding a C-terminal truncated α-Syn-EGFP (Δ155), which was previously shown to be a component of the α-Syn-insoluble species and able to form inclusion bodies (McLean et al., 2001). The effect of 14-3-3 on promoting ALLN-induced aggregation of α-Syn-EGFPΔ155 proteins was first confirmed by western blotting (supplementary material Fig. S1). To further characterize the function of 14-3-3 in α-Syn inclusion body formation, we co-transfected α-Syn-EGFPΔ155 with either exogenous 14-3-3 or pSCM138 in cells and examined inclusion body formation microscopically. In line with our biochemical data, a majority of the cells with overexpressed 14-3-3 bore large α-Syn inclusion bodies after a treatment of the proteasome inhibitor (ALLN), while few ALLN-induced inclusion bodies were observed in cells co-transfected with pSCM138 (Fig. 1C). Further quantification showed that overexpressing 14-3-3 in cells gave rise to significantly more frequent inclusion body formation than that in control cells. In contrast, co-transfection of pSCM138 led to a drastic decrease in the percentage of cells containing large inclusion bodies (Fig. 1B).
It is known that overexpression of α-Syn leads to formation of a specific type of inclusions termed aggresome, which is uniquely located in the vicinity of the centrosome and assembled by retrograde transport of dynein motors along the microtubule network (Opazo et al., 2008; Tanaka et al., 2004). Consistently, these perinuclear structures were enriched with vimentin, a known aggresome marker that displayed homogeneous distribution in cells without aggresome formation (Fig. 1C). Thus, the α-Syn-enriched inclusion bodies facilitated by 14-3-3 were aggresomes.
14-3-3 plays an important role in aggresome formation
To test whether 14-3-3 is able to promote aggresome formation of other aggregation-prone proteins, we utilized two other well-established aggresome formation models: GFP-250, which spontaneously forms aggresomes when expressed in cells; and a deletion mutant of cystic fibrosis transmembrane conductance regulator (CFTR), CFTRΔF508, which concentrates in aggresomes when proteasome activity is inhibited (García-Mata et al., 1999; Johnston et al., 1998). As shown in Fig. 2A, 14-3-3 overexpression increased the percentage of cells bearing large perinuclear GFP-250 aggresomes. In cells co-transfected with 14-3-3-binding antagonist pSCM138, however, aggresome formation was suppressed and GFP-250 appeared to exist in small aggregates in the cytoplasm. Similarly, GFP-CFTRΔF508 aggresome formation was enhanced in 14-3-3-overexpressed cells, but significantly suppressed in cells co-transfected with pSCM138 (Fig. 2B). These results show that 14-3-3 also promotes aggresome formation of other aggregation-prone proteins, suggesting a general role of 14-3-3 in the aggresome formation process.
Fig. 2.

14-3-3 promotes aggresome formation of GFP-250 and GFP-CFTRΔF508. (A,B) Representative confocal images of CHO cells in which 14-3-3γHA, pSCM138 or a control vector was co-transfected with GFP-250 (A, panels a–l) or GFP-CFTRΔF508 (B, panels a–l). Cells transfected with GFP-CFTRΔF508 were treated with ALLN (50 µM) for 24 h. All cells were stained using anti-vimentin antibody (A, panels d–f; B, panels d–f; red) and DAPI (A, panels g–i; B, panels g–i; blue). Superimposed (merged) images show colocalization of vimentin and aggresomes (visualized through GFP) as indicated by arrows (A, panels j–l; B, panels j–l). Scale bars: 10 µm. Percentages of cells bearing GFP-250 and CFTRΔF508 aggresomes are shown in the lower bar graphs (*P<0.05, n = 5). Note that the green signals in pSCM138-co-transfected cells (A, panels b and k; B panels b and k) include GFP-250 or GFP-CFTRΔF508, as well as spectrally overlapping YFP-difopein that is minimally present in the aggregates.
14-3-3 exhibits isoform specificity in promoting the formation of aggresomes
There are seven homologous 14-3-3 isoforms in mammals. To determine whether 14-3-3 has isoform specificity in promoting aggresome formation, we co-transfected GFP-CFTRΔF508 with each of the 14-3-3 isoforms in cells, and assessed their effects on aggresome formation by analyzing the percentage of cells containing aggresomes after proteasome inhibition. As shown in Fig. 3A, overexpression of 14-3-3γ, η, ε and ζ isoforms led to a statistically significant increase in the formation of GFP-CFTRΔF508-containing aggresome, but overexpression of 14-3-3τ, β and σ had minimal effects. Thus aggresome formation appears to be specifically facilitated by certain isoforms of 14-3-3.
Fig. 3.
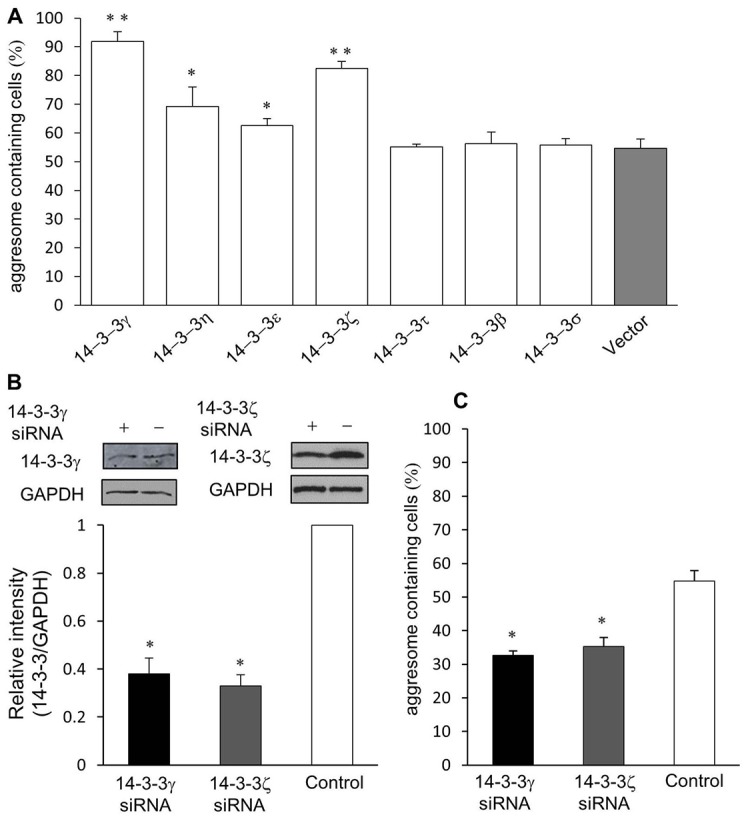
14-3-3 exhibits isoform specificity in promoting aggresome formation. (A) Quantification of GFP-CFTRΔF508-induced aggresome formation in HDAC6-knockdown A549 cells co-transfected with one of the seven 14-3-3 isoforms (γ, η, ε, ζ, τ, β and σ). (B) tsA201 cells were transfected with siRNA oligonucleotids specific for either 14-3-3γ or 14-3-3ζ, followed by western blot analysis using antibodies as indicated. Quantification of levels of endogenous 14-3-3γ and 14-3-3ζ was done by comparing the amount of endogenous 14-3-3 proteins normalized with levels of GAPDH in control cells or those transfected with siRNAs targeting 14-3-3. (C) Percentages of cells bearing CFTRΔF508 induced aggresomes in control tsA201 cells and in those transfected with 14-3-3γ or ζ (*P<0.05, **P<0.001, n = 5).
Given that 14-3-3γ overexpression resulted in the largest quantitative increase in aggresome formation (Fig. 3A), we further tested its ability in this process by examining aggresome formation in 14-3-3γ-knockdown cells, in which endogenous 14-3-3γ protein level was significantly reduced by transfection of siRNA targeting the 14-3-3γ gene (14-3-3 siRNA) (Fig. 3B). On average, 32% of the 14-3-3γ-knockdown cells transfected with GFP-CFTRΔF508 developed visible perinuclear aggresomes in response to proteasome inhibitor treatment, which is significantly less than that in control cells (55%) under the same treatment (Fig. 3C). Similarly, knockdown of 14-3-3ζ with 14-3-3 siRNA led to a significant decrease in the formation of GFP-CFTRΔF508-containing aggresomes (Fig. 3B,C). Together, these results confirm the effectiveness of specific isoforms of 14-3-3 proteins in promoting aggresome formation. Thus, we chose to use the γ isoform as exogenously overexpressed 14-3-3 in the subsequent experiments.
14-3-3 promotes the formation of aggresomes in an HDAC6-independent pathway
HDAC6 is a known protein adaptor that selectively promotes aggresome formation of ubiquitylated cargos such as CFTRΔF508, but has little effect on GFP-250-induced polyubiquitin-deficient aggresomes (Kawaguchi et al., 2003). Our data showed that 14-3-3 was positively involved in the aggresome formation of both GFP-250 and GFP-CFTRΔF508, raising a possibility that 14-3-3 functions in an HDAC6-independent pathway. To test this hypothesis, we first asked whether 14-3-3 physically interacts with HDAC6. As assessed by the co-immunoprecipitation assay, there was no apparent interaction between exogenous 14-3-3 and HDAC6 in co-transfected cells (Fig. 4A, lanes 2, 3). In addition, we evaluated the effects of 14-3-3 on CFTRΔF508 aggresome formation in HDAC6-knockdown cells. Similar to the previous report (Kawaguchi et al., 2003), aggresome formation efficiency was significantly reduced in HDAC6-knockdown cells at basal condition (Fig. 4B). However, overexpression of 14-3-3 was capable of promoting aggresome formation in HDAC6-knockdown cells, and the effect of exogenous 14-3-3 was more pronounced in HDAC6-knockdown cells than that in the control cells (Fig. 4B). On the other hand, co-transfection of the 14-3-3 binding antagonist pSCM138 led to a further reduction of aggresome formation in HDAC6-knockdown cells compared with control cells (Fig. 4B). Taken together, these results indicate that 14-3-3 promotes aggresome formation via a mechanism that is independent of HDAC6.
Fig. 4.

14-3-3 is independent of HDAC6 and localizes to aggresomes. (A) Western blot analyses of immunoprecipitates and cell lysates from tsA201 cells in which 14-3-3γHA was co-transfected with a control vector (lane 1), GFP-HDAC6 (lane 2), FL-HDAC6 (lane 3) or Raf-1 (lane 4). 14-3-3 co-immunoprecipitates with a known 14-3-3 binding partner, Raf-1, but not with the HDAC6 protein. (B) Quantification of GFP-CFTRΔF508 aggresome formation in HDAC6-knockdown and control cells in % (*P<0.05, **P<0.001, n = 5). (C) Representative confocal images of tsA201 cells in which 14-3-3γHA was co-transfected with α-Syn-EGFPΔ155 (a, b and c), GFP-250 (d, e and f) or GFP-CFTRΔF508 (g, h and i). Cells were immunostained with an antibody against 14-3-3γ (b, e and h; red). Superimposed images (merge, c, f and i) show colocalization of 14-3-3γHA with several aggresomes as indicated by arrows. Dashed lines indicate the borders of respective cells.
14-3-3 is a component of aggresomes
To determine the involvement of 14-3-3 in the aggresome formation process, we examined the subcellular localization of 14-3-3 in aggresome-bearing cells by immunocytochemistry. In these experiments, we co-transfected 14-3-3γ with α-Syn-EGFPΔ155, GFP-CFTRΔF508 or GFP-250. For α-Syn-EGFPΔ155 and GFP-CFTRΔF508 co-transfected cells, proteasome inhibitor was applied to induce aggresomes. As shown in Fig. 4C, 14-3-3γ was enriched in the aggresomes induced by all three aggregation-prone proteins. These results are consistent with previous findings that 14-3-3 proteins are components of pathological inclusion bodies and indicate that 14-3-3 is transported to aggresomal compartment together with misfolded proteins.
14-3-3 binds to the dynein intermediate chain
Dynein motors serve to transport associated cargos to the aggresomal compartment at the MTOC region. Using the Golgi morphology test (Palmer et al., 2009; Yadav and Linstedt, 2011), we verified that cytoplasmic dynein functions were not perturbed by the inhibition of 14-3-3 in cells (supplementary material Fig. S2). Given that 14-3-3 may function in parallel with HDAC6 to facilitate aggresome formation and it is a component of aggresomes, we hypothesized that 14-3-3 might act as a molecular adaptor to bridge the interaction of misfolded protein cargos and dynein motors. To test this, we first asked whether 14-3-3 associates with dynein. Results of the co-immunoprecipitation assay demonstrated that 14-3-3 interacted with both endogenous and exogenous DIC (Fig. 5A,B). To further determine the molecular details of this interaction, we generated a series of C-terminal truncated DIC constructs and tested their interactions with 14-3-3 by GST pull-down assay. As shown in Figs 5C,D, 14-3-3 binding to DIC was markedly reduced after a removal of 383 amino acids from its C-terminal end, and further deletion of 59 amino acids resulted in a loss of 14-3-3 binding. Similar results were obtained using the co-immunoprecipitation analyses of tsA 201 cell lysates co-expressing 14-3-3 and these truncated GFP-mDIC constructs (data not shown). Thus, the C-terminal region of DIC appears to be important for its interaction with 14-3-3.
Fig. 5.

14-3-3 interacts with dynein-intermediate chain. (A,B) 14-3-3 co-immunoprecipitates with both endogenous dynein-intermediate chain (DIC) (A) and exogenously transfected GFP-mDIC or GST-mDIC (B) in tsA201 cells. Exogenously expressed 14-3-3γHA was immunoprecipitated with an anti-HA antibody. (C) Deletion analyses reveal the region in DIC that is crucial for 14-3-3 binding. GFP-tagged full-length or C-terminal-truncated mDIC-deletion mutants were expressed in tsA201 cells. Their interactions with 14-3-3 were assessed in GST pull-down assays using GST-14-3-3, and in western blots (top panel). (D) Diagram that summarizes the effect of progressive truncation of the DIC C-terminus on its binding to 14-3-3.
Dimerization of 14-3-3 is a functional requirement in the formation of aggresomes
14-3-3 proteins exist as homo- or heterodimers in cells (Aitken, 2002). In some cellular pathways, 14-3-3 functions as a protein adaptor to bridge the interaction of two different proteins through its dimeric binding (Shen et al., 2003; Tzivion et al., 1998). To investigate whether 14-3-3 dimeric binding is required for promoting aggresome formation, we utilized several 14-3-3 dimerization-deficient mutants, including MM14-3-3, which bears site-directed mutations in both dimerization interfaces, and WM14-3-3 or MW14-3-3, which has mutations in one of the sites (Zhou et al., 2003). We have previously shown that all three mutants fail to form 14-3-3 dimers, but retain the ability to bind substrates (Zhou et al., 2003). When co-transfected with CFTRΔF508 into cells, none of the dimerization-deficient 14-3-3 mutants exhibited statistically significant effect on promoting aggresome formation (Fig. 6A), suggesting that 14-3-3 dimerization is a functional requirement for its role in aggresome formation.
Fig. 6.

Dimerization of 14-3-3 is required for aggresome formation. (A) Quantification of GFP-CFTRΔF508 induced aggresome formation in A549 HDAC6-knockdown cells that had been co-transfected with WT14-3-3, dimerization-deficient mutant MM14-3-3, MW14-3-3, WM14-3-3 or a control vector (**P<0.001, n = 5). (B) Expression of WT14-3-3 but not the dimerization-deficient mutant 14-3-3, suppresses the Htt103QP-induced growth defects in bmh1Δ cells. WT and bmh1Δ yeast cells with integrated PGALFLAG-Htt103QP-GFP plasmids were transformed with vector WT14-3-3 or MM14-3-3 plasmids. The saturated transformants were 10-fold diluted and then spotted on either glucose or galactose plates to examine the growth after 3-day incubation at 30 °C. (C) Representative fluorescence images of WT and bmh1Δ yeast cells with PGALFLAG-Htt103QP-GFP plasmids that were incubated in galactose medium. Transformation of WT14-3-3 (c), but not MM14-3-3 (d), restores aggresome formation in bmh1Δ cells. Aggresomes are shown as large green dots. Scale bar: 3 µm. (D) The yeast cells were incubated in galactose medium for 12 h, and separated into Triton X-100-soluble (supernatant) and -insoluble (pellet) fractions. Samples from both fractions were analyzed by western blotting using anti-FLAG (for Htt103QP) or anti-HA (for expressed WT and MM D14-3-3ζ–HA) antibody, respectively. Pgk1 was used as a loading control.
Furthermore, we assessed the role of 14-3-3 dimerization in aggresome formation in yeast cells. Bmh1, one of the two 14-3-3 homologs in yeast, is known to be essential for aggresome formation induced by the expression of Huntington's disease protein Htt103QP in yeast cells (Wang et al., 2009). Consistent with the report, we found that expression of Htt103QP-GFP led to formation of aggresomes and was not toxic in wild-type budding yeast cells (Fig. 6B,Ca). In contrast, Htt103QP failed to form aggresomes in the bmh1Δ mutant cells, which correlated with a slow growth phenotype (Fig. 6B,Cb). Interestingly, introduction of the wild-type Drosophila 14-3-3ζ (WT14-3-3) into bmh1Δ cells rescued the Htt103QP-induced defects on both growth and aggresome formation (Fig. 6B,Cc), but expression of a dimerization-deficient 14-3-3 mutant (MM14-3-3) was not effective in reducing the toxicity or restoring aggresome formation in the bmh1Δ mutant cells (Fig. 6B,Cd). As the levels of WT and MM14-3-3 proteins in bmh1Δ mutant cells were comparable (Fig. 6D, bottom panel), the difference in their rescuing abilities was not likely due to the instability of the MM14-3-3 protein, which has been observed in the fruit flies (Messaritou et al., 2010). In addition, we assessed the extent of aggresome formation by western blot analysis of the relative distribution of Htt103QP proteins in the pellet and supernatant fractions of the yeast cell extracts (Fig. 6D). Co-transformation of the WT14-3-3, but not the dimerization-deficient 14-3-3 mutant (MM14-3-3), in bmh1Δ cells shifted more Htt103QP proteins into the pellet fraction (Fig. 6D, top panel), which corresponds to the successful rescue of Htt103QP aggresome formation by wild-type 14-3-3. Taken together, these data demonstrate that dimerization of 14-3-3 is required to promote aggresome formation in both yeast and human cells.
14-3-3 interacts with the Hsp70 co-chaperone BAG3 in a phosphorylation-dependent manner
Subsequently, we addressed how 14-3-3 might associate with cargo proteins. Given that 14-3-3 promoted aggresome formation of several different aggregation-prone proteins, it is possible that the potential 14-3-3-cargo interaction is mediated by certain intermediate proteins, such as chaperones which are usually associated with misfolded proteins (Zhang and Qian, 2011).
A previous proteomic analysis has shown that 14-3-3 interacts with Hsp70 and its co-chaperone BAG3 (Ge et al., 2010a). Since both proteins are known to play important roles in aggresome formation (Gamerdinger et al., 2011; Zhang and Qian, 2011), we carried out studies to further characterize the binding of 14-3-3 to Hsp70 and BAG3. Our co-immunoprecipitation results demonstrated that 14-3-3 interacted with exogenous BAG3 and Hsp70 in co-transfected cells (Fig. 7A, lane 2; Fig. 7B, lane 2). However, exogenously expressed BAG3 significantly increased the amount of Hsp70 co-immunoprecipitated with 14-3-3 (Fig. 7B, lanes 1, 2), while overexpressing Hsp70 had little effect on the interaction between 14 and 3-3 and BAG3 (Fig. 7A, lanes 1, 2). Thus, the interaction between 14 and 3-3 and Hsp70 is likely bridged by BAG3.
Fig. 7.

14-3-3 interacts with chaperone proteins Hsp70 and BAG3. Western blot analyses of 14-3-3 immunoprecipitates and cell lysates from tsA201 cells expressing various combinations of proteins as indicated. (A) BAG3 and 14-3-3γHA co-immunoprecipitate from co-transfected tsA201 cells (lane 2), and the extent of BAG3-14-3-3 interaction is not altered by exogenously expressed Hsp70 (lane 1). (B) Hsp70 co-immunoprecipitates with 14-3-3γHA in co-transfected cells (lane 2), but its binding to 14-3-3 is increased when co-transfected with BAG3 (lane 1). (C) Residues S173 and S136 in BAG3 are necessary for 14-3-3 binding. Compared with WT (lane 5), the S136A BAG3 has reduced binding to 14-3-3 (lane 1), whereas the S173A mutation abolishes the interaction completely (lane 3). (D) Interaction between 14-3-3 and BAG3 is regulated by phosphorylation. (Left panels) co-immunoprecipitation of 14-3-3 and BAG3 is reduced by alkaline phosphatase (AP) treatment of cell lysates. (Right panels) In vitro phosphorylation of recombinant BAG3 with crude tsA201 cell extract significantly enhances its binding to 14-3-3γ, as assessed by co-immunoprecipitation and western blotting. For controls, either ATP (lane 2) or recombinant 14-3-3γ (lane 3) was omitted.
BAG3 contains two putative 14-3-3 binding motifs (RSQS136 and RSQS173) located upstream from its BAG domain (for Hsp70 binding). To define the molecular details of the interaction between 14 and 3-3 and BAG3, we assessed the participation of these putative motifs in 14-3-3 binding by site-directed mutagenesis. As shown in Fig. 7C, mutation of the key serine residue in the first motif (S136A) of BAG3 led to a decreased interaction with 14-3-3, while 14-3-3 binding was completely abrogated for the S173A BAG3 mutant. These results suggest that both sites are phosphoserine-containing 14-3-3 binding motifs (RSXpS) which contribute to the interaction between 14 and 3-3 and BAG3. Among them, the S173-containing motif seems to play a more important role.
To determine whether phosphorylation of BAG3 is important for its binding to 14-3-3, we treated the cell lysates of 14-3-3 and BAG3 co-transfected cells with alkaline phosphatase, and assessed its effect on the 14-3-3–BAG3 interaction by co-immunoprecipitation and western blot analyses. As shown in Fig. 7D, 14-3-3 binding to BAG3 was decreased by the phosphatase treatment. Moreover, we incubated recombinant BAG3 proteins with tsA201 cell extract in the presence of ATP and Mg2+, and subsequently mixed them with recombinant 14-3-3γ. Robust 14-3-3 binding was observed when BAG3 protein was subjected to the in vitro phosphorylation reaction by cell extract (Fig. 7D, right panel). Collectively, these results demonstrate that BAG3 phosphorylation is critical for 14-3-3 binding.
14-3-3 mediates the association of BAG3 with dynein
BAG3 has been reported to promote aggresome formation by coupling Hsp70-bound substrates to the dynein complex (Gamerdinger et al., 2011). Given that 14-3-3 interacts with both BAG3 and dynein, we investigated whether 14-3-3 plays a role in the association of BAG3 with dynein. For consistency, these experiments were conducted using approaches as described by Gamerdinger et al. As shown in Fig. 8A, exogenously expressed BAG3 was pulled down by co-transfected GST-DIC. Interestingly, overexpression of 14-3-3 increased the amount of BAG3 co-precipitated with GST-mDIC (Fig. 8A, lanes 1, 2), while the interaction between DIC and BAG3 was totally eliminated in the cells co-transfected with the 14-3-3 binding antagonist pSCM138 (Fig. 8A, lane 3). In addition, the DIC-BAG3 association was either eliminated or reduced for the two BAG3 mutants that were deficient in 14-3-3 binding (Fig. 8B), providing further evidence that the interaction between 14 and 3-3 and BAG3 is critical for the BAG3 and dynein association.
Fig. 8.

14-3-3 is crucial for associations of BAG3 and cargos with dynein. Western blot analyses of GST pull-down assays and cell lysates from tsA201 cells expressing various combinations of proteins as indicated. (A) The BAG3–DIC association is enhanced by exogenously expressed 14-3-3γHA (lane 2), but eliminated by co-transfection of pSCM138 (lane 3). (B) Compared with WT BAG3, the S136A BAG3 mutant has a reduced binding to DIC, and the S173A BAG3 mutant does not co-precipitate with GST-mDIC. (C) Binding of recombinant GST-mDIC and BAG3 is enhanced by in vitro phosphorylation of BAG3 with tsA201 cell extract (lane 1). This effect is not observed when 14-3-3 (lane 2) or ATP (lanes 3, 4) is absent. (D) The association of SODG85R-GFP with GST-mDIC is enhanced by exogenously expressed 14-3-3γHA (lane 3), and virtually abolished when pSCM138 (lane 1) is co-transfected.
To reveal the hierarchy of the 14-3-3–BAG3–DIC protein complex, we conducted in vitro binding assays using commercially acquired recombinant BAG3, 14-3-3γ and bacterially generated GST-mDIC fusion proteins. 14-3-3 co-precipitated with GST-mDIC (Fig. 8C, lanes 1, 3). However, only background level of recombinant BAG3 proteins was detected in the GST-mDIC beads when BAG3 was not subjected to in vitro phosphorylation (Fig. 8C, lanes 3, 4). Interestingly, GST pull-down of BAG3 was markedly enhanced after recombinant BAG3 was incubated with tsA201 cell extract along with ATP (Fig. 8C, lane 1). No such enhancement was observed in the absence of recombinant 14-3-3 (Fig. 8C, lane 2), suggesting that 14-3-3 mediates the BAG3–dynein interaction.
Next, we evaluated the involvement of 14-3-3–BAG3 interaction in recruiting misfolded proteins to the dynein motor. To correlate with the previous study (Gamerdinger et al., 2011), we used an amyotrophic lateral sclerosis (ALS)-linked mutant superoxide dismutase (SODG85R) as a model protein. Similar to the report, we found that overexpression of BAG3 strongly enhanced the association of dynein with SODG85R, but not with wild-type SOD in co-transfected cells (data not shown). Remarkably, the amount of dynein co-precipitated SODG85R was dramatically increased by exogenously expressed 14-3-3, but significantly reduced in the cells co-transfected with pSCM138 (Fig. 8D, lanes 1, 3). Taken together, these results indicate that 14-3-3 bridges the association of BAG3 with dynein, forming a molecular complex that is important for loading misfolded proteins to dynein motors.
The interaction between 14 and 3-3 and BAG3 is crucial for the formation of aggresomes
To investigate the role of 14-3-3 and BAG3 interaction in aggresome formation, we used siRNA targeting Bag3 to knockdown endogenous BAG3 protein in cells. Consistent with the previous report, aggresome formation was suppressed in cells treated with BAG3 siRNA (Fig. 9A, middle panels). Interestingly, overexpression of 14-3-3 no longer enhanced aggresome formation in cells transfected with BAG3 siRNA (Fig. 9A, left panels), suggesting that exogenous 14-3-3 requires sufficient BAG3 to further promote aggresome formation. In addition, virtually no aggresome formation was observed (less than 5%) in cells co-transfected with both BAG3 </emph>siRNA and the 14-3-3-binding antagonist pSCM138 (Fig. 9A, right panels).
Fig. 9.
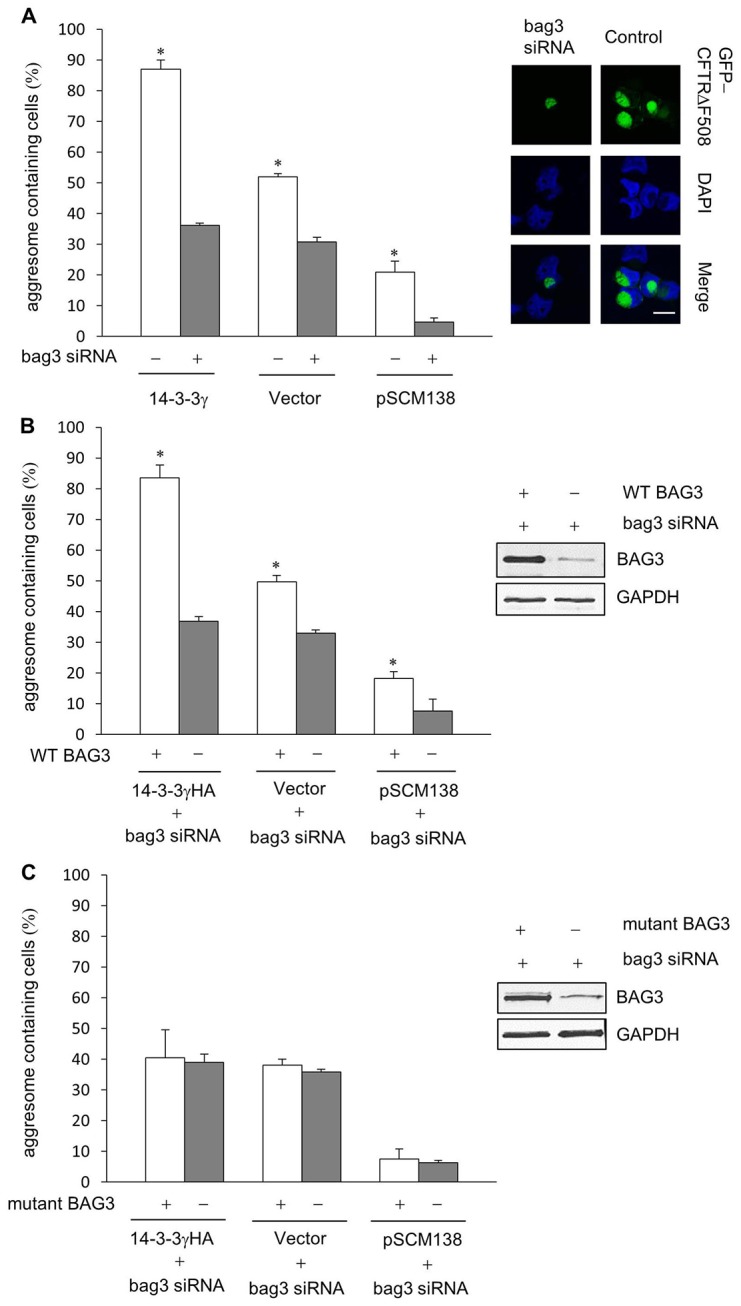
The interaction between 14-3-3 and BAG3 is required for aggresome formation. (A) Aggresome formation promoted by 14-3-3 is impaired in cells treated with siRNA targeting Bag3. The bar graph shows GFP-CFTRΔF508 aggresome formation in control cells (white bars) or BAG3-knockdown cells (gray bars) co-transfected with 14-3-3γHA, empty vector or pSCM138. Representative confocal images of aggresomes in control cells and cells treated with BAG3 siRNA are shown on the right. Scale bar: 10 µm. (B) Percentage of cells containing aggresomes in BAG3-knockdown cells that had BAG3 reintroduced through transfection of BAG3-siRNA-resistant wild-type BAG3 (WT BAG3). (C) Percentage of cells that contain aggresomes in cells that had been transfected with BAG3-siRNA (to knock down endogenous Bag3) and a 14-3-3-binding-deficient Bag3 mutant (S136A/S173A BAG3) that is resistant to BAG3 siRNA (mutant BAG3) (*P<0.05, n = 5). Insets in B and C show the levels of exogenously expressed BAG3-siRNA-resistant wild-type BAG3 (WT BAG3) and BAG3-siRNA-resistant 14-3-3-binding-deficient BAG3 (mutant BAG3), respectively, in cells treated with BAG3 siRNA.
Next, we directly tested whether binding to 14-3-3 is a functional requirement for BAG3 to promote aggresome formation. In these experiments, we reintroduced BAG3</emph>-siRNA-resistant wild-type or 14-3-3-binding-deficient Bag3 cDNAs into the BAG3-knockdown cells and assessed their effects on restoring aggresome formation. As shown in Fig. 9B, reintroduction of wild-type BAG3 led to a significant recovery of aggresome formation in BAG3-knockdown cells which were co-transfected with exogenous 14-3-3, control vector or pSCM138. In contrast, reintroduction of the 14-3-3-binding-deficient BAG3 mutant failed to enhance aggresome formation in BAG3-knockdown cells regardless of their levels of functioning 14-3-3 (Fig. 9C). Thus, these data provide strong evidence that 14-3-3 binding to BAG3 is crucial for aggresomal targeting of misfolded proteins.
Discussion
14-3-3 functions in an HDAC6-independent pathway
Loading misfolded protein aggregates onto the dynein–dynactin motor complex by protein adaptors is a key step for aggresome formation in cells (Olzmann et al., 2007). Previous studies have identified HDAC6 as one of the protein adaptors that couples ubiquitylated substrates to the dynein motor (Kawaguchi et al., 2003). Our data, however, suggest that 14-3-3 functions in a HDAC6-independent pathway to promote aggresome formation.
First, we identified the positive role of 14-3-3 in promoting aggresome formation of several different aggregation-prone proteins, including both ubiquitylated and non-ubiquitylated cargos such as CFTRΔF508 and GFP-250 respectively (Gamerdinger et al., 2009; García-Mata et al., 1999; Johnston et al., 1998). While HDAC6 is known to specifically promote aggresomal targeting of ubiquitylated cargos, 14-3-3 appears to have a broader effect than HDAC6 on aggresome formation.
Second, although 14-3-3 has been reported to interact with some members of the HDAC protein family, our co-immunoprecipitation analysis did not reveal any apparent interaction between 14 and 3-3 and HDAC6 (Fig. 4A). As 14-3-3 generally functions by binding to its target proteins, it is unlikely that 14-3-3 acts in conjunction with HDAC6 to promote aggresome formation.
Third, we found that aggresome formation is not only effectively promoted by exogenously expressed 14-3-3, but also significantly suppressed by co-transfection of 14-3-3 binding antagonist in HDAC6-knockdown cells, indicating that HDAC6 may not be required in 14-3-3-mediated aggresome formation pathway. We also showed that the aggresome formation efficiency in cells without both sufficient HDAC6 and functioning 14-3-3 is significantly lower than that in pSCM138 transfected control cells (Fig. 4B). This likely reflects the aggresome promoting effect of endogenous HDAC6 in the absence of 14-3-3. Taken together, these observations suggest that 14-3-3 and HDAC6 function in separate molecular pathways for aggresome formation.
14-3-3 bridges the association of misfolded proteins with the dynein motor
Our biochemical and functional studies support the hypothesis that 14-3-3 functions as a novel molecular adaptor to promote aggresome formation. Based on these data, we propose a working model for the 14-3-3-mediated aggresome formation pathway. As depicted in Fig. 10, misfolded proteins are usually refolded with the assistance of molecular chaperons and/or degraded by the ubiquitin-proteasome system. Under proteolytic stress, both ubiquitylated and non-ubiquitylated misfolded proteins are associated with 14-3-3 via its binding to BAG3. This allows the loading of the cargo complex onto the dynein motor through the interaction between 14 and 3-3 and DIC. In this pathway, dimeric binding of 14-3-3 is required for the recruitment of misfolded proteins to the dynein motor. The complex containing misfolded proteins, Hsp70, BAG3, 14-3-3 and dynein motors then transports along microtubule to the aggresome at the MTOC region for processing and degradation by macroautophagy.
Fig. 10.
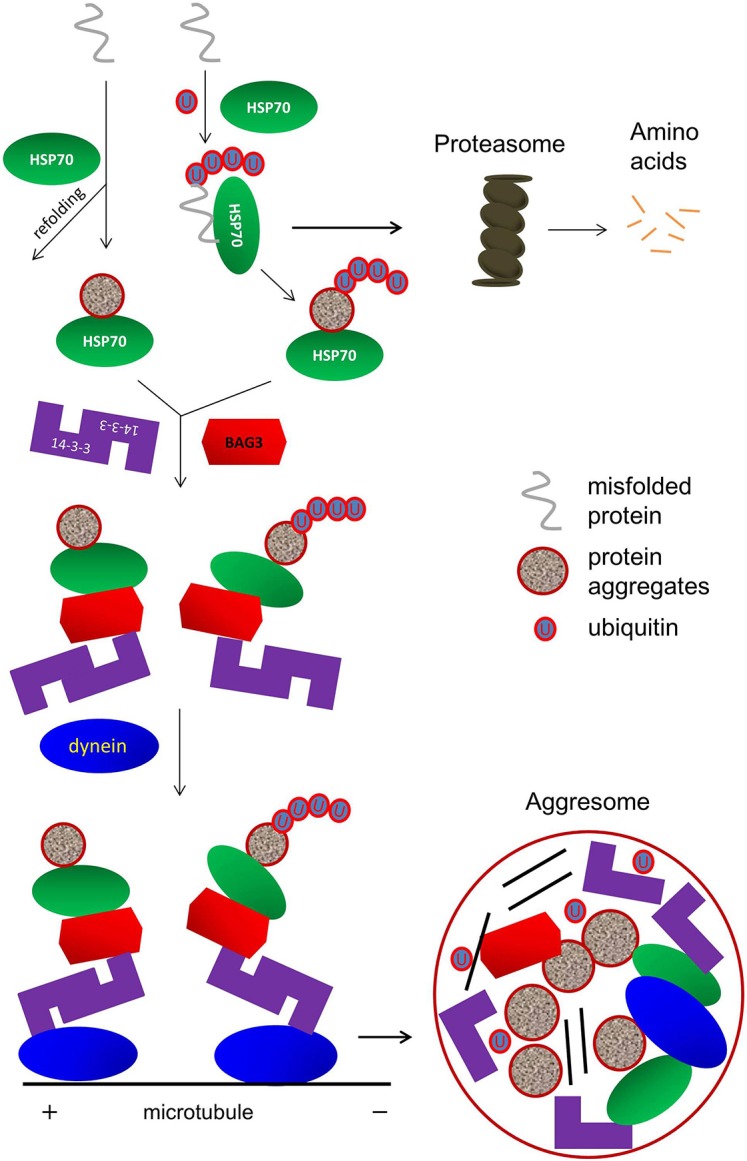
Model for 14-3-3-mediated aggresome-targeting pathway. Several steps of this process are described in the Discussion.
This model builds on a recently proposed aggresome-targeting pathway mediated by Hsp70 and BAG3 (Gamerdinger et al., 2011). We have provided several lines of evidence to demonstrate the important role of 14-3-3 proteins in this process. Firstly, we identified the interaction between 14 and 3-3 and DIC, and characterized the molecular complex consisting of 14-3-3, BAG3 and Hsp70. Secondly, we found that the BAG3-dynein association requires the intact 14-3-3 binding motifs in BAG3. We further showed that loading of both BAG3 and misfolded proteins onto the dynein motor is regulated by the level of functioning 14-3-3 proteins in the cell. Thirdly, we determined that exogenous 14-3-3 is ineffective in promoting aggresome formation without sufficient BAG3, while BAG3 mutants deficient in 14-3-3 binding fail to rescue the aggresome formation defect when reintroduced into BAG3-knockdown cells. Thus, 14-3-3 appears to be a key linker between chaperone associated misfolded proteins and the dynein motor.
In addition, we showed that 14-3-3 binding to BAG3 can be regulated by BAG3 phosphorylation. This is consistent with our site-directed mutagenesis studies demonstrating the importance of BAG3 phosphoserine containing motifs in mediating 14-3-3 binding. Based on a previous phosphoproteomic analysis, BAG3 is one of the many proteins that exhibit enhanced phosphorylations upon proteasome inhibition, but the phosphorylation of the two key serine residues (S136 and S173) was not identified in that study (Ge et al., 2010b). In view of the potential role of protein phosphorylation in regulating the formation and clearance of aggresomes (Watabe and Nakaki, 2011), it is important for future studies to reveal the cellular factors (e.g. protein kinases) that regulate phosphorylation of the putative 14-3-3 binding motifs in BAG3, particularly in response to misfolded protein stress.
On the other hand, we confirmed that 14-3-3 homolog Bmh1 is critical for aggresome formation in yeast cells (Wang et al., 2009). Similar to what we found in mammalian cells, dimerization of 14-3-3 is required to facilitate aggresome formation in yeast cells, suggesting that 14-3-3 may also function as a molecular adaptor in the yeast aggresome formation pathway. However, a BAG3 homolog is not found in yeast cells. Thus, it will be interesting to further define the molecular details of 14-3-3-mediated aggresome formation cascade in yeast cells, and particularly how 14-3-3 associates with misfolded proteins without a BAG3 homolog.
14-3-3 in the protein quality control system
In Drosophila cells, 14-3-3ζ level is upregulated by transcriptional activation in response to heat exposure, while overexpression of 14-3-3ζ led to the resolubilization of several protein aggregates under heat-induced condition (Yano et al., 2006). These behaviors of 14-3-3 are very similar to that of molecular chaperones such as heat shock proteins. In fact, 14-3-3 proteins have previously been referred as molecular chaperones (Vincenz and Dixit, 1996). In support of this notion, recent biophysical studies have also demonstrated that 14-3-3 prevents protein aggregation following chemical stress (Williams et al., 2011).
We report here that 14-3-3 plays a positive role in aggresomal targeting of various aggregation-prone proteins. This is consistent with previous studies showing that 14-3-3 promotes aggresome-like inclusion body formation when the chaperone and proteasome systems are compromised. Under such conditions, 14-3-3 likely functions as an adaptor protein rather than a molecular chaperone. It acts by binding to both the chaperone proteins (BAG3 and Hsp70) and the dynein motor, thereby promoting aggresome formation. In this molecular cascade, dimeric binding of 14-3-3 is key to bridging chaperone-bound misfolded proteins and the dynein motor. In contrast, 14-3-3 dimerization is not a functional requirement for its chaperone-like activity. In fact, monomeric 14-3-3 is reported to display higher chaperone-like activity than its dimeric counterpart or even a chaperone HspB6 (Sluchanko et al., 2012).
Aggresome has been recognized as a cytoprotective structure that facilitates the removal of toxic misfolded proteins and aggregates from the cytoplasm by concentrating them for efficient processing and degradation. Hence, 14-3-3 proteins appear to involve in different aspects of protein quality control system to prevent the accumulation of misfolded proteins. They can either facilitate resolubilization of protein aggregates under mild stress or promote aggresome-like inclusion body formation when chaperone and proteasome systems are overwhelmed.
14-3-3 and neurodegenerative diseases
14-3-3 proteins are known to colocalize with inclusion bodies found in a number of neurodegenerative diseases, which range from Parkinson's disease to Amyotrophic Lateral Sclerosis (Chen et al., 2003; Kawamoto et al., 2004; Kawamoto et al., 2002; Richard et al., 2003; Umahara et al., 2004). We showed here that 14-3-3 is a component of aggresomes induced by various aggregation-prone proteins, because it is one of the key factors in the aggresomal targeting cascade. Moreover, we found that 14-3-3 exhibits isoform specificity in promoting aggresome formation, and the 14-3-3 isoforms (γ, η, ε and ζ) which are effective in aggresomal targeting correspond to the particular isoforms previously identified in pathological inclusions such as Lewy bodies (Kawamoto et al., 2002; Ubl et al., 2002). Considering the similar characteristics between aggresomes and inclusion bodies, our findings provide a plausible explanation for the presence of 14-3-3 in pathological inclusion bodies and suggest a positive role of 14-3-3 in the formation of the aggresome-like structures during neurodegeneration.
It is well documented that 14-3-3 promotes cell survival via different signaling pathways such as suppressing apoptosis and reducing cytotoxicity of neurotoxins (Xing et al., 2000; Yacoubian et al., 2010; Zha et al., 1996). Our studies revealed another cytoprotective mechanism through which 14-3-3 facilitates the targeting of toxic protein aggregates into aggresome-like structures. As 14-3-3 proteins are highly enriched in the brain, they could potentially play important roles in neuroprotection by regulating responses to misfolded proteins commonly associated with neurodegenerative diseases.
Materials and Methods
Cell culture and transfection
CHO, tsA201 and A549 (wild-type and HDAC6-knockdown) cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Transfections were performed by using the Calcium Phosphate method, FuGENE 6 (Promega) reagent or X-tremeGENE 9 (Roche) reagent.
Plasmids and antibodies
The cDNAs encoding various isoforms of 14-3-3 were generated from a human brain cDNA library (Clontech) by PCR amplification, and then subcloned into pcDNA3 with a C-terminal hemagglutinin (HA) tag. GST-14-3-3 was constructed as reported previously (Zhou et al., 1999). The EGFP-fused α-Syn cDNA was constructed by fusing EGFP to the C-terminus of α-Syn coding region in the pEGFP-N1 vector. The plasmids for pSCM138 and pSCM174 were provided by Haian Fu, GFP-250 by Elizabeth S. Sztul, GFP-CFTRΔF508 by Ron R. Kopito, FL-HDAC6 and GFP-HDAC6 by Yi Qiu, and GFP-mDIC and GST-mDIC by Zuhang Sheng. The deletion mutants of the GFP-mDIC were constructed by introducing a stop codon into the cDNA at appropriate locations using GFP-mDIC as template by Quickchange strategy (Stratagene). A similar strategy was used to construct deletion mutant of α-Syn-EGFPΔ155. The mammalian GST-mDIC expression construct was generated by cloning the full length mouse dynein-intermediate chain 1 (Dynclil) cDNA into a pEBG vector. The His-HSPA1A construct was a gift of Harm Kampinga (Addgene plasmid # 19537). The BAG3 construct was generated by cloning a human BAG3 cDNA into the pCMV6-AC vector (Origene). Point mutants of BAG3 were generated by site-directed mutagenesis using Quickchange strategy (Stratagene). The S136A mutation in BAG3 (S136A BAG3) reduced binding to 14-3-3, the S173A mutation (S173A BAG3) abolished binding to 14-3-3 altogether.
The SODWT-GFP and SODG85R-GFP constructs were gifts of Elizabeth Fisher (Addgene plasmid # 26407 and 26410). All constructs were verified by DNA sequencing analysis.
The following antibodies were used in this study: polyclonal anti-α-Syn antibody (#2642, Cell Signaling Technology), monoclonal anti-vimentin antibody (#6630, Sigma-Aldrich), homemade monoclonal anti-HA antibody, monoclonal anti-flag M2 antibody (Sigma-Aldrich), monoclonal anti-dynein (intermediate chain) antibody (D5167, Sigma-Aldrich), monoclonal anti-GAPDH antibody (MCA-1D4, Stemcell Technologies), monoclonal anti-GFP antibody (NeuroMab), polyclonal anti-GFP antibody (sc-8334, Santa Cruz), polyclonal anti-BAG3 antibody (Proteintech), polyclonal anti-HSC70/HSP70 antibody (pAb, Enzo Life Sciences), polyclonal anti-14-3-3γ antibody (C-16, Santa Cruz), homemade monoclonal anti-1D4 antibody, polyclonal anti-Giantin antibody (Abcam), polyclonal IRDye 800CW Conjugated Goat anti-mouse IgG (H+L, LI-COR) and polyclonal IRDye 800CW Conjugated Goat anti-rabbit IgG (H+L, LI-COR).
Immunoprecipitation
Cells were lysed at 4°C using lysis buffer (20 mM Tris-HCl, pH 7.5, 120 mM NaCl, 25 mM NaF, 25 mM KCl, 10 mM EDTA, 1 mM Na3VO4, 1 mM DTT, protease inhibitor cocktail with 1 mM PMSF, 1 µg/ml aprotonin, l µg/ml leupeptin and 1 µg/ml pepstatin A) containing 1% Triton X-100. Cell lysates were separated by centrifugation at 14,000 pm for 10 min into Triton-X-100-soluble and -insoluble fractions. Supernatants were pre-cleared by incubating with protein A/G agarose beads at 4°C for 1 h. Followed by centrifugation at 4000 pm for 5 min, supernatants were transferred to new tubes and incubated with proper antibodies for 2 h at 4°C. 50 µl A/G beads were added into each tube and incubated overnight at the same temperature. Immunoprecipitates were washed three times with lysis buffer, followed by elution using 1×SDS-PAGE sample buffer.
SDS-PAGE and western blotting
For fractionation assay, 1/10 Triton X-100-soluble and total pellet fractions were dissolved in SDS-PAGE sample buffer (50 mM Tris-HCl, pH 6.8, 1% β-mercaptoethanol, 2% SDS, 0.1% bromophenol blue, 10% glycerol) and heated on a 95°C heater for 5 min. Proteins were resolved on SDS-PAGE and transferred to Nitrocellulose Membrane (Bio-rad). For western blots assay with enhanced chemiluminescence (ECL) solutions (GE Healthcare), membranes were blocked for 1 h in TBST containing 5% non-fat milk, followed by incubation with primary antibodies at 4°C overnight. The blots were washed three times (10 min each) in TBST. After incubation with horseradish peroxide-coupled secondary antibodies and three washes with TBST, immunoblots were developed using ECL on films. For western blots assay using the Odyssey Imaging System (LI-COR Biosciences), membranes were blocked for 1 h in TBS instead of TBST containing 5% non-fat milk. After incubation with proper primary antibodies at 4°C overnight and wash with Tween 20 mixed Tris-buffered saline (TBST) three times, blots were incubated with fluorescently-labeled secondary antibodies (LI-COR Biosciences) for 1 h at room temperature in the dark. Membranes were washed four times for 10 min each at room temperature in PBST and one extra time for 10 min in PBS with gentle shaking in the dark. Data were acquired on an Odyssey Imager (LI-COR Biosciences).
Phosphatase treatment of cell lysates
Cells were lysed in a phosphatase inhibitor-free lysis buffer (20 mM Tris-HCl, pH 7.5, 120 mM NaCl, 50 mM KCl, 10 mM MgCl2, 1 mM DTT, protease inhibitor cocktail with 1 mM PMSF, 1 µg/ml aprotonin, l µg/ml leupeptin and 1 µg/ml pepstatin A) containing 1% Triton X-100. 10 µl of calf intestinal alkaline phosphatase (New England BioLabs) was added to 500 µl lysates, followed by incubation at 37°C for 30 min.
In vitro phosphorylation and binding of BAG3 proteins
Phosphorylation of recombinant human BAG3 proteins (ProSpec) was carried out at 30°C for 1 h in 100 µl reactions containing 1 µg of BAG3, 20 µl of whole-cell extract from tsA201 cells, 20 mM Tris-HCL (pH, 7.5), 120 mM NaCl, 25 mM NaF, 25 mM KCl, 10 mM EDTA, 10 mM MgCl2 and 2 mM ATP. The mixtures were then added to 1 µg of recombinant 14-3-3γ proteins (ProSpec), and 14-3-3 immunoprecipitation and western blot analysis were carried out as described above. For in vitro binding assays, 30 µl of bacterially expressed GST-mDIC fusion proteins coupled to glutathione-agarose beads was added to the mixture and incubated overnight at 4°C. Proteins bound to the beads were subjected to western blotting with specific antibodies.
Immunocytochemistry and confocal microscopy
Cells were grown on Poly-L-lysine-coated glass coverslips, fixed in 4% paraformaldehyde for 30 min at room temperature, permeabilized and blocked in a PBS solution containing 0.1% Triton (PBST) and 1% normal goat serum for 30 min at room temperature. They were then incubated in the same solution containing appropriate primary antibodies at 4°C overnight. The next day, sections were washed with PBST 3×5 minutes, followed by incubation with Alexa Fluor® 647 donkey anti-rabbit or anti-mouse secondary antibodies (Invitrogen) for 2 h at room temperature. After several washes with PBST and PBS, the coverslips were counterstained with 5 µg/ml DAPI (Sigma-Aldrich, St. Louis, MO) in PBS for 5 min, washed in PBS for 5 min, and mounted with Vectashield to retard fluorescence fading. Cells were imaged on a Leica TCS SP2 SE laser scanning confocal microscope (Leica Microsystems, Bannockburn, IL) using a 63×objective at 1024×1024 resolution. Serial stack images were taken at 0.1 µm steps and all images were acquired by sequential scanning.
Yeast strains, cytological and protein techniques
Wild-type (WT) and bmh1Δ yeast strains are isogenic to Y300, a W303 derivative. A PGALFLAG-Htt103QP-GFP fragment was integrated into yeast genome, so that the expression of Huntington disease gene with 103 poly-glutamine (Htt103QP) was induced after growth in galactose medium. To express the Drosophila 14-3-3ζ protein in yeast cells, we constructed the plasmid by inserting the cDNA of WT D14-3-3ζ or dimerization-deficient mutant D14-3-3ζ (MM14-3-3) into a yeast expression plasmid under the control of yeast BMH1 promoter. Cells that express Htt103QP-GFP proteins were fixed with 3.7% formaldehyde for 5 min at room temperature and then resuspended in 1×PBS buffer for fluorescence microscopy. To analyze the Htt103QP protein levels, cells were resuspended in RAPI buffer (25 mM Tris pH 7.5, 10 mM EDTA, 150 mM NaCl and 0.05% Tween-20) supplemented with protease inhibitors, and then lysed by glass bead using a bead-beater. The lysates were centrifuged at 13,000 rpm for 30 min at 4°C. The supernatant and pellet fractions were used for SDS-PAGE and western blot analysis using anti-FLAG antibody.
RNA interference and rescue assay
A549 HDAC6-knockdown and tsA201 cells were transfected at 30–50% confluency using Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer's instructions, with 150 nM siRNAs targeting 14-3-3γ (sc-29582, Santa Cruz), 14-3-3ζ (sc-156019) or BAG3 (sc-72602, Santa Cruz). In the rescue assays, silent mutations (C1227A, T1230C, A1233C, G1341A, G1344A, T1347C) were introduced into cDNA of both wild-type and S136A/S173A BAG3 to make them resistant to BAG3 siRNA. They were then reintroduced into the cells that had been transfected with BAG3 siRNA 24 h earlier.
Quantification of aggresome-containing cells
For quantification analysis, five fields of each sample were randomly selected and over 100 cells were present in each field. The percentage of cells containing aggresomes was counted and averaged.
Statistical analysis
Student's t-test was conducted for statistical analysis of quantitative data (expressed as mean ± s.e.m.). The value of *P<0.05 was considered a statistically significant difference.
Supplementary Material
Acknowledgments
We thank Drs Elizabeth S. Sztul and Jianhua Zhang (University of Alabama at Birmingham) for providing us the GFP250 and α-Syn cDNA constructs, Dr Ron R. Kopito (Stanford University) for the GFPCFTRΔF508 construct, Drs Qian Cai (Rutgers Univeristy) and Zu-hang Sheng (NIH) for the GFP-mDIC constructs, and Dr Yi Qiu (University of Florida) for providing us with the HDAC6 constructs and knockdown cell line. We also thank Ruth Didier of the Confocal Facility in the FSU College of Medicine for her help with imaging, and Rani Dhanarajan and Cheryl Pye of the Molecular Core Facility at FSU for generating some of the mutant cDNA constructs.
Footnotes
Author contributions
Z.X. performed the majority of the experiments and analysed data; K.G., M.F., F.L. and R.R. performed experiments and analysed data; M.H., Y.W. and Y.W. provided expertise and helped in designing the experiments; Y.Z. designed the experiments and wrote the manuscript with Z.X. All coauthors contributed to the writing and correction of the manuscript.
Funding
This work was supported by the National Institutes of Health [grant number NS50355 to Y.Z.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.126102/-/DC1
References
- Aitken A. (2002). Functional specificity in 14-3-3 isoform interactions through dimer formation and phosphorylation. Chromosome location of mammalian isoforms and variants. Plant Mol. Biol. 50, 993–1010 10.1023/A:1021261931561 [DOI] [PubMed] [Google Scholar]
- Boston P. F., Jackson P., Thompson R. J. (1982). Human 14-3-3 protein: radioimmunoassay, tissue distribution, and cerebrospinal fluid levels in patients with neurological disorders. J. Neurochem. 38, 1475–1482 10.1111/j.1471-4159.1982.tb07928.x [DOI] [PubMed] [Google Scholar]
- Chen H. K., Fernandez-Funez P., Acevedo S. F., Lam Y. C., Kaytor M. D., Fernandez M. H., Aitken A., Skoulakis E. M., Orr H. T., Botas J. et al. (2003). Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 113, 457–468 10.1016/S0092-8674(03)00349-0 [DOI] [PubMed] [Google Scholar]
- Chin L. S., Olzmann J. A., Li L. (2010). Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 38, 144–149 10.1042/BST0380144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S. R., Katsov A., Hu L., Petros A., Fesik S. W., Yaffe M. B., Greenberg M. E. (2000). 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 6, 41–51 [PubMed] [Google Scholar]
- Fantl W. J., Muslin A. J., Kikuchi A., Martin J. A., MacNicol A. M., Gross R. W., Williams L. T. (1994). Activation of Raf-1 by 14-3-3 proteins. Nature 371, 612–614 10.1038/371612a0 [DOI] [PubMed] [Google Scholar]
- Foote M., Zhou Y. (2012). 14-3-3 proteins in neurological disorders. Int. J. Biochem Mol. Biol. 3, 152–164 [PMC free article] [PubMed] [Google Scholar]
- Fu H., Subramanian R. R., Masters S. C. (2000). 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647 10.1146/annurev.pharmtox.40.1.617 [DOI] [PubMed] [Google Scholar]
- Furukawa Y., Ikuta N., Omata S., Yamauchi T., Isobe T., Ichimura T. (1993). Demonstration of the phosphorylation-dependent interaction of tryptophan hydroxylase with the 14-3-3 protein. Biochem. Biophys. Res. Commun. 194, 144–149 10.1006/bbrc.1993.1796 [DOI] [PubMed] [Google Scholar]
- Gamerdinger M., Hajieva P., Kaya A. M., Wolfrum U., Hartl F. U., Behl C. (2009). Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 28, 889–901 10.1038/emboj.2009.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamerdinger M., Kaya A. M., Wolfrum U., Clement A. M., Behl C. (2011). BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 12, 149–156 10.1038/embor.2010.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Mata R., Bebök Z., Sorscher E. J., Sztul E. S. (1999). Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol. 146, 1239–1254 10.1083/jcb.146.6.1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata R., Gao Y. S., Sztul E. (2002). Hassles with taking out the garbage: aggravating aggresomes. Traffic 3, 388–396 10.1034/j.1600-0854.2002.30602.x [DOI] [PubMed] [Google Scholar]
- Ge F., Li W. L., Bi L. J., Tao S. C., Zhang Z. P., Zhang X. E. (2010a). Identification of novel 14-3-3ζ interacting proteins by quantitative immunoprecipitation combined with knockdown (QUICK). J. Proteome Res. 9, 5848–5858 10.1021/pr100616g [DOI] [PubMed] [Google Scholar]
- Ge F., Xiao C. L., Bi L. J., Tao S. C., Xiong S., Yin X. F., Li L. P., Lu C. H., Jia H. T., He Q. Y. (2010b). Quantitative phosphoproteomics of proteasome inhibition in multiple myeloma cells. PLoS ONE 5, e13095 10.1371/journal.pone.0013095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiguchi M., Sobue K., Paudel H. K. (2000). 14-3-3zeta is an effector of tau protein phosphorylation. J. Biol. Chem. 275, 25247–25254 10.1074/jbc.M003738200 [DOI] [PubMed] [Google Scholar]
- Johnston J. A., Ward C. L., Kopito R. R. (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898 10.1083/jcb.143.7.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko K., Hachiya N. S. (2006). The alternative role of 14-3-3 zeta as a sweeper of misfolded proteins in disease conditions. Med. Hypotheses 67, 169–171 10.1016/j.mehy.2006.01.019 [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y., Kovacs J. J., McLaurin A., Vance J. M., Ito A., Yao T. P. (2003). The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738 10.1016/S0092-8674(03)00939-5 [DOI] [PubMed] [Google Scholar]
- Kawamoto Y., Akiguchi I., Nakamura S., Honjyo Y., Shibasaki H., Budka H. (2002). 14-3-3 proteins in Lewy bodies in Parkinson disease and diffuse Lewy body disease brains. J. Neuropathol. Exp. Neurol. 61, 245–253 [DOI] [PubMed] [Google Scholar]
- Kawamoto Y., Akiguchi I., Nakamura S., Budka H. (2004). 14-3-3 proteins in Lewy body-like hyaline inclusions in patients with sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 108, 531–537 10.1007/s00401-004-0923-2 [DOI] [PubMed] [Google Scholar]
- Kopito R. R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530 10.1016/S0962-8924(00)01852-3 [DOI] [PubMed] [Google Scholar]
- Liu D., Bienkowska J., Petosa C., Collier R. J., Fu H., Liddington R. (1995). Crystal structure of the zeta isoform of the 14-3-3 protein. Nature 376, 191–194 10.1038/376191a0 [DOI] [PubMed] [Google Scholar]
- Masters S. C., Fu H. (2001). 14-3-3 proteins mediate an essential anti-apoptotic signal. J. Biol. Chem. 276, 45193–45200 10.1074/jbc.M105971200 [DOI] [PubMed] [Google Scholar]
- McLean P. J., Kawamata H., Hyman B. T. (2001). Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 104, 901–912 10.1016/S0306-4522(01)00113-0 [DOI] [PubMed] [Google Scholar]
- Messaritou G., Grammenoudi S., Skoulakis E. M. (2010). Dimerization is essential for 14-3-3zeta stability and function in vivo. J. Biol. Chem. 285, 1692–1700 10.1074/jbc.M109.045989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore B. W., Perez V. J. (1967). Specific acidic proteins of the nervous system. In Physiological and Biochemical Aspects of Nervous Integration (ed. F.D. Carlson) pp. 343–359Englewood Cliffs, NJ: Prentice-Hall. [Google Scholar]
- Muslin A. J., Tanner J. W., Allen P. M., Shaw A. S. (1996). Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889–897 10.1016/S0092-8674(00)81067-3 [DOI] [PubMed] [Google Scholar]
- Olzmann J. A., Li L., Chudaev M. V., Chen J., Perez F. A., Palmiter R. D., Chin L. S. (2007). Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 178, 1025–1038 10.1083/jcb.200611128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omi K., Hachiya N. S., Tanaka M., Tokunaga K., Kaneko K. (2008). 14-3-3zeta is indispensable for aggregate formation of polyglutamine-expanded huntingtin protein. Neurosci. Lett. 431, 45–50 10.1016/j.neulet.2007.11.018 [DOI] [PubMed] [Google Scholar]
- Opazo F., Krenz A., Heermann S., Schulz J. B., Falkenburger B. H. (2008). Accumulation and clearance of alpha-synuclein aggregates demonstrated by time-lapse imaging. J. Neurochem. 106, 529–540 10.1111/j.1471-4159.2008.05407.x [DOI] [PubMed] [Google Scholar]
- Ostrerova N., Petrucelli L., Farrer M., Mehta N., Choi P., Hardy J., Wolozin B. (1999). alpha-Synuclein shares physical and functional homology with 14-3-3 proteins. J. Neurosci. 19, 5782–5791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer K. J., Hughes H., Stephens D. J. (2009). Specificity of cytoplasmic dynein subunits in discrete membrane-trafficking steps. Mol. Biol. Cell 20, 2885–2899 10.1091/mbc.E08-12-1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C. Y., Graves P. R., Thoma R. S., Wu Z., Shaw A. S., Piwnica-Worms H. (1997). Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 277, 1501–1505 10.1126/science.277.5331.1501 [DOI] [PubMed] [Google Scholar]
- Ravikumar B., Acevedo-Arozena A., Imarisio S., Berger Z., Vacher C., O'Kane C. J., Brown S. D., Rubinsztein D. C. (2005). Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 37, 771–776 10.1038/ng1591 [DOI] [PubMed] [Google Scholar]
- Richard M., Biacabe A. G., Streichenberger N., Ironside J. W., Mohr M., Kopp N., Perret-Liaudet A. (2003). Immunohistochemical localization of 14.3.3 zeta protein in amyloid plaques in human spongiform encephalopathies. Acta Neuropathol. 105, 296–302 [DOI] [PubMed] [Google Scholar]
- Rittinger K., Budman J., Xu J., Volinia S., Cantley L. C., Smerdon S. J., Gamblin S. J., Yaffe M. B. (1999). Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol. Cell 4, 153–166 10.1016/S1097-2765(00)80363-9 [DOI] [PubMed] [Google Scholar]
- Rosenquist M., Sehnke P., Ferl R. J., Sommarin M., Larsson C. (2000). Evolution of the 14-3-3 protein family: does the large number of isoforms in multicellular organisms reflect functional specificity? J. Mol. Evol. 51, 446–458 [DOI] [PubMed] [Google Scholar]
- Ross C. A., Poirier M. A. (2005). Opinion: What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 6, 891–898 10.1038/nrm1742 [DOI] [PubMed] [Google Scholar]
- Sato S., Chiba T., Sakata E., Kato K., Mizuno Y., Hattori N., Tanaka K. (2006). 14-3-3eta is a novel regulator of parkin ubiquitin ligase. EMBO J. 25, 211–221 10.1038/sj.emboj.7600774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y. H., Godlewski J., Bronisz A., Zhu J., Comb M. J., Avruch J., Tzivion G. (2003). Significance of 14-3-3 self-dimerization for phosphorylation-dependent target binding. Mol. Biol. Cell 14, 4721–4733 10.1091/mbc.E02-12-0821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulakis E. M., Davis R. L. (1998). 14-3-3 proteins in neuronal development and function. Mol. Neurobiol. 16, 269–284 10.1007/BF02741386 [DOI] [PubMed] [Google Scholar]
- Sluchanko N. N., Artemova N. V., Sudnitsyna M. V., Safenkova I. V., Antson A. A., Levitsky D. I., Gusev N. B. (2012). Monomeric 14-3-3ζ has a chaperone-like activity and is stabilized by phosphorylated HspB6. Biochemistry 51, 6127–6138 10.1021/bi300674e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M., Kim Y. M., Lee G., Junn E., Iwatsubo T., Mouradian M. M. (2004). Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J. Biol. Chem. 279, 4625–4631 10.1074/jbc.M310994200 [DOI] [PubMed] [Google Scholar]
- Tyedmers J., Mogk A., Bukau B. (2010). Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 11, 777–788 10.1038/nrm2993 [DOI] [PubMed] [Google Scholar]
- Tzivion G., Avruch J. (2002). 14-3-3 proteins: active cofactors in cellular regulation by serine/threonine phosphorylation. J. Biol. Chem. 277, 3061–3064 10.1074/jbc.R100059200 [DOI] [PubMed] [Google Scholar]
- Tzivion G., Luo Z., Avruch J. (1998). A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature 394, 88–92 10.1038/27938 [DOI] [PubMed] [Google Scholar]
- Ubl A., Berg D., Holzmann C., Krüger R., Berger K., Arzberger T., Bornemann A., Riess O. (2002). 14-3-3 protein is a component of Lewy bodies in Parkinson's disease-mutation analysis and association studies of 14-3-3 eta. Brain Res. Mol. Brain Res. 108, 33–39 10.1016/S0169-328X(02)00510-7 [DOI] [PubMed] [Google Scholar]
- Umahara T., Uchihara T., Tsuchiya K., Nakamura A., Iwamoto T., Ikeda K., Takasaki M. (2004). 14-3-3 proteins and zeta isoform containing neurofibrillary tangles in patients with Alzheimer's disease. Acta Neuropathol. 108, 279–286 10.1007/s00401-004-0885-4 [DOI] [PubMed] [Google Scholar]
- Vincenz C., Dixit V. M. (1996). 14-3-3 proteins associate with A20 in an isoform-specific manner and function both as chaperone and adapter molecules. J. Biol. Chem. 271, 20029–20034 10.1074/jbc.271.33.20029 [DOI] [PubMed] [Google Scholar]
- Wang W., Shakes D. C. (1996). Molecular evolution of the 14-3-3 protein family. J. Mol. Evol. 43, 384–398 10.1007/BF02339012 [DOI] [PubMed] [Google Scholar]
- Wang Y., Meriin A. B., Zaarur N., Romanova N. V., Chernoff Y. O., Costello C. E., Sherman M. Y. (2009). Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J. 23, 451–463 10.1096/fj.08-117614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe M., Nakaki T. (2011). Protein kinase CK2 regulates the formation and clearance of aggresomes in response to stress. J. Cell Sci. 124, 1519–1532 10.1242/jcs.081778 [DOI] [PubMed] [Google Scholar]
- Williams D. M., Ecroyd H., Goodwin K. L., Dai H., Fu H., Woodcock J. M., Zhang L., Carver J. A. (2011). NMR spectroscopy of 14-3-3ζ reveals a flexible C-terminal extension: differentiation of the chaperone and phosphoserine-binding activities of 14-3-3ζ. Biochem. J. 437, 493–503 10.1042/BJ20102178 [DOI] [PubMed] [Google Scholar]
- Xiao B., Smerdon S. J., Jones D. H., Dodson G. G., Soneji Y., Aitken A., Gamblin S. J. (1995). Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature 376, 188–191 10.1038/376188a0 [DOI] [PubMed] [Google Scholar]
- Xing H., Zhang S., Weinheimer C., Kovacs A., Muslin A. J. (2000). 14-3-3 proteins block apoptosis and differentially regulate MAPK cascades. EMBO J. 19, 349–358 10.1093/emboj/19.3.349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoubian T. A., Slone S. R., Harrington A. J., Hamamichi S., Schieltz J. M., Caldwell K. A., Caldwell G. A., Standaert D. G. (2010). Differential neuroprotective effects of 14-3-3 proteins in models of Parkinson's disease. Cell Death Dis. 1, e2 10.1038/cddis.2009.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S., Linstedt A. D. (2011). Golgi positioning. Cold Spring Harb. Perspect. Biol. 3, a005322 10.1101/cshperspect.a005322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M., Nakamuta S., Wu X., Okumura Y., Kido H. (2006). A novel function of 14-3-3 protein: 14-3-3zeta is a heat-shock-related molecular chaperone that dissolves thermal-aggregated proteins. Mol. Biol. Cell 17, 4769–4779 10.1091/mbc.E06-03-0229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J., Harada H., Yang E., Jockel J., Korsmeyer S. J. (1996). Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87, 619–628 10.1016/S0092-8674(00)81382-3 [DOI] [PubMed] [Google Scholar]
- Zhang X., Qian S. B. (2011). Chaperone-mediated hierarchical control in targeting misfolded proteins to aggresomes. Mol. Biol. Cell 22, 3277–3288 10.1091/mbc.E11-05-0388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Schopperle W. M., Murrey H., Jaramillo A., Dagan D., Griffith L. C., Levitan I. B. (1999). A dynamically regulated 14-3-3, Slob, and Slowpoke potassium channel complex in Drosophila presynaptic nerve terminals. Neuron 22, 809–818 10.1016/S0896-6273(00)80739-4 [DOI] [PubMed] [Google Scholar]
- Zhou Y., Reddy S., Murrey H., Fei H., Levitan I. B. (2003). Monomeric 14-3-3 protein is sufficient to modulate the activity of the Drosophila slowpoke calcium-dependent potassium channel. J. Biol. Chem. 278, 10073–10080 10.1074/jbc.M211907200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.