

Abstract
Past work demonstrated that late‐life depression is associated with greater severity of ischemic cerebral hyperintense white matter lesions, particularly frontal lesions. However, these lesions are also associated with other neuropsychiatric deficits, so these clinical relationships may depend on which fiber tracts are damaged. We examined the ratio of lesion to nonlesioned white matter tissue within multiple fiber tracts between depressed and nondepressed elders. We also sought to determine if the AGTR1 A1166C and BDNF Val66Met polymorphisms contributed to vulnerability to lesion development in discrete tracts. The 3T structural MR images and blood samples for genetic analyses were acquired on 54 depressed and 37 nondepressed elders. Lesion maps were created through an automated tissue segmentation process and applied to a probabilistic white matter fiber tract atlas allowing for identification of the fraction of the tract occupied by lesion. The depressed cohort exhibited a significantly greater lesion ratio only in the left upper cingulum near the cingulate gyrus (F (1,86) = 4.62, P = 0.0344), supporting past work implicating cingulate dysfunction in the pathogenesis of depression. In the 62 Caucasian subjects with genetic data, AGTR1 C1166 carriers exhibited greater lesion ratios across multiple tracts including the anterior thalamic radiation and inferior fronto‐occipital fasciculus. In contrast, BDNF Met allele carriers exhibited greater lesion ratios only in the frontal corpus callosum. Although these findings did not survive correction for multiple comparisons, this study supports our hypothesis and provides preliminary evidence that genetic differences related to vascular disease may increase lesion vulnerability differentially across fiber tracts. Hum Brain Mapp, 2013. © 2011 Wiley Periodicals, Inc.
Keywords: depression, geriatrics, MRI, hyperintensities, fiber tracts, genetics, single nucleotide polymorphisms, brain‐derived neurotrophic factor, angiotensin receptor
INTRODUCTION
Cerebral white matter lesions (WMLs) are hyperintense, bright regions observed on T2‐weighted and FLAIR magnetic resonance imaging (MRI) brain scans that are strongly associated with increased age [Awad et al., 1986]. They are also associated with cerebrovascular risk factors including diabetes, cardiac disease, obesity, smoking, and hypertension [Bokura et al., 2008; Dufouil et al., 2001; Jokinen et al., 2009; Longstreth et al., 1996; Taylor et al., 2003a, 2005] and neuropathological studies demonstrate that many hyperintensities are ischemic in origin [Thomas et al., 2002a, b]. Depressed elders exhibit greater WML volumes than age‐matched comparison subjects [Herrmann et al., 2008; Taylor et al., 2005], and while worsening WML disease is associated with poorer course of depression [Taylor et al., 2003c], there remains substantial heterogeneity between these radiological findings and the clinical correlates. Beyond mood disorders, WMLs are additionally associated with cognitive decline [De Groot et al., 2002; Kramer et al., 2002, 2007] and gait disturbances [Blahak et al., 2009; Srikanth et al., 2009], but there are also many individuals with widespread WML disease observed on neuroimaging who do not exhibit clinically significant neurologic or psychiatric disturbances.
One explanation for this observed heterogeneity is that the clinical presentation depends upon the location of the WMLs, specifically what fiber tracts—and, by extension, what neural circuits—are affected. Most studies have found that WMLs occurring in the frontal white matter are most strongly associated with late‐life depression (LLD) [Firbank et al., 2004; MacFall et al., 2005; O'Brien et al., 2006; Taylor et al., 2003b]. This conclusion is supported by studies utilizing diffusion tensor imaging (DTI) or magnetization transfer ratio (MTR) imaging which associate LLD with altered structural integrity of frontal white matter [Bae et al., 2006; Gunning‐Dixon et al., 2008; Taylor et al., 2004]. In an attempt to further localize WML location, Sheline et al. found that individuals with LLD are more likely to exhibit WMLs in specific white matter tracts including the superior longitudinal fasciculus, the inferior longitudinal fasciculus, the inferior fronto‐occipital fasciculus, and the uncinate fasciculus [Sheline et al., 2008]. This work builds on previous reports associating depression with increased likelihood of ischemic lesions in the dorsolateral prefrontal cortex [Thomas et al., 2002a], where some components of the superior longitudinal fasciculus terminate. It also extends that work to frontotemporal tracts, which have previously been implicated in LLD [Taylor et al., 2007b]. Importantly, this work built on the hypothesis that ischemic injury to specific tracts may contribute to the development of LLD, presumptively by affecting function of these networks.
In a parallel series of investigations, there has been substantial work examining associations between genetic variation and WML severity. Such work has examined a wide range of candidates, including genes implicated in ischemic stroke, dementia, cardiac disease, and hypertension [Paternoster et al., 2009]. Despite heritability of WML volume being estimated between 55% and 80% [Atwood et al., 2004; Carmelli et al., 1998; Turner et al., 2004], these candidate gene studies and genome‐wide association studies have had mixed results that are difficult to interpret. There may be many reasons for this discrepancy, including insufficient sample sizes or epistatic effects [Paternoster et al., 2009]. However, if genetic differences contribute to regional rather than global vulnerability to WML development, and if different genetic influences increase vulnerability to ischemia in different brain regions, it is possible that the phenotype of examining whole‐brain WML volume may be too broad.
In this study we hypothesized that in LLD subjects, fiber tracts involved in mood regulation would have a greater proportion of their volume occupied by WMLs, defined as an increased fiber tract‐white matter lesion (ftWML) ratio. We further hypothesized the AGTR1 A1166C and BDNF Val66Met polymorphisms, which have previously been associated with greater cerebral WML severity [Taylor et al., 2008, 2010], may have differential regional influences on WML development. We first examined if a diagnosis of Major Depressive Disorder in this elderly cohort was associated with greater ftWML ratios in discrete frontal tracts. In subsequent exploratory analyses, we then examined if the AGTR1 and BDNF polymorphisms were associated with greater tract‐specific ftWML ratios.
METHODS
Depressed participants were age 60 or over and were enrolled in the National Institute of Mental Health‐sponsored Conte Center for the Neuroscience of Depression in Late Life at Duke University Medical Center (DUMC). Subjects were subsequently enrolled in the Neurocognitive Outcomes of Depression (NCODE) study, where they received antidepressant treatment. At time of enrollment, all depressed participants met DSM‐IV criteria for Major Depressive Disorder as diagnosed using the Diagnostic Interview Schedule [Robins et al., 1981] and confirmed via a clinical evaluation by a geriatric psychiatrist. Exclusion criteria for study entry included: (1) another major psychiatric illness, including bipolar disorder, schizophrenia, or dementia; (2) history of alcohol or drug abuse or dependence; (3) primary neurologic illness, including stroke or dementia; (4) medical illness, medication use, or disability that would prevent study participation; (5) metal in the body precluding magnetic resonance imaging (MRI) and (6) Mini Mental State Exam score less than 25. Depressed participants were primarily recruited from clinical referrals and limited advertisements.
Nondepressed participants were community‐dwelling Conte Center participants recruited primarily through advertisements and from the Aging Center Subject Registry at Duke University. Eligible participants met similar entry criteria, but additionally had no evidence of a current or past psychiatric disorder based on the Diagnostic Interview Schedule.
Assessment of hypertension consisted of self‐report and followed a format used in the NIMH Epidemiological Catchment Area Study [Regier et al., 1984]. The study protocol was approved by the Duke University Medical Center Institutional Review Board. All participants provided written informed consent before beginning study procedures.
Antidepressant Treatment
Although depressed participants were required to meet DMS‐IV criteria for MDD at study entry, not all subjects continued to be depressed at time of imaging. All depressed participants received antidepressant treatment through the Duke STAGED approach [Steffens et al., 2002]. This algorithm mimics “real world” treatment options, accounting for past treatments and current depression severity rather than adhering to a rigid clinical trial design. All marketed antidepressant treatments are allowed and most participants start on a selective serotonin reuptake inhibitor. Although electroconvulsive therapy is allowed, none of the subjects in the current study had received ECT prior to imaging.
MRI Acquisition
Cranial MRI was performed using the eight‐channel parallel imaging head coil on a 3 T whole‐body MRI system (Trio, Siemens Medical Systems, Malvern, PA). Proton density (PD), T1‐weighted, T2‐weighted, and fluid‐attenuated inversion recovery (FLAIR) images were acquired. Parallel imaging was employed with an acceleration factor of 2. All pulse sequences used a square field‐of‐view of 256 mm and Nex = 1. The T1‐weighted image set was acquired using a three‐dimensional axial TURBOFLASH sequence with TR/TE = 22/7 msec, flip angle = 25°, a 100 Hz/pixel bandwidth, a 256 × 256 matrix, 160 slices with a 1‐mm slice thickness, yielding an image with 1 mm cubic voxels. The T2‐weighted acquisition used a two‐dimensional turbo spin‐echo pulse sequence with TR/TE = 7,580/86 msec, turbo factor = 7, a 210 Hz/pixel bandwidth, a 256 × 256 matrix, 100 slices with a 1.5 mm slice thickness, yielding a 1 × 1 × 1.5 mm voxel. The PD weighted acquisition used a two‐dimensional turbo spin‐echo pulse sequence with TR/TE = 7,580/17 msec, turbo factor = 3, a 210 Hz/pixel bandwidth, a 256 × 256 matrix, 100 slices with a 1.5 mm slice thickness, yielding a 1 × 1 × 1.5 mm voxel. Finally, the FLAIR image was acquired with TR/TI/TE = 9,000/2,400/101 msec, a 210 Hz/pixel bandwidth, a turbo factor of 11, a 256 × 256 matrix, 75 slices with a 2 mm slice thickness, yielding a 1 × 1 × 2 mm voxel.
MRI Processing: Whole Brain Segmentation
The MR images were transferred to the Duke Neuropsychiatric Imaging Research Laboratory (NIRL) where images were resliced to a common geometry of 1 × 1 × 1.5 mm voxels and all analyses performed. An automated four‐channel tissue and lesion segmentation, utilizing FLAIR images for WML detection, was performed to assess gray matter, white matter, cerebrospinal fluid, and WMLs. The algorithm used was a variation on the fully automated Expectation Maximization Segmentation (EMS) method [Van Leemput et al., 1999, 2001, 2003] optimized for WML assessment [Chang et al., 2011]. The software assigns a probability that each pixel should be classified as gray matter, white matter, cerebrospinal fluid, lesion, or nonbrain in the following manner. First, images are aligned to an atlas of tissue probability images using the mutual information registration tool (MIRIT) [Maes et al., 1997]. The probability atlas provides spatial priors for each tissue that are used to initialize the tissue intensity histograms for the segmentation algorithm. The tissue probabilities are then derived in an iterative process using the intensity distributions of the different tissues for each of the input image contrasts. The process also evaluates and compensates for spatial distributions of intensity that could be due to various magnetic resonance imaging artifacts such as radiofrequency inhomogeneity (bias correction). Lesions are detected as “outliers” to the normal tissue distributions.
MRI Processing: Fiber Tract Identification
White matter fiber tracts were identified with a probabilistic white matter tract atlas including 20 fiber tracts (Table I; all are present bilaterally except for CCF and CCO) developed from the manually traced maps of 27 neurologically normal subjects (atlas provided by S. Mori and the Johns Hopkins Medical Institute Laboratory of Brain Anatomical MRI). The reproducibility of such tracing techniques has been evaluated and shown to have main variability from biological differences between subjects, with interrater reliability contributing only minor variance [Wakana et al., 2007]. Thus the probabilistic atlas approach reflects the biological variability of the course of white matter fiber tracts.
Table I.
White Matter Fiber Tract Abbreviations
| Anterior thalamic radiation | ATR |
| Cingulum, cingulate gyrus region | CgC |
| Cingulum, hippocampus region | CgH |
| Corpus Callosum, frontal (forceps minor) | CCF |
| Corpus Callosum, occipital (forceps major) | CCO |
| Corticospinal tract | CST |
| Inferior fronto‐occipital fasciculus | IFO |
| Inferior longitudinal fasciculus | ILF |
| Superior longitudinal fasciculus, body | SLFb |
| Superior longitudinal fasciculus, temporal | SLFt |
| Uncinate fasciculus | UNC |
The atlas was constructed using the T1‐weighted images associated with the manually traced fiber tracts for each of the 27 individuals which were transformed to align with a template image (Wrap 5 Atlas of ICBM452 T1 atlas (LONI, ICBM 452 T1 Atlas; available at: http://www.loni.ucla.edu/ICBM/Downloads/Downloads_452T1.shtml.), using an initial 12th‐order affine transformation followed by a high‐order nonrigid registration algorithm [Denton et al., 1999; Rueckert et al., 1999; Schnabel et al., 2001] which uses a b‐spline based transformation based on normalized mutual information. The same transformation was then applied to the tract map to align with the template image. For each fiber tract, in each pixel, the fraction of the maps of all subjects that had the tract pass through that pixel was then interpreted as the probability that the tract passed through the pixel in the atlas space. The lesion maps of our population were aligned with the template in a similar fashion using a nonlinear transformation derived using the T1‐weighted image and applying it to the lesion map for that subject. After this procedure, each subject's T1‐weighted MRI scan and lesion map were aligned with the probability maps of fiber tracts in the atlas space.
All images were visually examined for quality of alignment to the template. Due to use of the high order transformation, only 5% of the images processed were considered to have unsatisfactory alignment. Those five scans with unsatisfactory alignment were not included in study analyses. There were no scans exhibiting severe encephalomalacia requiring removal of individual tract data or entire scans from the dataset, although mild or moderate diffuse hyperintense lesions intersecting tracts were allowed given the study hypotheses.
The fraction of lesion in each tract, F, was calculated as:
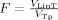 , where
, where
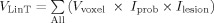 and
and
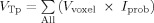 ,
,
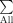 means to sum up over all voxels in the space of the template image; V
voxel is the volume of a single voxel; I
prob is the probability value of the voxel in the probability map of the given tract; I
lesion is the binary “intensity value” of the voxel in the lesion map of the given subject, where 1 means a lesion is present in that voxel and 0 means a lesion is not present. F is an extension from the standard idea of an overlap integral between two binary maps to the case of overlap between a probability map and a binary map. This metric F, the fraction of lesion in the tract, illustrates the proportion of voxels within a tract consisting of lesions, among voxels projected as being within that fiber tract modulated by the probability of the voxel being in the tract. This was used as our primary measure, defined as the fiber tract‐white matter lesion (ftWML) ratio.
means to sum up over all voxels in the space of the template image; V
voxel is the volume of a single voxel; I
prob is the probability value of the voxel in the probability map of the given tract; I
lesion is the binary “intensity value” of the voxel in the lesion map of the given subject, where 1 means a lesion is present in that voxel and 0 means a lesion is not present. F is an extension from the standard idea of an overlap integral between two binary maps to the case of overlap between a probability map and a binary map. This metric F, the fraction of lesion in the tract, illustrates the proportion of voxels within a tract consisting of lesions, among voxels projected as being within that fiber tract modulated by the probability of the voxel being in the tract. This was used as our primary measure, defined as the fiber tract‐white matter lesion (ftWML) ratio.
Genetic Analyses
As previously described [Taylor et al., 2010], single nucleotide polymorphism (SNP) genotyping was performed by TaqMan, using “Assays‐on‐Demand” SNP genotyping products (Applied Biosystems, Foster City, CA). For all assays, quality control measures were applied, including genotyping a series of blinded duplicate samples and Centre d'Etude du Polymorphism Humain (CEPH) controls. The genotypes of all duplicate samples had to match 100% in order for the assay to pass quality control. Further, we required that each assay achieve 95% efficiency (the genotypes of at least 95% of the samples could be called with certainty) before statistical analysis. PCR was performed on the ABI 9700 dual 384‐well Geneamp PCR system and genotypes analyzed using an ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). Each reaction contained 2.7 ng of total genomic DNA which had been extracted from whole blood using the Pure Gene Method by Gentra.
Statistical Analyses
All analyses were conducted using SAS 9.2 (Cary, NC). Based on our prior approaches and frequency of the less common alleles [Taylor et al., 2007c, 2010], subjects were categorized as being BDNF Val66 homozygous or Met66 carriers, and AGTR1 A1166 homozygous or C1166 carriers. Initial comparisons between diagnostic cohorts used χ2 tests for categorical variables and pooled, two‐tailed t‐tests for continuous variables.
Initial general linear models including all subjects with available data were created examining ftWML ratio as the dependent variable. These models examined differences between diagnostic cohorts, while controlling for age, sex, race, and presence or absence of hypertension, but to maximize sample size, did not include genetic polymorphism data. Subsequent models were limited to those Caucasian subjects with genetic data; models again examined ftWML ratio as the dependent variable, while including the independent variables of diagnostic cohort, AGTR1 genotype, BDNF genotype, age, sex, and hypertension. Finally, these same models were again examined after including an AGTR1‐BDNF interaction term. We report uncorrected P values from these models for this report. A conservative Bonferroni correction for the 20 fiber tracts assessed across both hemispheres would have resulted in a nominal P value of 0.0025 to achieve statistical significance.
RESULTS
This study examined 54 depressed and 37 nondepressed subjects who completed clinical assessments and 3T MRI, and whose scans exhibited satisfactory alignment to the atlas‐based template image. The only difference in demographics between cohorts was in age (Table II), where the nondepressed participants were significantly older. Univariate analyses of total cerebral WML volume showed no significant difference between depressed and nondepressed subjects. Of note, the depressed cohort included individuals who were currently symptomatic as well as individuals remitted at the time of MRI. Using a MADRS <8 as a definition of remission, 21 depressed subjects were remitted, and 33 were nonremitted.
Table II.
Demographic Differences Between Diagnostic Cohorts
| Depressed (N = 54) | Nondepressed (N = 37) | Test value | P | |
|---|---|---|---|---|
| Age (yr) | 68.9 (5.6) | 73.8 (5.8) | t = 4.25 | 0.0001 |
| Sex, % Female (N) | 64.8 (35) | 64.9 (24) | χ2 = 0.00 | 0.9961 |
| Race, % Caucasian (N) | 83.3 (45) | 94.6 (35) | Fisher's exact | 0.1888 |
| MMSE | 28.9 (1.3) | 29.2 (0.9) | t = 0.89 | 0.3785 |
| MADRS | 10.1 (7.8) | — | — | — |
| Cerebral WML volume (ml) | 8.7 (9.3) | 8.8 (12.7) | t = 0.06 | 0.9497 |
| Total cerebral volume (ml) | 1,238.1 (145.6) | 1,259.5 (126.1) | t = 0.73 | 0.4690 |
| Cerebral WML ratio | 0.0070 (0.0072) | 0.0068 (0.0090) | t = 0.10 | 0.9200 |
| Genetic cohort | N = 29 | N = 33 | ||
| AGTR1 genotype | ||||
| A/A | 58.6% (17) | 57.6% (19) | χ2 = 0.01 | 0.9337 |
| C allele carrier | 41.4% (12) | 42.4% (14) | ||
| BDNF genotype | ||||
| Val/Val | 58.6% (17) | 72.7% (24) | χ2 = 1.37 | 0.2416 |
| Met allele carrier | 41.4% (12) | 27.3% (9) | ||
All continuous variables presented as mean (standard deviation) and were compared using pooled, two‐tailed t‐tests with 89 degrees of freedom. Categorical variables presented as percentage (N) and compared using a χ2 test (for sex, AGTR1, and BDNF) or Fisher's exact test (for race), with 1 degree of freedom.
MADRS = Montgomery Asberg Depression Rating Scale; MMSE = Mini‐mental state exam; WML = white matter lesion.
Fiber Tract WML Volume Differences Between Diagnostic Cohorts
We tested for differences in ftWML ratio between depressed and nondepressed cohorts after controlling for age, presence of hypertension, and racial background (Table III). There was a significant difference between cohorts in the ftWML measure in the left CgC, where the depressed cohort exhibited a significantly higher mean ftWML ratio than did the nondepressed cohort.
Table III.
Cohort Differences in Individual Fiber Tract WML (ftWML) Ratios
| Cohort | ATR, L | ATR, R | CCF | CCO | CgH, L | CgH, R | CgC, L | CgC, R | CST, L | CST, R |
|---|---|---|---|---|---|---|---|---|---|---|
| Nondepressed (N = 37) | 0.0107 (0.0147) | 0.0136 (0.0211) | 0.0036 (0.0057) | 0.0112 (0.0162) | 0.0008 (0.0014) | 0.0037 (0.0050) | 0.0003 (0.0006) | 0.0011 (0.0006) | 0.0063 (0.0.0137) | 0.0091 (0.0160) |
| Depressed (N = 54) | 0.0127 (0.0204) | 0.0132 (0.0206) | 0.0049 (0.0094) | 0.0148 (0.0171) | 0.0007 (0.0012) | 0.0048 (0.0057) | 0.0012 (0.0038) | 0.0013 (0.0031) | 0.0063 (0.0194) | 0.0086 (0.0189) |
| F (1,86) value | 1.44 | 0.80 | 1.23 | 3.08 | 0.10 | 0.82 | 4.62 | 1.03 | 0.20 | 0.83 |
| P | 0.2339 | 0.3721 | 0.2713 | 0.0829 | 0.7565 | 0.3683 | 0.0344 | 0.3138 | 0.6598 | 0.3657 |
| IFO, L | IFO, R | ILF, L | ILF, R | SLFb, L | SLFb, R | SLFt, L | SLFt, R | UNC, L | UNC, R | |
| Nondepressed (N = 37) | 0.0060 (0.0100) | 0.0093 (0.0143) | 0.0064 (0.0117) | 0.0067 (0.0086) | 0.0051 (0.0171) | 0.0060 (0.0171) | 0.0054 (0.0188) | 0.0057 (0.0214) | 0.0027 (0.0062) | 0.0052 (0.0053) |
| Depressed (N = 54) | 0.0078 (0.0108) | 0.0119 (0.0167) | 0.0093 (0.0131) | 0.0085 (0.0092) | 0.0056 (0.0148) | 0.0065 (0.0167) | 0.0057 (0.0164) | 0.0059 (0.0173) | 0.0019 (0.0032) | 0.0047 (0.0072) |
| F (1,86) value | 2.35 | 1.86 | 3.02 | 0.83 | 0.35 | 0.63 | 0.21 | 0.12 | 0.03 | 0.56 |
| P | 0.1288 | 0.1757 | 0.0859 | 0.3639 | 0.5540 | 0.4308 | 0.6446 | 0.7252 | 0.8572 | 0.4552 |
Data presented as mean ratio (standard deviation). Statistical test results derive from models predicting ftWML ratio, controlling for diagnostic cohort, age, sex, presence of hypertension, and race. See Table I for key to tract abbreviations.
Influence of Genetic Polymorphisms on Fiber Tract WML Volumes
For the genetic analyses, we restricted the sample to Caucasian participants, as both the AGTR1 A1166C polymorphism and BDNF Val66Met polymorphism have been associated with differences in allele frequency across individuals of different racial backgrounds [Hindorff et al., 2002; Taylor et al., 2007c]. Moreover, genetic data were unavailable for 18 participants, resulting in genetic analyses being conducted on a subset of 62 individuals. Results of testing for Hardy‐Weinberg equilibrium have previously been reported [Taylor et al., 2007c, 2010]. There were no significant differences in SNP frequencies between the depressed and nondepressed cohorts (Table II).
Statistical models predicted ftWML ratio in each fiber tract and included the independent variables age, sex, hypertension, diagnostic cohort, AGTR1 and BDNF genotypes. In these models with a reduced sample, the left CgC continued to exhibit a significant difference in ftWML ratio between diagnostic cohorts (F (1,55) = 5.39, P = 0.0240).
In these models, the ftWML ratio significantly differed by AGTR1 A1166C genotype in multiple fiber tracts (Table IV), including: the ATR and IFO bilaterally; the right CgC, CST, and SLFb; the left ILF; and the CCO. In all cases, the at‐risk AGTR1 C1166 allele carriers exhibited significantly greater ftWML volumes in these regions than did A1166 homozygous individuals. However, in contrast to the widespread associations between AGTR1 and multiple ftWML ratios, BDNF Val66Met genotype was significantly associated only with ftWML ratio of the CCF. In this analysis, BDNF Met66 allele carriers exhibited greater CCF ftWML ratios (Met carriers = 0.0082 (0.0146)) than did Val/Val homozygous individuals (Val/Val = 0.0032 (0.0046); F (1,55) = 4.54, P = 0.0375).
Table IV.
Differences in Fiber Tract WML (ftWML) Ratios by AGTR1 A1166C Genotype
| Cohort | ATR, L | ATR, R | CCF | CCO | CgH, L | CgH, R | CgC, L | CgC, R | CST, L | CST, R |
|---|---|---|---|---|---|---|---|---|---|---|
| AGTR1 aa (N = 36) | 0.0084 (0.0126) | 0.0085 (0.0081) | 0.0030 (0.0056) | 0.0094 (0.0092) | 0.0007 (0.0012) | 0.0037 (0.0049) | 0.0006 (0.0015) | 0.0005 (0.0010) | 0.0036 (0.0049) | 0.0041 (0.0066) |
| AGTR1 c/* (N = 26) | 0.0205 (0.0277) | 0.0240 (0.0328) | 0.0075 (0.0128) | 0.0199 (0.0250) | 0.0009 (0.0016) | 0.0045 (0.0057) | 0.0016 (0.0051) | 0.0030 (0.0056) | 0.0125 (0.0307) | 0.0159 (0.0281) |
| F (1,55) value | 4.82 | 6.37 | 3.28 | 5.39 | 0.26 | 0.72 | 0.89 | 7.26 | 2.51 | 4.94 |
| P | 0.0323 | 0.0145 | 0.0755 | 0.0240 | 0.6116 | 0.4006 | 0.3492 | 0.0093 | 0.1191 | 0.0303 |
| IFO, L | IFO, R | ILF, L | ILF, R | SLFb, L | SLFb, R | SLFt, L | SLFt, R | UNC, L | UNC, R | |
| AGTR1 aa (N = 36) | 0.0043 (0.0043) | 0.0069 (0.0076) | 0.0049 (0.0054) | 0.0063 (0.0071) | 0.0024 (0.0058) | 0.0026 (0.0055) | 0.0025 (0.0069) | 0.0027 (0.0077) | 0.0016 (0.0027) | 0.0041 (0.0044) |
| AGTR1 c/* (N = 26) | 0.0123 (0.0161) | 0.0164 (0.0215) | 0.0141 (0.0195) | 0.0091 (0.0109) | 0.0115 (0.0256) | 0.0123 (0.0252) | 0.0122 (0.0285) | 0.0115 (0.0294) | 0.0037 (0.0073) | 0.0068 (0.0069) |
| F (1,55) value | 7.75 | 5.27 | 7.31 | 1.87 | 3.73 | 4.43 | 3.39 | 2.78 | 1.60 | 2.00 |
| P | 0.0073 | 0.0256 | 0.0091 | 0.1775 | 0.0587 | 0.0399 | 0.0712 | 0.1012 | 0.2111 | 0.1633 |
Sample limited to Caucasian subjects only. Data presented as mean ratio (standard deviation). Statistical test results derive from models predicting ftWML volume, including independent variables of AGTR1 genotype, BDNF genotype, age, sex, presence of hypertension, and diagnostic cohort. See Table I for key to tract abbreviations.
Finally, we incorporated a gene‐gene interaction term into these models. This interaction term did not achieve statistical significance in any model, although a trend was observed for the right CST ftWML ratio (F (1,54) = 3.31, P = 0.0742). This trend appeared to be driven by the cohort of subjects who carried both the BDNF Met66 and AGTR1 C1166 alleles (N = 8, mean CST ftWML ratio = 0.0286 (0.0423)), compared with the mean ftWML ratios for other genotype combinations (Met*, A/A: N = 13, 0.0030 (0.0041); Val/Val, C*: N = 18, 0.0103 (0.0179); Val/Val, A/A: N = 23, 0.0047 (0.0076)).
DISCUSSION
Although WMLs are observed even in healthy aging, it is a mistake to assume they are benign. WMLs are visible signs of ischemic injury to white matter tracts, which likely is the mechanism behind the observed relationships between WMLs and depression, cognitive decline [De Groot et al., 2002; Kramer et al., 2002, 2007], and gait disturbances [Blahak et al., 2009; Srikanth et al., 2009]. Such injury to fiber tracts would hypothetically result in impairment in neural network communication and function, whether they are networks involved in emotion, cognition, or motor function. The current study lends credence to the concept that the presence of clinically relevant mood symptoms is associated with WMLs occurring in specific fiber tracts.
In our primary analysis comparing depressed and nondepressed elders, we found that the only statistically significant difference between diagnostic groups was in the ftWML ratio of the cingulum, around the region of the cingulate gyrus (CgC). This finding is supported by a substantial amount of structural and functional neuroimaging data implicating the anterior cingulate cortex (ACC) in Major Depressive Disorder (reviewed in [Pizzagalli, 2011]). Moreover, our finding was limited to the left hemisphere CgC, which is concordant with much work implicating left hemispheric lesions in the generation of depression symptoms [Narushima et al., 2003]. Thus our work supports and builds on these two bodies of literature, although it is important to note our findings differ from a prior study, where LLD was associated with greater WML volume in multiple fiber tracts, including the SLF, IFO, ILF, and UNC [Sheline et al., 2008]. These discrepant findings may have been related to either methodological differences in determining which fiber tracts were affected by WMLs or related to the underlying heterogeneity of LLD.
It is also important to acknowledge that WMLs, regardless of location, are only one factor of many that contributes to the pathogenesis of LLD [Alexopoulos, 2005]. Psychosocial stressors, disability, and genetic differences have substantial influences on vulnerability to depression. Moreover, cortical atrophy and other neuroanatomical findings may have independent but complementary effects on depression vulnerability [Kumar et al., 2000].
In analyses examining genetic influences, we found widespread associations between AGTR1 genotype and ftWML ratios, including the ATR, IFO, ILF, and CCO. By comparison, in these same models BDNF genotype was significantly associated only with the CCF ftWML ratio. These findings support our initial hypothesis that if genetic differences contribute to ischemia or the development of WMLs through different mechanisms, they may also exhibit different influences on where the WMLs develop. We have previously hypothesized that AGTR1 contributes to the development of WMLs through its relationship with hypertension and cardiac disease [Deinum et al., 2001; Hindorff et al., 2002; Miyaki et al., 2006; Rubattu et al., 2004; Taylor et al., 2010], so it is reasonable it would have a broad relationship with multiple ftWML measures. We have also hypothesized that BDNF may be important in the tissue response to transient ischemia [Taylor et al., 2008], as increased BDNF expression may facilitate neuroprotection and be associated with improved functional outcomes [Dmitrieva et al., 2010; Nomura et al., 2005; Yanamoto et al., 2004]. In this study, BDNF appears to have more of a localized effect, although it's also possible it has a weaker effect than AGTR1 variants, resulting in this study being underpowered to fully detect its effect.
Despite limited power, we conducted analyses testing for a gene‐gene interaction between the AGTR1 and BDNF SNPs. Although exploratory, this decision was not atheoretical as we hypothesized these genes may contribute to tissue vulnerability to ischemia through different but complementary mechanisms, so individuals with both risk genes would be expected to exhibit the highest ftWML volumes. Although we identified a trend in the right CST for individuals who carried both the BDNF Met66 and AGTR1 C1166 alleles to exhibit higher ftWML ratios, this did not reach statistical significance and could not support our hypothesis. However, recognizing we were underpowered to detect such a difference, this is worth exploring in future studies with larger samples.
It should be noted that visible WMLs do not occupy an extensive proportion of the fiber tracts we examined. When we examined differences between diagnostic cohorts (Table III) and between AGTR1 genotypes (Table IV), mean ftWML ratios are not greater than 0.03, with many ftWML ratios being less than 0.01. However, these visible hyperintense regions may only be the “tip of the iceberg,” as we have previously demonstrated a strong association between WML volumetric measures and fractional anisotropy as measured by diffusion tensor imaging (DTI) [Taylor et al., 2007a], so a relatively small proportion of a tract being occupied by WMLs may represent broader loss of tract integrity. In turn, this would hypothetically affect communication and function of the connected regions.
The study has several limitations that need to be considered. The primary issue is that we examined a relatively small and heterogeneous sample, particularly for a study examining genetic associations, a problem compounded by making multiple comparisons. We examined 20 fiber tracts and using a Bonferroni correction this would result in an adjusted alpha of 0.0025. At this adjusted alpha, none of our reported findings would have reached statistical significance, so this raises concerns about Type I error and necessitates replication. Additionally, the functional implication of these findings is unclear as with our current imaging data we are unable to link ftWML ratios with measures of brain activity. Finally, our depressed population was a mixture of individuals who were and were not clinically remitted at the time of MRI, although it is unlikely that remission from depression would alter our measure WMLs.
Future studies should endeavor to link these fiber tract WML ratios with measures of brain activation to determine the functional significance of these findings. These efforts can be combined with methods of analyzing DTI fiber tract data to provide multiple measures of tract integrity. Such approaches would inform us not only of the mechanism by which WMLs may contribute to the pathogenesis of depression, but could be applied to treatment outcome studies to determine if treatment nonresponse in LLD is related to structural changes of fiber tracts influencing the function of the ACC and its connected regions.
Acknowledgements
The authors acknowledge Mr. Kulpreet Singh and Mr. Brian D. Boyd of the Duke Neuropsychiatric Imaging Research Laboratory for their assistance with image preprocessing.
REFERENCES
- Alexopoulos GS ( 2005): Depression in the elderly. Lancet 365: 1961–1970. [DOI] [PubMed] [Google Scholar]
- Atwood LD, Wolf PA, Heard‐Costa NL, Massaro JM, Beiser A, D'Agostino RB, DeCarli C ( 2004): Genetic variation in white matter hyperintensity volume in the Framingham Study. Stroke 35: 1609–1613. [DOI] [PubMed] [Google Scholar]
- Awad IA, Spetzler RF, Hodak JA, Awad CA, Carey R ( 1986): Incidental subcortical lesions identified on magnetic resonance imaging in the elderly. I. Correlation with age and cerebrovascular risk factors. Stroke 17: 1084–1089. [DOI] [PubMed] [Google Scholar]
- Bae JN, MacFall JR, Krishnan KR, Payne ME, Steffens DC, Taylor WD ( 2006): Dorsolateral prefrontal cortex and anterior cingulate cortex white matter alterations in late‐life depression. Biol Psychiatry 60: 1356–1363. [DOI] [PubMed] [Google Scholar]
- Blahak C, Baezner H, Pantoni L, Poggesi A, Chabriat H, Erkinjuntti T, Fazekas F, Ferro JM, Langhorne P, O'Brien J, Visser MC, Wahlund LO, Waldemar G, Wallin A, Inzitari D, Hennerici MG ( 2009): Deep frontal and periventricular age related white matter changes but not basal ganglia and infratentorial hyperintensities are associated with falls: cross sectional results from the LADIS study. J Neurol Neurosurg Psychiatry 80: 608–613. [DOI] [PubMed] [Google Scholar]
- Bokura H, Yamaguchi S, Iijima K, Nagai A, Oguro H ( 2008): Metabolic syndrome is associated with silent ischemic brain lesions. Stroke 39: 1607–1609. [DOI] [PubMed] [Google Scholar]
- Carmelli D, DeCarli C, Swan GE, Jack LM, Reed T, Wolf PA, Miller BL ( 1998): Evidence for genetic variance in white matter hyperintensity volume in normal elderly male twins. Stroke 29: 1177–1181. [DOI] [PubMed] [Google Scholar]
- Chang CC, Yu SC, McQuoid DR, Messer DF, Taylor WD, Singh K, Boyd BD, Krishnan KR, MacFall JR, Steffens DC, Payne ME. ( 2011): Reduction of dorsolateral prefrontal cortex gray matter in late‐life depression. Psychiatry Res 193: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot JC, De Leeuw FE, Oudkerk M, Van Gijn J, Hofman A, Jolles J, Breteler MM ( 2002): Periventricular cerebral white matter lesions predict rate of cognitive decline. Ann Neurol 52: 335–341. [DOI] [PubMed] [Google Scholar]
- Deinum J, van Gool JMG, Kofflard MJM, ten Cate FJ, Jan Danser AH ( 2001): Angiotensin II type 2 receptors and cardiac hypertrophy in women with hypertrophic cardiomyopathy. Hypertension 38: 1278–1281. [DOI] [PubMed] [Google Scholar]
- Denton ER, Sonoda LI, Rueckert D, Rankin SC, Hayes C, Leach MO, Hill DL, Hawkes DJ ( 1999): Comparison and evaluation of rigid, affine, and nonrigid registration of breast MR images. J Comput Assit Tomogr 23: 800–805. [DOI] [PubMed] [Google Scholar]
- Dmitrieva VG, Povarova OV, Skvortsova VI, Limborska SA, Myasoedov NF, Dergunova LV ( 2010): Semax and Pro‐Gly‐Pro activate the transcription of neurotrophins and their receptor genes after cerebral ischemia. Cell Mol Neurobiol 30: 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufouil C, de Kersaint‐Gilly A, Besancon V, Levy C, Auffray E, Brunnereau L, Alperovitch A, Tzourio C ( 2001): Longitudinal study of blood pressure and white matter hyperintensities. The EVA MRI cohort. Neurology 56: 921–926. [DOI] [PubMed] [Google Scholar]
- Firbank MJ, Lloyd AJ, Ferrier N, O'Brien JT ( 2004): A volumetric study of MRI signal hyperintensities in late‐life depression. Am J Geriatr Psychiatry 12: 606–612. [DOI] [PubMed] [Google Scholar]
- Gunning‐Dixon FM, Hoptman MJ, Lim KO, Murphy CF, Klimstra S, Latoussakis V, Majcher‐Tascio M, Hrabe J, Ardekani BA, Alexopoulos GS ( 2008): Macromolecular white matter abnormalities in geriatric depression: A magnetization transfer imaging study. Am J Geriatr Psychiatry 16: 255–262. [DOI] [PubMed] [Google Scholar]
- Herrmann LL, Le Masurier M, Ebmeier KP ( 2008): White matter hyperintensities in late life depression: A systematic review. J Neurol Neurosurg Psychiatry 79: 619–624. [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Heckbert SR, Tracy R, Tang Z, Psaty BM, Edwards KL, Siscovick DS, Kronmal RA, Nazar‐Stewart V ( 2002): Angiotensin II type 1 receptor polymorphisms in the cardiovascular health study: Relation to blood pressure, ethnicity, and cardiovascular events. Am J Hypertens 15: 1050–1056. [DOI] [PubMed] [Google Scholar]
- Jokinen H, Kalska H, Ylikoski R, Madureira S, Verdelho A, Gouw A, Scheltens P, Barkhof F, Visser MC, Fazekas F, Schmidt R, O'Brien J, Hennerici M, Baezner H, Waldemar G, Wallin A, Chabriat H, Pantoni L, Inzitari D, Erkinjuntti T ( 2009): MRI‐defined subcortical ischemic vascular disease: Baseline clinical and neuropsychological findings. The LADIS Study. Cerebrovasc Dis 27: 336–344. [DOI] [PubMed] [Google Scholar]
- Kramer JH, Mungas D, Reed BR, Wetzel ME, Burnett MM, Miller BL, Weiner MW, Chui HC ( 2007): Longitudinal MRI and cognitive change in healthy elderly. Neuropsychology 21: 412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JH, Reed BR, Mungas D, Weiner MW, Chui HC ( 2002): Executive dysfunction in subcortical ischaemic vascular disease. J Neurol Neurosurg Psychiatry 72: 217–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Bilker W, Jin Z, Udupa J ( 2000): Atrophy and high intensity lesions: complementary neurobiological mechanisms in late‐life depression. Neuropsychopharmacology 22: 264–274. [DOI] [PubMed] [Google Scholar]
- Longstreth WTJ, Manolio TA, Arnold A, Burke GL, Bryan N, Jungreis CA, Enright PL, O'Leary D, Fried L ( 1996): Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people: the cardiovascular health study. Stroke 27: 1274–1282. [DOI] [PubMed] [Google Scholar]
- MacFall JR, Taylor WD, Rex DE, Pieper S, Payne ME, McQuoid DR, Steffens DC, Kikinis R, Toga AW, Krishnan KR ( 2005): Lobar distribution of lesion volumes in late‐life depression: The Biomedical Informatics Research Network (BIRN). Neuropsychopharmacology 31: 1500–1507. [DOI] [PubMed] [Google Scholar]
- Maes F, Collignon A, Vandermeulen D, Marchal G, Suetens P ( 1997): Multimodality image registration by maximization of mutual information. IEEE Trans Med Imaging 16: 187–198. [DOI] [PubMed] [Google Scholar]
- Miyaki K, Hara A, Araki J, Zhang L, Song Y, Kimura T, Omae K, Muramatsu M ( 2006): C3123A polymorphism of the angiotensin II type 2 receptor gene and salt sensitivity in healthy Japanese men. J Hum Hypertens 20: 467–469. [DOI] [PubMed] [Google Scholar]
- Narushima K, Kosier JT, Robinson RG ( 2003): A reappraisal of poststroke depression, intra‐ and inter‐hemispheric lesion location using meta‐analysis. J Neuropsychiatry Clin Neurosci 15: 422–430. [DOI] [PubMed] [Google Scholar]
- Nomura T, Honmou OH, K., Houkin K, Hamada H, Kocsis JD ( 2005): I.V. infusion of brain‐derived neurotrophic factor gene‐modified human mesenchymal stem cells protects against injury in a cerebral ischemia model in adult rat. Neuroscience 136: 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JT, Firbank MJ, Krishnan MS, van Straaten EC, van der Flier WM, Petrovic K, Pantoni L, Simoni M, Erkinjuntti T, Wallin A, Wahlund LO, Inzitari D ( 2006): White matter hyperintensities rather than lacunar infarcts are associated with depressive symptoms in older people: The LADIS study. Am J Geriatr Psychiatry 14: 834–841. [DOI] [PubMed] [Google Scholar]
- Paternoster L, Chen W, Sudlow CL ( 2009): Genetic determinants of white matter hyperintensities on brain scans: A systematic assessment of 19 candidate gene polymorphisms in 46 studies in 19,000 subjects. Stroke 40: 2020–2026. [DOI] [PubMed] [Google Scholar]
- Pizzagalli DA ( 2011): Frontocingulate dysfunction in depression: Toward biomarkers of treatment response. Neuropsychopharmacology 36: 183–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regier DA, Myers JK, Kramer M, Robins LN, Blazer DG, Hough RL, Eaton WW, Locke BZ ( 1984): The NIMH Epidemiologic Catchment Area program: Historical context, major objectives, and study population characteristics. Arch Gen Psychiatry 41: 934–941. [DOI] [PubMed] [Google Scholar]
- Robins LN, Helzer JE, Croughan J, Ratcliff KS ( 1981): National Institute of Mental Health Diagnostic Interview Schedule. Its history, characteristics, and validity. Arch Gen Psychiatry 38: 381–389. [DOI] [PubMed] [Google Scholar]
- Rubattu S, Di Angelantonio E, Stanzione R, Zanda B, Evangelista A, Pirisi A, De Paolis P, Cota L, Brunetti E, Volpe M ( 2004): Gene polymorphisms of the renin‐angiotensin‐aldosterone system and the risk of ischemic stroke: A role of the A1166C/AT1 gene variant. J Hypertens 22: 2129–2134. [DOI] [PubMed] [Google Scholar]
- Rueckert D, Sonoda LI, Hayes C, Hill DL, Leach MO, Hawkes DJ ( 1999): Nonrigid registration using free‐form deformations: Application to breast MR images. IEEE Trans Med Imaging 18: 712–721. [DOI] [PubMed] [Google Scholar]
- Schnabel JA, Rueckert D, Quist M, Blackall JM, Castellano‐Smith AD, Hartkens T, Gerritsen FA, Hill DLG, Hawkes DJ. ( 2001): A Generic Framework for Non‐rigid Registration Based on Non‐uniform Multi‐level Free‐Form Deformations. Lecture Notes in Computer Science. Heidelberg, Germany: Springer Berlin; pp 573–581. [Google Scholar]
- Sheline YI, Price JL, Vaishnavi SN, Mintun MA, Barch DM, Epstein AA, Wilkins CH, Snyder AZ, Couture L, Schechtman K, McKinstry RC ( 2008): Regional white matter hyperintensity burden in automated segmentation distinguishes late‐life depressed subjects from comparison subjects matched for vascular risk factors. Am J Psychiatry 165: 524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth V, Beare R, Blizzard L, Phan T, Stapleton J, Chen J, Callisaya M, Martin K, Reutens D ( 2009): Cerebral white matter lesions, gait, and the risk of incident falls: A prospective population‐based study. Stroke 40: 175–180. [DOI] [PubMed] [Google Scholar]
- Steffens DC, McQuoid DR, Krishnan KRR ( 2002): The Duke Somatic Treatment Algorithm for Geriatric Depression (STAGED) approach. Psychopharmacol Bull 36: 58–68. [PubMed] [Google Scholar]
- Taylor WD, Bae JN, MacFall JR, Payne ME, Provenzale JM, Steffens DC, Krishnan KR ( 2007a): Widespread effects of hyperintense lesions on cerebral white matter structure. Am J Roentgenol 188: 1695–1704. [DOI] [PubMed] [Google Scholar]
- Taylor WD, MacFall JR, Gerig G, Krishnan KR ( 2007b): Structural integrity of the uncinate fasciculus in geriatric depression: Relationship with age of onset. Neuropsychiatr Dis Treat 3: 669–674. [PMC free article] [PubMed] [Google Scholar]
- Taylor WD, MacFall JR, Payne ME, McQuoid DR, Provenzale JM, Steffens DC, Krishnan KRR ( 2004): Late‐life depression and microstructural abnormalities in dorsolateral prefrontal cortex white matter. Am J Psychiatry 161: 1293–1296. [DOI] [PubMed] [Google Scholar]
- Taylor WD, MacFall JR, Payne ME, McQuoid DR, Steffens DC, Provenzale JM, Krishnan KR ( 2005): Greater MRI lesion volumes in elderly depressed subjects than in control subjects. Psychiatry Res 139: 1–7. [DOI] [PubMed] [Google Scholar]
- Taylor WD, MacFall JR, Provenzale JM, Payne ME, McQuoid DR, Steffens DC, Krishnan KRR ( 2003a): Serial MR imaging of hyperintense white matter lesion volumes in elderly subjects: Correlation with vascular risk factors. Am J Roentgenol 181: 571–576. [DOI] [PubMed] [Google Scholar]
- Taylor WD, MacFall JR, Steffens DC, Payne ME, Provenzale JM, Krishnan KRR ( 2003b): Localization of age‐associated white matter hyperintensities in late‐life depression. Prog Neuropsychopharmacol Biol Psychiatry 27: 539–544. [DOI] [PubMed] [Google Scholar]
- Taylor WD, Steffens DC, Ashley‐Koch A, Payne ME, MacFall JR, Potocky C, Krishnan KR ( 2010): Angiotensin receptor gene polymorphisms and 2‐year change in cerebral hyperintense lesion volume in men. Mol Psychiatry 15: 816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor WD, Steffens DC, MacFall JR, McQuoid DR, Payne ME, Provenzale JM, Krishnan KRR ( 2003c): White matter hyperintensity progression and late‐life depression outcomes. Arch Gen Psychiatry 60: 1090–1096. [DOI] [PubMed] [Google Scholar]
- Taylor WD, Zuchner S, McQuoid DR, Payne ME, MacFall JR, Steffens DC, Speer M, Krishnan KR ( 2008): The brain‐derived neurotrophic factor Val66Met polymorphism and cerebral white matter hyperintensities in late‐life depression. Am J Geriatr Psychiatry 16: 263–271. [DOI] [PubMed] [Google Scholar]
- Taylor WD, Zuchner S, McQuoid DR, Steffens DC, Speer MC, Krishnan KR ( 2007c): Allelic differences in the BDNF val66met polymorphism in late‐life depression. Am J Geriatr Psychiatry 15: 850–857. [DOI] [PubMed] [Google Scholar]
- Thomas AJ, O'Brien JT, Davis S, Ballard C, Barber R, Kalaria RN, Perry RH ( 2002a): Ischemic basis for deep white matter hyperintensities in major depression. Arch Gen Psychiatry 59: 785–792. [DOI] [PubMed] [Google Scholar]
- Thomas AJ, Perry R, Barber R, Kalaria RN, O'Brien JT ( 2002b): Pathologies and pathological mechanisms for white matter hyperintensities in depression. Ann NY Acad Sci 977: 333–339. [DOI] [PubMed] [Google Scholar]
- Turner ST, Jack CR, Fornage M, Mosley TH, Boerwinkle E, de Andrade M ( 2004): Heritability of leukoaraiosis in hypertensive sibships. Hypertension 43: 483–487. [DOI] [PubMed] [Google Scholar]
- Van Leemput K, Maes F, Vandermeulen D, Colchester A, Suetens P ( 2001): Automated segmentation of multiple sclerosis lesions by model outlier detection. IEEE Trans Med Imaging 20: 677–688. [DOI] [PubMed] [Google Scholar]
- Van Leemput K, Maes F, Vandermeulen D, Suetens P ( 1999): Automated model‐based tissue classification of MR images of the brain. IEEE Trans Med Imaging 18: 897–908. [DOI] [PubMed] [Google Scholar]
- Van Leemput K, Maes F, Vandermeulen D, Suetens P ( 2003): A unifying framework for partial volume segmentation of brain MR images. IEEE Trans Med Imaging 22: 105–119. [DOI] [PubMed] [Google Scholar]
- Wakana S, Caprihan A, Panzenboeck MM, Fallon JH, Perry M, Gollub RL, Hua K, Zhang J, Jianga H, Dubey P, Blitz A, Zijl Pv, Mori S ( 2007): Reproducibility of quantitative tractography methods applied to cerebral white matter. Neuroimage 36: 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamoto H, Xue JH, Miyamoto S, Nagata I, Nakano Y, Murao K, Kikuchi H ( 2004): Spreading depression induces long‐lasting brain protection against infarcted lesion development via BDNF gene‐dependent mechanism. Brain Res 1019: 178–188. [DOI] [PubMed] [Google Scholar]