

Abstract
Cell lines are an important tool in understanding all aspects of cancer growth, development, metastasis, and tumor cell death. There has been a dramatic increase in the number of cell lines and diversity of the cancers they represent; however, misidentification and cross-contamination of cell lines can lead to erroneous conclusions. One method that has gained favor for authenticating cell lines is the use of short tandem repeats (STR) to generate a unique DNA profile. The challenge in validating cell lines is the requirement to compare the large number of existing STR profiles against cell lines of interest, particularly when considering that the profiles of many cell lines have drifted over time and original samples are not available. We report here methods that analyze the variations and the proportional changes extracted from tetra-nucleotide repeat regions in the STR analysis. This technique allows a paired match between a target cell line and a reference database of cell lines to find cell lines that match within a user designated percentage cut-off quality matrix. Our method accounts for DNA instability and can suggest whether the target cell lines are misidentified or unstable.
Keywords: Short Tandem Repeat (STR), Cell Line Validation
Introduction
Cell line cross-contamination and misidentification has been a problem ever since the establishment of the first cell culture lines. Even though many researchers were aware of the problem and sought solutions,1–4 it has not been until the last five years that concerted efforts have been made to require the use of validated cell lines in grants and publications.5, 6 Using misidentified cell lines not only affects researchers who have had to retract papers but also has implications on data generated in the past.7 The use of misidentified cell lines has set back research in Mesenchymal stem cell transplantation, thyroid cancer, leukemia, and esophageal cancers (see websites from American Type Culture Collection (ATCC), German Collection of Microorganisms and Cell Cultures (DMSZ), Japanese Collection of Research Bio-resources (JCRB), and RIKEN8–11). Misidentified cell lines have also had an effect on clinical practice; data from cell lines of the wrong tumor type have been used to justify clinical trials, which then failed to demonstrate benefit in patients. Because of these high-profile failures, many journals now require that a cell line be validated prior to publication.12
Although there are several methods that can be used to authenticate a cell line, the one that is most commonly used for human cell lines is based on short tandem repeat (STR) profiling.13–15 STR repeats are regions of microsatellite instability with defined tri- or tetra-nucleotide repeats that are located across multiple chromosomes. PCR reactions using primers on non-repetitive flanking regions will generate PCR products of different sizes based on the number of repeats in the region; the size of these PCR products are determined by capillary electrophoresis. By combining between 8 and 16 STR loci, such as D5S818, D13S317, D7S820, D16S539, vWA, TH01, TPOX, CSF1PO, it is possible to uniquely identify a sample. This is the same method that is used in forensics to match biological samples. The biggest advantage of using STR to validate cell lines is that it is quick and relatively inexpensive. Much work has been done to determine the characteristics of the STR loci that are currently in use. The loci must be variable enough so that a unique pattern can be discerned but stable enough so that the PCR products generated fall within a size range that can be detected by standard capillary electrophoresis approaches. There are several initiatives that are gathering STR profiles so that researchers can directly compare their STR profiles against reference sequences. 16, 17
However, STR profiling has several limitations. Unless original patient tissue is available there is no absolute way to guarantee that the STR profile generated is from the expected source. This is less of a problem for new cell lines that are being established since patient tissue is often available, but the historical cell line STR profiles must be inferred by comparing cell lines with the same name across researchers and across institutions, using the lowest passages available. The STR profiling method is also not useful for determining inter-species contamination especially if human DNA is present since any small amount of human DNA would be amplified and no non-human DNA would be detected. Another problem is that STR profiles can change depending on the stresses to which a cell line has been subjected. Stresses that can alter an STR profile include passage over time, viral contamination, and exposure to drugs or passage through mice to generate a better mouse model.14 Most of the time these stresses result in loss of heterozygosity or genomic rearrangements and these DNA aberrations can affect the STR profile. Because of these issues, some leeway must be allowed to say that a cell line is of the expected linage. Another issue unique to cancer cell lines is that many cell lines have defects in DNA repair that can cause microsatellite instability. Since STR regions are microsatellite regions, the STR profile in such lines can be unstable. Knowledge of whether the cell line has certain mutations in the DNA repair pathway can help infer instability in the STR profile.
We present here an automated system that can be used to compare a target or list of target STR profiles against an STR database consisting of a variable numbers of reference loci. Our matching algorithm takes into account the variability that can occur within cell lines that are used in cancer research and can suggest whether there are mixtures of cell lines. Since the algorithm is based on the number of repeats in a locus and not the PCR length, we can use our method across different STR platforms as long as the entire STR variable regions are included. The method can accommodate multiple input target STR profiles matched against different reference STR profile collections, thus taking advantage of the work of many companies and institutions that are generating the reference datasets.
Materials and Methods
I. Cell Lines
HCT-116 (NCI-60), IGROV-1, TK-10 and CAKI1 were obtained as part of the NCI-60 cell line collection from the National Cancer Institute. NCI-60 cell lines were grown in RPMI (Cellgrow 10–040-CV) media containing 10% fetal bovine serum (Gibco HiFBS 10438–024 lot 804875).
II. Cell Line authentication
Cell lines were grown to 70–80% confluence in a T-75 flask, trypsinized and the cell pellets were washed once with 1X phosphate buffered saline. DNA was extracted from cell pellets using QiaAMP mini preps (Qiagen cat 51306) and DNA was quantitated by Nanodrop spectrometry. Cell lines were validated by STR DNA fingerprinting using the AmpFℓSTR Identifiler kit according to manufacturer instructions (Applied Biosystems cat 4322288). A higher DNA amount of 0.15 ng DNA was used so that lower level mixing of cell lines could be visible. The samples were run on an Applied Biosystems 96-well 3730 Genetic Analyzer. Data were analyzed using GeneMapper (Applied Biosystems) and exported for STR comparisons. For the purposes of this paper, we did not change any of the automated calls. The STR profiles were compared to known ATCC fingerprints (ATCC.org), to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (http://bioinformatics.istge.it/clima/),18 to the complete 16 loci reference listed in Lorenzi, et al.,19 and to the MD Anderson fingerprint database. The STR profiles matched known DNA fingerprints (see Table 1 and Supplemental Table 1). All cell lines were also tested by Giemsa-banding and silver staining of metaphase chromosomes to verify that the cell lines were not cross-contaminated.
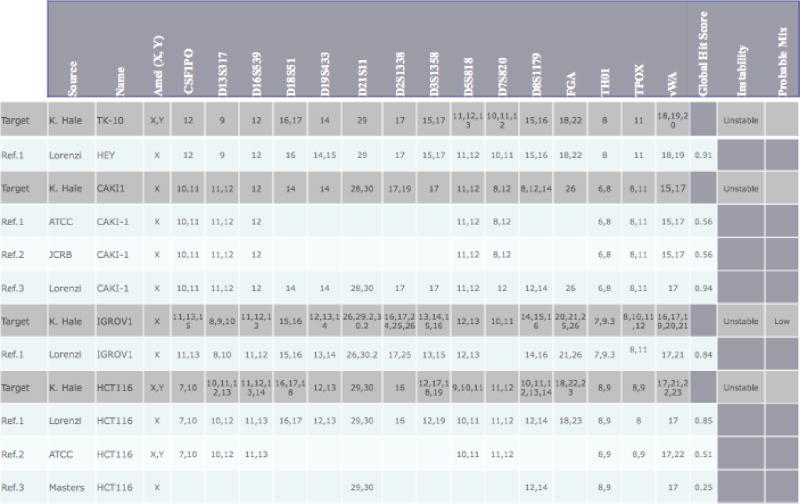
Table 1.
Results of STR profiles generated vs Database of known STR reference profiles
|
Matching algorithm
Our matching algorithm calculates two different scores to determine the percentage identity and instability (or potential cross-contamination) for each target line. These scores are calculated from the number of repeats in each allele at each STR locus. The percentage identity is determined by comparing between the reference sequences and the target sequence, while the degree of instability and potential cross-contamination are intrinsic to the cell line itself and do not depend on any information from an external reference.
I. Identification
A global weighted hit score (η) is calculated for each target-reference pair by comparing all loci in common between the target set A, and the reference set B. This is done by first defining a hit score for each locus, μi The variable μi is calculated for each locus by counting the number of alleles that appear in common between the target aij in set A and reference bij in set B, where we define aij as allele j at locus i, Ai as the set of target alleles at locus i and Bi as the set of reference alleles at locus i. Each loci is corrected to a local weight by dividing by the total distinct number of alleles in both target and reference (m), where aij is given the score 1 if the allele is present in both reference and target and 0 otherwise (eq. 1.1).
| (1.1) |
| (1.2) |
An intermediate hit score, μ, is calculated by summing μi across all loci (see eq. 1.2); if the target and reference are a perfect match and have the same number of loci, μ would equal the total number of loci.
To correct for the discordance between the number of loci in the target and the reference, we define two limits using the intermediate hit score μ. The upper hit score limit ℓ ul is defined as μ divided by the number of loci in the target (Ia,), and the lower hit score limit ℓll is defined as μ divided by the number of loci in the reference (Ib) (see eq. 2.1).
| (2.1) |
To further correct for the missing loci information, we calculate a global hit correction factor μc (eq. 2.2). The global weight, ω, is calculated by counting the number of locus with matched information (i.e. not blank,) and dividing by the total number of locus found with information.
| (2.2) |
Finally, the global hit score η is calculated in eq. 2.3. This final step is used to correct the average for the loci with zero information no = abs (Ia–Ib). This decision was made to correct for samples were the bounds are at wider limits due to missing information; i.e. if only one or two loci have information, the percentage match may be 100%, but the confidence that the target and reference are the same is very low. The comparison between Test HCT-116 and the ATCC HCT-116 samples is illustrated in Supplemental Table 2.
| (2.3) |
In Table 2, the upper hit score in percentage for CAKI1 matching ATCC CAKI1 is 100% because all of the STR regions that are present match at 100% but the global hit score is only 0.5625 since ATCC reports fewer loci than are present in the Test CAKI1 sample. The percentage match between our test sample CAKI1 and the reference from Lorenzi, et al. is only 88.54% due to the overall drifts in loci vWA and D2S1338. Scores for all cell lines are shown in Supplemental Table 1.
Table 2.
A target cell line (CAKI1) with selected matched references
| Name | Source | Influence | Stability | Min. Hit | Global Hit Score | τ | |
|---|---|---|---|---|---|---|---|
| Target | CAKI1_100 | MDACC | (16/16) | Unstable | 0.283 | ||
| Hit Reference | Cdki-1 | ATCC | (9/16) | Stable | 100 | 0.563 | |
| Target | CAKI1_100 | MDACC | (16/16) | Unstable | 0 283 | ||
| Hit Reference | CaMi-1 | JCRB | (9/16) | Stable | 100 | 0.563 | |
| Target | CAKI1_100 | MDACC | (16/16) | Unstable | 0.283 | ||
| Hit Reference | CAKI-1 | NCI-6Q | (16/16) | Stabile | 88.542 | 0.943 | |
| Target | CAKI1_100 | MDACC | (16/16) | Unstable | 0.283 | ||
| Hit Reference | MCAS | JCRB | (9/16) | Stable | 77.778 | 0.500 | |
| Target | CAKI1_100 | MDACC | (16/16) | Unstable | 0.283 | ||
| Hit Reference | Pane 03.27 | ATCC | (9/16) | Stable | 72.222 | 0.484 | |
| Target | CAKI-1_100 | MDACC | (16/16) | Unstable | 0.283 | ||
| Hit Reference | DcGin | JCRB | (9/16) | Stable | 72.222 | 0.484 | |
| Target | HCT-116_100 | MDACC | (16/16) | Unstable | 35 | ||
| Hit Reference | HCT-116 | ATCC | (9/16) | Stable | 81.482 | 0.510 | |
| Target | TK-10_100 | MDACC | (16/16) | Unstable | 0.6 | ||
| Hit Reference | TK-10 | NCI-60 | (16/16) | Stable | 83.333 | 0.917 |
II. Instability
Instability is an intrinsic property of a cell line sample and does not depend on values from any other cell lines. For normal chromosomes, there can exist one (homozygous) or two (heterozygous) alleles per locus. If relative peak height for all alleles is available, then heterozygous calls should be at half the height when compared to single allele locus. Instability is often seen as a halo effect where the major allele has the highest peak and minor alleles including non-integer repeats have lower peak heights (Figure 1, loci D8S1179, D351358, D135317, D165639, vWA, D18s51 and D55818). However, most public references do not have the height information as they only report the number of tri- or tetra- repeats. For this reason, our algorithm does not check for instability until there are three or more alleles per locus. We also cannot rely on relative peak height (halo effect) to determine whether an external reference has locus instability.
Figure 1. STR profiles for representative cell lines.

Shown are the STR profiles generated using the GeneMapper (Applied Biosystems) software using the AmpFℓSTR Identifiler kit. The Y-axis shows relative peak heights for the PCR products that are generated. On the X-axis the PCR length are shown. The grey columns indicate the length of the known PCR products within a locus. (A) STR profile for CAKI-1. (B) STR profile for a mixture of two cell lines, CAKI1 at 95% and TK-10 at 5%. (C) STR profile for HCT-116.
An instability score is calculated for each locus within the STR profile and the overall score is summed over all loci. The formula used in this calculation was obtained from our training set of approximately 1500 STR profiles and tested on 2000 additional samples. The formula is as follows:
| (3) |
Where,
j = the allele number in any one locus.
m = the total number of alleles at that locus.
xj = the number of tetra- repeats for that allele, starting with the second allele length.
For the test HCT-116, τ is calculated as: 1.0 for loci CSF1PO, D13S317 and D16S539, 0.2 for loci D18S51 and D19S433, 0.5 for loci D3S1358, 0.2 for loci D5S818, 0.9 for loci D8S1179, 0.7 for loci FGA and vWA and zero otherwise. Our empirical cutoff for the instability was experimentally determined by looking at a number of known unstable lines; any locus greater than τ> 0.2 are flagged as potentially unstable.
In our set of four test samples, three of the four samples are flagged as potential unstable since three or more loci had an increased τ. Two of these, IGROV1 and HCT-116, are known to have microsatellite instability due to mutations in mismatch repair genes. 20 The TK-10 when run at a higher DNA concentration also shows three loci that have more than two alleles.
III. Cross-Contamination
Any cell line that is a mixture of two cell lines will also appear as unstable. Again, if peak height is available, and if the cell lines are mixed in unequal parts, there will be one set of alleles that have higher peak heights than the other, indicating a mixture (see Figure 1 with CAKI1 at 95% and TK-10 at 5%). However, we cannot rely on two cell lines being present in disequilibrium and we do not always have relative peak heights to compare. Therefore, we flag cell lines with more than three regions of instability as possible mixtures of cell lines. In Supplementary Table 1, we have mixtures of the four cells lines, CAKI1, TK-10, HCT-116 and IGROV1. For our test we set the minimum reported match at 70% so we could display the matches. Even CAKI1 when present at 95% of the sample was flagged when mixed with any of the cell lines.
Results
The STR profile for CAKI1 is shown in Figure 1. The 16 loci STR allele fragment patterns for TK-10, CAKI1, HCT-116 and IGROV1 are listed in Table 1. For CAKI1, reference STR from three sources were available: ATCC and JCRB which report the regions D5S818, D13S317, D7S820, D16S539, vWA, TH01, TPOX, CSF1PO plus gender amelogenin, and the work of Lorenzi et al.19 that use the same Identifiler assay set as in our study. Even from this relatively stable cell line, the STR profile differs slightly between the four samples. Lorenzi et al. shows that one locus, vWA, is missing one allele (15,17 for ATCC, JCRB and our test sample vs. 17 in Lorenzi et al.) when compared to the published data. Another locus, D2S1338, is missing an allele when compared to our data; however, our STR profile agrees with the STR profile on both the ATCC and JCRB websites. This discrepancy can either be due to a technical failure of the PCR reaction in Lorenzi et al. where one allele was preferentially amplified and the other alleles were below the level of detection, or due to a biological change where the NCI-60 cells from Lorenzi et al. have lost an allele. Since we do not have access to the original sequencing traces or the original DNA from Lorenzi et al., we cannot determine whether there was a technical failure or a real biological change. Overall, all four cell lines matched and should be considered CAKI1.
As shown in Table 1, both HCT-116 and IGROV1 have multiple alleles per loci and there is a great deal of differences between the different sources. Both cell lines are known to have microsatellite instability and this alone can generate different profiles. Another factor that can cause fluctuations in the STR profile is chromosome rearrangements or loss of chromosomes. This is one of the reasons HCT-116 is described in different databases as having the AMEL locus as either X or XY (See Table 1 and see websites from ATCC and DMSZ). As can be seen in Figure 1, the Y peak is very close to background levels. If lower amounts of DNA are use so as to make the spectra appear to have fewer alleles, the Y band will fall below the level of detection. The G-band karyotype shows that not all cells contain the Y chromosome; different sources have reported from 16–50% of cells without a Y chromosome.21
Discussion
In order to ensure that research conducted with human cell lines is of the highest caliber, cell lines must be validated. While the tools for validation are inexpensive, the task of comparing a test set of STR profiles for several cell lines against the large number of existing cell line STR profiles is very time consuming since current methods can only compare one cell line at a time and in most cases manual review is required. In addition, as is seen with the unstable cell lines IGROV1 and HCT-116 in this paper, the reference profiles do not always match, especially when the DNA concentration is increased in the analysis to allow detection of mixtures of cell lines.
Implementation of the algorithm in this paper will allow researchers to set up automated comparisons of target STR profiles with current reference STR profiles that can be downloaded from several sources. The current requirement that a cell line must match by at least 80% may not be achievable for cell lines with mismatch repair defects as can be seen with IGROV1 in our test samples. In these cases, our algorithm allows the user to lower the stringency threshold of reported matches so that the best hit will be displayed allowing manual comparison. Since no one technique can answer all questions, if researchers are unsure whether a cell line is unstable or a mixture of clones, there are several other methods that can be used in addition to STR profiling to ascertain whether the cell line is a mixture of more than one cell line. A simple method to verify whether a cell line is mixed is to select for single clones and re-test the STR profile. Alternate approaches used in this study were Giemsa-banding and silver staining of metaphase chromosomes in addition to STR profiling to validate the unstable cell lines IGROV1 and HCT-116.
Although the purpose of this paper is to provide the scientific community with a tool to validate cell lines, it does highlight some of the problems when using unstable cell lines. In highly unstable cell lines like HCT-116, the major allele can drift even over the course of a week which might account for different biological data generated in different laboratories.22, 23 Even if misidentification of a cell line is ruled out, there remains the possibility that mutations or rearrangements may alter the phenotype of the cell line between cell passages. As more cell lines become available and in order to enhance consistency in comparing data between laboratories, it may be that researchers also “drift” away from unstable cell lines unless the observation under investigation is genomic instability in cancer.
Supplementary Material
Supplementary Table 1: Complete data and scores for all samples. Shown are mixtures of stable cell lines with stable cell lines, and unstable cell lines with unstable cell lines.
Supplemental Table 2: Example calculation of test HCT-116 and ATCC HCT-116. The value for μ is calculated as 7.16. The upper hit score limit (ℓul) and lower hit score limit (ℓll) for the target/reference pair are calculated as: 7.16/9 = 0.7955 (79.55%) and 7.16/16 = 0.4475 (44.75%). Since the test (i.e. target) has 16 loci and the ATCC (i.e. reference) sample has 9 loci, the valueω is calculated as 9/16 = 0.5625 and μc is 7.16* 9/16 = 4.0275. The upper and lower hit scores are further adjusted by subtracting μc from both limits (ℓul × 100–μc = 79.6 – 4.03=75.57, and ℓll × 100–μc = 44.75 – 4.03 = 40.72). The average of these adjusted limits is 58.2 and the number of alleles that were missing information is subtracted (58.2 – 7=51.2) for a final global hit score of 0.512.
Novelty and Impact.
Validation of cell lines is now required for many journals, including the International Journal of Cancer. The most common method used for human cell line identification is short tandem repeat profiling (STR). The work in this paper presents a novel automated detection algorithm that can match target STR profiles against multiple reference STR databases and that takes into account cell line instability and potential cross-contamination.
Acknowledgments
The research in this grant was supported by the Kleberg Center for Molecular Markers. STR DNA fingerprinting and Giemsa-banding were done by the Cancer Center Support grant funded Characterized Cell Line core and the Molecular Cytogenetics Facility, NCI # CA16672. NE, XL, KBR, GBM and KSH were partially supported by the Kleberg Center for Molecular Markers and NE, XL, VG and KSH were partly supported by the Characterized Cell Line core, NCI #CA16672.
Footnotes
GBM has potential conflict of interests based on his consulting work for Asuragen, Aushon, Catena, Diichi Pharmaceuticals, Foundation Medicine and The Komen Foundation, his stock ownership in Catena and PTV Ventures and his sponsored research with AstraZeneca, Celgene, CeMines, Exelixis, GlaxoSmithKline, LPath, Roche, and Wyeth/Pfizer. The algorithm will be available for download and testing for a simple comparison on our website.
References
- 1.Nelson-Rees WA, Flandermeyer R. Inter-and intraspecies contamination of human breast tumor cell lines HBC and BrCa5 and other cell cultures. Science. 1977;195:1343–44. doi: 10.1126/science.557237. [DOI] [PubMed] [Google Scholar]
- 2.Bubenik J. Cross-contamination of cell lines in culture. Folia Biologica. 2000;46:163–4. [PubMed] [Google Scholar]
- 3.Drexler HG, Dirks WG, MacLeod RA. False human hematopoietic cell lines: cross-contaminations and misinterpretations. Leukemia. 1999;13:1601–7. doi: 10.1038/sj.leu.2401510. [DOI] [PubMed] [Google Scholar]
- 4.MacLeod RA, Dirks WG, Matsuo Y, Kaufmann M, Milch H, Drexler HG. Widespread intraspecies cross-contamination of human tumor cell lines arising at source. Int J Cancer. 1999;83:555–63. doi: 10.1002/(sici)1097-0215(19991112)83:4<555::aid-ijc19>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 5.Nardone RM. Eradication of cross-contaminated cell lines: a call for action. Cell Biology & Toxicology. 2007;23:367–72. doi: 10.1007/s10565-007-9019-9. [DOI] [PubMed] [Google Scholar]
- 6.Lacroix M. Persistent use of “false” cell lines. International Journal of Cancer. 2008;122:1–4. doi: 10.1002/ijc.23233. [DOI] [PubMed] [Google Scholar]
- 7.Lucey DJ, Walsh MA, Costello R. Impostor cell lines. Laryngoscope. 2006;116:161–2. doi: 10.1097/01.mlg.0000191475.23030.78. [DOI] [PubMed] [Google Scholar]
- 8.Boonstra JJ, van Marion R, Beer DG, Lin L, Chaves P, Ribeiro C, Pereira AD, Roque L, Darnton SJ, Altorki NK, Schrump DS, Klimstra DS, et al. Verification and unmasking of widely used human esophageal adenocarcinoma cell lines. J National Cancer Institute. 2010;102:271–4. doi: 10.1093/jnci/djp499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogel G. Cell biology. To scientists’ dismay, mixed-up cell lines strike again. Science. 2010;329:1004. doi: 10.1126/science.329.5995.1004. [DOI] [PubMed] [Google Scholar]
- 10.Drexler HG, Dirks W, Matsuo Y, MacLeod RA. False Leukemia-lymphoma cell lines: an update on over 500 cell lines. Leukemia & Lymphoma. 2003;17:416–26. doi: 10.1038/sj.leu.2402799. [DOI] [PubMed] [Google Scholar]
- 11.MacLeod RAF, Dirks WG, Drexler HG. One falsehood leads easily to another. Int J Cancer. 2008;122:2165–8. doi: 10.1002/ijc.23327. [DOI] [PubMed] [Google Scholar]
- 12.Identity crisis. Nature. 2009;457:935–6. doi: 10.1038/457935b. [DOI] [PubMed] [Google Scholar]
- 13.Masters JR, Thomson JA, Daly-Burns B, Reid YA, Dirks WG, Packer P, Toji LH, Ohno T, Tanabe H, Arlett CF, Kelland LR, Harrison M, et al. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc Natl Acad Sci USA. 2001;98:8012–7. doi: 10.1073/pnas.121616198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parson W, Kirchebner R, Muhlmann R, Renner K, Kofler A, Schmidt S, Kofler R. Cancer cell line identification by short tandem repeat profiling: power and limitations. FASEB J. 2005;19:434–6. doi: 10.1096/fj.04-3062fje. [DOI] [PubMed] [Google Scholar]
- 15.Barallon R, Bauer SR, Butler J, Capes-Davis A, Dirks WG, Elmore E, Furtado M, Kline MC, Kohara A, Los GV, MacLeod RAF, Masters JRW, et al. Recommendation of short tandem repeat profiling for authenticating human cell lines, stem cells, and tissues. In Vitro Cell & Dev Biol 2010; Animal. 46:727–32. doi: 10.1007/s11626-010-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dirks WG, MacLeod RAF, Nakamura Y, Kohara A, Reid Y, Milch H, Drexler HG, Mizusawa H. Cell line cross-contamination initiative: an interactive reference database of STR profiles covering common cancer cell lines. INT J Cancer. 2010;126:303–4. doi: 10.1002/ijc.24999. [DOI] [PubMed] [Google Scholar]
- 17.Dirks WG, Drexler HG. Online verification of human cell line identity by STR DNA typing. Methods in Mol Biol. 2011;731:45–55. doi: 10.1007/978-1-61779-080-5_5. [DOI] [PubMed] [Google Scholar]
- 18.Romano P, Manniello A, Aresu O, Armento M, Cesaro M, Parodi B. Cell Line Data Base: structure and recent improvements towards molecular authentication of human cell lines. Nucleic Acids Research. 2009;37:D925–32. doi: 10.1093/nar/gkn730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorenzi PL, Reinhold WC, Varma S, Hutchinson AA, Pommier Y, Chanock SJ, Weinstein JN. DNA fingerprinting of the NCI-60 cell line panel. Mol Cancer Ther. 2009;8:713–24. doi: 10.1158/1535-7163.MCT-08-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taverna P, Liu L, Hanson AJ, Monks A, Gerson SL. Characterization of MLH1 and MSH2 DNA mismatch repair proteins in cell lines of the NCI anticancer drug screen. Cancer Chemotherapy & Pharm. 2000;46:507–16. doi: 10.1007/s002800000186. [DOI] [PubMed] [Google Scholar]
- 21.Masramona L, Ribasb M, Cifuentesb P, Arribasa R, Garcia F, Egozcueb J, Peinadoa MA, Miro R. Cytogenetic characterization of two colon cell lines by using conventional G-banding, comparative genomic hybridization, and whole chromosome painting. Cancer Genetics and Cytogenetics. 2000;121:17–21. doi: 10.1016/s0165-4608(00)00219-3. [DOI] [PubMed] [Google Scholar]
- 22.Shureiqi I, Wu Y, Chen D, Yang XL, Guan B, Morris JS, Yang P, Newman RA, Broaddus R, Hamilton SR. The critical role of 15-lipoxygenase-1 in colorectal epithelial cell terminal differentiation and tumorigenesis. Cancer Res. 2005;65:11486. doi: 10.1158/0008-5472.CAN-05-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narayan S. Curcumin, a multi-functional chemopreventive agent, blocks growth of colon cancer cells by targeting -catenin-mediated transactivation and cell-cell adhesion pathways. J Mol Histology. 2004;35:301–07. doi: 10.1023/b:hijo.0000032361.98815.bb. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Complete data and scores for all samples. Shown are mixtures of stable cell lines with stable cell lines, and unstable cell lines with unstable cell lines.
Supplemental Table 2: Example calculation of test HCT-116 and ATCC HCT-116. The value for μ is calculated as 7.16. The upper hit score limit (ℓul) and lower hit score limit (ℓll) for the target/reference pair are calculated as: 7.16/9 = 0.7955 (79.55%) and 7.16/16 = 0.4475 (44.75%). Since the test (i.e. target) has 16 loci and the ATCC (i.e. reference) sample has 9 loci, the valueω is calculated as 9/16 = 0.5625 and μc is 7.16* 9/16 = 4.0275. The upper and lower hit scores are further adjusted by subtracting μc from both limits (ℓul × 100–μc = 79.6 – 4.03=75.57, and ℓll × 100–μc = 44.75 – 4.03 = 40.72). The average of these adjusted limits is 58.2 and the number of alleles that were missing information is subtracted (58.2 – 7=51.2) for a final global hit score of 0.512.