

Abstract
The 70kD heat shock proteins (Hsp70s) are ubiquitous molecular chaperones essential for cellular protein folding and proteostasis. Each Hsp70 has two functional domains: a nucleotide-binding domain (NBD) that binds and hydrolyzes ATP, and a substrate-binding domain (SBD) that binds extended polypeptides. NBD and SBD interact little when in ADP; however, ATP binding allosterically couples the polypeptide- and ATP-binding sites. ATP binding promotes polypeptide release; polypeptide rebinding stimulates ATP hydrolysis. This allosteric coupling is poorly understood. Here we present the crystal structure of an intact Hsp70 from Escherichia coli in an ATP-bound state at 1.96 Å resolution. NBD-ATP adopts a unique conformation, forming extensive interfaces with a radically changed SBD that has its α-helical lid displaced and the polypeptide-binding channel of its β-subdomain restructured. These conformational changes together with our biochemical tests provide a long-sought structural explanation for allosteric coupling in Hsp70 activity.
Maintaining protein homeostasis (proteostasis) is a fundamental task for every living organism, and failure contributes to destructive human diseases such as cancers and neurodegenerative disorders1,2. Through assisting protein folding, assembly, degradation and transport across membranes, the 70kD heat shock proteins (Hsp70s) are key players in proteostasis.
Hsp70s are highly conserved. Each contains two functional domains, a nucleotide binding domain (NBD) and a substrate binding domain (SBD) joined by a short linker segment (Fig. 1a)3–5. NBD is an ATPase and SBD substrates are extended hydrophobic polypeptides as from unfolded proteins6. The first NBD structure was of the isolated NBD from bovine Hsc70, having ADP bound at a site between two lobes7, and all subsequent Hsp70 structures were either also ADP-bound or nucleotide-free8–11. The first SBD structure was of the isolated SBD from the Escherichia coli Hsp70 DnaK in complex with a short peptide12. Other known SBD structures have essentially the same conformation10,11,13–15 with the polypeptide-binding site more dynamic in solution in the absence of peptide substrate14.
Figure 1. Characteristics of DnaK locked in an ATP-bound state.

a, Schematic of Hsp70 domain structure. Structured subdomains are given color assignments and positions of key elements are identified.
b, Ribbon representation of the DnaK SBD-
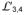 structure. Domain coloring is as in a.
structure. Domain coloring is as in a.
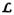 stands for loop. The Linker segment from a symmetry mate (in purple) binds to the polypeptide-binding site between
stands for loop. The Linker segment from a symmetry mate (in purple) binds to the polypeptide-binding site between
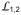 and
.
and
.
c, Superposition of SBDβ from the DnaK SBD-
structure (colored as in b) with that of the WT SBD structure (orange; PDB 1DKZ). The peptide substrate NR in the WT SBD structure is colored in cyan.
d, Tryptophan-fluorescence emission spectra for assaying the ATP-induced allosteric coupling of DnaK.
Although NBD and SBD can each bind their substrates independently, the chaperone activity of Hsp70s strictly requires the tight coupling of these two domains upon ATP binding. Various biochemical and structural studies, mainly on DnaK as a model Hsp70, have shown that in ADP-bound or nucleotide-free states there is little contact between NBD and SBD; each domain acts independently as if isolated11,16–18. In contrast, upon ATP binding, the affinity of SBD for its substrates is reduced by two to three orders of magnitude19,20. Both binding and release rates are greatly accelerated, more for release than binding. Reciprocally, polypeptide binding to SBD stimulates the ATP hydolysis3,20. This allosteric coupling ensures that energy from ATP hydrolysis is efficiently used to control peptide substrate binding and release for efficient chaperone activity. In spite of extensive efforts, the basic mechanism of this ATP-induced allosteric coupling is still ill defined.
Crystal structures of full-length Hsp70s containing both functional domains in the ATP-bound state have been highly sought in anticipation of insights into Hsp70 allostery. This goal has proved to be challenging, however, because of ATP lability and the transience of allosteric coupling. In fact, even structures of isolated ATP-bound NBDs are elusive7,8,21,22. Hsp110s are distant homologs of Hsp70s, and our structure of Sse1-ATP, a yeast Hsp110, provided an invaluable starting point for mechanistic understanding of allosteric coupling in Hsp70s4,5,23–27; however, relatively low sequence conservation, especially between SBD regions23, has limited its applicability to Hsp70s. To explore the molecular mechanism of this essential ATP-induced allosteric coupling, we solved a crystal structure of an intact Hsp70 from E. coli in complex with ATP. Our biochemical analysis inspired by this structure provides a mechanistic understanding of how ATP binding allosterically opens the polypeptide-binding site.
RESULTS
Hsp70 DnaK locked in an ATP-bound state
To study a functionally complete Hsp70 in the ATP-bound state, we incorporated two previously reported mutations into E. coli DnaK: T199A and loop
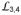 alternative
(Fig. 1a and Supplementary Fig. 1). T199A is a well-characterized mutation that abolishes ATPase activity but maintains allosteric coupling16,28; it helped to stabilize a relevant ATP-bound state. DnaK tends to self-associate into flexible oligomers29, which complicates crystallization. Recently, we produced a peptide-binding-deficient DnaK mutation having peptide-binding loop
replaced by the shortened
sequence (TAEDNQS→MGG)30; this solved the self-association problem. Furthermore, to facilitate crystallization, we removed 28 residues at the extreme C-terminal region, which is disordered18,31.
alternative
(Fig. 1a and Supplementary Fig. 1). T199A is a well-characterized mutation that abolishes ATPase activity but maintains allosteric coupling16,28; it helped to stabilize a relevant ATP-bound state. DnaK tends to self-associate into flexible oligomers29, which complicates crystallization. Recently, we produced a peptide-binding-deficient DnaK mutation having peptide-binding loop
replaced by the shortened
sequence (TAEDNQS→MGG)30; this solved the self-association problem. Furthermore, to facilitate crystallization, we removed 28 residues at the extreme C-terminal region, which is disordered18,31.
To check on structural impact of the
modification, we solved the crystal structure of the isolated SBD-
variant at 1.62Å resolution (Fig. 1b and Table 1). SBD comprises two subdomains: SBDβ forms a single polypeptide-binding channel between loops
and
; and SBDα functions as a lid covering the polypeptide-binding site on SBDβ (Fig. 1c)12. Except for the shortening of
by four residues, SBD-
is almost identical to the previously solved WT SBD structure12 (0.45 Å root-mean-squared deviation [rmsd] in SBDβ Cα atoms). SBDα is rotated slightly (Fig. 1c), consistent with the intrinsic flexibility of this region12,15. Substrate peptides bind too weakly to the DnaK-
mutant to be detected by fluorescence anisotropy assays30; however, the Linker segment from a symmetry mate does bind to SBD-
at the high concentrations in crystals, and it does so in a similar fashion as for NR peptide binding to WT SBD (Figs. 1b,c).
Table 1.
Data collection, phasing and refinement statistics (molecular replacement and SAD)
| DnaK SBD-
Native |
DnaK-ATP Native | DnaK-ATP SAD | |
|---|---|---|---|
| Data collection | |||
| Space group | P3121 | I422 | I422 |
| Cell dimensions | |||
| a, b, c (Å) | 67.0, 67.0, 128.8 | 290.7, 290.7, 99.3 | 291.7, 291.7, 99.5 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 30-1.62 (1.65-1.62) * | 30-1.96 (2.01-1.96) * | 30-2.30 (2.36-2.30) * |
| Rsym or Rmerge | 0.047 (0.261) | 0.070 (0.358) | 0.125 (0.498) |
| I/σI | 54.0 (5.1) | 21.7 (5.1) | 64.5 (16.2) |
| Completeness (%) | 98.9 (86.8) | 99.8 (97.4) | 100.0 (100.0) |
| Redundancy | 8.9 (5.0) | 8.0 (5.8) | 148.5 (87.1) |
| Refinement | |||
| Resolution (Å) | 30-1.62 (1.66-1.62) | 30-1.96 (1.98-1.96) | 30-2.30 (2.33-2.30) |
| No. reflections | 381,376 | 1,204,143 | 12,331,244 |
| Rwork / Rfree | 0.191 / 0.216 | 0.172 / 0.201 | 0.164 / 0.197 |
| No. atoms | 1923 | 10,677 | 10,366 |
| Protein | 1600 | 9100 | 9100 |
| Ligand/ion | - | 118/62 | 118/62 |
| Water | 323 | 1397 | 1086 |
| B-factors | 28.9 | 32.5 | 32.4 |
| Protein | 27.2 | 31.4 | 31.7 |
| Ligand/ion | - | 42.1 | 35.2 |
| Water | 37.5 | 39.4 | 38.1 |
| r.m.s. deviations | |||
| Bond lengths (Å) | 0.006 | 0.005 | 0.005 |
| Bond angles (°) | 0.844 | 0.916 | 0.889 |
| PDB | 4JNF | 4JNE | 4JN4 |
One crystal was used for each native data set, and 5 crystals were used for the SAD data set.
Values in parentheses are for highest-resolution shell.
To check on functional impact of the T199A and
mutations, we used a well-established tryptophan fluorescence assay of allosteric coupling. DnaK has a single tryptophan, Trp102 in NBD, whose intrinsic fluorescence emission spectrum is sensitive to the nucleotide binding state. Relative to the apo and ADP-bound forms, ATP binding induces a blue-shift and fluorescence quenching, but only in the context of full-length DnaK when there is allosteric coupling16. Tryptophan-fluorescence changes with ATP addition to DnaK-T199A
were almost identical to those for WT DnaK (Fig. 1d). Thus, this variant has a WT-like conformation in the ATP-bound state.
Crystals of DnaK-T199A
grew only in the presence of ATP. Henceforth, we call the complex DnaK-ATP. The crystals diffract well and have two DnaK chains per asymmetric unit (Table 1). The structure was solved by the method of multi-crystal native single-wavelength anomalous diffraction (SAD)32. Resulting electron-density maps were of high quality, showing well-defined Mg2+-ATP ligands (Supplementary Fig. 2) and allowing automated model building. Separately collected native data were used for refinement at 1.96Å resolution. The final model has excellent stereochemistry (no Ramachandran outliers).
The two crystallographically unique DnaK-ATP protomers are almost identical (Cα rmsd = 0.40Å; all-atom rmsd = 0.82Å), related by near-dyad symmetry (rotation χ=179.9°; translation tχ=0.15Å) (Fig. 2a). Each protomer is composed of familiar NBD, Linker, SBDβ and SBDα domains and ATP; however, domains are disposed uniquely (Fig. 2b) with extensive interfaces and unanticipated conformational changes. Thus, the DnaK-ATP structure gives a first view of a full-length, ATP-bound Hsp70.
Figure 2. Overall structure of ATP-bound DnaK (DnaK-ATP).

Domain coloring in the ribbon diagrams is as in Fig. 1a throughout.
a, Ribbon diagrams of the DnaK dimer. Left: View with dyad axis (orange rod) vertical. Domains are labeled for protomer A; protomer B is on the right. Right: Orthogonal view from below, along the dyad axis (orange dot).
b, Ribbon diagram of a DnaK protomer in the canonical, front-face NBD view (left) and in an orthogonal side view (right). The NBD lobes are labeled as I and II. The bound ATP molecule is in stick representation.
c, Comparison with a DnaK-ADP structure. Left: ribbon diagram of the DnaK-ADP structure in the front-face NBD view (PDB 2KHO). Right: Side view of DnaK-ATP as in Fig. 2b right panel, but in backbone worm representation and superimposed onto DnaK-ADP based on NBD Lobe-I Cαs. Domain coloring is NBD from DnaK-ATP (blue), SBD from DnaK-ATP (red), NBD from DnaK-ADP (cyan) and SBD from DnaK-ADP (green).
d, Comparison with the Sse1-ATP structure. Left: ribbon diagram of the Sse1-ATP structure in the front-face NBD view (PDB 2QXL). Right: Side view of DnaK-ATP as in Fig. 2b right panel, but in backbone worm representation and superimposed onto Sse1-ATP based on NBD Lobe-I Cαs. Domain coloring is NBD from DnaK-ATP (blue), SBD from DnaK-ATP (red), NBD from Sse1-ATP (cyan) and SBD from Sse1-ATP (green).
Comparisons to Hsp70-ADP and Hsp110-ATP
The DnaK-ATP structure is radically different from ADP-bound and nucleotide-free Hsp70 structures10,11,18 wherein NBD and SBD are mostly independent, structurally and functionally; however, it shares similarities with the Sse1-ATP structure. An NMR analysis of DnaK-ADP found a distribution of orientations for SBD relative to NBD and produced an average solution structure using X-ray crystal structures for the domains (Fig. 2c)18.
Various biochemical and NMR studies have suggested that NBD and SBD do contact one another in the presence of ATP3,4,5,16,17,33. Consistent with these observations, there are extensive contacts between domains of DnaK-ATP (Fig. 2b), which involve substantial conformational change within both NBD and SBD as detailed below. In summary, SBDα is peeled away from covering the polypeptide binding site on SBDβ as seen in the isolated SBD structure (and adopted in the DnaK-ADP model)18. Instead, subdomain SBDα docks onto the Lobe-I side of NBD and subdomain SBDβ binds between Lobe-I and Lobe-II at the lower backside of NBD, in the canonical view (Fig. 2b). The two lobes of NBD move apart such that the Linker fits into the cleft between them; and, consistent with its essential role in allosteric coupling17,34–36, the Linker then extends directly into SBDβ. Thus, the overall conformation of DnaK-ATP is completely different from that of DnaK-ADP (Fig. 2c).
The NBD-SBD interfaces and relative orientation of the domains in DnaK-ATP agree well with many previous biochemical and biophysical studies, including a number of recent studies using NMR and FRET24–27,35–37. Notably, this structure provides a direct explanation for the well-established tryptophan fluorescence assay described above. The Sse1-ATP structure already hinted at this explanation, but the NBD-SBDα interface of DnaK-ATP actually has SBDα residues Leu507 and Met515 directly contacting Trp102 (Supplementary Fig. 3a); in ADP-bound or nucleotide-free states, Trp102 is exposed. This more hydrophobic environment for Trp102 explains the spectral shift and quenching upon ATP binding. In addition, cysteine residues introduced strategically at structure-inspired NBD-SBDα interfaces formed disulfide bonds specifically in the presence of ATP (Supplementary Fig. 3), supporting the existence of a crystal-like conformation of DnaK-ATP in solution.
As much as DnaK-ATP differs from DnaK-ADP (and related Hsp70 structures), DnaK-ATP is similar to Sse1-ATP (Fig. 2d). This is as expected from biochemical analyses based on the yeast Hsp110 structure, Sse1-ATP23, and it supports the hypothesis that Sse1-ATP is an evolutionary vestige of the Hsp70-ATP state23 with some shared characteristics of allosteric coupling30. Most notably, the Linker disposition is highly conserved between the two structures (Supplementary Fig. 4a). Although DnaK-ATP and Sse1-ATP are superficially similar, there are many important differences. When the NBDs are aligned, both SBDα and SBDβ are rotated substantially in DnaK compared to Sse1, 41.8° and 24.0°, respectively (Fig. 2d), due to marked changes in the NBD-SBDα and NBD-SBDβ domain interfaces (see details below). The most remarkable difference is the conformation of the SBDβ (Supplementary Fig. 4b). Furthermore, SBDα is also rotated 45.1° relative to SBDβ.
ATP-elicited opening of the polypeptide-binding site
DnaK when bound to ATP assumes a markedly different conformation than that seen in any known Hsp70 structure, both with respect to NBD and to SBD. Changes initiate at the ATP-binding site; they propagate through NBD to interfaces for Linker and SBD subdomains; and they transform SBDβ into a state that releases its polypeptide substrate. Other Hsp70 NBD structures are in either ADP-bound or nucleotide-free states4,5,7–11,21, and NBD of DnaK-ATP is substantially different from those (Fig. 3a). It does resemble the ATP-bound conformation of Hsp110 Sse1-ATP23 (Cα-rmsd=1.54 Å; Fig. 3b). Indeed, the Mg2+-ATP complexes are virtually identical in the two structures (Supplementary Fig. 4c). ATP interacts intimately with DnaK, contacting 24 residues of which 21 are identical across a range of Hsp70s (Supplementary Fig. 1). When modeled into the structure, the hydroxyl group of Thr199 is poised to play its role in ATP hydrolysis. Upon ATP binding, consistent with NMR studies35,37, Lobe-I rotates by 25.5° relative to Lobe-II (Fig. 3a). This rotation opens a slot between subdomains IA and IIA that binds the Linker segment; however, this Linker binding is such that it can accommodate SBD only when SBDα is displaced from SBDβ (Supplementary Fig. 4d).
Figure 3. Unique conformations of NBD and SBD in DnaK-ATP.

a, Comparison of NBD conformations in DnaK-ATP and human Hsp70-ADP (PDB 1S3X). NBDs are superimposed based on Cαs in lobe-II. Coloring has in DnaK-ATP Lobe-I (red) and Lobe-II (blue); human Hsp70-ADP Lobe-I (grey) and Lobe-II (green). The left panel shows the canonical front-face view, and the right panel shows the view from above the left panel with IB and IIB removed to show the IA-IIA changes.
b, Comparison of DnaK-ATP and Sse1-ATP. Superpositions and coloring are as in a, except for replacement of human Hsp70-ADP by Sse1-ATP. Conformational differences between the two ATP-bound structures are slight.
c, Comparison of SBDβ conformations. The left and middle panels are ribbon diagram of SBDβs from the DnaK-ATP and the isolated DnaK-SBD (PDB 1DKZ) structures, respectively. SBDβs are superimposed based on Cαs in
and
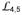 . Glycine residues Gly461 and Gly468 on loop
. Glycine residues Gly461 and Gly468 on loop
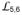 are highlighted as purple and black balls, respectively. The Cα atoms of Arg467 are highlighted as orange balls. The peptide substrate NR is in cyan. The right panel is the superposition of the left and middle panels.
are highlighted as purple and black balls, respectively. The Cα atoms of Arg467 are highlighted as orange balls. The peptide substrate NR is in cyan. The right panel is the superposition of the left and middle panels.
d, Comparison of the polypeptide-binding sites. The left and middle panels are the details of the polypeptide-binding sites in the DnaK-ATP and isolated DnaK-SBD structures, respectively. The two structures are superimposed based on Cαs in
and
. Residues in van der Waals contacts with the three leucine residues of the NR peptide in the isolated DnaK-SBD structure are shown in sticks. The backbone of the NR peptide is in cyan, and the side chains of the three leucine residues in the NR peptide are highlighted in orange. The NR peptide in the left panel is from the isolated DnaK-SBD structure. The right panel is the superposition of the left and middle panels. The side chains of the NR peptide are not shown.
The highly conserved, DVLLLD-Linker segment of DnaK runs directly into SBD, whereby the proximate end of SBDβ interacts intimately with NBD. Interfacial contacts made from loops
 ,
,
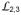 and
of SBD keep the ADP-state conformation of the isolated SBD-peptide complex12 (Fig. 3c and Supplementary Fig. 4d-f); in contrast, other contacts mediated by
and
of SBD keep the ADP-state conformation of the isolated SBD-peptide complex12 (Fig. 3c and Supplementary Fig. 4d-f); in contrast, other contacts mediated by
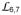 and
and
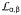 generate substantial movements in those loops and β8 relative to that fixed base (Fig. 3c). These changes happen as SBDα necessarily separates from SBDβ (Supplementary Fig. 4d–f). Helices αA and αB fuse into one long helix that binds alongside NBD Lobe-I (Supplementary Fig. 4e), and this displacement ‘pulls’
upwards (relating the middle panel to the left panel in Fig. 3c), moving strand β8 from the lower to the upper β-sheet.
also moves (although its conformation does not change, Supplementary Fig. 4g) against the fixed fulcrum of
,
and
contacts, appearing to exert a torque that ‘pushes’ distal elements of SBDβ outward and downward. As a result, the β-sandwich of the SBDβ domain in DnaK-ATP is twisted relative to that in the SBD-peptide complex (Fig. 3c) such that the distal loops (both the shortened
and
) project from the β-strand core, out and away from the peptide site (Fig. 3c,d). Consequently, the polypeptide-binding channel is flipped open on the distal side, with shifts of up to 22.8 Å (
Arg467 Cα). The proximal side of the binding site,
and
shows very little change. In the isolated SBD-peptide structure, leucine residues Leu3, Leu4 and Leu5 from the NR peptide reside in the center of the polypeptide-binding site, mediating numerous van der Waals contacts (Fig. 3d)12. The flipping open of
and
in DnaK-ATP abolishes contacts from that side of the binding site (Fig. 3d). Thus, the SBDβ conformation in DnaK-ATP matches well with the reduced peptide affinity, and accelerated binding and dissociation rates in ATP-bound Hsp70s. This ATP-bound conformation of
and
is also consistent with increased solvent accessibility of these loops in solution17,38. This conformational change that accompanies ATP binding to DnaK, whereby the polypeptide-binding channel is opened up, could be the structural basis for allosteric coupling in Hsp70 chaperones generally.
generate substantial movements in those loops and β8 relative to that fixed base (Fig. 3c). These changes happen as SBDα necessarily separates from SBDβ (Supplementary Fig. 4d–f). Helices αA and αB fuse into one long helix that binds alongside NBD Lobe-I (Supplementary Fig. 4e), and this displacement ‘pulls’
upwards (relating the middle panel to the left panel in Fig. 3c), moving strand β8 from the lower to the upper β-sheet.
also moves (although its conformation does not change, Supplementary Fig. 4g) against the fixed fulcrum of
,
and
contacts, appearing to exert a torque that ‘pushes’ distal elements of SBDβ outward and downward. As a result, the β-sandwich of the SBDβ domain in DnaK-ATP is twisted relative to that in the SBD-peptide complex (Fig. 3c) such that the distal loops (both the shortened
and
) project from the β-strand core, out and away from the peptide site (Fig. 3c,d). Consequently, the polypeptide-binding channel is flipped open on the distal side, with shifts of up to 22.8 Å (
Arg467 Cα). The proximal side of the binding site,
and
shows very little change. In the isolated SBD-peptide structure, leucine residues Leu3, Leu4 and Leu5 from the NR peptide reside in the center of the polypeptide-binding site, mediating numerous van der Waals contacts (Fig. 3d)12. The flipping open of
and
in DnaK-ATP abolishes contacts from that side of the binding site (Fig. 3d). Thus, the SBDβ conformation in DnaK-ATP matches well with the reduced peptide affinity, and accelerated binding and dissociation rates in ATP-bound Hsp70s. This ATP-bound conformation of
and
is also consistent with increased solvent accessibility of these loops in solution17,38. This conformational change that accompanies ATP binding to DnaK, whereby the polypeptide-binding channel is opened up, could be the structural basis for allosteric coupling in Hsp70 chaperones generally.
Consistent with the DnaK-ATP structure, an NMR study of isolated apo SBDβ, DnaK (393–507) without peptide substrate, has shown peptide-binding loops
and
are flexible and dynamic14. Compared to the DnaK-ATP and isolated SBD-peptide structures, the positions of
and
in the NMR structure of apo SBDβ are somewhere in between (Supplementary Fig. 5a–d). The polypeptide-binding loops in DnaK-ATP make contacts with symmetry mates in the crystal lattice (Supplementary Fig. 5e,f). Thus, it is possible that loops
and
are dynamic in the ATP-bound state, as in isolated apo SBDβ, and that lattice contacts stabilize a flipped-out member of an ensemble of ATP-bound conformations. To estimate how much crystal contacts might have affected the conformation of these loops, we carried out molecular dynamic (MD) simulations without crystal contacts. The MD results showed SBDβ to be the most stable structural element in the entire DnaK-ATP structure and to be much more stable than other SBDβ structures; apo SBDβ was the least stable (Supplementary Fig. 5g). Stabilizing contacts permeate the β-strands of SBDβ, which differ markedly from those in isolated SBD structures (Fig. 3c). Loops
and
emanate in an opened trajectory from these strands as stabilized by NBD-SBD interfaces, and we conclude that flexibility is limited to tips of the loops. Disulfide cross-linking experiments confirm that DnaK-ATP in solution is similar to the crystal structure (Supplementary Fig. 3). Thus, NBD-SBD interfaces generated upon binding of ATP fix the bases of
and
in SBDβ in an opened conformation, incompatible with strong peptide binding. The tips of loops may remain flexible, consistent with previous NMR studies14,39; however, unlike those of apo or peptide-bound SBDβ domains, the ensemble of states for DnaK-ATP is predisposed toward open conformations.
Conformational impact of two highly conserved glycine residues on
To test the hypothesis that opening of the polypeptide-binding channel upon ATP binding could be responsible for polypeptide release, we examined residues on
. We found that two highly conserved glycine residues, Gly461 and Gly468, changed backbone conformations dramatically (Fig. 4a and Supplementary Fig. 1), whereas other residues showed only moderate changes. We made glycine-to-proline mutations G461P and G468P since proline residues allow the phi/psi conformation angles of DnaK-ATP but not of isolated SBD40, the ADP-state (Fig. 2c); Thus, these mutants are designed to locally restrict SBDβ to its NBD(ATP)-bound conformation independent of ATP-induced domain contacts. Mutating either glycine residue individually compromised in vivo DnaK function modestly, but mutating both together (PP mutant) abolished DnaK function (Fig. 4b and Supplementary Fig. 6a) despite WT-levels of expression (Supplementary Fig. 6e). Tests with yeast Hsp70 Ssa1 gave similar results, suggesting a universal importance in Hsp70s (Fig. 4b and Supplementary Fig. 6a).
Figure 4. Two highly conserved glycine residues in
.
a, Comparison of phi and psi conformational angles for Gly461 and Gly468 in DnaK-ATP and isolated DnaK-SBD structures.
b, Growth tests for glycine mutants in Hsp70-deficient E. coli and yeast backgrounds. Serial dilutions of fresh cultures were spotted onto plates and incubated at 37°C. Functional DnaK and Ssa1 are required for growth at 37°C.
c, Refolding activity of WT and G461P G468P (PP) mutant DnaK. Refolding of the heat-denatured luciferase was started by adding the DnaK chaperone system (DnaK, DnaJ and GrpE) for the corresponding WT or PP DnaK.
d, Fluorescence anisotropy assay of peptide substrate binding affinity. Serial dilutions of DnaK proteins were incubated with F-NR, a model peptide substrate. Fluorescence anisotropy measurements were made after binding reached equilibrium.
e, Peptide substrate binding kinetics determined by fluorescence anisotropy assay. Immediately after mixing DnaK proteins with the F-NR peptide, peptide binding as a function of time was measured kinetics by fluorescence anisotropy.
To test impact of the PP mutation on specific characteristics of DnaK activity, we purified the mutant protein for in vitro activity assays. We found that the chaperone activity of WT DnaK in refolding of heat-denatured luciferase is lost for the PP mutant (Fig. 4c). Moreover, we found that peptide binding to DnaK PP is aberrant. Unlike the normal behavior in ADP or absence of nucleotides, where binding affinity is high and binding kinetics are slow, DnaK PP binds substrate peptides with reduced affinity (Fig. 4d) and remarkably fast binding kinetics (Fig. 4e) as happens for WT DnaK in ATP19. The PP mutant does undergo normal ATP-induced NBD-SBD associations as measured by tryptophan fluorescence. These results are consistent with the hypothesis that SBDβ of the PP mutant is locked locally into an ATP-like conformation irrespective of ATP or ADP status of the NBD. Thus, the two
glycine residues are critical for ATP-induced conformational changes in SBDβ. In summary, the results from MD simulations, disulfide cross-linking and the PP mutant suggest that a flipped out conformation of
and
in DnaK-ATP is physiologically important even though crystal contacts may stabilize one of an ensemble of states.
Domain interfaces in allosteric coupling
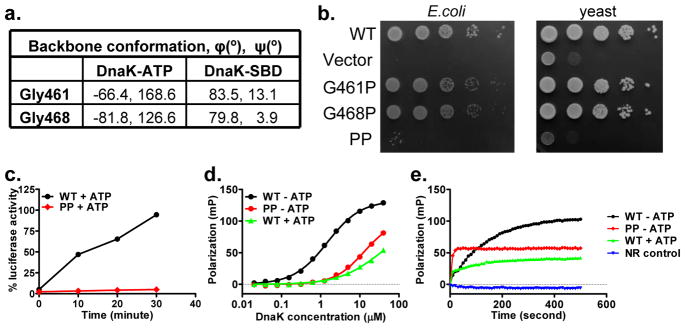
The unique conformation of DnaK-ATP arises from extensive interfaces that form between domains as they adapt to ATP binding. There are three major inter-domain contacts: NBD-Linker, NBD-SBDβ, and NBD-SBDα (Fig. 2b). The interfacial surfaces are conservative among Hsp70s (Fig. 5), and especially so in the critical NBD-SBDβ interface. SBDβ has additional conservation at the polypeptide-binding channel (Fig. 5b). Large surface areas are buried into the interfaces; however, shape complementarity is low as compared to similar interfaces in Sse1-ATP (Supplementary Table 1), which might relate to the transient nature of allosteric coupling in Hsp70s.
Figure 5. Sequence conservation at domain interfaces in DnaK-ATP.
The top panels are sequence conservation at the surface of NBD (a) and SBD (b). Conservation and surface mapping was calculated by ConSurf (http://consurf.tau.ac.il/). The conservation key is shown above b. The dotted yellow circle in b locates the polypeptide-binding site. The bottom panels are mappings of domain interfaces onto the surfaces of NBD (a) and SBD (b). In the bottom panel of a, the surface of NBD is blue, and the imprints of contacts are colored by domain: SBDα (red), SBDβ (green), Linker (purple). In the bottom panel of b, The surfaces of Linker, SBDβ and SBDα are purple, green, and red, respectively, and the imprints of contacts from NBD are colored by the sub-domain receiving that contact: SBDβ (blue), SBDα (marine), and Linker (purple, as it is buried entirely into the interface with NBD).
The NBD-SBD interaction in DnaK-ATP originates at the Linker segment connecting the two domains, and it involves intimate contacts between both the Linker-proximate end of the SBDβ β-sandwich and the displaced SBDα α-helical lid as in Sse1-ATP (Fig. 6a and Supplementary Fig. 7a,b). Besides its contacts to the Linker and SBDα, NBD also interacts with the three proximate SBDβ loops that join β-strands (
,
and
) and with those at each end (
joining Linker to β1 and
joining β8 to SBDα). Consistent with a recent NMR study35, the Linker makes extensive contacts with NBD (Fig. 2b and Supplementary Fig. 1,7c). These interactions are similar to those in Sse1-ATP, as previous mutational tests at Asn170, Thr173 and Val389 had indicated23.
Figure 6. Characteristics of domain interfaces in DnaK-ATP.

a, Identification of NBD-contacting loops from SBDβ (upper left; domain coloring as in Fig. 1a), and associated details of the individual contacts as labeled. Residues targeted for mutagenesis are featured in the details. Residues that were tested in the Sse1-ATP structure paper are labeled in black.
b, Growth tests on interfacial mutant variants. Serial dilutions of fresh cultures, transformed with the respective mutant genes, were spotted onto plates and grown at 37°C.
c, ATP-induced tryptophan-fluorescence shifts. Shifts in the maximum of Trp102 fluorescence are shown in the absence of peptide substrates (black bars) and after addition of 30μM substrate peptide NR (red bars). For each protein, the blue shift was averaged from five independent measurements from at least two purifications. Error bars are standard errors (n=5).
Conformations of SBDβ loops
,
and
in DnaK-ATP differ from those in Sse1-ATP, and this generates a number of novel contacts (Supplementary Fig. 7a,b). On
, Val394 and Leu397 make hydrophobic contacts with NBD (Fig. 6a and Supplementary Fig. 7g); and both Lys414 and Asn415 on
form hydrogen bonds with Asp326 and Thr221 from NBD (Fig. 6a and Supplementary Fig. 7h), consistent with previous studies on essentiality for allosteric coupling24,41. Moreover, Ile418 from
mediates hydrophobic interaction with the Linker backbone. Gln442 on
forms hydrogen bonds with Arg151 and Asp148 on NBD (Fig. 6a and Supplementary Fig. 7i). Mutational studies have implicated both Arg151 and Asp148 in allosteric coupling42,43. Loop
is heavily involved in the NBD-SBDβ interface (Fig. 6a and Supplementary Fig. 7d) through Hsp70-conservative residues Asp481 and Ile483 interacting with highly conserved counterparts on NBD (Arg151, Gln152, Lys155 and Ile168). The functional importance of the former two was confirmed by our previous mutagenesis23 and that of Gln152, Arg151 and Lys155 was shown by others34,43,44.
Both
and SBDα of DnaK change radically upon ATP binding, and the NBD-
especially is quite different in DnaK-ATP than in Sse1-ATP. Whereas Asp159 hydrogen bonds to the α-β juncture in Sse1-ATP23, a similar role is played by Asp100 in DnaK-ATP (Fig. 6a and Supplementary Fig. 7e). In keeping, Asp156 (cf. Sse1 D159) was the only one of nine tested DnaK counterparts of Sse1-ATP interfacial residues having a negligible phenotype. NBD-SBDα interactions also differ substantially from those in Sse1-ATP (Supplementary Fig. 7f). Residue Ile160 is still central, interacting with several αA hydrophobes (Ile512, Met515, Leu550 and Ala519; Supplementary Fig. 7f) in keeping with mutational sensitivity at I160 (ref. 23); however, structural adjustments also engage Ala11, Trp102 and Pro113 with Leu507 and Met515 (Fig. 6a and Supplementary Fig. 7j).
To test the importance of previously unidentified contacts in allosteric coupling, we mutated the key residues Val394, Leu397, Ile418, Gln442, Leu507, Met515, and Asp100, individually, in DnaK. These residues are highly conserved across Hsp70s except for Asp100, which is conserved in prokaryotes and mitochondria (Supplementary Fig. 1). As expected, all of these mutations compromised the in vivo activity of DnaK in growth tests (Fig. 6b and Supplementary Fig. 6b,e). To test whether the ATP-induced allosteric coupling is disrupted in these mutants, we purified each mutant variant (except D100L, which showed a moderate growth phenotype) and biochemically characterized allosteric coupling using the tryptophan fluorescence assay. Each of the mutant proteins showed smaller blue-shifts than the WT DnaK (Fig. 6c), confirming defects in ATP-induced allosteric coupling. Moreover, consistent with previous observations, addition of peptide substrate reduced the blue-shifts. In contrast, peptide substrate binding activity was largely intact except for I418D even though all the mutations are to SBD residues (Supplementary Table 2). Moreover, the importance of these residues is mostly preserved in Ssa1 (Supplementary Fig. 6c,d). Thus, as for eight of nine putative DnaK-ATP interfacial contacts identified in Sse1-ATP, these additional contact residues also prove essential for allosteric coupling.
Potential role of dimeric DnaK-ATP
The two almost symmetric DnaK-ATP molecules in the crystal are associated with face-to-face contacts between NBDs and with each SBDα extending alongside NBD Lobe-II from its partner (Fig. 2a). This mode of association resembles that in the Sse1-hHsp70 nucleotide-exchange heterodimer45. The interfacial area between protomers is large (>3000Å2) and shape complementarity is high (0.71) (Supplementary Table 1), indicating that the structure reflects a molecular dimer; however, dimerization of DnaK in solution is weak with a dissociation constant (Kd) of 0.90±0.16 mM from sedimentation equilibrium analysis using analytical ultracentrifugation. The function of dimerization needs to be explored by future work.
DISCUSSION
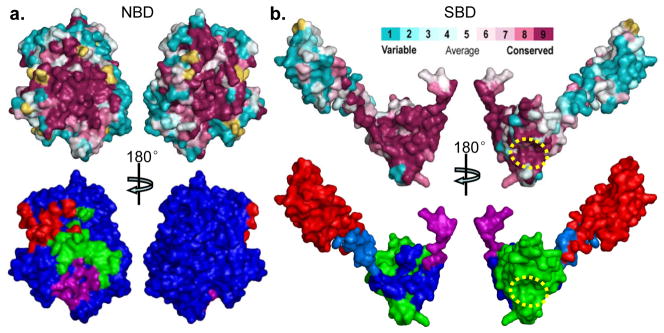
The structure presented here for an intact Hsp70 in the ATP-bound state shows how ATP binding to NBD induces an opening of the polypeptide-binding channel in SBD (Fig. 7). In the ADP-bound state, Hsp70 has high affinity for polypeptide substrates. When substrates bind, the polypeptide-binding channel closes and SBDα covers the polypeptide-binding site. Upon ATP-ADP exchange, Hsp70 is converted into the ATP-bound state, and the NBD and SBD form extensive contacts, which lead to radical conformational changes in SBD. The NBD pulls SBDα away from SBDβ, holds onto the
side of the polypeptide-binding channel on SBDβ through
,
, and
, and then twists
, and
. The distortions in
and
propagate to the other side of the SBDβ, and then the
side of the polypeptide-binding channel is flipped open to make the peptide binding site more accessible. Subsequently, bound polypeptide substrate is released either to fold or to enter another round of the chaperone cycle. Associated biochemical and cellular tests support such a mechanism whereby ATP binding causes release of polypeptide substrates. Conformational changes are transmitted through domain interfaces that are highly conserved across Hsp70s from different species and different cellular compartments. Moreover, our previous mutational tests showed essentialities in common between cytoplasmic yeast Hsp70 Ssa1 and bacterial Hsp70 DnaK, and this is also true here (Fig. 6b and Supplementary Fig. 6c,d). Thus, the proposed allosteric mechanism for Hsp70 activity seems universal.
Figure 7. Schematic model for allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP.
Hsp70 domain coloring is the same as in Fig. 1a. Polypeptide substrates are highlighted in black.
The similarities between the DnaK-ATP and Sse1-ATP structures further confirmed that Hsp70s and Hsp110s are homologs. However, there are also substantial differences in both relative domain orientations and domain interfaces, which could underlie their functional differences. Although peptide substrate binding in both Hsp70s and Hsp110s are ATP-sensitive, most likely they use different mechanisms since the conformations of SBDβ are quite different in these two structures.
Allosteric coupling is a two-way street in Hsp70s: ATP binding effects polypeptide release, as we now better understand, but polypeptide re-binding strongly stimulates ATP hydrolysis. We anticipate further characterization of Hsp70-ATP in an ATPase-stimulating state (Q.L., R. Fan, J.Y., J.L.W., Q.L., and W.A.H., unpublished).
ONLINE METHODS
Protein expression and purification
The DnaK-T199A
(residues 2–610) and SBD-
(residues 388–610) constructs were cloned into a pSMT3 vector and expressed as Smt3 fusion proteins in BL21(DE3) Gold. pSMT vector is a generous gift from Dr. Chris Lima (Structural Biology Program, Sloan-Kettering Institute, New York, NY 10065). The fusion proteins were first purified on a HisTrap column. After the Smt3 tag was removed by Ulp1 protease, the DnaK proteins were separated from the Smt3 tag using a HisTrap column, and further purified using a HiTrap Q and Superdex 200 16/60 column. The Ulp1 cleavage introduces a serine residue, effectively generating a M1S mutation. All the columns are from GE Healthcare. The purified proteins were concentrated to > 30 mg/ml in a buffer containing 5 mM Hepes-KOH, pH7.5 and 10 mM KCl, and flash frozen in liquid nitrogen.
All of the DnaK mutant proteins used in the biochemical assays were cloned into a dnak expression plasmid pBB46, and expressed in a dnak deletion strain BB205 (camRkanR)46. To facilitate purification, a hexahistidine tag was fused to the C-terminus, and addition of this tag has no observable influence on the in vivo function of DnaK. Proteins were purified as described previously36. Briefly, after a HisTrap column, each protein was further purified on a HiTrap Q column. Before flash freezing in liquid Nitrogen, each protein was concentrated to > 10mg/ml. The purification of DnaJ and GrpE was essentially the same as published before30,37.
Crystallization, data collection and model building
For the DnaK-T199A
protein, crystals were grown at 4°C in 1.8–2.0M (NH4)2SO4, 0.1M Hepes-NaOH, pH7.5, and 2–3% PEG400. Crystals of SBD-
were grown at 20°C in 2.0M (NH4)2SO4, and 0.1M Tris-HCl, pH8.5. Both crystals were cryo-protected with 15% glycerol in mother liquor and flash frozen in liquid nitrogen. All diffraction datasets were collected at the X4A and X4C beamlines of NSLS, Brookhaven National Laboratory. To enhance anomalous signals from native DnaK-T199A
crystals, the wavelength of beamline X4A was tuned to the Fe K edge (λ=1.743 Å, E=7.112keV) as verified by fluorescence scan. Anomalous diffraction data sets from 5 crystals were integrated by XDS47 and scaled by SCALA48. A higher resolution native data set was collected at wavelength 0.979 Å using beamline X4C and processed. Data collection and reduction statistics for native and multi-crystal Single-wavelength anomalous diffraction (SAD) data sets are listed in Table 1.
The Multi-crystal native-SAD method32 was used for structure determination based on S, P and Mg anomalous scatterers (51 sites for 1212 residues). Briefly, anomalous signals from 5 crystals were analyzed, scaled and merged to enhance anomalous signals. Anomalous scatters substructure was determined by SHELXD49. The substructure was refined and completed for phase calculation using Phaser50. The phases were then density modified by DM51 to break the phase ambiguity, resulting in sufficient quality of electron density maps for automated model building with Arp/wArp52. Although non-crystallographic 2-fold symmetry is present, such information was not required for native-SAD phasing. Further refinement and model building were carried out using Phenix53 and COOT54. Both multi-crystal SAD data and the 1.96 Å resolution native data were used for refinements. The refined model was validated by Procheck55 and Molprobity56 to ensure good geometry. All residues are in favored regions with no Ramachandran outliers.
To solve the structure of SBD-
, a diffraction data set at 1.62 Å resolution from a single crystal was collected at Beamline X4C at wavelength 0.979 Å, integrated and scaled by HKL2000 (ref. 57). The structure was solved by molecular replacement using the structure of isolated DnaK SBD (PDB code: 1DKZ) as search model. Model building, refinement and validation of SBD-
followed the same protocol as for the full-length DnaK.
Tryptophan fluorescence assay
The assay was performed as described previously36. Briefly, DnaK proteins were diluted to 1 μM in buffer A (25mM Hepes-KOH, pH7.5, 100mM KCl, 10 mM Mg(OAc)2, 10% glycerol, and 2 mM DTT). Right before measurement, ATP or ADP was added, and incubated for 2 minutes. Emission spectra were collected from 310 to 400 nm on a PC1 Photon Counting Spectrofluorimeter from ISS Inc. (Illinois, USA) with excitation wavelength at 295 nm. The peak reading of the ADP spectrum of each protein was set as 1, and the relative fluorescence was plotted versus wavelength. The blue shift for each protein was calculated as the difference of emission maximums between the spectra in the presence and absence of ATP.
Site-directed mutagenesis and growth tests
All DnaK and Ssa1 mutations were introduced using the QuikChange Lightening Site-directed Mutagenesis kit (Stratagene). Growth tests were performed as described previously23 with modifications. For growth tests in DnaK, pBB46 plasmids (ampR) carrying mutant dnak were transformed into a dnak deletion strain BB205 (camRkanR)46. Since E. coli growth is dependent on a functional DnaK at 37°C but not at 30°C, growth tests were carried out at 37°C with control tests performed at 30°C. For each mutant, a fresh overnight culture was prepared from a fresh transformation. 5 μl serial dilutions of the overnight cultures were spotted on LB plates containing 50μg/ml ampicillin, 25μg/ml kanamycin, 25μg/ml chloramphenicol, and 20 μM IPTG, and grown one overnight at 37°C to evaluate growth.
All of the Ssa1 mutants were cloned into an Ssa1-expressing plasmid, pRS313-SSA1, and transformed into a SSA1 mutant yeast strain JB67 (ref. 58). Besides harboring deletions of SSA2-4, JB67 carries a temperature-sensitive SSA1 mutant, ssa1-45, and thus is unable to grow at 37°C. 10 μl serial dilutions of fresh overnight cultures from fresh transformations were spotted onto yeast minimum media lacking histidine. Growth was scored after incubating at 37°C for two to three days. Growth tests at 30°C were used as controls.
Luciferase refolding assay
Purified firefly luciferase was purchased from Promega. Before denaturing at 42°C for 20 min, luciferase was diluted in buffer B (25mM Hepes-KOH, pH7.5, 100mM KCl, 10 mM Mg(OAc)2, 2 mM DTT, and 3 mM ATP) to a final concentration of 100nM in the presence of indicated DnaK proteins (3 μM). To start refolding reactions, a reaction mixture containing DnaK (3 μM), DnaJ (0.67 μM) and GrpE (0.33 μM) was added to heat-denatured luciferase. At indicated time points, a 2 μl of refolding reaction was mixed with 50 μl luciferase substrate, and luciferase activity was read in a Berthold LB9507 Luminometer.
Fluorescence anisotropy assay of peptide substrate binding affinity and kinetics
F-NR, NR peptide (sequence: NRLLLTG) with Fluorescein labeled at the N terminus, was ordered from NEOBioscience with greater than 95% purity. To determine binding affinity, serial dilutions of DnaK proteins were incubated with F-NR (20 nM final concentration) in buffer A for at least three hours to reach equilibrium. Fluorescence anisotropy measurements were carried out on a Beacon 2000 instrument (Invitrogen). To calculate dissociation constants (Kd), data were fitted to a one-site binding equation using PRISM (GraphPad). Each sample was read at least three times, and each protein was repeated at least two times with more than two different protein preparations. For binding kinetics measurements, 10 μM of DnaK proteins were incubated with indicated nucleotide for 2 min, then F-NR was added to start the binding reaction. Anisotropy was read every 10 s.
Molecular dynamic (MD) simulation analysis
MD simulations were carried out using GROMACS59. Simulations for each structure were carried out for 20 ns without consideration of crystal contacts and symmetry mates. Principal component analysis with the G_COVAR program60 on the conformations collected by MD simulations was used to calculate the raw covariance matrix for each pair of Cα atom in three dimensions. To systematically survey the strength of all contacts, we used the normalized covariance factor (scale: 0–1), which is also called cross-correlation61. The higher the normalized covariance factor, the more stable a contact is.
Supplementary Material
Acknowledgments
We thank E. Craig, D. Logothetis, G. Tseng, L. Avery, L. Greene, J. Rife, and C. Fox for critically reading the manuscript and providing insightful suggestions. We are grateful to staff at Brookhaven National Laboratory Beamline X4A and X4C, J. Schwanof, R. Abramowitz, and X. Yang, for their assistance in collecting diffraction data. We thank D. Kumar for technical support, and C. Escalante for the PC1 Photon Counting Spectrofluorimeter. This work was supported by startup funds from the Virginia Commonwealth University School of Medicine (Q.L. (Qinglian)), a New Scholar Award in Aging from the Ellison Medical Foundation (AG-NS-0587-09 to Q.L. (Qinglian)) and a Grant-In-Aid Award from the American Heart Association (11GRNT7460003 to Q. L. (Qinglian)).
Footnotes
Note: Supplementary information is available in the online version of the paper.
Accession codes: Atomic coordinates and structure factors have been deposited in the RSCB Protein Data Bank under the accession number 4JNF, 4JNE and 4JN4.
AUTHOR CONTRIBUTIONS
Q.L. (Qinglian) designed the study and experiments. Q.L. (Qinglian) and L.Z. screened, identified and carried out the cloning for the crystallization construct of full-length DnaK. R.Q. and E.B.S. prepared the crystals for full-length DnaK and isolated SBD, respectively. Q.L. (Qun) and Q.L. (Qinglian) performed the structure determination and modeling. R.Q., E.B.S., K.Q.L., X.X., H.X., J.Y., J.L.W., and C.V. carried out the cloning of the mutations for biochemical analysis, protein purification, biochemical and genetic analysis. L.Z. performed computational analysis on the structures. Q.L. (Qinglian), W.A.H., and L.Z. analyzed the structures, carried out data analysis, and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475 (7356):324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 2.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319 (5865):916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 3.Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92 (3):351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 4.Mayer MP. Gymnastics of molecular chaperones. Mol Cell. 2010;39 (3):321–331. doi: 10.1016/j.molcel.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Zuiderweg ER, et al. Allostery in the Hsp70 Chaperone Proteins. Top Curr Chem. 2012 doi: 10.1007/128_2012_323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudiger S, Buchberger A, Bukau B. Interaction of Hsp70 chaperones with substrates. Nat Struct Biol. 1997;4 (5):342–349. doi: 10.1038/nsb0597-342. [DOI] [PubMed] [Google Scholar]
- 7.Flaherty KM, DeLuca-Flaherty C, McKay DB. Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature. 1990;346 (6285):623–628. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- 8.Sriram M, Osipiuk J, Freeman B, Morimoto R, Joachimiak A. Human Hsp70 molecular chaperone binds two calcium ions within the ATPase domain. Structure. 1997;5 (3):403–414. doi: 10.1016/s0969-2126(97)00197-4. [DOI] [PubMed] [Google Scholar]
- 9.Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science. 1997;276 (5311):431–435. doi: 10.1126/science.276.5311.431. [DOI] [PubMed] [Google Scholar]
- 10.Jiang J, Prasad K, Lafer EM, Sousa R. Structural basis of interdomain communication in the Hsc70 chaperone. Mol Cell. 2005;20 (4):513–524. doi: 10.1016/j.molcel.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang YW, Sun YJ, Wang C, Hsiao CD. Crystal structures of the 70-kDa heat shock proteins in domain disjoining conformation. J Biol Chem. 2008;283 (22):15502–15511. doi: 10.1074/jbc.M708992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X, et al. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272 (5268):1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, et al. NMR solution structure of the 21 kDa chaperone protein DnaK substrate binding domain: a preview of chaperone-protein interaction. Biochemistry. 1998;37 (22):7929–7940. doi: 10.1021/bi9800855. [DOI] [PubMed] [Google Scholar]
- 14.Pellecchia M, et al. Structural insights into substrate binding by the molecular chaperone DnaK. Nat Struct Biol. 2000;7 (4):298–303. doi: 10.1038/74062. [DOI] [PubMed] [Google Scholar]
- 15.Liebscher M, Roujeinikova A. Allosteric coupling between the lid and interdomain linker in DnaK revealed by inhibitor binding studies. J Bacteriol. 2009;191 (5):1456–1462. doi: 10.1128/JB.01131-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buchberger A, et al. Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J Biol Chem. 1995;270 (28):16903–16910. doi: 10.1074/jbc.270.28.16903. [DOI] [PubMed] [Google Scholar]
- 17.Swain JF, et al. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26 (1):27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci U S A. 2009;106 (21):8471–8476. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmid D, Baici A, Gehring H, Christen P. Kinetics of molecular chaperone action. Science. 1994;263 (5149):971–973. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- 20.Flynn GC, Chappell TG, Rothman JE. Peptide binding and release by proteins implicated as catalysts of protein assembly. Science. 1989;245 (4916):385–390. doi: 10.1126/science.2756425. [DOI] [PubMed] [Google Scholar]
- 21.Flaherty KM, Wilbanks SM, DeLuca-Flaherty C, McKay DB. Structural basis of the 70-kilodalton heat shock cognate protein ATP hydrolytic activity. II. Structure of the active site with ADP or ATP bound to wild type and mutant ATPase fragment. J Biol Chem. 1994;269 (17):12899–12907. [PubMed] [Google Scholar]
- 22.Jiang J, et al. Structural basis of J cochaperone binding and regulation of Hsp70. Mol Cell. 2007;28 (3):422–433. doi: 10.1016/j.molcel.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Q, Hendrickson WA. Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell. 2007;131 (1):106–120. doi: 10.1016/j.cell.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smock RG, et al. An interdomain sector mediating allostery in Hsp70 molecular chaperones. Mol Syst Biol. 2010;6 (414):1–9. doi: 10.1038/msb.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mapa K, et al. The conformational dynamics of the mitochondrial Hsp70 chaperone. Mol Cell. 2010;38 (1):89–100. doi: 10.1016/j.molcel.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 26.Marcinowski M, et al. Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat Struct Mol Biol. 2011;18 (2):150–158. doi: 10.1038/nsmb.1970. [DOI] [PubMed] [Google Scholar]
- 27.Schlecht R, Erbse AH, Bukau B, Mayer MP. Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat Struct Mol Biol. 2011;18 (3):345–351. doi: 10.1038/nsmb.2006. [DOI] [PubMed] [Google Scholar]
- 28.McCarty JS, Walker GC. DnaK as a thermometer: threonine-199 is site of autophosphorylation and is critical for ATPase activity. Proc Natl Acad Sci U S A. 1991;88 (21):9513–9517. doi: 10.1073/pnas.88.21.9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osipiuk J, Georgopoulos C, Zylicz M. Initiation of lambda DNA replication. The Escherichia coli small heat shock proteins, DnaJ and GrpE, increase DnaK’s affinity for the lambda P protein. J Biol Chem. 1993;268 (7):4821–4827. [PubMed] [Google Scholar]
- 30.Xu X, et al. The unique peptide substrate binding properties of 110 KDA heatshock protein (HSP110) determines its distinct chaperone activity. J Biol Chem. 2012;287 (8):5661–5672. doi: 10.1074/jbc.M111.275057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smock RG, Blackburn ME, Gierasch LM. Conserved, disordered C terminus of DnaK enhances cellular survival upon stress and DnaK in vitro chaperone activity. J Biol Chem. 2011;286 (36):31821–31829. doi: 10.1074/jbc.M111.265835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Q, et al. Structures from anomalous diffraction of native biological macromolecules. Science. 2012;336 (6084):1033–1037. doi: 10.1126/science.1218753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16 (1):102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Vogel M, Mayer MP, Bukau B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J Biol Chem. 2006;281 (50):38705–38711. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- 35.Zhuravleva A, Gierasch LM. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc Natl Acad Sci U S A. 2011;108 (17):6987–6992. doi: 10.1073/pnas.1014448108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar DP, et al. The four hydrophobic residues on the Hsp70 inter-domain linker have two distinct roles. J Mol Biol. 2011;411 (5):1099–1113. doi: 10.1016/j.jmb.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhattacharya A, et al. Allostery in Hsp70 chaperones is transduced by subdomain rotations. J Mol Biol. 2009;388 (3):475–490. doi: 10.1016/j.jmb.2009.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rist W, Graf C, Bukau B, Mayer MP. Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. J Biol Chem. 2006;281 (24):16493–16501. doi: 10.1074/jbc.M600847200. [DOI] [PubMed] [Google Scholar]
- 39.Swain JF, Schulz EG, Gierasch LM. Direct comparison of a stable isolated Hsp70 substrate-binding domain in the empty and substrate-bound states. J Biol Chem. 2006;281 (3):1605–1611. doi: 10.1074/jbc.M509356200. [DOI] [PubMed] [Google Scholar]
- 40.Lovell SC, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50 (3):437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 41.Montgomery DL, Morimoto RI, Gierasch LM. Mutations in the substrate binding domain of the Escherichia coli 70 kDa molecular chaperone, DnaK, which alter substrate affinity or interdomain coupling. J Mol Biol. 1999;286 (3):915–932. doi: 10.1006/jmbi.1998.2514. [DOI] [PubMed] [Google Scholar]
- 42.Gassler CS, et al. Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc Natl Acad Sci U S A. 1998;95 (26):15229–15234. doi: 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogel M, Bukau B, Mayer MP. Allosteric regulation of Hsp70 chaperones by a proline switch. Mol Cell. 2006;21 (3):359–367. doi: 10.1016/j.molcel.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 44.Davis JE, Voisine C, Craig EA. Intragenic suppressors of Hsp70 mutants: interplay between the ATPase- and peptide-binding domains. Proc Natl Acad Sci U S A. 1999;96 (16):9269–9276. doi: 10.1073/pnas.96.16.9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Polier S, Dragovic Z, Hartl FU, Bracher A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell. 2008;133 (6):1068–1079. doi: 10.1016/j.cell.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 46.Burkholder WF, et al. Mutations in the C-terminal fragment of DnaK affecting peptide binding. Proc Natl Acad Sci U S A. 1996;93 (20):10632–10637. doi: 10.1073/pnas.93.20.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabsch W. Xds. Acta Crystallogr D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 50.Read RJ, McCoy AJ. Using SAD data in Phaser. Acta Crystallogr D. 2011;67:338–344. doi: 10.1107/S0907444910051371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cowtan KD, Zhang KYJ. Density modification for macromolecular phase improvement. Prog Biophys Mol Bio. 1999;72:245–270. doi: 10.1016/s0079-6107(99)00008-5. [DOI] [PubMed] [Google Scholar]
- 52.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams PD, et al. The Phenix software for automated determination of macromolecular structures. Methods. 2011 doi: 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 55.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 56.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 58.Becker J, Walter W, Yan W, Craig EA. Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol Cell Biol. 1996;16 (8):4378–4386. doi: 10.1128/mcb.16.8.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. Journal of Molecular Modeling [online computer file] 2001;7 (8):306–317. [Google Scholar]
- 60.Balsera MA, Wriggers W, Oono Y, Schulten K. Principal component analysis and long time protein dynamics. Journal of Physical Chemistry. 1996;100 (7):2567–2572. [Google Scholar]
- 61.Ichiye T, Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1991;11 (3):205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.