

Summary
Plectin is a versatile cytolinker of the plakin family conferring cell resilience to mechanical stress in stratified epithelia and muscles. It acts as a critical organizer of the cytoskeletal system by tethering various intermediate filament (IF) networks through its C-terminal IF-binding domain (IFBD). Mutations affecting the IFBD cause devastating human diseases. Here, we show that serine 4642, which is located in the extreme C-terminus of plectin, is phosphorylated in different cell lines. Phosphorylation of S4642 decreased the ability of plectin IFBD to associate with various IFs, as assessed by immunofluorescence microscopy and cell fractionation studies, as well as in yeast two-hybrid assays. Plectin phosphorylated at S4642 was reduced at sites of IF network anchorage along cell-substrate contacts in both skin and cultured keratinocytes. Treatment of SK-MEL-2 and HeLa cells with okadaic acid increased plectin S4642 phosphorylation, suggesting that protein phosphatase 2A dephosphorylates this residue. Moreover, plectin S4642 phosphorylation was enhanced after cell treatment with EGF, phorbol ester, sorbitol and 8-bromo-cyclic AMP, as well as during wound healing and protease-mediated cell detachment. Using selective protein kinase inhibitors, we identified two different kinases that modulate the phosphorylation of plectin S4642 in HeLa cells: MNK2, which is downstream of the ERK1/2-dependent MAPK cascade, and PKA. Our study indicates that phosphorylation of S4642 has an important regulatory role in the interaction of plectin with IFs and identifies a novel link between MNK2 and the cytoskeleton.
Key words: Cytoskeleton, Intermediate filaments, Plakin, Plectin, Protein phosphorylation
Introduction
Among the seven members of the plakin family in mammals (Sonnenberg and Liem, 2007), plectin is one of the best characterized (Winter and Wiche, 2013). Plectin is a ubiquitous structural cytoskeletal element, critical for the maintenance of the cytoarchitecture and cell resilience, particularly in tissues exposed to mechanical stress, such as the epidermis, skeletal and cardiac muscles. In the skin, plectin is a component of hemidesmosomes, junctional membrane complexes that promote dermo-epidermal adhesion linked to the cytokeratin network (Borradori and Sonnenberg, 1999). In skeletal and cardiac muscles, various isoforms of plectin are specifically localized at Z-lines, intercalated disks and costameres (Konieczny et al., 2008), where the desmin intermediate filament (IF) network is anchored (Rezniczek et al., 2003). Plectin-null mice die soon after birth and show widespread epithelial detachment and muscular dystrophy with necrotic muscle fibers, streaming of Z-lines, focal ruptures of the sarcolemmal membrane, and subsarcolemmal accumulation of mitochondria (Andrä et al., 1997). Similar findings are also observed in tissue-restricted conditional knockout mice (Konieczny et al., 2008). In humans, pathogenic mutations in the plectin gene cause distinct types of epidermolysis bullosa simplex that are variably associated with muscular dystrophy, pyloric atresia, central nervous manifestations and neuropathy (Winter and Wiche, 2013).
Plectin binds to several types of IF proteins, including epidermal and simple cytokeratins, vimentin, desmin, GFAP and neurofilaments, via its C-terminal tail. This part of plectin is composed of five type B and one type C globular plakin repeat domains (PRDs) that are connected by intervening sequences (Winter and Wiche, 2013). Specifically, a 50-amino-acid stretch in the linker region between the fifth type B PRD and the last PRD of type C contains sequences critical for IF binding (Nikolic et al., 1996) (Fig. 1A). Nevertheless, our laboratory recently found evidence that the PRD-C associated with the C-terminal extremity was also able to interact with IFs (Favre et al., 2011) (B.J.-E., unpublished results). Strikingly, non-sense mutations in the PRD C or the C-terminal extremity that are downstream the above mentioned linker region lead to devastating effects in humans with skin, skeletal muscle and central nervous system manifestations (Schröder et al., 2002; Winter and Wiche, 2013). These observations indicate that the C-terminal extremity is critical for IF-plectin linkage and tissue homeostasis.
Fig. 1.
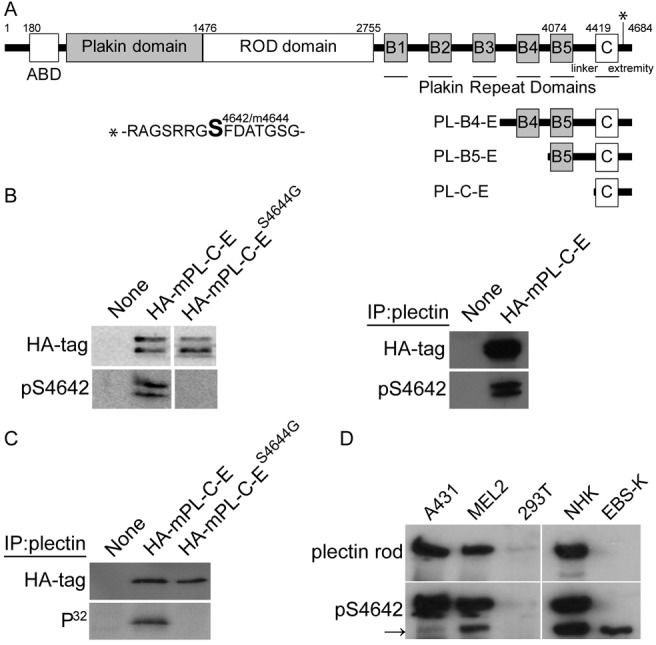
Phosphorylation of plectin S4642 in cultured cells. (A) Schematic representation of human plectin isoform 1. The different domains, with the corresponding amino acid positions, are depicted (ABD: actin-binding domain). The C-terminal region consists of six plakin repeat domains (PRDs) (boxes B1 to B5 and C) separated by connecting segments, with an important role of the linker, bridging B5 to C, for the binding of plectin to IFs. The serine residue, equivalent to desmoplakin S2849, is located within the C-terminal extremity at position 4642 [residues 4644 in mouse (m) plectin isoform 1]. (B) Extracts and plectin immunoprecipitates from PtK2 cells, transfected with plasmids encoding HA–mPL-C-E or HA–mPL-C-ES4644G (see A), were analyzed by western blotting (WB, ECL) with anti-HA and anti-pS4642 antibodies as indicated. (C) Transfected PtK2 cells were 32P-radiolabeled in vivo prior to immunoprecipitation of recombinant proteins from cell extracts with the anti-plectin antibodies GP21. Immunoprecipitates were analyzed by WB with anti-HA antibodies and autoradiography. (D) Anti-pS4642 and anti-plectin rod antibodies recognized a protein with the same electrophoretic mobility on WB (ECL) prepared with extracts from A431, SK-MEL-2 and NHK cells (HEK 293T cells express very low levels of plectin). The rod-less isoform(s) of plectin whose migration is indicated by the arrow, was also detected by the anti-pS4642 antibodies [MD-EBS keratinocytes only express rod-less plectin (Koster et al., 2004) whereas desmoplakin migrates below plectin rod-less isoform(s)].
The IF family consists of six sub-types of highly insoluble proteins (Herrmann and Aebi, 2004), which are differentially expressed in tissues according to the cell type and differentiation state. Previous studies have indicated that plectin binds to the rod domain of vimentin and desmin (Favre et al., 2011). As for plectin, inherited pathogenic or engineered mutations in the genes encoding keratin 5 and keratin 14 in human or mouse, respectively cause epidermolysis bullosa simplex with cell fragility and disorganization of the IF cytoskeleton (Porter and Lane, 2003). In humans and mice, loss of desmin also disturbs myofiber cytoarchitecture and integrity with disruption of the contractile apparatus (van Spaendonck-Zwarts et al., 2010).
A number of fundamental biological processes, such as cell division, migration and differentiation, require profound cytoskeletal reorganizations involving IFs remodeling and the regulation of the interaction of plectin with IFs (Omary et al., 2006). Evidence has been provided that phosphorylation of plectin affects its binding to IFs. Specifically, the phosphorylation of the sixth PRD of plectin at threonine 4542 by CDK1 during M-phase is associated with a concomitant dissociation of plectin from the vimentin network (Foisner et al., 1996). Phosphorylation of plectin by either PKA or PKC has a different impact on its binding to vimentin, but the phosphorylation sites have not been characterized (Foisner et al., 1991).
Here we have found that phosphorylation of S4642 in the C-terminal extremity of plectin is increased in cell wound healing, and is associated with a weakening of its binding to IFs. In cultured HeLa cells, plectin S4642 phosphorylation is under the control of protein phosphatase 2A, cyclic AMP-dependent protein kinase (PKA) and mitogen-activated protein kinase-interacting kinase 2 (MNK2). Until now, the latter has only been involved in the modulation of protein synthesis by phosphorylating the eukaryotic initiation factor 4E (Parra et al., 2005). Our findings provide new insight into the dynamic regulation of the association of plectin with IFs during cytoskeletal reorganization and reveal a new role for MNK2 in the regulation of the cytoskeleton.
Results
Recombinant and endogenous plectin proteins are phosphorylated at S4642 in various cell lines
Phosphorylation of a S2849 in the C-terminal extremity of desmoplakin, a plakin member very similar to plectin, weakens its affinity for IFs (Stappenbeck et al., 1994; Meng et al., 1997; Fontao et al., 2003; Godsel et al., 2005). Mammalian plectin has an identical sequence surrounding S4642 in its C-terminal extremity (position corresponding to the human plectin isoform 1) (Fontao et al., 2003). Large-scale phosphoproteomic studies have indicated that this residue is phosphorylated in various cells and tissues (Dephoure et al., 2008; Hornbeck et al., 2012). To corroborate these results and further study the role of this phosphorylation, we raised in rabbits antibodies against a desmoplakin peptide phosphorylated at S2849 (pS2849) and purified them by affinity chromatography. These antibodies not only recognized desmoplakin pS2849 (unpublished data) but also plectin phosphorylated at S4642 (pS4642). Western blotting (WB) of extracts from PtK2 (Fig. 1B) and HEK 293T cells (supplementary material Fig. S1A,B), transiently expressing recombinant proteins encompassing the C-terminal domains of plectin, and various non-transfected human cell lines (Fig. 1D), demonstrate that the anti-plectin pS4642 antibodies recognize bands with the same electrophoretic mobility as the anti-HA or anti-plectin antibodies. Substitution of S4642 with alanine or glycine (phospho-deficient) abrogated the recognition of the recombinant proteins by the anti-pS4642 antibodies (Fig. 1B and supplementary material Fig. S1A) and alkaline phosphatase treatment of WB membranes or transfected and fixed cells reduced the immunoreactivity of recombinant plectin proteins to the anti-pS4642 antibodies indicating phospho-specificity of the antibodies (supplementary material Fig. S1B,C). Immunoprecipitation of HA mouse (m) PL-C-E and HA–mPL-C-ES4644G (S4642 in human plectin corresponds to S4644 in mouse plectin) from transfected and metabolically 32P-radiolabelled PtK2 cells revealed that only the wild-type recombinant protein had incorporated phosphate (Fig. 1C), suggesting that S4644 was the major phosphorylated site within mPL-C-E.
Recombinant pS4642 plectin proteins are not associated with the IF proteins in transfected cells
To study the effect of plectin pS4642 on the ability of recombinant plectin IFBDs to localize to IFs, we transfected PtK2 cells, in which the negative impact of phosphorylation at S2849 in the desmoplakin C-terminal extremity was much more pronounced than in other cell lines (Stappenbeck et al., 1994). Expression of the phospho-deficient HA-mPL-C-ES4644G mutant resulted in the reorganization and collapse of the K8/K18 network two times more frequently than HA–mPL-C-E (Fig. 2). Analogous to studies of the C-terminal region of desmoplakin (Stappenbeck et al., 1994), significant differences in the co-alignment potential between HA–mPL-C-E and HA–mPL-C-ES4644G with various IF networks was not observed in other cell lines such as HaCat (K5/14), HeLa (K8/18 and vimentin), HEK293T (vimentin and neurofilaments), and SW13 (vimentin) (data not shown). Larger plectin C-terminal recombinant proteins, encompassing PL-B5-E and PL-B4-E (see Fig. 1A), co-aligned with IFs in all transfected cell lines including PtK2 (supplementary material Fig. S2 and data not shown). Furthermore, we could not discern noticeable difference of localization pattern among these recombinant proteins bearing a serine, glycine, alanine, aspartate or glutamate at position 4642 (supplementary material Fig. S2 and data not shown).
Fig. 2.
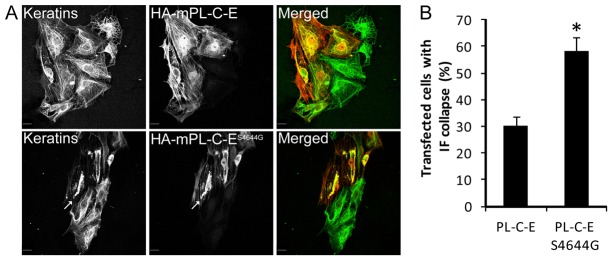
Frequency of IF collapse is higher in transfected PtK2 cells expressing HA–mPL-C-ES4644G than HA–mPL-C-E. (A) Immunofluorescence microscopy images of transfected PtK2 cells, expressing HA–mPL-C-E or HA–mPL-C-ES4644G, with anti-HA antibodies (red) and anti-panKeratin antibodies (green). (B) Quantification of bundling and collapse of the IF network (arrows) in transfected cells (means ± s.d.). Scale bars: 10 µm. More than 100 transfected cells were counted per slide. *P<0.01.
To assess whether phosphorylation of plectin S4642 affects binding to IFs, we performed Triton X-100 (TX-100)-based cell fractionation of HeLa cells expressing various plectin recombinant proteins. In transfected HeLa cells, EGFP–PL-B4-E (S4642 or S4642A) and to a lesser extent HA–mPL-C-E (S4644 or S4644G) localized to IFs (supplementary material Fig. S3). Nevertheless, quantitative WB analyses showed that the pS4642 forms of recombinant plectin-IFBD proteins were mainly soluble in TX-100 cell lysates, while the TX-100 insoluble fractions contained a larger proportion of total recombinant plectin-IFBD proteins (Fig. 3A). Moreover, phospho-deficient plectin S4642 mutants were more associated with the cytoskeletal fractions (70±10%) than their wild-type equivalents (Fig. 3A,B). In TX-100 extraction, endogenous plectin and vimentin were only found in the insoluble fraction (Fig. 3C). However, with an extraction buffer containing 5 mM EDTA (TX-100-2), plectin became partly soluble and pS4642 plectin was 1.8 times more soluble than total plectin (Fig. 3D). A similar difference (1.5×) between the solubility of pS4642 plectin and plectin was observed in a high salt buffer (data not shown).
Fig. 3.
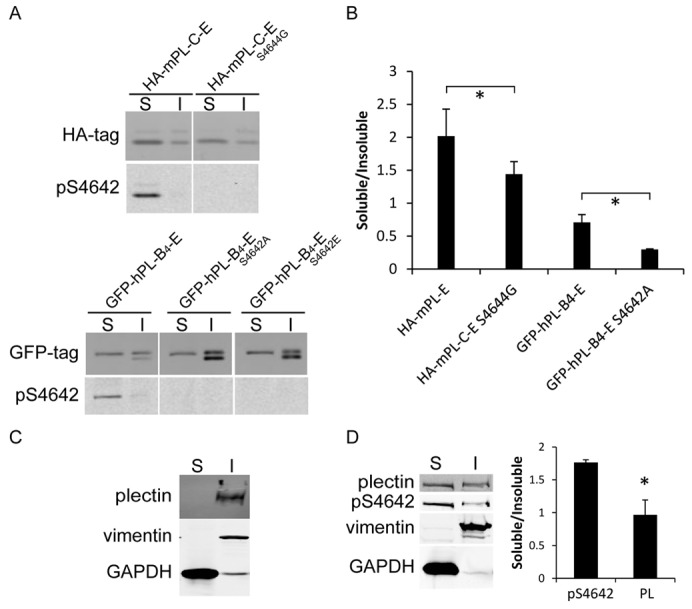
pS4642/4 plectin-IFBD proteins are predominantly found in the TX-100-soluble fraction from transfected HeLa cells. (A) HeLa cells, transfected with the indicated plectin constructs (see Fig. 1A) fused to HA or GFP tag, were fractionated in TX-100 buffer. Soluble (S) and insoluble (I) fractions were analyzed by WB with anti-tag and anti-pS4642 plectin antibodies (the cause for the presence of a doublet in some fractions is unknown). (B) WB quantitative analysis. *P<0.05 (n = 3). (C) WB analysis of the solubility of endogenous plectin, vimentin and GAPDH in HeLa cell extracts lyzed in TX-100 buffer. (D) Cells lyzed in TX-100-2 buffer. Graphs show means ± s.d. *P<0.05 (n = 3).
Lastly, we tested in yeast two-hybrid and three-hybrid assays the ability of mPL-C-E and the S4644G mutant, fused to GAL4-AD, to bind to several IF proteins of different types fused to GAL4-BD; K5/14 and K8/18 (type I and II), vimentin and desmin (type III) and NFL/NFH (type IV). In contrast to mPL-C-ES4644G, mPL-C-E did not support yeast growth (supplementary material Fig. S4A). WB from transformed yeast extracts indicated that mPL-C-E was phosphorylated (supplementary material Fig. S4B), suggesting that phosphorylation of mPL-C-E at S4644 inhibited its interaction with IF proteins. Altogether, these findings strongly support the conclusion that phosphorylation of S4642 weakens the association of plectin with IFs.
Endogenous pS4642 plectin has an altered cytoplasmic distribution with the IF network in cultured cells
To assess the impact of S4642 phosphorylation on the cytoplasmic distribution of endogenous plectin, we carried out confocal laser immunofluorescence microscopy studies with the anti-pS4642 antibodies and the GP21 anti-plectin antibodies, raised against a recombinant plectin comprising the PRD-C and the C-terminal extremity. To exclude cross-reactivity of the anti-pS4642 antibodies with pS2849 desmoplakin, we used two cell lines with negligible or lacking desmoplakin expression respectively, the metastatic melanocytic SK-MEL-2 (Wu et al., 2009) and the immortalized desmoplakin-deficient keratinocyte cells, LAEB (Hobbs and Green, 2012). In SK-MEL-2 cells, plectin co-localized with the vimentin (a type III IF protein) network, whereas the staining of pS4642 plectin was less filamentous throughout the cytoplasm (Fig. 4A). To confirm this difference in staining, we analyzed plectin and pS4642 intensity signals after plectin knockdown with siRNA. Plectin silencing similarly decreased the signals obtained by both the anti-pS4642 and GP21 antibodies in immunofluorescence as well as in WB studies (supplementary material Fig. S5), confirming that both antibodies have a similar specificity for plectin. Determination of the Pearson's coefficient (Pc) (Adler and Parmryd, 2010) for quantifying co-distribution of two signal intensities indicated that pS4642 plectin was significantly less associated with IFs than total plectin (Fig. 4A). A similar albeit weaker difference in localization between pS4642 and total plectin was also observed in LAEB keratinocytes, which express a dense type I and type II keratin 5/14 IF network (Pc pS4642 PL-IF: 0.5, Pc PL-IF: 0.7, P<0.05, data not shown).
Fig. 4.
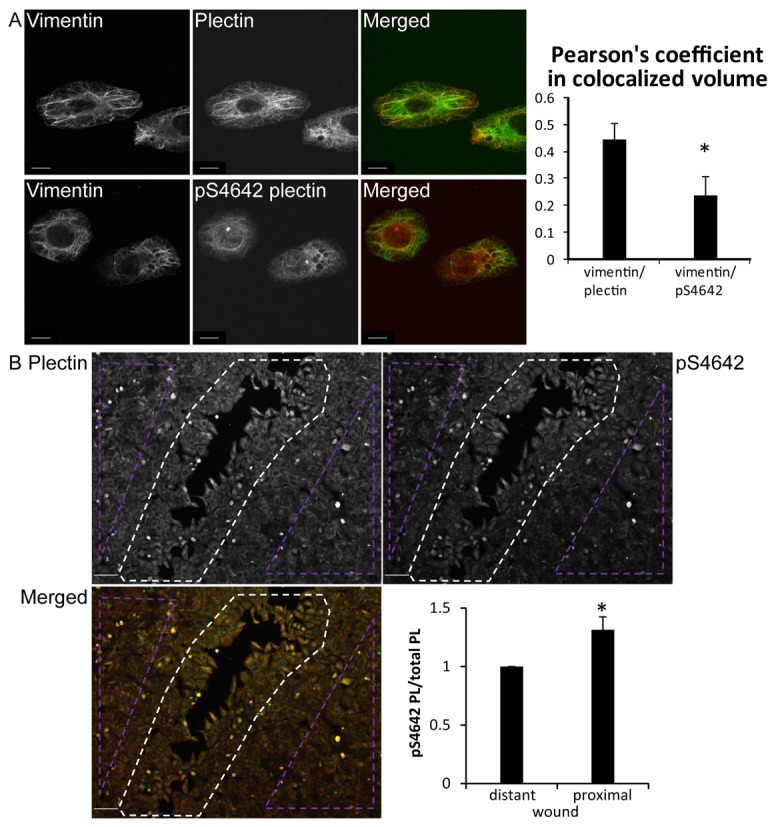
pS4642 plectin has an altered cytoplasmic distribution and increases during wound healing in SK-MEL-2 cells. (A) Confocal immunofluorescence microscopy images of SK-MEL-2 cells, immunostained with antibodies against plectin or pS4642 and vimentin as indicated. Degree of colocalization was determined by measuring the Pearson's coefficient using the Imaris software. Scale bar: 10 µm. More than 100 cells counted per slide. *P<0.01. (B) Confluent SK-MEL-2 cells were scratched, fixed 20 hours later and immunostained with anti-plectin and pS4642 antibodies. Scale bar: 100 µm. The ratio pS4642 plectin to plectin was determined in wound-proximal (white dotted area) and scratch-distant cells (violet dotted areas). Graphs show means ± s.d. *P<0.05 (n = 3).
pS4642 plectin levels are increased during wound healing
Cell migration and wound healing are affected by the presence of the IF cytoskeleton and plectin. During these processes, IFs and plectin are redistributed suggesting that their interaction is regulated (Osmanagic-Myers et al., 2006; Ivaska et al., 2007; McInroy and Määttä, 2011). Therefore, we scratched confluent cultures of SK-MEL-2 cells and analyzed by immunofluorescence microscopy the levels of pS4642 plectin during wound closure. The amount of pS4642 plectin was higher in wound-proximal cells than in those distant from site of scratching (Fig. 4B), suggesting that cell migration enhances phosphorylation of plectin at S4642.
Plectin S4642 is mainly unphosphorylated in the epidermis and PAJEB-β4 keratinocytes at sites of cell-substrate contact
Since plectin is a key component of hemidesmomes by anchoring the keratin IFs to the basal cell membrane in basal keratinocytes (Borradori and Sonnenberg, 1999), we analyzed the phosphorylation state of plectin in skin. WB analysis of epidermal protein extracts showed that plectin was phosphorylated at S4642 (Fig. 5B). In immunofluorescence microscopy studies on cryosections of normal human skin, the polyclonal GP21 antibodies predominantly stained the epidermal basal membrane zone, where hemidesmosomes are localized (Borradori and Sonnenberg, 1999). Some cytoplasmic and cell membrane labeling was also observed in basal and suprabasal cell layers as previously described (Andrä et al., 2003) (Fig. 5A). By contrast, the anti-pS4642 antibodies labeled only weakly the epidermal basement membrane zone, whereas there was a strong membranous and cytoplasmic labeling in the suprabasal layers (Fig. 5A). The latter is likely due to the cross-reactivity with pS2849 desmoplakin based on the distribution of desmoplakin in human epidermis. Since desmoplakin is not localized along the epidermal basal membrane (Getsios et al., 2004) (Fig. 5A), the obtained results indicate that, in contrast to unphosphorylated plectin, pS4642 plectin is almost absent along the epidermal basement membrane with hemidesmosomes.
Fig. 5.
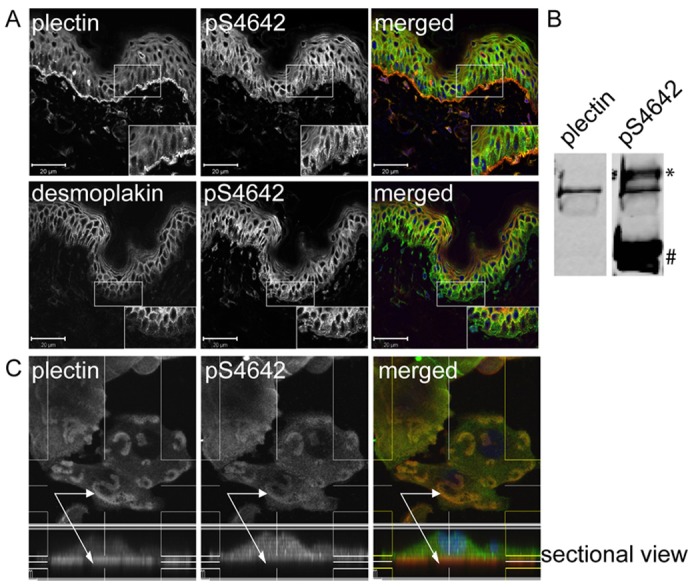
Plectin is less phosphorylated on S4642 at sites of cell-substrate contact in human epidermis and PAJEB-β4 keratinocytes. (A) Acetone-fixed cryosections of human skin were co-immunostained for plectin and pS4642 or for desmoplakin and pS4642. High-power views of the boxed areas are depicted in the insets. In the basal layer of the epidermis, there was no correlation between plectin and pS4642 signals whereas the immunostained patterns of desmoplakin and pS4642 were similar showing that plectin is weakly phosphorylated in hemidesmosomes. Scale bars: 20 µm. (B) Epidermal cell extracts were immunoblotted with anti-plectin and anti-pS4642 antibodies, indicating plectin S4642 phosphorylation in the skin (*, immunoreactive protein migrating above plectin; #, p2849 desmoplakin). (C) PAJEB-β4 keratinocytes, cultured in high Ca2+ for 24 hours, were fixed with formaldehyde and co-immunostained for plectin (red) and pS4642 (green) as indicated. Z-stack sectional view of plectin (red) and pS4642 (green) showing the dichotomy between the two signals in hemidesmosome-like structures (arrows indicate cell-substrate contact/hemidesmosome-like structures with a prevalence of unphosphorylated plectin in the lower part).
The phosphorylation state of plectin S4642 at sites of cell-substrate contact was further investigated in cultured PAJEB-β4 keratinocytes. By immunofluorescence analyses of Z-stack sections of PAJEB-β4 keratinocytes cultured in high Ca2+, both the integrin β4-subunit and plectin had a similar distribution pattern, typical for hemidesmome-like structures or stable anchoring contacts (Fig. 5C and supplementary material Fig. S6) (Borradori et al., 1997). By contrast, the labeling obtained with anti-pS4642 antibodies was weak at cell-substrate attachment sites but increased on the cytoplasmic side where integrin β4 signals faded away (Fig. 5C and supplementary material Fig. S6).
Collectively, these findings indicate that plectin is negligibly phosphorylated at S4642 when associated with hemidesmosomes in the epidermis and in hemidesmosome-like structures in cultured keratinocytes, at sites where plectin is known to anchor the IF network (Borradori and Sonnenberg, 1999).
Treatment of HeLa cells with EGF, phorbol ester, sorbitol, proteases or okadaic acid increases the level of pS4642 plectin
According to prediction programs (Blom et al., 2004; Xue et al., 2008), several protein kinases (PKs) are potentially able to phosphorylate plectin S4642/4. In vitro, both PKA and PKC phosphorylated purified, recombinant H6-tagged mPL-C-E protein (supplementary material Fig. S7). Trypsin digestion, phospho-peptide enrichment by affinity chromatography and phospho-site identification by mass spectrometry revealed that S4644 was the only residue phosphorylated by both PKA and PKC within mPL-C-E (data not shown).
To identify the PK(s) phosphorylating plectin S4642 in vivo, we next tested various stimuli for their ability to increase pS4642 including EGF, phorbol-12-myristate-13-acetate (PMA, an activator of conventional and novel PKC isozymes), sorbitol (hyperosmotic shock) and anisomycin (a protein synthesis inhibitor). pS4642 plectin in HeLa cells rapidly increased after EGF, PMA and sorbitol treatments (Fig. 6A). By contrast, anisomycin decreased the phosphorylation level of pS4642 (Fig. 6A). Upon increasing incubation times with anisomycin and sorbitol there was a progressive degradation of plectin, most likely reflecting proteolysis during apoptosis (Stegh et al., 2000) induced by these compounds (Marfe et al., 2009; Liu et al., 2013). In agreement with previous reports (Raingeaud et al., 1995; Kayali et al., 2000; Bagowski et al., 2003), we observed a rapid and long lasting stimulation of ERK1/2 in HeLa cells incubated with EGF, PMA and sorbitol, whereas anisomycin was a poor stimulator of ERK1/2. By contrast, the stimulation of p38 by EGF and PMA treatment was weak and short compared with the effects of sorbitol and anisomycin (Fig. 6A). Therefore, our results suggest that phosphorylation of plectin S4642 is selectively dependent on the ERK1/2 pathway.
Fig. 6.

Cell treatment with PMA, EGF, sorbitol, trypsin or OA increases plectin S4642 phosphorylation. (A) Starved HeLa cells were treated with 100 nM PMA, 100 ng/ml EGF, 400 mM sorbitol or 1 µg/ml anisomycin for the indicated times and the levels of plectin, pS4642, tubulin, pERK1/2 and p-p38 in whole cell extracts were assessed by WB. Ratios of plectin pS4642/total plectin in response to these treatments are depicted in the graphs (# indicate the onset of plectin proteolysis). (B) Starved HeLa cells were pretreated with 100 nM Gö6983 or DMSO (control) for 1 hour and then treated or not for 15 minutes with 100 nM PMA or 100 ng/ml EGF as indicated. WB analysis was carried out as in A. *P<0.05 (n≥3). (C) HeLa cells were detached by trypsinization and levels of plectin and pS4642 analyzed as in A. *P <0.05 (n = 3). (D) SK-MEL-2 and HeLa cells were treated with 100 nM OA for 3 hours and analyzed as in A. Graphs show means ± s.d.
Since PKC is stimulated by PMA and EGFR (Boonstra et al., 1995), we next tested whether a PKC inhibitor could inhibit both EGF- and PMA-induced phosphorylation of plectin S4642 in HeLa cells. Pretreatment of cells with the specific PKC inhibitor, Gö6983 completely abrogated the PMA-induced but not the EGF-induced phosphorylation of plectin S4642 (Fig. 6B). These results confirm that PKC is involved in the phosphorylation of plectin S4642 and reveals that the action of EGF is independent of PKC.
Detachment of HeLa cells by trypsinization, which is known to stimulate the ERK1/2 pathway (Myatt and Hill, 2005; Riteau et al., 2006), also increased the levels of plectin pS4642 (Fig. 6C). A similar phenomenon was observed when HeLa cells were detached from substratum with dispase II treatment but not with EDTA (data not shown). The effect of trypsin was reversible when cells were re-plated. During the first hour, the levels of plectin pS4642 decreased, followed a few hours later by a slight increase, probably corresponding to cell spreading (supplementary material Fig. S8).
Finally, in all tested cell lines (A431, SK-MEL-2 and HeLa), treatment with okadaic acid, a protein phosphatase 2A (PP2A) inhibitor (Favre et al., 1997), increased phosphorylation of plectin S4642 (Fig. 6D and not shown) suggesting that PP2A directly dephosphorylates pS4642 and/or inhibits signaling pathways leading to phosphorylation of plectin S4642.
EGF and PMA stimulate the phosphorylation of plectin S4642 via MNK2
As EGFR and PKC predominantly activate the MAPKs ERK1 and/or ERK2 (ERK1/2) and ERK5, we pretreated HeLa cells with U0126, a specific inhibitor of their upstream stimulators MEK1/2 and MEK5 (Cargnello and Roux, 2011), before challenging them with EGF or PMA. U0126 completely blocked the action of both stimuli on plectin pS4642 and pERK1/2 levels (Fig. 7A), indicating that PKC does not directly phosphorylate plectin S4642 in response to PMA treatment. Phosphorylation of plectin S4642 was hence dependent on ERK1/2 and/or ERK5. Nevertheless, they cannot directly phosphorylate plectin S4642, because of their substrate consensus motif incompatible with S4642 (Lewis et al., 1998). ERK1/2 and ERK5 activate several downstream kinases encompassing 90 kDa ribosomal S6 kinases (p90RSKs), mitogen and stress activated kinases (MSKs), and MAP-kinase-interacting kinases (MNKs) (Cargnello and Roux, 2011). MSKs were excluded as candidate kinases because they are almost exclusively nuclear, are activated by p38 (Cargnello and Roux, 2011), and are inhibited by H-89, a broad range PK inhibitor (Davies et al., 2000). H-89 treatment could not inhibit the EGF-induced phosphorylation of plectin S4642 (supplementary material Fig. S9). MNK1 was also excluded because it is activated by p38 (Cargnello and Roux, 2011). Thus, the four p90RSKs and MNK2, represented the main candidates for the phosphorylation of plectin S4642 upon EGF and PMA treatment. We treated HeLa cells with BI-D1870, a specific inhibitor of all p90RSKs (Sapkota et al., 2007), which actually increased plectin pS4642 and pERK1/2 (Fig. 7B), excluding p90RSKs from the list of candidate kinases. Cell-dependent ERK1/2 activation by BI-D1870 has been previously reported (Sapkota et al., 2007) and seems to be due to the inhibition of RSK4, which negatively regulates ERK1/2 activation (Myers et al., 2004).
Fig. 7.
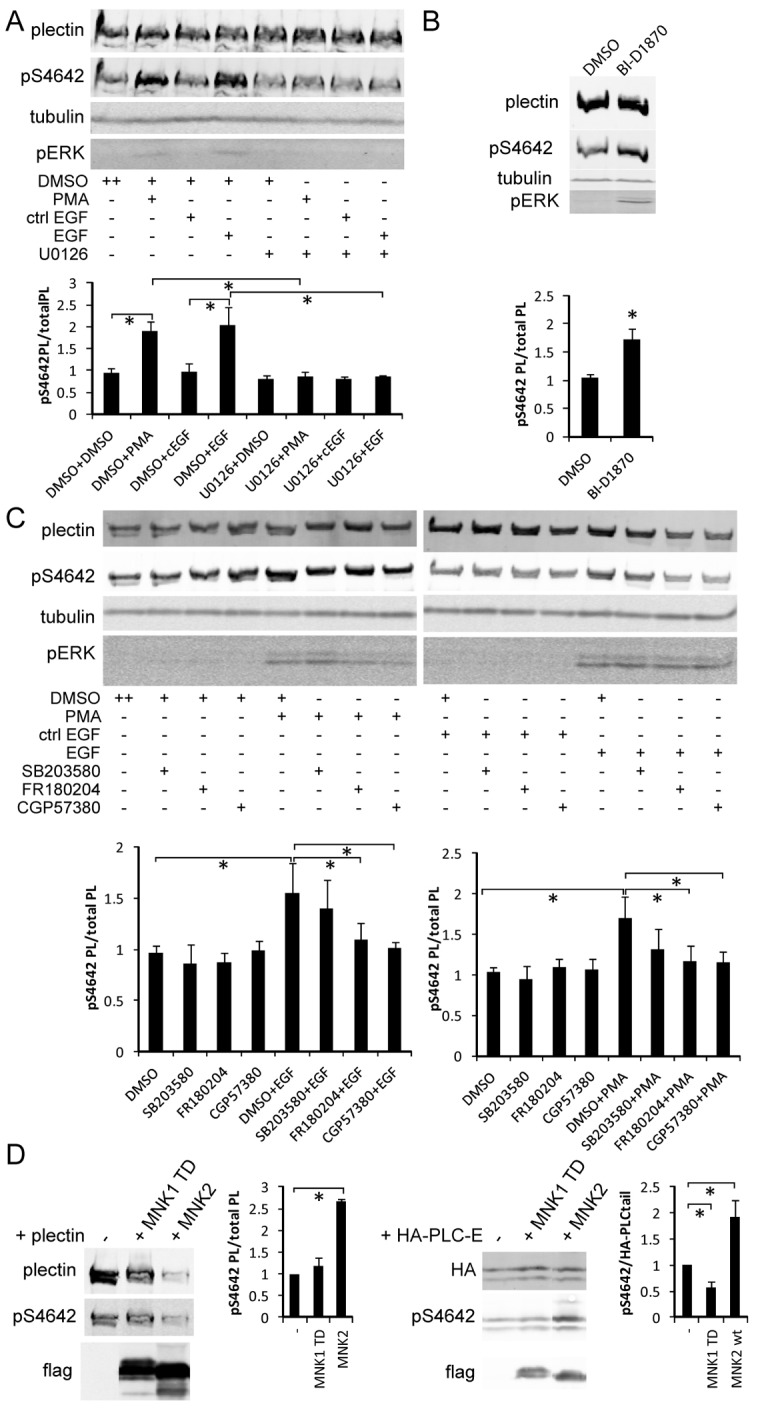
Inhibitors of the ERK1/2 MAPK pathway and MNKs block EGF-induced and PMA-induced S4642 phosphorylation. (A) Starved HeLa cells were pretreated for 1 hour with 10 µM U0126 (MEK1/2 and 5 inhibitor) or DMSO (control) and then treated or not with PMA or EGF (100 nM or 100 ng/ml respectively for 15 minutes). WB analysis was performed as in Fig. 6. *P<0.05 (n≥3). (B) Starved HeLa cells were treated with 10 µM BI-D1870 (RSKs inhibitor) for 1 hour and pERK and pS4642 levels analyzed by WB. *P<0.05 (n = 3). (C) Starved HeLa cells were pretreated for 1 hour with 10 µM FR 180204 (ERK1/2 inhibitor), 40 µM CGP 57380 (MNK inhibitor), 10 µM SB 203580 (p38 inhibitor) or DMSO (control) and then treated or not with PMA or EGF (100 nM or 100 ng/ml respectively for 15 minutes), WB analysis as in A. *P<0.05 (n≥3). (D) WB analysis of whole extracts from HEK 293T cells co-transfected with plasmids encoding either HA–plectin or HA–plectin-C-E and pcDNA3, pcDNA3–FLAG–MNK2, or pcDNA3–FLAG–MNK1-T344D. *P<0.05 (n = 3). For unclear reasons, the amount of HA–plectin in cells co-transfected with pcDNA3–FLAG–MNK2 was systematically lower than with the empty vector or pcDNA3–FLAG–MNK1-T344D. All graphs show means ± s.d.
The sole remaining candidate kinase was therefore MNK2. HeLa cells were pretreated with an inhibitor of MNKs CGP 57380, with an ERK1/2 inhibitor FR 180204, or with a p38 inhibitor SB 203580 prior to PMA or EGF treatment (Knauf et al., 2001; Ohori et al., 2005). WB analyses showed that ERK1/2 and MNK inhibitors prevented the EGF- and PMA-dependent increase of pS4642 while the p38 inhibitor did not (Fig. 7C). As expected, none of these compounds inhibited the activation of ERK1/2 by PMA and EGF (Fig. 7C). Furthermore, we co-transfected HEK 293T cells with cDNA constructs encoding either HA–plectin or HA–mPL-C-E with a plasmid coding for 1) FLAG–MNK2, which is constitutively active in HEK 293T cells; 2) a constitutively active mutant of MNK1, FLAG–MNK1-T332D (TD); or 3) an empty vector as control (Knauf et al., 2001). Both recombinant plectin proteins were more strongly phosphorylated on S4642 when FLAG–MNK2 was co-expressed compared with FLAG–MNK1 TD or control, confirming that MNK2 is involved in the phosphorylation of plectin S4642 (Fig. 7D).
Plectin S4642 is phosphorylated by PKA
Sorbitol induces MNK2 activation via the MEK1/2 MAPK module (Cargnello and Roux, 2011). Nevertheless, pretreatment of HeLa cells with either U0126 or H-89 did not block the positive effect of sorbitol on phosphorylation of S4642. However, H-89 decreased the pS4642 level in serum-starved HeLa cells (Fig. 8A). These findings raised the possibility that, in addition to MNK2, an H-89-sensitive PK is also involved in plectin S4642 phosphorylation. To confirm the involvement of a second kinase stimulated by sorbitol, we pretreated HeLa cells with combinations of PK inhibitors. Sorbitol-induced phosphorylation of S4642 was inhibited when cells were preincubated with H-89 and U0126 or CGP 57380 (Fig. 8B), demonstrating that sorbitol stimulates MNK2 and an H-89-sensitive PK that is independent from the MEK1/2 cascade. As treatment of cells with sorbitol is able to stimulate PKA (Mao et al., 2004), purified PKA phosphorylates recombinant plectin S4642 in vitro (see above), and PKA is sensitive to H-89 (Davies et al., 2000), we next incubated HeLa cells with a potent cell permeable PKA stimulator, 8-bromo-cyclic AMP (8-Br-cAMP) and observed a strong increase in the phosphorylation of plectin S4642 (Fig. 8C). This stimulation was inhibited by pretreatment of cells with H-89 (Fig. 8D), suggesting that PKA is likely to directly phosphorylate plectin S4642 in serum-starved HeLa cells. HeLa cells treatment with 8-Br-cAMP increased the soluble pool of plectin after cell fractionation with TX100-2 buffer (data not shown).
Fig. 8.

PKA stimulation increases plectin S4642 phosphorylation. (A) Starved HeLa cells were pretreated for 1 hour with DMSO (control) or the following PK inhibitors: 10 µM U0126 (MEK 1/2, 5), 40 µM CGP 57380 (MNKs) or 10 µM H-89 (PKA) prior to hyperosmotic shock induction with 400 mM sorbitol for 30 minutes. Levels of plectin, pS4642, tubulin and pERK1/2 were analyzed by WB. H-89 decreased pS4642 levels in unstimulated cells but none of the inhibitors blocked the sorbitol-induced increase in pS4642 levels. *P<0.05 (n≥3). (B) Starved HeLa cells were pretreated for 1 hour with combinations of PK inhibitors as indicated and then treated and analyzed as in A. Sorbitol-induced pS4642 increase was blocked by combining H-89 with U0126 or with CGP 57380. *P<0.05 (n≥3). (C) Starved HeLa cells were incubated with 0.5 mM 8-Br-cAMP for the indicated time periods and pS4642/total plectin ratios were measured by WB. *P<0.05 (n = 3). (D) Starved HeLa cells were pretreated for 1 hour with 10 µM H-89 or DMSO (control) prior to 30 minutes of stimulation with 0.5 mM 8-Br-cAMP and processed as in C. Graphs show means ± s.d. *P<0.05 (n = 3).
Discussion
Proper regulation of plakin-IF connections is important in the maintenance of the cytoarchitecture and in biological processes requiring a dynamic reorganization of the cytoskeletal system, such as cell division, differentiation and stress responses (Suozzi et al., 2012). Phosphorylation is one of the major regulatory posttranslational modifications. We here demonstrate that (1) S4642 in the C-terminal extremity of plectin is phosphorylated in vivo and in vitro; (2) phosphorylation of S4642 affects the interaction of plectin with various IFs, including epidermal and simple cytokeratins; (3) the phosphorylation level of S4642 plectin is low at sites of IF anchorage along the basal cell membrane of either basal keratinocytes in the epidermis or of cultured keratinocytes in vivo; (4) pS4642 level is increased during cell wound healing; (5) plectin pS4642 levels are specifically modulated by PP2A, PKA and MNK2; and finally, (6) MNK2 has a newly recognized function in the regulation of cytoskeletal reorganization.
Identification of S4642 as an important phosphosite within the C-terminal extremity of plectin
Our results, obtained with generated anti-pS2849 desmoplakin antibodies crossreacting with pS4642 plectin, demonstrate that endogenous and ectopically expressed recombinant plectin proteins are phosphorylated at S4642. Phosphoproteomic analyses have identified S4642 as a phosphosite and have revealed that plectin PRD-C domain is flanked by two dense clusters of phosphorylated residues with at least 18 phosphosites (Hornbeck et al., 2012). However, our 32P autoradiography studies of immunoprecipitated mPL-C-E and mPL-C-ES4644G from PtK2 cells demonstrated that only the wild-type recombinant protein mPL-C-E was phosphorylated under the tested conditions. The other sites are hence either negligibly or not phosphorylated in interphase or their phosphorylation is dependent on prior phosphorylation of S4642 (Fiol et al., 1988). Therefore, S4642 appears to be an important phosphorylation site within the plectin C-terminal extremity in unsynchronized PtK2 cells. Since treatment of HeLa cells with OA, sorbitol or 8-Br-cAMP strongly increased the phosphorylation level of plectin S4642, only a portion of plectin is phosphorylated at S4642 in growing cells (Fig. 6A,D). CDK1 was reported to phosphorylate plectin in the PRD-C at T4542 during mitosis (Foisner et al., 1996; Malecz et al., 1996). However, this phosphorylation site has not been identified yet in phosphoproteomic analyses (Hornbeck et al., 2012) and is not conserved in mammalian plectin sequences.
Phosphorylation of S4642 inhibits the interaction of the C-terminal extremity portion of plectin with IFs
We have previously shown that mutation of S2849 in desmoplakin has a positive impact on its association with IF proteins (Stappenbeck et al., 1994; Fontao et al., 2003; Lapouge et al., 2006), inhibits its cellular trafficking (Godsel et al., 2005) and promotes desmosome adhesive strength (Hobbs and Green, 2012). Our present findings reveal that phosphorylation of plectin S4642 also inhibits the association of plectin with various types of IFs, including epidermal K5/K14, simple K8/K18 keratins, type III IF proteins and type IV neurofilaments. PL-B4-E, phosphorylated at S4642, was highly soluble in transfected HeLa cells, an observation supporting the idea that pS4642 impairs the association of plectin with IF cytoskeletal network. Although previous studies have shown that the C-terminal extremity of plectin is dispensable for its binding to IFs (Nikolic et al., 1996), recent data indicate that both the PRD-C and the C-terminal extremity strengthen the binding of plectin to distinct IFs (Favre et al., 2011) (B.J.-E., unpublished results). The key physiological role of this region in vivo is best attested by the severe phenotype with epidermolysis bullosa simplex and myopathy observed in a patient with a plectin mutation (13803ins16/13803ins16) leading to the truncation of the last 129 amino acids of the C-terminal extremity that includes S4642 (Schröder et al., 2002).
The finding that substitution of plectin S4642 by an aspartic or glutamic acid did not have a phospho-mimetic effect, as previously observed for desmoplakin S2849 (Fig. 3A and data not shown) (Fontao et al., 2003), suggests that this phosphorylation event does not simply exert a charge effect. It is conceivable that sequential phosphorylation of additional sites is required, as described for the modulation of the interaction between plectin and the integrin β4 subunit (Frijns et al., 2010), which may trigger more pronounced conformational changes.
In contrast to what we observed with the recombinant pS4642 PL-B4-E protein, endogenous plectin was exclusively found in the TX100-insoluble fraction in PBS (Fig. 3C). Since plectin is dimeric and is able to form oligomers by lateral association (Walko et al., 2011), incompletely phosphorylated plectin oligomers could remain associated with IFs. Moreover, a vimentin-binding site has been identified in the N-terminal ABD of plectin (Sevcik et al., 2004) and plectin interacts with other insoluble proteins than IFs, including microfilaments, microtubules and large membrane protein complexes. Nevertheless, in more stringent extraction conditions, partially solubilizing plectin, pS4642 plectin was more abundant than total plectin in the soluble fraction (Fig. 3D), providing additional support to the idea that phosphorylation of plectin at S4642 is associated with a weaker interaction of plectin with the cytoskeletal fraction.
S4642 is predominantly unphosphorylated in plectin at sites of cell-substrate contact and is increased during cell wound healing and in protease-mediated cell detachment
Plectin gene mutations in epidermolysis bullosa simplex lead to a disorganization and collapse of the IF network, attesting the importance of plectin for the anchorage of IFs to hemidesmosomes in basal keratinocytes (Winter and Wiche, 2013). Our immunofluorescence microscopy studies showed that the phosphorylation level of plectin S4642 along the epidermal basement membrane of skin as well as in hemidesmosome-like structures of cultured keratinocytes was significantly reduced compared with total plectin (Fig. 5A,C and supplementary material Fig. S6). It is unlikely that the weak labeling obtained with anti-pS4642 antibodies is due to an accessibility problem, since total plectin was detected by GP21 antibodies, also directed against a C-terminal portion of plectin, and immunoreactivity with desmoplakin pS2849 at cell membrane sites in suprabasal cell layers of skin was maintained. Hence, these observations corroborate the idea that unphosphorylated plectin is concentrated in hemidesmosomes, where it mediates linkage of IFs to the basal membrane.
The cell morphology changes that occur during cell migration, detachment and spreading imply significant cytoskeleton reorganization. These highly coordinated processes are known to be influenced by the expression and/or localization of IFs and plectin (Osmanagic-Myers et al., 2006; Ivaska et al., 2007; McInroy and Määttä, 2011). Our findings that pS4642 is affected during wound healing and protease-mediated detachment (Fig. 4B and Fig. 6C) suggest that plectin phosphorylation participates in increasing cell plasticity required for morphologic changes. Since these latter processes are dynamic, phosphorylation of pS4642 by different stimuli (Figs 6, 7 and 8) may represent a mean by which plectin-IFs association and dissociation are rapidly modulated.
Plectin S4642 is phosphorylated by MNK2 and PKA
Stimulation with EGF or phorbol ester increased plectin pS4642 levels in HeLa cells (Fig. 6A,B and supplementary material Fig. S9). The results, obtained using several inhibitors of PKs along the MAPK pathways, demonstrate that both stimuli independently converge to the same MEK1/2-dependent PK cascades, with MNK2 as the final effective kinase, since no PK downstream MNK2 has been identified so far (Cargnello and Roux, 2011). MNK2 bears a binding site for ERK1/2, which in contrast to the ERK1/2 binding site of MNK1, is not affected by phosphorylation of ERK1/2 by MEK1/2 (Cargnello and Roux, 2011). The unique, so far well characterized substrate of MNK2 is the eukaryotic initiation factor 4E (Cargnello and Roux, 2011). Hence, our findings link, for the first time, MNK2 to a cytoskeletal protein and potential regulation of the cellular architecture.
MNK2 is co-activated with other MAPK-activated PKs like p90RSKs after MEK1/2-ERK1/2 stimulation. Interestingly, PKC and EGF also independently promote hemidesmosome disassembly via the activation of the ERK1/2 MAPK pathway. During this process, ERK1/2, p90RSKs and PKD1 (PKCμ) phosphorylate the integrin β4 subunit on multiple sites, inducing its dissociation from plectin (Frijns et al., 2010; Frijns et al., 2012). Therefore, our results suggest that plectin S4642 and integrin β4 subunit phosphorylation could occur in parallel in response to EGF. Although the association of plectin with keratins was not found to be essential for hemidesmosome assembly (Geerts et al., 1999), recent studies indicate that the keratin cytoskeleton is essential for the proper incorporation and/or maintenance of plectin in hemidesmosomes and their clustering (Seltmann et al., 2012). Consequently, the phosphorylation of the C-terminal extremity of plectin may represent a mechanism regulating not only IF–plectin association but also hemidesmosome assembly and organization.
The observation that sorbitol-induced phosphorylation of plectin S4642 was only inhibited by combinations of unrelated PK inhibitors and that H-89 decreased the basal level of pS4642 in serum-starved HeLa cells, suggested the involvement of a second kinase. The implication of PKA is pointed by its sensitivity to H-89 and the stimulation of plectin S4642 by 8-Br-cAMP. In this context, treatment of HeLa cells by forskolin, an activator of adenylate cyclase, stimulates the phosphorylation of desmoplakin S2849 (Stappenbeck et al., 1994; Godsel et al., 2005), while PKC activity promotes desmoplakin assembly into desmosomes (Bass-Zubek et al., 2008).
Early analyses showed that the plectin COOH tail is a substrate for both PKA and PKC in vitro (Foisner et al., 1991). Plectin, phosphorylated in vitro by PKA, had a higher affinity for IFs than PKC-phosphorylated or PPase-treated plectin, suggesting that PKA and PKC phosphorylate different sites with opposite effects (Foisner et al., 1991). Our study with MALDI-TOF and recombinant proteins demonstrated that S4642 is the major target within PL-C-E for both PKA and PKC in vitro (supplementary material Fig. S7). The observation that S4642 is phosphorylated in vitro by PKC, but not in HeLa cells (Fig. 6B), is most likely due to the fact that PKs are less specific in vitro than in vivo (Hubbard and Cohen, 1993).
Based on a previously published algorithm (Neuberger et al., 2007), two sites along the whole primary sequence of plectin isoform 1 have a very high probability of being phosphorylated by PKA: S4642 and the residue S4268, located in the linker region critical for IF binding (Nikolic et al., 1996). The latter has also been identified as a phospho-site in numerous phopho-proteomic analyses. Since a previous study suggested that phosphorylation of additional residues by PKA in the C-terminus of plectin promotes its association with IF proteins (Foisner et al., 1991), we mutated S4268 to a Glu residue in PL-B5-linker. This phospho-mimic substitution had no effect on the partition of the recombinant protein in cytoskeletal fractionation experiments using transfected cells (unpublished data).
Finally, our results show that cell treatment with OA, a selective inhibitor of PP2A both in vitro and in vivo (Favre et al., 1997), also increased the cellular level of pS4642, suggesting that PP2A dephosphorylates pS4642. Since PP2A is able to interact with and dephosphorylate IFs (Turowski et al., 1999), the spatial proximity of PP2A to both IFs and plectin may facilitate pS4642 dephosphorylation, contributing to the fine-tuning of the pS4642 level.
Phosphorylation of IFs has been extensively studied (Izawa and Inagaki, 2006; Sihag et al., 2007). Both PKA and PKC phosphorylate IFs, while in response to growth and stress signals, the ERK1/2 and p38-dependent MAPK pathways affect the organization of IF networks. Therefore, regulation of the interaction of plectin with IFs could also be influenced by IF phosphorylation.
In summary, remodeling of IF networks and dynamic regulation of IFs-associated proteins such as plectin take place in a variety of cellular processes. Our results show that stimulation of various signaling pathways regulating cell cytoarchitecture, differentiation, migration or detachment enhance the phosphorylation level of plectin S4642 by MNK2 and/or PKA (Fig. 9) which inhibits the binding of the C-terminal region of plectin to various IF proteins. At sites of cell-substrate contact, such as hemidesmosomes, plectin is predominantly dephosphorylated to ensure proper anchorage of IFs critical for maintenance of cytoarchitecture and cell resilience. Together, these findings give new insights into the regulation of the association of plectin with IFs.
Fig. 9.
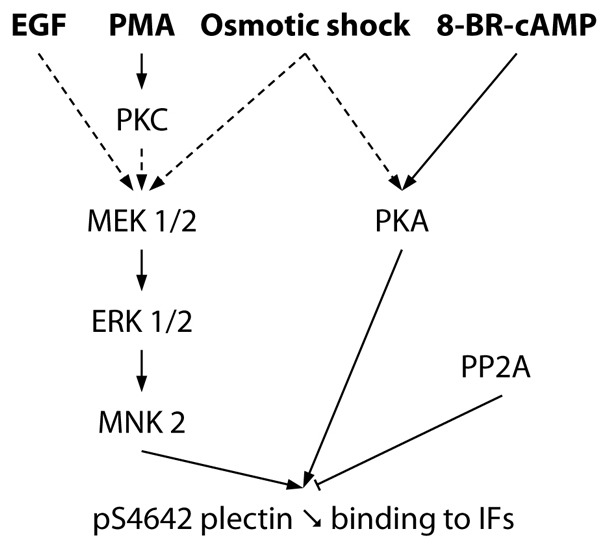
Signaling pathways regulating the phosphorylation of plectin S4642 in HeLa cells. EGF, PMA and hyperosmotic shock (sorbitol) stimulate the phosphorylation of plectin S4642 via the ERK1/2 MAPK pathway (dotted arrows indicate indirect activations). EGF effect is independent of PKC. Sorbitol-induced hyperosmotic stress also stimulates plectin S4642 phosphorylation in a PKA-dependent manner. Phosphorylation of plectin S4642 decreases its binding to IFs. Protein Phosphatase 2A (PP2A) directly or indirectly mediates pS4642 dephosphorylation.
Materials and Methods
Cloning
Plasmid inserts were generated by restriction enzyme digestion or PCR using Pfu DNA polymerase (Promega) and cDNA-specific primers containing restriction sites. Open reading frames of human vimentin, desmin, keratins 5, 8, 14, 18 and rat heavy neurofilament (NF-H), except for light NF (NF-L, res. 24-542) (GenBank accession number: NM_003380.3, NM_001927.3, NM_000424, NM_002273, NM_000526, NM_000224, NM_031783, NM_012607, respectively) were cloned in frame with the yeast GAL4-activation domain (AD) encoded by pACT2, pGAD (Clontech) or pACT2-URA (Fontao et al., 2003). Fragments of human or mouse (m) plectin (GenBank accession number: NM_201380 and NM_201389, respectively): PL-B4-E (res. 3655-4684), PL-C-E (res. 4387-4684), mPL-B5-E (res. 4067-4686) and mPL-C-E (res. 4405-4686) were cloned in various vectors for expression in yeast pAS2-1 (Clontech), mammalian cells pEGFP-C (Clontech) and pcDNA3-HA (Fontao et al., 2003), or bacteria pET15b (Novagen). Mutagenesis of plectin S4642/mS4644 into G, A, D or E was carried out as previously described (Zheng et al., 2004). FLAG-tagged MNK2 and MNK1-TD cloned into pcDNA3 were obtained from Hermann Gram (Novartis, Basel, Switzerland) (Knauf et al., 2001) and HA–plectin-1C in pcDNA3 from Arnoud Sonnenberg (NKI, Amsterdam, The Netherlands) (Koster et al., 2003). All PCR-amplified constructs were sequenced.
Cell culture, transfection and immunofluorescence microscopy
HaCat, HEK 293T, HeLa, PtK2 and SK-MEL-2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Sigma), 100 U/ml penicillin and 10 µg/ml streptomycin (Sigma); MD-EBS, NHK and PAJEB-β4 keratinocytes (Frijns et al., 2010) in keratinocyte serum-free medium (SFM with human keratinocyte growth supplements, Invitrogen); LAEB keratinocytes (Hobbs and Green, 2011) in keratinocyte SFM M-154 containing 0.07 mM Ca2+ and supplemented with human keratinocyte growth supplements (Invitrogen). For hemidesmosome formation, PAJEB-β4 cells were switched to 1 mM Ca2+ for 24 hours. HEK 293T cells were transfected by standard calcium phosphate method, the other cell lines with lipofectamine 2000 (Invitrogen) according to manufacturer's protocol. Where mentioned, 90–95% confluent HeLa cells were serum-starved for 15 hours prior to treatments. Cells were treated with EGF, PMA, anisomycin, 8-Br-cAMP, U0126, BI-D1870, SB203580, FR180204, CGP57380 and H-89, (all from Enzo Life Science), D-sorbitol (Sigma), Gö6983 and OA (LC Laboratories) at the specified concentrations. Chemicals were dissolved according to the manufacturer's recommendations in DMSO or water. For cell detachment experiments, confluent HeLa cells were washed with PBS, and incubated at 37°C with trypsin solution (Invitrogen) for 5 minutes, dispase II (Roche), 2.4 U/ml in PBS for 10 minutes or EDTA, 0.5 mM in PBS for 10 minutes. Trypsin was blocked with two volumes of complete medium or soybean trypsin inhibitor dissolved in PBS (Gibco). Detached cells were collected by centrifugation at 180g for 5 minutes and lyzed in SDS-sample buffer for WB analysis. For re-adhesion, trypsinized cells were replated in the culture medium and adhered cells were lyzed after various incubation periods for WB analysis.
For immunofluorescence microscopy, cells were grown on glass slides and usually fixed with methanol at −20°C for 10 minutes or occasionally with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 minutes followed by permeabilization with 0.1% TX-100. Skin biopsy cryosections, 6 µm thick, were fixed in acetone for 10 minutes at −20°C. Incubation with primary and secondary antibodies were performed as previously described (Favre et al., 2011). Antibodies used are given in the supplementary material Table S1. Fluorescence was viewed with a microscope Eclipse 80i (Nikon) and LSM 510 confocal inverted laser-scanning microscope (Carl Zeiss MicroImaging). Colocalization analysis was performed by using Imaris (Bitplane) and Image J software and fluorescent intensity profiles were obtained using LSM 510 software. IF collapse, defined as thick bundles or aggregates around the nucleus, was counted in ≥100 cells per transfection. For immunofluorescence analysis of wound healing, SK-MEL-2 cells were grown until confluence on glass coverslips and scratched in the middle with a 200 µl tip. After 20 hours, cells were washed and fixed with 4% paraformaldehyde for 10 minutes and immunostained for plectin and pS4642. Quantification of the signal intensities were performed with the NIS-Element (Nikon) software on five wounds for each experiment.
Metabolic 32P labeling and HA-tagged protein immunoprecipitation
Transfected PtK2 cells were incubated for 3 hours in phosphate-free DMEM supplemented with 32P (4.6×106 Bq/ml medium). Immunoprecipitations were performed as previously described (Fontao et al., 2003). Cells were lyzed in 50 mM Tris-HCl (pH 8), 150 mM NaCl, 1% NP40, 1 mM EGTA (300 µl/9 cm2). Soluble extracts were precleared with 50 µl of 10% (vol/vol) protein A Sepharose 4B (GE Healthcare), incubated with 5 µl GP21 antibodies (Progen). Immunoprecipitates were analyzed by WB and autoradiography.
Western blot analysis
Cells grown on 4 cm2 were lyzed in 150 µl SDS-sample buffer (SB) or SDS-SB containing 6 M urea. Extracts were boiled for 5 minutes (except for samples containing urea) and DNA fragmented by repetitive up and down pipetting with a microsyringe. For TX-100-based cytoskeletal fractionation, cells were scraped on ice in 150 µl/4 cm2 TX-100 buffer: PBS 0.1% TX-100, 50 mM β-glycerophosphate (Sigma), 10 mM NaF (Sigma) and 1∶100 protease inhibitor cocktail (Sigma P8340). TX-100-2 buffer was PBS 0.5% TX-100, 5 mM β-glycerophosphate, 10 mM NaF, 5 mM EDTA (Gibco 15575), 2 mM dithiothreitol (DTT), 100 µM OA and 1∶100 protease inhibitor cocktail. Lysates were centrifuged for 30 minutes at 4°C at 21,000 RCF. To the soluble fractions 1/4 volume of 5× SDS-SB was added. Pellets were washed twice in lysis buffer, dissolved in SDS-SB with 6M urea (same final volume as soluble extracts in SDS-SB) by vortexing and brief sonication. WB was performed as previously described (Fontao et al., 2003). Skin extracts were prepared from pieces of skin explants which were frozen in liquid nitrogen, boiled in SDS-SB for 5 minutes, then disrupted with a cell disruptor (Disruptor Genie) with 0.5 mm glass beads for 5 minutes and stored at −20°C. WBs decorated with HRP-conjugated secondary antibodies were developed with ECL Plus reagents (GE Healthcare), whereas those decorated with fluorescent secondary antibodies were directly scanned and quantified with Typhoon 9200 (GE Healthcare) for Alexa Fluor 488 and Odyssey (Li-Cor) for infrared IRDye 680 and 800CW (Li-Cor), see supplementary material Table S1. For accurate quantitative analyses of the phosphorylation level of recombinant or endogenous plectin proteins, the same membrane was first blotted with mouse anti-tag (GFP or HA) or anti-plectin antibodies (10F6) and rabbit anti-pS4642 antibodies, and then with both secondary Alexa Fluor 488 anti-mouse and IRDye 800CW anti-rabbit antibodies, which did not crossreact with the other species (see supplementary material Table S1).
Recombinant protein expression, purification, in vitro phosphorylation and mass spectrometry analysis
Escherichia coli BL21 (DE3) were transformed with pET15b-mPL-C-E. Expression of recombinant protein was induced by adding 0.5 mM IPTG into the broth medium at 37°C for 2 hours. Bacteria were lyzed with a sonicator in PBS, 0.5 M NaCl, 10 mM imidazole, 1% TX-100, 10 µg/ml leupeptin, aprotinin, and pepstatin (20 ml for 200 ml of culture) and centrifuged for 10 minutes at 10,000 RCF. H6-tagged-mPL-C-E was purified by adding 150 µl HIS-Select Nickel Affinity Gel (Sigma) for 20 ml of lysates. After centrifugation, beads were washed three times with PBS. Protein concentration was determined with Bradford protein assay reagent (Bio-Rad), using BSA as standard. Protein purity was analyzed by SDS-PAGE, followed by Coomassie Brilliant Blue staining (Bio-Rad).
H6-mPL-C-E (0.1 µg/µl) was phosphorylated in vitro by the active catalytic subunit of PKA (0.5 U/100 µl) and PKC (0.04 U/100 µl) (Sigma) in 25 mM Tris HCl, pH 7.5, 2.5 mM MgCl2, 1 mM NaF, 0.5 mM DTT, 10 mM β-glycerophosphate with 100 µM ATP, containing 185 kBq [γ-32P]ATP for radiolabeling for 2 hours at 37°C. Phosphorylation was analyzed by autoradiography after SDS-PAGE. For identification of phosphorylated residue(s), phosphorylated protein samples were subjected to chymotrypsin digestion, desalting and TiO2 affinity chromatography, followed by MALDI and LC-MS/MS analysis by the protein analysis facility of the Center for Integrative Genomics at the University of Lausanne, Switzerland.
Yeast three-hybrid assays
Yeast three-hybrid (Y3H) assays were performed as previously described (Fontao et al., 2003). To ascertain phosphorylation of recombinant plectin in yeast, cell extracts were prepared by the trichloroacetic acid method (Fontao et al., 2003) and subjected to WB.
Phosphatase treatment
WBs or glass coverslips with fixed cells, both blocked with BSA, were incubated with or without (mock-treated) calf intestine alkaline phosphatase (CIAP) 10 U/ml (Promega) in the manufacturer's buffer for 2 hours at 37°C. Treated membranes and fixed cells were then extensively washed and incubated with antibodies as usual.
Supplementary Material
Acknowledgments
The authors are kindly indebted to A. Sonnenberg (The Netherlands Cancer Institute, Amsterdam) for providing PAJEB-β4, MD-EBS keratinocytes and HA–plectin-1C clone and for critical reading of the manuscript; to S. Yousefi (Institute of Pharmacology, Bern, Switzerland) for helpful advice about confocal microscopy, S. Yousefi, G. van der Goot (Global Health Institute-EPFL, Lausanne, Switzerland) and P. Janscak (Institute of Molecular Cancer Research, Zurich, Switzerland) for free access to their Typhoon 9400 scanners, to E. Müller and A. Galichet for anti-phospho-p38 antibodies, to M. Constantinescu for providing skin biopsies and to H. Gram for MNK-encoding plasmids. The technical support of U. Läderach in immunohistochemistry experiments was greatly appreciated.
Footnotes
Author contributions
J.-E.B., Y.S., L.F., B.F. and L.B. designed the study; J.-E.B. and Y.S. performed the experiments that constitute the main body of this work; N.B., R.H., P.L., L.F. and B.F. performed complementary experiments; K.G. and L.B. provided expertise and intellectual input; L.F., B.F. and L.B. coordinated the project; J.-E.B. wrote the manuscript with the help of B.F. and L.B.; R.H. and K.G. thoroughly edited the manuscript.
Funding
This work was supported by a grant from the Swiss National Foundation for Research [grant numbers 3100A0-121966 to L.B.]; Swiss Foundation for Research on Muscle Diseases (to L.B.); and the U.S. National Institutes of Health [grant numbers R37AR043380 to K.J.G.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.127779/-/DC1
References
- Adler J., Parmryd I. (2010). Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry 77A, 733–742 10.1002/cyto.a.20896 [DOI] [PubMed] [Google Scholar]
- Andrä K., Lassmann H., Bittner R., Shorny S., Fässler R., Propst F., Wiche G. (1997). Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 11, 3143–3156 10.1101/gad.11.23.3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrä K., Kornacker I., Jörgl A., Zörer M., Spazierer D., Fuchs P., Fischer I., Wiche G. (2003). Plectin-isoform-specific rescue of hemidesmosomal defects in plectin (-/-) keratinocytes. J. Invest. Dermatol. 120, 189–197 10.1046/j.1523-1747.2003.12027.x [DOI] [PubMed] [Google Scholar]
- Bagowski C. P., Besser J., Frey C. R., Ferrell J. E., Jr (2003). The JNK cascade as a biochemical switch in mammalian cells: ultrasensitive and all-or-none responses. Curr. Biol. 13, 315–320 [DOI] [PubMed] [Google Scholar]
- Bass-Zubek A. E., Hobbs R. P., Amargo E. V., Garcia N. J., Hsieh S. N., Chen X., Wahl J. K., III, Denning M. F., Green K. J. (2008). Plakophilin 2: a critical scaffold for PKC alpha that regulates intercellular junction assembly. J. Cell Biol. 181, 605–613 10.1083/jcb.200712133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N., Sicheritz-Pontén T., Gupta R., Gammeltoft S., Brunak S. (2004). Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 10.1002/pmic.200300771 [DOI] [PubMed] [Google Scholar]
- Boonstra J., Rijken P., Humbel B., Cremers F., Verkleij A., van Bergen en Henegouwen P. (1995). The epidermal growth factor. Cell Biol. Int. 19, 413–430 10.1006/cbir.1995.1086 [DOI] [PubMed] [Google Scholar]
- Borradori L., Sonnenberg A. (1999). Structure and function of hemidesmosomes: more than simple adhesion complexes. J. Invest. Dermatol. 112, 411–418 10.1046/j.1523-1747.1999.00546.x [DOI] [PubMed] [Google Scholar]
- Borradori L., Koch P. J., Niessen C. M., Erkeland S., van Leusden M. R., Sonnenberg A. (1997). The localization of bullous pemphigoid antigen 180 (BP180) in hemidesmosomes is mediated by its cytoplasmic domain and seems to be regulated by the beta4 integrin subunit. J. Cell Biol. 136, 1333–1347 10.1083/jcb.136.6.1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargnello M., Roux P. P. (2011). Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 10.1128/MMBR.00031-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies S. P., Reddy H., Caivano M., Cohen P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 10.1042/0264-6021:3510095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008). A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 105, 10762–10767 10.1073/pnas.0805139105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre B., Turowski P., Hemmings B. A. (1997). Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J. Biol. Chem. 272, 13856–13863 10.1074/jbc.272.21.13856 [DOI] [PubMed] [Google Scholar]
- Favre B., Schneider Y., Lingasamy P., Bouameur J. E., Begré N., Gontier Y., Steiner-Champliaud M. F., Frias M. A., Borradori L., Fontao L. (2011). Plectin interacts with the rod domain of type III intermediate filament proteins desmin and vimentin. Eur. J. Cell Biol. 90, 390–400 10.1016/j.ejcb.2010.11.013 [DOI] [PubMed] [Google Scholar]
- Fiol C. J., Haseman J. H., Wang Y. H., Roach P. J., Roeske R. W., Kowalczuk M., DePaoli-Roach A. A. (1988). Phosphoserine as a recognition determinant for glycogen synthase kinase-3: phosphorylation of a synthetic peptide based on the G-component of protein phosphatase-1. Arch. Biochem. Biophys. 267, 797–802 10.1016/0003-9861(88)90089-6 [DOI] [PubMed] [Google Scholar]
- Foisner R., Traub P., Wiche G. (1991). Protein kinase A- and protein kinase C-regulated interaction of plectin with lamin B and vimentin. Proc. Natl. Acad. Sci. USA 88, 3812–3816 10.1073/pnas.88.9.3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisner R., Malecz N., Dressel N., Stadler C., Wiche G. (1996). M-phase-specific phosphorylation and structural rearrangement of the cytoplasmic cross-linking protein plectin involve p34cdc2 kinase. Mol. Biol. Cell 7, 273–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontao L., Favre B., Riou S., Geerts D., Jaunin F., Saurat J. H., Green K. J., Sonnenberg A., Borradori L. (2003). Interaction of the bullous pemphigoid antigen 1 (BP230) and desmoplakin with intermediate filaments is mediated by distinct sequences within their COOH terminus. Mol. Biol. Cell 14, 1978–1992 10.1091/mbc.E02-08-0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijns E., Sachs N., Kreft M., Wilhelmsen K., Sonnenberg A. (2010). EGF-induced MAPK signaling inhibits hemidesmosome formation through phosphorylation of the integrin beta4. J. Biol. Chem. 285, 37650–37662 10.1074/jbc.M110.138818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijns E., Kuikman I., Litjens S., Raspe M., Jalink K., Ports M., Wilhelmsen K., Sonnenberg A. (2012). Phosphorylation of threonine 1736 in the C-terminal tail of integrin β4 contributes to hemidesmosome disassembly. Mol. Biol. Cell 23, 1475–1485 10.1091/mbc.E11-11-0957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerts D., Fontao L., Nievers M. G., Schaapveld R. Q., Purkis P. E., Wheeler G. N., Lane E. B., Leigh I. M., Sonnenberg A. (1999). Binding of integrin alpha6beta4 to plectin prevents plectin association with F-actin but does not interfere with intermediate filament binding. J. Cell Biol. 147, 417–434 10.1083/jcb.147.2.417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getsios S., Huen A. C., Green K. J. (2004). Working out the strength and flexibility of desmosomes. Nat. Rev. Mol. Cell Biol. 5, 271–281 10.1038/nrm1356 [DOI] [PubMed] [Google Scholar]
- Godsel L. M., Hsieh S. N., Amargo E. V., Bass A. E., Pascoe-McGillicuddy L. T., Huen A. C., Thorne M. E., Gaudry C. A., Park J. K., Myung K. et al. (2005). Desmoplakin assembly dynamics in four dimensions: multiple phases differentially regulated by intermediate filaments and actin. J. Cell Biol. 171, 1045–1059 10.1083/jcb.200510038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann H., Aebi U. (2004). Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu. Rev. Biochem. 73, 749–789 10.1146/annurev.biochem.73.011303.073823 [DOI] [PubMed] [Google Scholar]
- Hobbs R. P., Green K. J. (2012). Desmoplakin regulates desmosome hyperadhesion. J. Invest. Dermatol. 132, 482–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. (2012). PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40 Database issue, D261–D270 10.1093/nar/gkr1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard M. J., Cohen P. (1993). On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem. Sci. 18, 172–177 10.1016/0968-0004(93)90109-Z [DOI] [PubMed] [Google Scholar]
- Ivaska J., Pallari H. M., Nevo J., Eriksson J. E. (2007). Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 313, 2050–2062 10.1016/j.yexcr.2007.03.040 [DOI] [PubMed] [Google Scholar]
- Izawa I., Inagaki M. (2006). Regulatory mechanisms and functions of intermediate filaments: a study using site- and phosphorylation state-specific antibodies. Cancer Sci. 97, 167–174 10.1111/j.1349-7006.2006.00161.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayali A. G., Austin D. A., Webster N. J. (2000). Stimulation of MAPK cascades by insulin and osmotic shock: lack of an involvement of p38 mitogen-activated protein kinase in glucose transport in 3T3-L1 adipocytes. Diabetes 49, 1783–1793 10.2337/diabetes.49.11.1783 [DOI] [PubMed] [Google Scholar]
- Knauf U., Tschopp C., Gram H. (2001). Negative regulation of protein translation by mitogen-activated protein kinase-interacting kinases 1 and 2. Mol. Cell. Biol. 21, 5500–5511 10.1128/MCB.21.16.5500-5511.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny P., Fuchs P., Reipert S., Kunz W. S., Zeöld A., Fischer I., Paulin D., Schröder R., Wiche G. (2008). Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J. Cell Biol. 181, 667–681 10.1083/jcb.200711058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster J., Geerts D., Favre B., Borradori L., Sonnenberg A. (2003). Analysis of the interactions between BP180, BP230, plectin and the integrin alpha6beta4 important for hemidesmosome assembly. J. Cell Sci. 116, 387–399 10.1242/jcs.00241 [DOI] [PubMed] [Google Scholar]
- Koster J., van Wilpe S., Kuikman I., Litjens S. H., Sonnenberg A. (2004). Role of binding of plectin to the integrin beta4 subunit in the assembly of hemidesmosomes. Mol. Biol. Cell 15, 1211–1223 10.1091/mbc.E03-09-0697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapouge K., Fontao L., Champliaud M. F., Jaunin F., Frias M. A., Favre B., Paulin D., Green K. J., Borradori L. (2006). New insights into the molecular basis of desmoplakin- and desmin-related cardiomyopathies. J. Cell Sci. 119, 4974–4985 10.1242/jcs.03255 [DOI] [PubMed] [Google Scholar]
- Lewis T. S., Shapiro P. S., Ahn N. G. (1998). Signal transduction through MAP kinase cascades. Adv. Cancer Res. 74, 49–139 10.1016/S0065-230X(08)60765-4 [DOI] [PubMed] [Google Scholar]
- Liu Y., Ge J., Li Q., Gu L., Guo X., Ma Z. G., Zhu Y. P. (2013). Anisomycin induces apoptosis of glucocorticoid resistant acute lymphoblastic leukemia CEM-C1 cells via activation of mitogen-activated protein kinases p38 and JNK. Neoplasma. 3 60, 101–110 10.4149/neo_2013_014 [DOI] [PubMed] [Google Scholar]
- Malecz N., Foisner R., Stadler C., Wiche G. (1996). Identification of plectin as a substrate of p34cdc2 kinase and mapping of a single phosphorylation site. J. Biol. Chem. 271, 8203–8208 10.1074/jbc.271.14.8203 [DOI] [PubMed] [Google Scholar]
- Mao X., Bravo I. G., Cheng H., Alonso A. (2004). Multiple independent kinase cascades are targeted by hyperosmotic stress but only one activates stress kinase p38. Exp. Cell Res. 292, 304–311 10.1016/j.yexcr.2003.09.012 [DOI] [PubMed] [Google Scholar]
- Marfe G., Pucci B., De Martino L., Fiorito F., Di Stefano C., Indelicato M., Aventaggiato M., Russo M. A., Tafani M. (2009). Heat-shock pretreatment inhibits sorbitol-induced apoptosis in K562, U937 and HeLa cells. Int. J. Cancer 125, 2077–2085 [DOI] [PubMed] [Google Scholar]
- McInroy L., Määttä A. (2011). Plectin regulates invasiveness of SW480 colon carcinoma cells and is targeted to podosome-like adhesions in an isoform-specific manner. Exp. Cell Res. 317, 2468–2478 10.1016/j.yexcr.2011.07.013 [DOI] [PubMed] [Google Scholar]
- Meng J. J., Bornslaeger E. A., Green K. J., Steinert P. M., Ip W. (1997). Two-hybrid analysis reveals fundamental differences in direct interactions between desmoplakin and cell type-specific intermediate filaments. J. Biol. Chem. 272, 21495–21503 10.1074/jbc.272.34.21495 [DOI] [PubMed] [Google Scholar]
- Myatt A., Hill S. J. (2005). Trypsin stimulates the phosphorylation of p42,44 mitogen-activated protein kinases via the proteinase-activated receptor-2 and protein kinase C epsilon in human cultured prostate stromal cells. Prostate 64, 175–185 10.1002/pros.20205 [DOI] [PubMed] [Google Scholar]
- Myers A. P., Corson L. B., Rossant J., Baker J. C. (2004). Characterization of mouse Rsk4 as an inhibitor of fibroblast growth factor-RAS-extracellular signal-regulated kinase signaling. Mol. Cell. Biol. 24, 4255–4266 10.1128/MCB.24.10.4255-4266.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuberger G., Schneider G., Eisenhaber F. (2007). pkaPS: prediction of protein kinase A phosphorylation sites with the simplified kinase-substrate binding model. Biol. Direct 2, 1 10.1186/1745-6150-2-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic B., Mac Nulty E., Mir B., Wiche G. (1996). Basic amino acid residue cluster within nuclear targeting sequence motif is essential for cytoplasmic plectin-vimentin network junctions. J. Cell Biol. 134, 1455–1467 10.1083/jcb.134.6.1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohori M., Kinoshita T., Okubo M., Sato K., Yamazaki A., Arakawa H., Nishimura S., Inamura N., Nakajima H., Neya M. et al. (2005). Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem. Biophys. Res. Commun. 336, 357–363 10.1016/j.bbrc.2005.08.082 [DOI] [PubMed] [Google Scholar]
- Omary M. B., Ku N. O., Tao G. Z., Toivola D. M., Liao J. (2006). ‘Heads and tails’ of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem. Sci. 31, 383–394 10.1016/j.tibs.2006.05.008 [DOI] [PubMed] [Google Scholar]
- Osmanagic-Myers S., Gregor M., Walko G., Burgstaller G., Reipert S., Wiche G. (2006). Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J. Cell Biol. 174, 557–568 10.1083/jcb.200605172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra J. L., Buxadé M., Proud C. G. (2005). Features of the catalytic domains and C termini of the MAPK signal-integrating kinases Mnk1 and Mnk2 determine their differing activities and regulatory properties. J. Biol. Chem. 280, 37623–37633 10.1074/jbc.M508356200 [DOI] [PubMed] [Google Scholar]
- Porter R. M., Lane E. B. (2003). Phenotypes, genotypes and their contribution to understanding keratin function. Trends Genet. 19, 278–285 10.1016/S0168-9525(03)00071-4 [DOI] [PubMed] [Google Scholar]
- Raingeaud J., Gupta S., Rogers J. S., Dickens M., Han J., Ulevitch R. J., Davis R. J. (1995). Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 270, 7420–7426 10.1074/jbc.270.13.7420 [DOI] [PubMed] [Google Scholar]
- Rezniczek G. A., Abrahamsberg C., Fuchs P., Spazierer D., Wiche G. (2003). Plectin 5′-transcript diversity: short alternative sequences determine stability of gene products, initiation of translation and subcellular localization of isoforms. Hum. Mol. Genet. 12, 3181–3194 10.1093/hmg/ddg345 [DOI] [PubMed] [Google Scholar]
- Riteau B., de Vaureix C., Lefèvre F. (2006). Trypsin increases pseudorabies virus production through activation of the ERK signalling pathway. J. Gen. Virol. 87, 1109–1112 10.1099/vir.0.81609-0 [DOI] [PubMed] [Google Scholar]
- Sapkota G. P., Cummings L., Newell F. S., Armstrong C., Bain J., Frodin M., Grauert M., Hoffmann M., Schnapp G., Steegmaier M. et al. (2007). BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 401, 29–38 10.1042/BJ20061088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder R., Kunz W. S., Rouan F., Pfendner E., Tolksdorf K., Kappes-Horn K., Altenschmidt-Mehring M., Knoblich R., van der Ven P. F., Reimann J. et al. (2002). Disorganization of the desmin cytoskeleton and mitochondrial dysfunction in plectin-related epidermolysis bullosa simplex with muscular dystrophy. J. Neuropathol. Exp. Neurol. 61, 520–530 [DOI] [PubMed] [Google Scholar]
- Seltmann K., Roth W., Kroger C., Loschke F., Lederer M., Huttelmaier S., Magin T. M. (2012). Keratins mediate localization of hemidesmosomes and repress cell motility. J. Invest. Dermatol. 133, 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevcik J., Urbanikova L., Kost'an J., Janda L., Wiche G. (2004). Actin-binding domain of mouse plectin. Crystal structure and binding to vimentin. FEBS 271, 1873–1884 [DOI] [PubMed] [Google Scholar]
- Sihag R. K., Inagaki M., Yamaguchi T., Shea T. B., Pant H. C. (2007). Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp. Cell Res. 313, 2098–2109 10.1016/j.yexcr.2007.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg A., Liem R. K. (2007). Plakins in development and disease. Exp. Cell Res. 313, 2189–2203 10.1016/j.yexcr.2007.03.039 [DOI] [PubMed] [Google Scholar]
- Stappenbeck T. S., Lamb J. A., Corcoran C. M., Green K. J. (1994). Phosphorylation of the desmoplakin COOH terminus negatively regulates its interaction with keratin intermediate filament networks. J. Biol. Chem. 269, 29351–29354 [PubMed] [Google Scholar]
- Stegh A. H., Herrmann H., Lampel S., Weisenberger D., Andrä K., Seper M., Wiche G., Krammer P. H., Peter M. E. (2000). Identification of the cytolinker plectin as a major early in vivo substrate for caspase 8 during CD95- and tumor necrosis factor receptor-mediated apoptosis. Mol. Cell. Biol. 20, 5665–5679 10.1128/MCB.20.15.5665-5679.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suozzi K. C., Wu X., Fuchs E. (2012). Spectraplakins: master orchestrators of cytoskeletal dynamics. J. Cell Biol. 197, 465–475 10.1083/jcb.201112034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turowski P., Myles T., Hemmings B. A., Fernandez A., Lamb N. J. (1999). Vimentin dephosphorylation by protein phosphatase 2A is modulated by the targeting subunit B55. Mol. Biol. Cell 10, 1997–2015 10.1091/mbc.10.6.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spaendonck-Zwarts K., van Hessem L., Jongbloed J. D., de Walle H. E., Capetanaki Y., van der Kooi A. J., van Langen I. M., van den Berg M. P., van Tintelen J. P. (2010). Desmin-related myopathy: a review and meta-analysis. Clin. Genet. 80, 354–366 [DOI] [PubMed] [Google Scholar]
- Walko G., Vukasinovic N., Gross K., Fischer I., Sibitz S., Fuchs P., Reipert S., Jungwirth U., Berger W., Salzer U. et al. (2011). Targeted proteolysis of plectin isoform 1a accounts for hemidesmosome dysfunction in mice mimicking the dominant skin blistering disease EBS-Ogna. PLoS Genet. 7, e1002396 10.1371/journal.pgen.1002396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter L., Wiche G. (2013). The many faces of plectin and plectinopathies: pathology and mechanisms. Acta Neuropathol. 125, 77–93 10.1007/s00401-012-1026-0 [DOI] [PubMed] [Google Scholar]
- Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C. L., Haase J., Janes J., Huss J. W. III et al(2009). BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10, R130 10.1186/gb-2009-10-11-r130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y., Ren J., Gao X., Jin C., Wen L., Yao X. (2008). GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 7, 1598–1608 10.1074/mcp.M700574-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L., Baumann U., Reymond J. L. (2004). An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.