

The discovery of a lead compound is an essential process in early drug discovery, hopefully eventually resulting into a clinical candidate and a drug for the treatment of a disease. Besides affinity and selectivity for the target, however, other target unrelated compound properties are equally important for the fate of drug candidate, e.g. water solubility, lipophilicity and molecular weight since they determine important aspects such as oral bioavailability, dosing schedule and side effects. The parallel discovery and early development of several leads is therefore now pursued whenever possible, an approach that takes into account the high attrition rate of early drug discovery projects. Currently, hits as starting points for medicinal chemistry optimisations are mostly found by high-throughput screening (HTS) campaigns and to a much less extent by structure-based approaches including fragment-based and computational drug discovery. For certain target classes, however, HTS often yields very low numbers of hits.[1] For example, protein-protein interactions (PPIs) are notoriously difficult to hit with drug-like small molecules.[2] This has been assigned to the unusual structure, topology and flexibility of protein-protein interfaces.[3] The recent advancement of several drugs into clinical development clearly shows that certain PPIs, e.g., Bcl-x and XIAP, can be efficiently targeted by small molecules.[4] Here, we describe a complementary process that led to the parallel discovery of several compounds belonging to seven different scaffold classes, amenable to synthesis by efficient multicomponent reaction (MCR) chemistry in just one step, that antagonize the PPI between the transcription factor p53 and its negative regulator Hdm2.
Protein-protein interactions are often mediated by only a few key amino acid side chains and the terms “hot spot” and “anchor” have been introduced for such locally constrained areas and amino acids on the surface of interacting proteins.[2,5,8] In a first approximation, the depth into which a specific amino acid side chain of the donor protein is buried in the acceptor protein is indicative of its energetic importance. We reasoned that this “anchor” amino acid side chain might serve as reasonable starting point for the design of (ant)agonists of a PPI. Thus, we use this particular amino acid side chain as an initial “anchor” in virtual libraries of low molecular weight scaffolds. Virtual compounds containing anchor side chains are selected for the synthesis and screening based on docking into the protein-protein interaction interface. The starting pose for the docking/energy minimisation procedure is forced in such a way that an overlap between the anchor and the template amino acid side chain is ensured. In order to rapidly test these ideas, we chose an efficient and fast but versatile synthesis approach, MCR (Fig. 1).[6] MCR allows the assembly of many diverse and complex scaffolds in a one-step/one-pot manner, thus saving time and resources, and potentially increasing the success rate of lead discovery.
Figure 1.
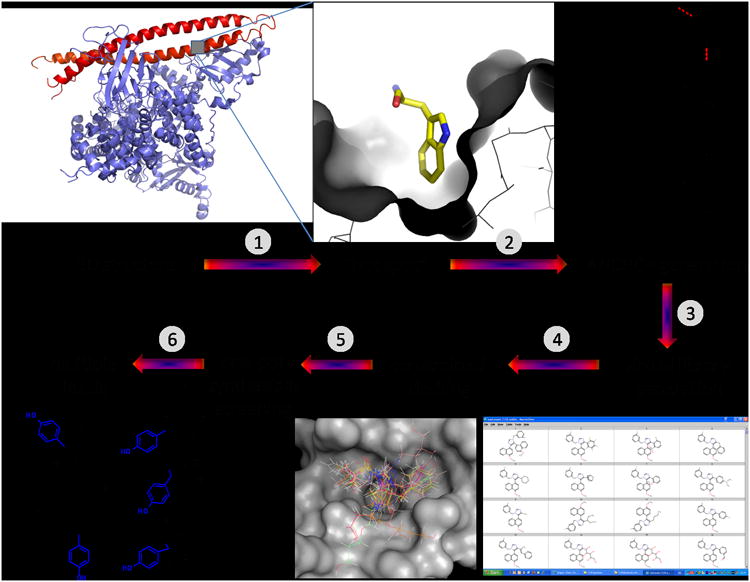
Graphical representation of the workflow in the discovery procedure for the rapid generation of small molecular weight (ant) agonists of protein-protein interactions. The process relies on high resolution structural target data (1), the presence and identification of a highly buried amino acid (2), a fragmentation/anchor generation step (3), virtual chemistry employing the anchor and based on multicomponent reactions (4), constrained docking forcing the anchor fragment into the binding site (5), and synthesis and screening (6). (FG = functional group).
The PPI interface of p53/Hdm2 has been characterized in molecular detail by X-ray structure analysis.[7] It relies on the steric complementarity between the Hdm2 cleft and the hydrophobic face of the p53 α -helix and, in particular, on a triad of p53 amino acids Phe19, Trp23, and Leu26, which insert deep into the Hdm2 cleft (SI Fig 1A). We chose the indole side chain of Trp23 as the “anchor” residue in our approach for three reasons: 1) it is the central amino acid of the triad, thus facilitating addressing by the antagonists the crucial Phe19 and Leu26 binding sites; 2) it is deeply buried in Hdm2; and 3) it also features, in addition to extensive van der Waals contacts, a hydrogen bond to the Leu54 backbone carbonyl of Hdm2. In fact, calculation of the solvent-accessible surface areas of all amino acids in the p53/Hdm2 interaction ranks Trp23>Phe19>Leu26 the highest.[8] Next, from our in-house database of several hundred MCR scaffolds, we selected 40 MCR scaffolds to create virtual compound libraries.[6] By design, each of the compounds from the different scaffolds incorporated the anchor. We used indole and bioistosteric 4-chlorophenyl derivatives[24 a,b] endued with the corresponding functional groups as anchors to act as starting materials for the MCRs (SI Fig. 1B); for example, we used unsubstituted indole and oxindole to perform a Friedel-Crafts type alkylation and an Ugi-four component condensation, respectively (Fig. 2, SI Table 1).
Figure 2.
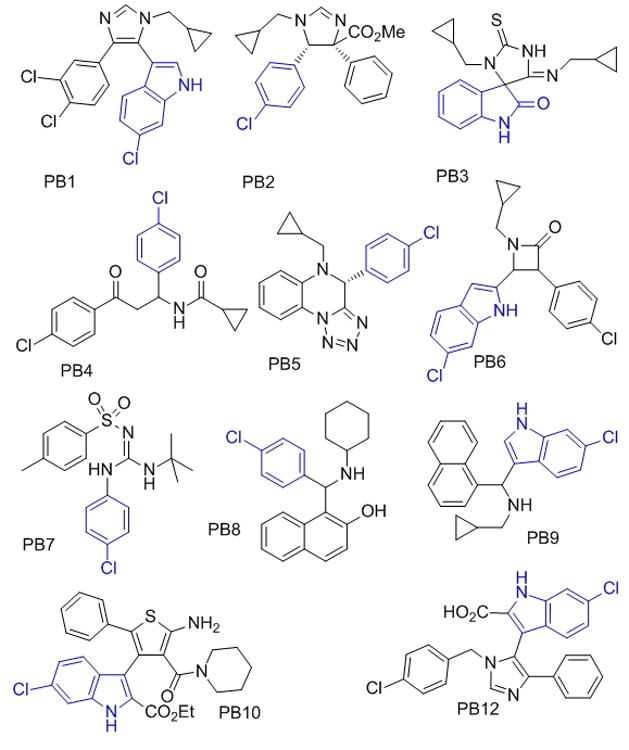
Different predicted and found p53-Hdm2 antagonists based on multiple MCR scaffolds (the anchor moiety is marked blue, PB = Pittsburgh).
The virtual library of compounds comprising all possible stereoisomers was generated using REACTOR software.[9] Different aliphatic and aromatic starting materials to represent different shape and electrostatic features were chosen for the MCR starting material classes. The virtual compound libraries incorporating the anchor side chain were docked into a rigid model of the p53 binding site in the Hdm2 receptor (PDB identifier 1YCR) using the modelling/docking software package Moloc.[7,10] Assuming that the anchor residue predefines the binding site of the molecules and in order to avoid non productive docking poses, we forced the anchor part (indole or 4-chlorophenyl) of the compounds to overlap with the respective Trp23 anchor as a starting point for energy minimization. As an encouraging evidence for the anchor approach we solved the crystal structure of the MCR compound PB12 bound to Hdm2 (Fig. 3).[11] This structure shows an almost perfect alignment of the Trp23-indole anchor with the indole moiety of PB12 with an RMSD (indole) of 0.34 Å.
Figure 3.
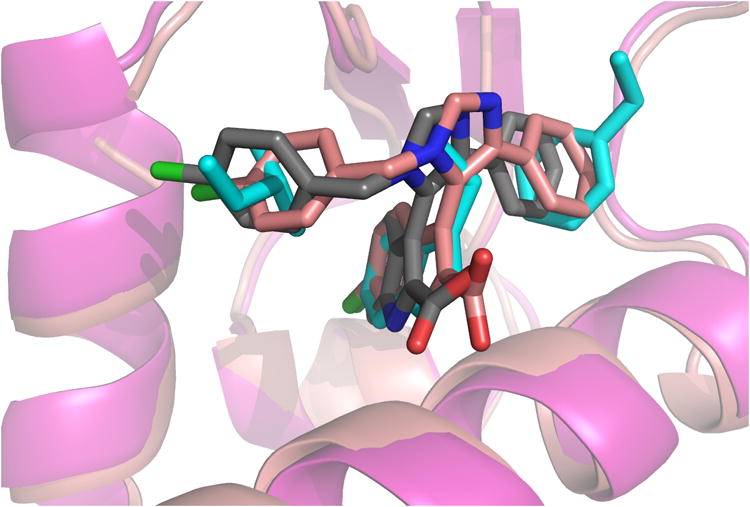
Alignment of the anchor p53 residues Phe19, Trp23 and Leu26 (cyan sticks) bound to Hdm2 (pink cartoon, PDB ID: 1YCR) with a van Leusen imidazole PB12 based antagonist (pink sticks) bound to Hdm2 (pink cartoon, PDB ID: 3LBK) and the corresponding predicted docking pose of PB12 (grey sticks).
From the highest-ranking docked compounds, which addressed all three binding sites of p53 (Phe19, Trp23 and Leu26), we chose several compounds for synthesis and screening based on different features (Fig. 2). First, the compounds were chosen so as to belong to different types of scaffolds to ensure variation in backbones and chemistries. Second, we chose reactions where the corresponding starting materials were commercially available or readily accessible by synthesis. As a third criterion, we preferentially chose MCRs we had previous experienced.[6a] Products were synthesized according to two different Ugi MCR variations (PB3, PB5),[12] the van Leusen three component reaction (PB1, PB12),[13] the Orru three component reaction (PB2) followed by an amidation step (PB11),[14] the Passerini three component reaction (PB9),[15] the Betti three component reaction (PB8),[16] the Staudinger three component reaction (PB6),[17] an MCR that formed an amidinosulfone amide (PB7),[18] the Gewald three component reaction to an aminothiophene (PB10),[19] and an α -amidoalkylation (PB4).[20] Among the 11 different MCR series prepared, seven scaffolds showed binding activity <60 μM (see Fig. 2 and Table 1).
Table 1.
Activities of predicted, synthesized and tested compounds and known optimized compounds.
| Compound | Kd measured by NMR (FP [c]) |
|---|---|
| PB1 | 40±15[a] |
| syn-PB2 | 3±1[b] |
| anti-PB2 | 40±10[a] |
| PB2a | 2±1[b] |
| PB3 | 20± 7[a,b] |
| PB4 | 30± 10[a] |
| PB5 | 30±10[b] |
| PB6 | precipitation |
| PB7 | >100[a] |
| PB8 | 60±20[a,b] |
| PB9 | 60±30[a] |
| PB10 | 5±2[b] (11±0.5) |
| PB11 | 0.8±0.4[b] |
| PB12 | 1.1[b](0.7± 0.04) |
| Nutlin-3 [d] | 0.09±0.03[b] (0.04±0.01) |
| MI-63 [d] | 0.01±0.01[b] (0.003±0.002) |
bin. titr., the binary titration of a ligand with the apo-15N-Hdm2 protein using 15N-HSQ.[22]
AIDA (antagonist induced dissociation assay),[23] the competition NMR experiment between ligand and the p53-Hdm2 complex.
FP, fluorescent polarization assay.
nutlin-3 and MI-63 are described in[24] and [25,11], respectively; the Kd's are our measurements, in agreement with[11, 24, 25].
The experimental error of Kd's measured by NMR was assumed to be ca. 30%; [a][b] both a and b gave the same values within the error boundaries; errors for FP values are determined by fit confidence.
Representative docking poses of the predicted p53/Hdm2 antagonist compounds are shown in Fig. 4 and SI Fig. 2. Next, these compounds were screened for their ability to bind to Hdm2 and to antagonize the p53/Hdm2 PPI. We chose NMR spectroscopy to test the compounds ability to bind and antagonize the p53/Hdm2 complex as it can provide a wealth of information that otherwise cannot be assessed in high-throughput assays. Importantly, use of NMR-based screening allowed for determination of both compound affinity to Hdm2 and the ability of compounds to dissociate preformed p53/Hdm2 complex. Other important questions about the compound properties can also be answered with an NMR-based approach. These include: Is the compound sufficiently water soluble? Does the compound bind to Hdm2 or to p53? Does the compound cause precipitation of any of the proteins? Does the compound lead to major conformational changes in the binding protein? What is the binding site of the compound on the protein surface?
Figure 4.
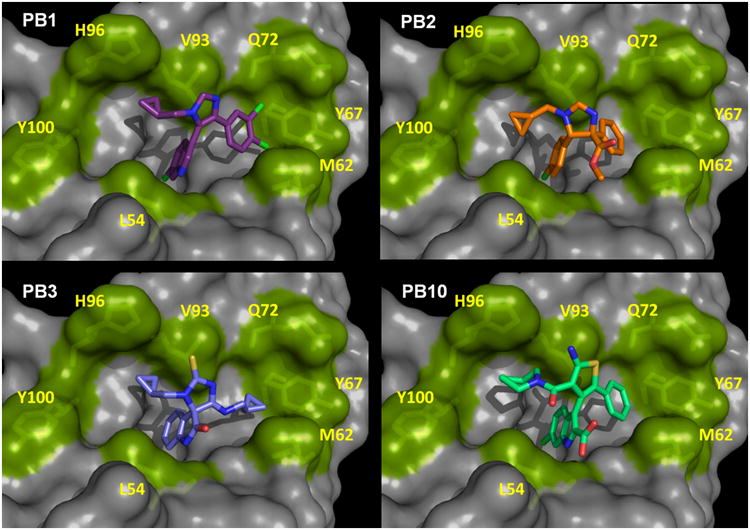
Binding models of four different p53/Hdm2 antagonists by constrained docking into PDB 1YCR. 5-indolo-imidazole PB1; B: imidazoline PB2; C: thiohydantoin-imide PB3; and D: 2-aminothiophene-3-acetamide PB10. Several rim key amino acids of the receptor Hdm2 are shown as sticks and green surface and numbered. The docking models are in accordance with the observed Hdm2 HSQC-NMR shifts obtained from the compound titrations. All poses show the receptor in the same orientation shown in Fig.3.
All of the information is instrumental in order to optimize a compound series not only for potency and selectivity but also for, e.g., water solubility and protein binding. Moreover the usage of screening techniques based on independent physical methods prevents from discovering PAIN compounds (Pan Assay Interference Compounds) and covalent binders.[21] The different compounds were tested for binding to Hdm2 by performing a series of NMR titrations with isotopically enriched 15N-Hdm2.[22] Strong binding of a compound to its target is indicated by appearance of splitting of the signals in a heteronuclear single quantum coherence (HSQC) spectrum, whereas a shift of signals indicates weaker binding. Figure 5A and SI Fig. 3 show 15N-HSQC spectra of Hdm2 titrated with PB12, as an example of the slow chemical exchange, and diastereomers of PB2 as an example of the fast chemical exchange, respectively. Additionally we used our recently introduced antagonist induced dissociation assay (AIDA), a fast 1D-NMR method to determine the KD (Fig. 5C and SI Fig. 4).[23]
Figure 5.
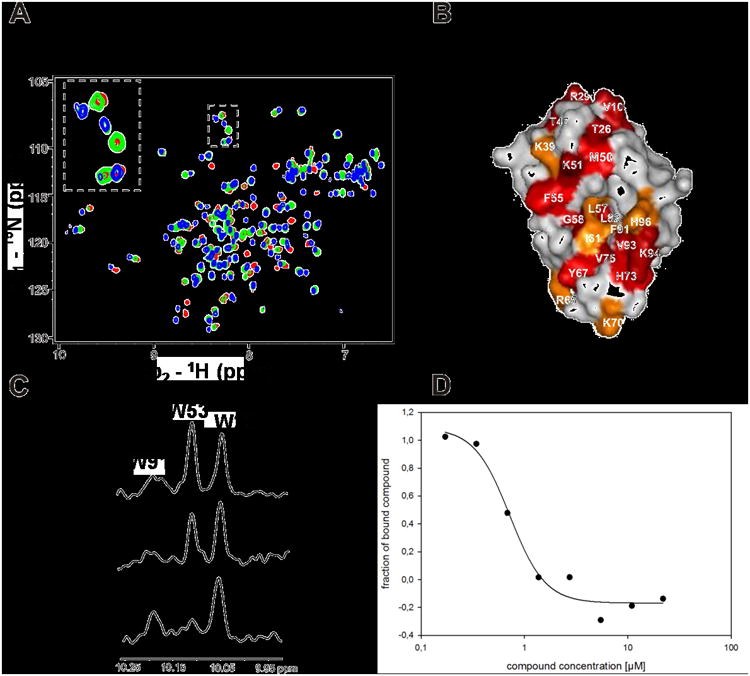
Screening activities of PB12. A. HSQC1H-15N HSQC spectra of Hdm2 (1-125) titrated with increasing amount of PB12. Red spectrum is a reference of apo-Hdm2, green spectrum corresponds to approximately 40% saturation of Hdm2 with PB12; the spectrum shows slow chemical exchange that is typical for strong interactions with submicromolar Kds. Blue spectrum corresponds to Hdm2 fully saturated with PB12. B: perturbation plot of PB12; C: AIDA-NMR experiment: the concentration of the p53 released from the Hdm2/p53 complex by the antagonist is proportional to the height of the HN indole peak of W23(p53). The bottom spectrum shows the downfield shifted NMR signals of 20 μM Hdm2/p53 complex, the middle one shows approximately 70% of dissociation of the complex upon addition of PB12 to the complex in 1:1 molar ratio, the upper spectrum shows signals of the free p53.; D: study of the PB12 by the fluorescent polarization assay.[11,25]
From the 15N-HSQC spectra, we calculated the KDNMR values for syn-PB2 and anti-PB2 to be 3 ± 1 μM and ca. 40 μM, respectively. Table 1 shows a selection of other Hdm2-binding compounds with their KD values, ranging from 60 μM down to the best compound showing less than 1 μM. Due to the inherent requirements of the Hdm2 binding pocket, the PB compounds are highly hydrophobic and most of the active PB compounds show a low total polar surface area (TPSA, SI Table 2) most probably resulting in low water solubility, an unfavourable quality of compounds designed to become drugs. According to our binding model of PB2, the methyl ester group is not involved in the binding site, but rather remains exposed to solvent (Fig. 4). Therefore, we reasoned that derivatization of the ester bond by amidation could potentially improve water solubility and binding affinity. Indeed, conversion of the carboxymethyl group of PB2 derivative PB2a to amide PB11 improved the potency of the imidazoline scaffold, as well as its water solubility as judged by the NMR experiments and the PSA (Scheme 1, Table 1 and SI Table 2, SI Fig. 6).
Scheme 1.
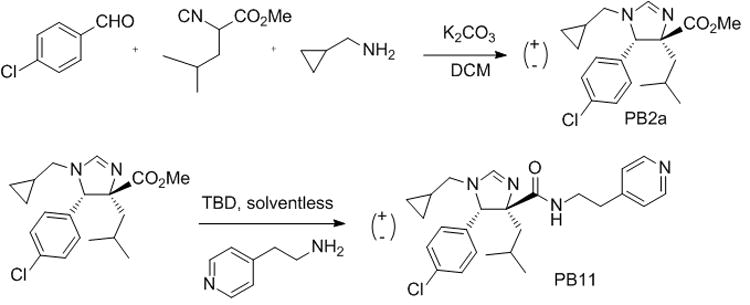
One-pot Orru-3CR of imidazoline PB2a and optimization by amidation yielding derivative PB11 with improved affinity and water solubility. (TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene).
We have introduced a new process for predicting and producing antagonists of the cancer relevant PPI p53/Hdm2. The approach differs substantially from currently used techniques, including high throughput screening, fragment-based approaches, structure-based design or computational screening of compound libraries. For example classical fragment based drug discovery approaches are strong in detecting weakly however efficiently binding small molecular units which serve as starting points to assemble more potent drug-like compounds, however with no straight forward synthetic pathway.[27] Herein the chemistry to more potent leads starting from the fragment (=anchor) is predefined by the MCR chemistry. Thus a seamless pathway for the optimization of affinity and other important drug properties is predefined. E.g. nutlin-3 is synthesized in a sequential > 8-step synthesis whereas PB11 is accessible in only two steps from commercial available starting materials.[28] The herein found compounds are drug-like and can be optimized regarding potency and physicochemical properties (SI Fig. 6). Certainly, this new approach, successful in one target example, demands further validation with different types of PPIs. Extensive optimization and in vitro and in vivo testing of optimized compounds of the different scaffolds reported herein are under way and will be reported in due course.
Supplementary Material
Footnotes
We thank Dr. Ulli Rothweiler for fruitful discussion. This work was supported by the NIH grant 1R21GM087617-01 (to AD), Deutsche Krebshilfe, Grant 108354 (to TH) and is part of a NCI-RAND program. We thank Haixia Liu for the synthesis of PB3.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Anna Czarna, Max-Planck-Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany
Dr. Barbara Beck, Departments of Pharmaceutical Sciences and Chemistry, University of Pittsburgh, Biomedical Science Tower 3, 3501 Fifth Avenue, Pittsburgh, PA 15261, USA, Fax: (+) 412-383-5298
Dr. Stuti Srivastava, Departments of Pharmaceutical Sciences and Chemistry, University of Pittsburgh, Biomedical Science Tower 3, 3501 Fifth Avenue, Pittsburgh, PA 15261, USA, Fax: (+) 412-383-5298
Grzegorz Popowicz, Max-Planck-Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany.
Siglinde Wolf, Max-Planck-Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany.
Yijun Huang, Departments of Pharmaceutical Sciences and Chemistry, University of Pittsburgh, Biomedical Science Tower 3, 3501 Fifth Avenue, Pittsburgh, PA 15261, USA, Fax: (+) 412-383-5298.
Michal Bista, Max-Planck-Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany.
Tad A. Holak, Max-Planck-Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany
Alexander Dömling, Email: asd30@pitt.edu, Departments of Pharmaceutical Sciences and Chemistry, University of Pittsburgh, Biomedical Science Tower 3, 3501 Fifth Avenue, Pittsburgh, PA 15261, USA, Fax: (+) 412-383-5298.
References
- 1.a) Spencer RW, Spencer RW. Biotech Bioeng Comb Chem. 1998;61:61–67. [Google Scholar]; b) Jacoby E, Boettcher A, Mayr LM, Brown N, Jenkins JL, Kallen J, Engeloch C, Schopfer U, Furet P, Masuya K, Lisztwan J. Chemogenomics, Methods in Molecular Biology. In: Jacoby E, editor. Series: Methods in Molecular Biology. Vol. 575. 2009. pp. 173–194. [DOI] [PubMed] [Google Scholar]
- 2.Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 3.Nooren IMA, Thornton JM. EMBO J. 2003;22:3466–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]; b) Zobel K, Wang L, Varfolomeev E, Franklin MC, Elliott LO, Wallweber HJ, Okawa DC, Flygare JA, Vucic D, Fairbrother WJ, Deshayes K. ACS Chem Biol. 2006;1:525–33. doi: 10.1021/cb600276q. [DOI] [PubMed] [Google Scholar]
- 5.a) Bogan AA, Thorn KS. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]; b) Moreira IS, Fernandes PA, Ramos MJ. Proteins. 2007;68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 6.a) Dömling A. Chem Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; b) Orru RVA, de Greef M. Synthesis. 2003:1471–1499. [Google Scholar]; c) Weber L, Illgen K, Almstetter M. Synlett. 1999:366–374. [Google Scholar]; d) Hulme C, Lee YS. Mol Divers. 2008;12:1–15. doi: 10.1007/s11030-008-9072-1. [DOI] [PubMed] [Google Scholar]; e) Ugi I, Dömling A, Hörl W. Endeavour. 1994;18:115–122. [Google Scholar]; f) Zhu J, Bienaymé H, editors. Multicomponent Reactions. Wiley; Weinheim: 2005. [Google Scholar]; g) Ganem B. Acc Chem Res. 2009;42:463–472. doi: 10.1021/ar800214s. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Sunderhaus JD, Martin SF. Chem Eur J. 2009;15:1300–1308. doi: 10.1002/chem.200802140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 8.Meireles L, Dömling A, Camacho CJ. 2010 doi: 10.1093/nar/gkq502. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pirok G, Mate N, Varga J, Szegezdi J, Vargyas M, Dorant S, Csizmadia F. J Chem Inf Model. 2006;46:563–568. doi: 10.1021/ci050373p. [DOI] [PubMed] [Google Scholar]
- 10.a) Gerber PR, Müller K. J Comp-Aided Mol Des. 1995;9:251–68. doi: 10.1007/BF00124456. [DOI] [PubMed] [Google Scholar]; b) Gerber PR. J Comp-Aided Mol Des. 1998;12:37–51. doi: 10.1023/a:1007902804814. [DOI] [PubMed] [Google Scholar]
- 11.Popowicz GM, Czarna A, Wolf S, Wang K, Wang W, Dömling A, Holak TA. Cell Cycle. 2010;9:1104–1111. doi: 10.4161/cc.9.6.10956. [DOI] [PubMed] [Google Scholar]
- 12.Ugi I. In: Isonitrile Chemistry. Ugi I, editor. Academic Press; New York: 1971. pp. 149–155. [Google Scholar]
- 13.Kalinski C, Umkehrer M, Gonnard S, Jäger N, Ross G, Hiller W. Tetrahedron Lett. 2006;47:2041–2044. [Google Scholar]
- 14.van Leusen D D. Org React. 2001;57:417–666. [Google Scholar]
- 15.a) Bon RS, Hong C, Bouma MJ, Schmitz RF, de Kanter FJJ, Lutz M, Spek AL, Orru RVA. Org Lett. 2003;5:3759–3762. doi: 10.1021/ol035521g. [DOI] [PubMed] [Google Scholar]; b) Bon RS, Sprenkels NE, Koningstein MM, Schmitz RF, Kanter FJJ, Dömling A, Groen MB, Orru RVA. Biomol Chem. 2008;6:130–137. doi: 10.1039/b713065a. [DOI] [PubMed] [Google Scholar]
- 16.Passerini M, Bonciani T. Gazz Chim Ital. 1933;63:138–144. [Google Scholar]
- 17.Betti M. Gazz Chim Ital. 1900;30:306–316. [Google Scholar]
- 18.Staudinger H. Liebigs Ann Chem. 1907;356:51–123. [Google Scholar]
- 19.a) Gewald K, Schinke E, Böttcher H. Chem Ber. 1966;99:94–100. [Google Scholar]; b) Huang Y, Dömling A. Mol Divers. 2010 doi: 10.1007/s11030-010-9229-6. [DOI] [PubMed] [Google Scholar]
- 20.Bossio R, Marcaccini S, Pepino R. Tetrahedron Lett. 1995;36:2325–2326. [Google Scholar]
- 21.a) Baell JB, Holloway GA. J Med Chem. 2010 doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]; b) Reed D, Shen Y, Shelat A, Arnold A, Ferreira A, Zhu F, Mills N, Smithson D, Regi C, Bashford D, Cicero S, Schulman B, Jochemsen AG, Guy K, Dyer MA. J Biol Chem. 2010 doi: 10.1074/jbc.M109.056747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]; b) Pellecchia M, Bertini I, Cowburn D, Dalvit C, Giralt E, Jahnke W, James TL, Homans SW, Kessler H, Luchinat C, Meyer B, Oschkinat H, Peng J, Schwalbe H, Siegal G. Nat Rev Drug Disc. 2008;7:738–745. doi: 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Krajewski M, Rothweiler U, D'Silva L, Majumdar S, Klein C, Holak TA. J Med Chem. 2007;50:4382–4387. doi: 10.1021/jm070365v. [DOI] [PubMed] [Google Scholar]; b) Bista M, Kowalska K, Janczyk W, Dömling A, Holak T. J Am Chem Soc. 2009;131:7500–7501. doi: 10.1021/ja901863h. [DOI] [PubMed] [Google Scholar]
- 24.a) Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;6:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]; b) Grassberger BL, et al. J Med Chem. 2005;48:909–912. doi: 10.1021/jm049137g. [DOI] [PubMed] [Google Scholar]
- 25.Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. J Med Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 26.Czarna A, Popowicz G, Pecak A, Wolf S, Dubin G, Holak T. Cell Cycle. 2009;8:1176–1184. doi: 10.4161/cc.8.8.8185. [DOI] [PubMed] [Google Scholar]
- 27.Murray CW, Rees DC. Nat Chem. 2009;1:187–192. doi: 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- 28.a) Fry DC, Emerson SD, Palme S, Vu BT, Liu CM, Podlaski F. J Biomol NMR. 2004;30:163–173. doi: 10.1023/B:JNMR.0000048856.84603.9b. [DOI] [PubMed] [Google Scholar]; b) Wang Z, Malgorzata J, Lambros T, Ferguson S, Goodnow R. J Pharm Biomed Anal. 2007;45:720–729. doi: 10.1016/j.jpba.2007.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.