

Summary
Polycomb Group (PcG) proteins play an essential role in the epigenetic maintenance of repressive chromatin states. The gene silencing activity of the Polycomb Repressive Complex 2 (PRC2) depends on its ability to tri-methylate lysine 27 of histone H3 (H3K27) via the catalytic SET domain of the EZH2 subunit, and at least two other subunits of the complex: Suz12 and Eed. We show that the C-terminal domain of Eed specifically binds to histone tails carrying tri-methyl lysine residues associated with repressive chromatin marks and that this leads to the allosteric activation of the methyltransferase activity of PRC2. Mutations in Eed that prevent it from recognising repressive trimethyl-lysine marks abolish activation of PRC2 in vitro and, in Drosophila, reduces global methylation and disrupts development. These findings suggest a model for the propagation of the H3K27me3 mark that accounts for the maintenance of repressive chromatin domains and for the transmission of a histone modification from mother to daughter cells.
Introduction
A cell’s fate is specified by its gene expression profile, often set early in development and maintained throughout the life-time of the cell by epigenetic mechanisms. The PcG group of proteins function by silencing inappropriate expression by maintaining a repressive epigenetic state1. It is thought that the PRC2-mediated trimethylation of lysine 27 on histone H3 plays a key role in marking repressive chromatin domains, while PRC1 is important for effecting transcriptional repression. Thus, once established, H3K27 trimethylation is the epigenetic mark for maintaining transcriptional repression. Mechanisms are therefore required to maintain faithfully this mark in repressed chromatin domains in non-dividing cells and to restore it after the two-fold dilution caused by DNA replication in dividing cells. However, it is not yet clear how PRC2 complexes recognize previously marked sites and how they propagate accurately these repressive marks to unmodified nucleosomes deposited during DNA replication.
The histone lysine methyltransferase (HKMT) activity of the PRC2 complex resides in the SET-domain containing protein Ezh22–5 but activity requires the other subunits of the core complex; the zinc-finger containing Suz12 and the WD40 repeat proteins Eed and RbAp48. In certain contexts, the PHD domain containing protein PHF1 plays an important role in modulating the HKMT activity of PRC26, 7. In this work we have examined the structure and biochemistry of Eed, and determined the role of its homologue ESC in Drosophila development. From this we have established that the Eed subunit of PRC2 binds to repressive methyl-lysine marks ensuring the propagation of H3K27 trimethylation on nucleosomes by allosterically activating the methyltransferase activity of the complex (see Supplementary Fig. S1).
Results
Eed Contains an Aromatic Cage that binds to ‘repressive chromatin’ marks
We crystallised a truncated version of Eed (residues 77 to 441, hereafter ΔEed) and used selenomethionine-substituted ΔEed to solve the structure. The WD40-repeats of ΔEed fold into a seven-bladed β-propeller domain with a central pocket on either end (Fig. 1), as seen previously8. We noticed unaccounted electron density in one of these pockets; our crystallisation mixture included a non-detergent sulfobetaine additive, NDSB-195, which we were able to build into the extra electron density. Since the quarternary amine of the sulfobetaine resembled a trimethylated lysine side chain9 we reasoned that Eed might bind to trimethylated lysine residues on the N-terminal tails of histones.
Figure 1. Trimethyl-lysine binding to an aromatic cage on Eed.

Ribbons representation of the Eed/H3K27me3 complex where Eed is coloured grey and the histone peptide is coloured yellow with its methyl-lysine side chain shown in stick representation. The Cα positions of the aromatic cage are shown as blue circles, and the Cα position of tyrosine 358 by a red circle. The bottom panel shows the methyl-lysine binding site with 2fo-fc electron density for the four cage residues and the H3K27me3 peptide. Designed mutations to the cage are shown in red in parentheses. The side-chain of methionine 256 is also shown; this is equivalent to Met-236 in esc which has been identified from classical genetic screens in Drosophila as essential for the function of Eed.
Histone lysine residues methylated in vivo include lysine 4 of histone H3 (H3K4), H3K9, H3K27, H3K36, H3K79, H4K20 and H1K26. We measured the binding affinity of ΔEed to trimethylated versions of these lysine residues using synthetic peptides by fluorescence competition assays. ΔEed bound to H3K9me3, H4K20me3, H1K26me3 and H3K27me3 peptides with Kd values ranging from 10 to 45 μM and the binding became approximately 4-fold weaker for each successive loss of a methyl group from the methyl-lysine (Supplementary Table S1). Notably, ΔEed did not bind appreciably to H3K4me3, H3K36me3 or H3K79me3, “marks” associated with active transcription10. We validated these results by Isothermal Titration Calorimetry (Supplementary Table S1 & Fig. S2b) and there is good agreement between the two independent methods.
Next, we solved the structure of ΔEed co-crystallised with H1K26me3, H3K27me3, H3K9me3 and H4K20me3 peptides (Supplementary Table S2 & Fig. S3). The peptides in the four co-crystal structures adopt similar, largely extended structures and all exploit the aromatic cage of ΔEed to recognize the tri-methyl lysine residue (Fig. 1 & Supplementary Fig. S4). This is the first example of such a binding site on a β-propeller domain and it consists of three aromatic side-chains, Phe-97, Tyr-148 and Tyr-365 (Fig. 1). The tri-methylammonium group of the lysine is inserted into this cage and is stabilised by van der Waals and cation-π interactions. A fourth aromatic side-chain (Trp-364) interacts with the aliphatic moiety of the lysine side-chain via hydrophobic interactions (Figs. 1, 2 & S5). Adjacent to the methyl-lysine pocket, ΔEed makes two hydrogen bond interactions with carbonyls on the peptides (Fig. 2A). First, the main-chain carbonyl of the methyl-lysine residue hydrogen bonds to the side-chain of Arg-414. Second, the main-chain carbonyl of the residue immediately N-terminal of the methyl-lysine on the peptide makes a hydrogen bond with the main-chain amide of Trp-364. The residues flanking the methyl-lysine residue, at the −1 and +1 positions, are oriented away from the protein whereas the next residues at the −2 and +2 positions make important contacts (Fig. 2). Comparison of the four complexes (Fig. 2 & Figs. S2A, S4), suggests an important role for two distinct hydrophobic interaction sites (Fig. 2B). H1K26, H3K9 and H3K27 each have an alanine residue, two amino-acids N-terminal to the lysine (−2), which fits into a small pocket on the surface of Eed formed by the hydrophobic moieties of Trp-364, Tyr-308 and Cys-324 (Fig. 2C). The size of this pocket is sufficient to accommodate an alanine residue but not larger hydrophobic residues. In the case of H4K20 peptide, the only one of the four that bound to ΔEed and lacks an alanine at −2, its binding is facilitated by an alternative hydrophobic interaction between the leucine residue in the +2 position of the peptide with a second hydrophobic pocket formed by residues Ile-363, Ala-412 and Gln-382 (CG) of Eed (Fig. 2D). It appears that the ability to exploit one or other of these two small hydrophobic pockets is an important component of the specificity of Eed towards the methyl-lysine marks associated with repressive chromatin. However, the affinity of Eed for these modified peptides is relatively modest and it is likely that this interaction only becomes physiologically relevant in association with the histone binding activity of other components of the PRC2 complex, as suggested by earlier work on Drosophila PRC2 11.
Figure 2. Interactions between Eed and trimethylated histone peptides.

(A) Schematic representation of the interaction between Eed and H3K27me3. For clarity, the aromatic methyl-lysine binding cage has been omitted and the methylated lysine side-chain shown as a yellow circle. Hydrogen bonds from the main-chain carbonyl of the methyl-lysine, and the residue immediately N-terminal to it, with Eed are shown as dashed lines. The green hatching indicates the hydrophobic pocket on EED which accommodates the alanine side chain two residues before the methyl-lysine. (B) Eed is shown in surface representation with a composite of two of its cognate peptides shown in sticks representation and coloured yellow (H3K9me3) and pink (H4K20me3). (C) shows the pocket on Eed that accommodates Ala (−2) from the H3K9me3 peptide while (D) shows the other pocket that contains and Leu (+2) from the H4K20me3 peptide. The Eed surface is coloured according to atom type.
To probe the physiological role of the aromatic cage of Eed, site-directed mutants of a number of the cage residues were created. Mutations of Phe-97, Trp-364 and Tyr-365 to alanine produced well-behaved protein and competition experiments showed that Trp-364Ala and Tyr-365Ala, had no detectable binding to H1K26Me3 peptides, while Phe-97Ala ΔEed bound about eight-fold more weakly than wild-type ΔEed to histone peptides (Supplementary Table 1). As a control for the effect of mutation of an aromatic residue on the Eed structure that is not involved in the aromatic cage, we also generated the mutation Tyr-358Ala (Fig. 1); binding by this mutant was reduced by about two-fold (Supplementary Table S1).
PRC2-Ezh2 and Eed specifically bind nucleosomes trimethylated on H3K27
Next, we used nucleosome arrays reconstituted with chemically modified histones that carry a single modification of the four possible methylation states of H3K27, H3K36 or H3K912. The nucleosome arrays were incubated with full-length His-tagged Eed protein followed by NiNta pull-down assays. Western blotting for H3 and Eed demonstrated interaction between Eed and nucleosomes containing trimethylated H3K27 (Fig. 3). This interaction was specific as Eed was not able to interact with chromatin reconstituted with histones containing the different levels of H3K36 methylation, (Supplementary Fig. S8) but did bind to chromatin trimethylated on H3K9 (data not shown). Interestingly, the truncated ΔEed protein tested in the peptide binding experiments also failed to interact with nucleosomes (Supplementary Fig. S8). Presumably, the diminished binding is due to the absence of a previously characterized H3 binding site within the N terminus of Eed 13, which may act together with the methyllysine binding site to achieve stable binding.
Figure 3. Eed and PRC2 interaction with Chromatin.
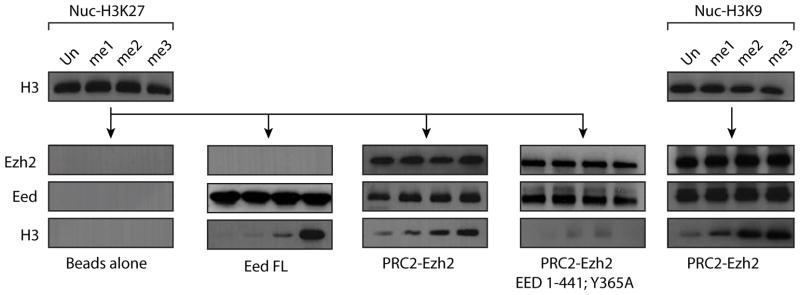
Pull-down experiment to analyze the interaction between Eed full-length, PRC2-Ezh2 wild type or reconstituted with Eed Tyr-365Ala and H3K27 modified chromatin (left) or between PRC2-Ezh2 wild type and H3K9 modified chromatin (right). Note that “beads only” control for interaction H3K9 modified chromatin is not shown but was identical to the control shown in the figure.
Given that other subunits of PRC2 contact histones and thus modulate chromatin binding, we repeated the nucleosome binding experiment using a PRC2 complex purified from insect cells co-infected with baculovirus expressing each of the subunits (Fig. 4A left). Although, as expected, the reconstituted PRC2 complex displayed some binding to unmodified chromatin, the complex bound significantly tighter to chromatin carrying the H3K27me3 or the H3K9me3 modification (Fig. 3). Interestingly, PRC2 reconstituted with Eed carrying the Phe-97Ala or Tyr-365Ala substitution does not show binding to chromatin under these conditions, with either methylated or unmodified nucleosomes (Fig. 3 and data not shown). Together, our results demonstrate that the aromatic cage in Eed is critical for the PRC2 complex to bind to repressive marks, through its specific recognition of defined (repressive) trimethylated-lysine residues.
Figure 4. Peptide mimicking repressive marks stimulates PRC2 activity.

(A) Left, coomassie blue staining of reconstituted PRC2-Ezh2 complex. Right, HMT assay with PRC2-Ezh2 alone or in the presence of 10 and 40 μM H3K27, unmodified, mono, di or tri methylated peptides. (B) Titration of the methyl donor (S-adenosyl-Methionine) in the presence of H3K27me0/1/2/3 peptides (C) Nucleosome titration in the presence of H3K27me0/3 peptides. (D) Left, Coomassie blue staining of reconstituted PRC2-Ezh2 Tyr365Ala complex and right, HMT assay with the corresponding complex in the same condition as (A) (E) Relative PRC2 histone methyltransferase activity in the presence of various peptides as indicated in the legend. (F) Table indicating the peptides used for the stimulation study as well as their Kd values (μM) for ΔEed binding (G) Relative PRC2 histone methyltransferase activity in the presence of various peptides as indicated in the legend.
Trimethylated repressive marks stimulate PRC2 enzymatic activity
Since a likely function for the binding of PRC2 to trimethylated lysine would be to contribute to the propagation of the H3K27me3 mark, we performed histone lysine methyltransferase (HKMT) assays using recombinant oligonucleosomes in the presence of methylated peptides. Addition of unmodified or monomethylated H3K27 peptides did not significantly affect the enzymatic activity of PRC2 but trimethylated peptides activated it about seven-fold (Fig. 4A). Stimulation of enzymatic activity by H3K27me3 peptide reached a plateau around 100μM while half maximum stimulation is achieved around 30-40μM (Fig. S7), which is in good agreement with the dissociation constant determined for ΔEed and H3K27me3 peptide (Supplementary Table S1) and gives us strong confidence that the binding event we observe with purified, truncated Eed is closely correlated with the allosteric activation mechanism. We also determined the Michaelis parameters for PRC2 in the presence of variously methylated H3K27 peptides (Fig. 4B and C). During titrations of SAM we observed a dramatic increase in the maximum reaction rate (Vm) in the presence of H3K27me3 peptide. A similar result was observed with titration of nucleosomes (Fig. 4C). Importantly, in both cases the substrate concentration required to achieve the half-maximal reaction rate (Km) is not significantly affected by the incubation with peptides.
To ascertain if the observed stimulation was Eed-mediated, mutant PRC2 complexes containing Eed (Phe-97Ala) or (Tyr-365Ala) were reconstituted (Fig. 4D and Supplementary S8). These mutant rPRC2 complexes retain a similar basal activity to wild-type but neither mutant rPRC2 was stimulated by the addition of H3K27me3 peptides (Fig. 4D). Our data also show that H3K9me3, H4K20me3 and H1K26me3 peptides were all able to stimulate PRC2 activity to some extent whereas H3K4me3 and H3K36me3 peptides were ineffectual (Fig. 4E). However, we noticed that binding affinity to Eed and stimulation of PRC2 activity do not strictly correlate (i.e. H3K9me3 has a good binding affinity for Eed but stimulates PRC2 activity relatively poorly). To investigate the role of histone sequence in binding/activation we first mutated the arginine residue at the −1 position, present in all four histone peptides that activate the methyltransferase activity of PRC2, to alanine (R26A of H3K27me3 in Fig. 4F & G). Remarkably, whilst the binding of this mutant peptide to ΔEed is only reduced about 1.5-fold (Fig. 4F), it is no longer able to activate PRC2 HKMT activity (Fig. 4G), demonstrating that repressive histone peptide binding to the aromatic cage of Eed is necessary, but not sufficient for PRC2 activation. To further test this model we made a series of chimeric and mutant peptides that show that the lysine at (−4), the alanine at (−3) and the arginine at (−1) are not important for binding to Eed but are key to the activation of PRC2. We envision that these are the residues that mediate an interaction with another part of the PRC2 complex that leads to its activation.
Recognition of repressive trimethylated marks is required for PRC2 function
To evaluate the importance of Eed binding to trimethylated marks in vivo, we turned to ESC, the Eed homolog in Drosophila, and tested the effect of mutating its aromatic cage. We reconstituted the dPRC2 complex and showed that addition of H3K27me2-3 peptides to the HKMT assay resulted in a robust stimulation of dPRC2 enzymatic activity (Fig. 5A). ESC is required throughout development but in the early embryo the maternal stock of esc product is critical, as evidenced by the resultant derepression of homeotic genes in embryos produced by esc− mothers 14, 15. At later stages of development, PRC2 activity is sustained through the overlapping participation of ESC and its close homologue ESCL 15, 16. Overexpression of ESC in the ovaries (e.g. in a female with one extra esc copy) can supply enough function to allow development of esc embryos, producing flies that are virtually normal except for the eponymous extra sex combs in males. We constructed mutations affecting the aromatic cage: Phe-77Ala (equivalent to Eed Phe-97) and Phe-345Ala (equivalent to Eed Tyr-365), as well as Tyr-338Ala (equivalent to Eed Tyr-358Ala) just preceding the aromatic cage (Fig. 5B), and expressed MYC-tagged wild type or mutant esc transgenes under the control of the esc promoter (Fig. 5B). While the wild type transgene rescued the extra sex comb phenotype almost completely (217 out of 218 males counted), the aromatic cage mutant transgenes were ineffectual (no rescue in several hundred males examined) (Fig. 5C). Flies lacking both zygotic ESC and ESCL in these crosses produce larvae with poorly developed brain and imaginal discs, which die when they pupate. This lethality is completely rescued by one copy of the wild-type esc>MYC-ESC transgene. In contrast, none of the aromatic cage mutant transgenes were able to rescue the lethality even when present in two copies (Fig. 5C), although the esc>MYC-ESC Phe-77Ala transgene alleviated the brain and imaginal disc phenotypes (data not shown). Of note, zygotic expression of the Phe-345Ala transgene impaired the contribution of a wild type esc indicating that this mutant acts as a dominant negative. The failure of the mutant Myc-ESC to rescue is not due to instability or to inability to be incorporated into a PRC2 complex: the ESC mutants were expressed at levels comparable to that of the wild type. Furthermore, immunoprecipitation experiments revealed that the mutant ESCs co-immunoprecipitated with endogenous E(Z) as efficiently as the wild type protein (Fig. 5D). To determine whether the mutant ESCs affected PRC2 function with respect to its gene targeting or activity, we performed chromatin immunoprecipitation followed by qPCR with the primer sets indicated in Figure 5E. Immunoprecipitation using anti-E(Z) shows that wild type Myc-ESC is nearly as effective as endogenous ESC (compare with the esc+ escl− chromatin) while PRC2 complex with Myc-ESC bearing mutations in the aromatic pocket is recruited less efficiently to the Ubx Polycomb Response Element (PRE) (Fig. 5F). Chromatin immunoprecipitation with anti-H3K27me3 antibodies also shows that wild type Myc-ESC is nearly as effective as endogenous ESC (yw) in trimethylating H3K27 in the Ubx upstream enhancer region (PBX, -30 kb), in the vicinity of the PRE (FM1, FM6, -23 kb) or at the Ubx promoter. Importantly, the mutant ESCs are deficient in the extent of H3K27me3 (Fig. 5G) and this decrease correlates with the phenotypes described in Figure 5C. Importantly, the observed effects are due to the aromatic cage, as a mutation of Tyr-338Ala, which is not important for cage formation, had no effect (Fig 5G). Finally, we analysed the global levels of H3K27 methylation by western blot (Fig. 5H). We observed an almost complete loss of H3K27me3 in extracts from esc− escl− larvae expressing the mutant ESCs. Perhaps surprisingly, the H3K27me2 levels were equally strongly affected.
Figure 5. The aromatic cage in Drosophila ESC is important for its in vivo function.

(A) Left, Coomassie blue staining of reconstituted dPRC2-Ez complex. Right, HMT assay with dPRC2-Ez and H3K27, unmodified, mono, di or tri methylated peptides. (B) Top, amino acid residues Phe-77, Tyr-338 and Phe-345 that were mutated to alanine in Drosophila ESC and corresponding residues in Eed. Bottom, schematic representation of transposon constructs. The Myc-tagged ESC or its mutants are expressed under the control of the esc gene promoter. (C) Rescue experiment. Details of the crossing schemes are shown in Supplementary Figures S9 and S10. Several independent lines were examined for each transgenic construct and showed the same phenotype except for one line of Myc-ESC Tyr338Ala. In the case of Myc-ESC Phe345Ala, transgenes were inserted at •C31 att sites at 68E and 86Fb, respectively. For direct comparison, wild type Myc-ESC lines were also established at the same chromosomal location and showed the same results as other wild type Myc-ESC lines established by conventional P-element transformation (D) ESC aromatic cage mutation does not impair binding to E(Z). Both Myc-ESC and Myc-ESC Phe-77Ala co-immunoprecipitated E(Z) from ovarian extracts of heterozygous esc−escl− flies. The double Myc-ESC bands are caused by the well known phosphorylation of ESC. (E) Scheme indicating the genomic location of primers used for ChIP. (F) ChIP analysis of E(Z) binding to the bxd PRE (FM4) in homozygous esc6, escld01514 carrying the same Myc-ESC transgenes. In the presence of the aromatic cage mutations, E(Z) binding is strongly reduced. yw indicates the wild type stock with endogenous wild type ESC and ESCL. (G) ChIP analysis of the H3K27me3 distribution at four sites in the Ubx gene. H3K27me3 is drastically reduced in the presence of the aromatic cage mutations. (H) Histone H3K27 methylation in esc6, escld01514 double mutant larvae expressing Myc-ESC transgenes. Total protein lysates from wild type and homozygous esc6, escld01514 3rd instar larvae expressing Myc-ESC transgenes were used for western blot analysis with anti-H3, anti-H3K27me2 and anti-H3K27me3 antibodies. The aromatic cage mutations Myc-ESC Phe77Ala and Myc-ESC Phe345Ala cause an almost complete loss of histone H3K27 di- and trimethylation while wild type Myc-ESC fully restores the loss of H3K27 di- and tri-methylation in esc6, escld01514 double mutant larvae.
Discussion
Chromatin domains are distinguished by the presence of a characteristic set of marks. When these marks are used to sustain an epigenetic state, eukaryotic cells must have the means of propagating these marks through cellular division and of ensuring that they obey appropriate boundaries during development. That PRC2 might recognize the chromatin mark it sets was anticipated by Hansen et al. 17 who reported that PRC2 binds to H3K27me3, although this study did not address the mechanism for the propagation of H3K27me3. Our work shows the structural and functional basis for epigenetic self-renewal and leads us to conclude that PRC2 readout of H3K27me3 (and to a lesser extent other “repressive” marks) is key to the propagation of this repressive mark.
A combination of aromatic and hydrophobic residues is commonly used by proteins that recognise methylated lysine residues and has been found in chromo-, tudor- and plant homeo-domains (PHD)18–20, but no such arrangement has previously been described for any WD40-repeat-containing protein (e.g. Supplementary Fig. 7 and 21). Sequence analysis across the family of β-propeller domains leads us to conclude that the ability of Eed to specifically recognise repressive methyl-lysine marks is a feature, limited amongst WD40 proteins, to Eed-related molecules (Supplementary Fig. S6).
This methyl-lysine interaction provides an additional contribution to nucleosome binding that is mainly driven by a combination of contacts from other subunits of PRC2; RbAp binds to histone H422, 23, and the N-terminal domain of Eed binds to H313 and it may well be that these different interactions act cooperatively. In Drosophila, recruitment of PRC2 may also be facilitated by certain DNA-binding factors 24, 25. Our Drosophila experiments show that when the Drosophila Eed orthologue ESC bears mutations in the aromatic cage, the recruitment of PRC2 to the PRE is less effective, as shown by the drop in E(Z) binding to the bxd PRE, the massive reduction in the global level of H3K27me2/3 and by the phenotype of the Phe-77Ala and Phe-345Ala mutants. Our chromatin modification assays suggest that a major effect of Eed binding to repressive methyl-lysine marks is the stimulation of PRC2 methyltransferase activity, thus providing a mechanism for the propagation of this mark. Thus, when PRC2 is recruited to appropriate chromatin domains, the presence of pre-existing H3K27me3 marks on neighbouring nucleosomes activates the complex to carry out further methylation of unmodified H3K27 (Supplementary Fig. 1). Accordingly, a PcG target gene that had been repressed in one cell cycle will tend to be repressed again in the following cell cycle, and previously active genes will be left unmodified at H3K27. We propose that the ability to recognize a previously established mark that triggers its renewal is a feature that will be found in other epigenetic mechanisms mediated by histone modifications.
On-line methods
Protein Expression and Purification
Residues 78-441 of Eed (ΔEed) were cloned into pGEX-4T vector (Amersham Biosciences) and expressed in E. coli. Proteins were prepared as N-terminal glutathione-S-transferase (GST) fusion proteins and cleaved from GST with human α-thrombin (Haematologic Technologies, Inc.). Proteins were purified further using size exclusion chromatography (Superdex 200, GE Healthcare) in buffer containing 50 mM Tris-HCl pH 8.7, 150 mM NaCl and 3 mM TCEP. Site-directed mutants of ΔEed were generated with the ExSite protocol (Stratagene) and purified in a similar manner. Crystallographic and binding studies were carried out using a construct containing the mutation Met-370Thr, however the binding properties of this construct are identical to those of the ‘wild type’ construct. Peptides were synthesised and purified by reversed phase HPLC at the University of Bristol Peptide Synthesis Facility. Peptide masses were verified by mass spectrometry.
Crystallography
For crystallisation trials, protein solutions were prepared as either ΔEed alone at 2.5 mg/ml or as a complex solution at 1.5 mg/ml with peptide at a 7-fold higher molar ratio. All protein solutions contained TCEP at 15 mM concentration. Crystals were grown at 18°C using the vapour diffusion technique in hanging drops. Drops were prepared by mixing equal volumes of ΔEed protein alone with reservoir solution containing 4.0–4.1 M formate and 0.6–0.7 M NDSB-195, or by mixing equal volumes of ΔEed protein complex with 3.7–3.9 M formate solution. Crystals were transferred into mother liquor with 5 to 10% glycerol prior to flash cooling in liquid nitrogen. Diffraction data for the ΔEed-only native and selenomethionine crystals were collected at the Daresbury synchrotron on beamline 10.1 at the peak wavelength for selenium. Diffraction data for the H1K26me3, H3K9me3 and H4K20me3 protein complex crystals were collected using an in-house MicroMax 007HF rotating anode coupled to a RaxisIV++ detector. Data for H3K27me3 was collected at Diamond Light Source on beamline I04 at a wavelength 0.97 A. Data were integrated using Denzo and scaled with Scalepack28. Phases for the selenomethionine-substitued ΔEed structure were generated and extended using the single wavelength anomalous dispersion (SAD) method and SOLVE29 and RESOLVE30 programs. Phases from RESOLVE were used to autobuild a model with ARP/wARP31 in warpNtrace mode. The protein complex crystal structures were solved by molecular replacement using AMoRe32 and the selenomethionine-substitued ΔEed structure as the search model. Standard refinement was carried out with refmac533 and CNS34 together with manual model building with O35 and Coot36. Figures were created with Pymol (DeLano Scientific; http://pymol.sourceforge.net/).
Binding studies
Histone peptide binding experiments were performed by competition fluorescence spectroscopy and Isothermal Titration Calorimetry (ITC). All fluorescence emission spectra were measured using a dansyl labelled peptide (sequence: KKKARK(Me3)SAGAAK-dansyl) at 20¼ C in 50 mM Tris-HCl pH 8.7, 150 mM NaCl and 3 mM TCEP. Measurements were recorded using a SPEX FluoroMax fluorimeter (excitation wavelength 330nm, emission wavelength 537nm). Binding of dansyl peptide to Eed was monitored by titrating excess Eed into 5 μM peptide. Dissociation constants for the unlabelled histone peptides were determined using a competition assay by adding excess unlabelled peptide to a complex of 35 uM Eed with 35 uM dansyl peptide and monitoring the subsequent reduction in fluorescence. ITC measurements were carried out by injecting peptide at 400–1000 μM into the ITC cell containing Δ77Eed at 40–100 μM. Experiments were performed at 20¼ C in 50 mM Tris-HCl pH 8.7, 150 mM NaCl and 3 mM BME.
Methyl lysine analog production
Pseudo-lysine (ϕK) containing histones were generated by a modification of known literature methods12. In brief; proteins to be modified (5–10mg) were weighed into 1.5 ml siliconized Eppendorf microcentrifuge tubes and 950 μl alkylation buffer (4 M guanadinium chloride, 1 M HEPES, 10mM D/L-methionine pH 7.8, the solution is passed through a 0.22 micron filter and purged with argon prior to use) was added. Proteins that do not readily dissolve were sonicated for 10–15 min in a Branson 1510 ultrasonic cleaning bath at ambient temperature to effect dissolution. The resultant clear colorless solutions were treated with 20 μl of a 1 M DTT solution in alkylation buffer prepared just prior to use, and agitated at 37°C for 1hr. At the end of this period the fully reduced proteins were treated as indicated below.
Pseudo-lysine (ϕK-NH2): to the reduced histone was added 100 μl of a 1 M 2-chloroethylamine monohydrochloride solution in alkylation buffer prepared just prior to use. The mixture was agitated in the dark at 45°C for 2.5 hr. At the end of this period the mixture was treated with a second portion of DTT (10 μl of the above 1 M solution) and heated with agitation at 45°C for an additional 2.5 hr. At reaction was then quenched with BME (50 μl) and cooled to room temperature prior to purification as outlined below.
Pseudo-monomethyl lysine (ϕK-Me1): to the reduced histone was added 100 μl of a 1 M N-methylaminoethyl chloride hydrochloride solution in alkylation buffer prepared just prior to use. The mixture was agitated in the dark at 45°C for 2.5 hr. At the end of this period the mixture was treated with a second portion of DTT (10 μl of the above 1 M solution) and heated with agitation at 45°C for an additional 2.5 hr. At reaction was then quenched with BME (50 μl) and cooled to room temperature prior to purification as outlined below.
Pseudo-dimethyl lysine (ϕK-Me2): to the reduced histone was added 50 μl of a 1 M 2-(dimethylamine)ethyl chloride hydrochloride solution in alkylation buffer prepared just prior to use. The mixture was agitated in the dark at 25°C for 2 hr. At the end of this period the mixture was treated with additional DTT (10 μl of the above 1 M solution) and agitated at 25°C for 30 min prior to addition of 50 μl of the 1 M 2-(dimethylamine)ethyl chloride hydrochloride solution. The reaction was allowed to proceed at ambient temperature for an additional 2 hr, quenched with BME (50 μl), and cooled to room temperature prior to purification as outlined below.
Pseudo-trimethyl lysine (ϕK-Me3): to the reduced histone was added 100 μl of a 1 M (2-bromoethyl) trimethylammonium bromide solution in alkylation buffer prepared just prior to use. The mixture was agitated in the dark at 50°C for 2.5 hr. At the end of this period the mixture was treated with a second portion of DTT (10 μl of the above 1 M solution) and heated with agitation at 50°C for an additional 2.5 hr. At reaction was then quenched with BME (50 μl) and cooled to room temperature prior to purification as outlined below.
Purification Scheme
A PD-10 column was pre-equilibrated with 0.1% BME in 18 water. This was loaded with the reaction mixture, the reaction tube was rinsed with an additional 1 ml of alkylation buffer and this was also added to the top of the column. The proteins were then eluted according to the manufacturers protocol for centrifugal isolation. The eluent was frozen and lyophilized prior providing the modified histones as crispy foams. A portion of each (~0.1 mg) was analyzed by RP-HPLC and MALDI-TOF mass spectrometry to insure product identity and homogeneity.
Chromatin and interaction experiment
Histone H3 variants with the respective point mutations (Lys to Cys at the position to be modified and Cys to Ala at position 110) were expressed in E. coli, purified from inclusion bodies, and solubilized in 7 M guanidine hydrochloride, 20 mM Tris pH 8, 10 mM DTT. After dialysis to replace guanidine hydrochloride with 7 M urea, histones were further purified by sequential anion and cation chromatography. Histone-containing fractions were pooled, dialyzed against 5 mM beta-mercaptoethanol, and lyophilized. Histones were reconstituted into octamer as previously described37 and chromatin was formed by salt dialysis. To prevent unspecific binding to free histone in the pull-down experiment, chromatin was further purified on an agarose2 column. For interaction, 2 ug of chromatin was incubated with 2 ug of protein or complex of interest in Buffer A (50mM Tris pH8.0, 50mM NaCl, EDTA 1mM, NP40 0.1%) for 2 hours at 4¼C in the presence of NiNta beads (Eed) or M2-Beads (PRC2). Beads were extensively washed, eluted with 1X SDS-Page loading buffer and analyzed by western blot.
HKMT assay
HKMT assays were performed as previously described38. For autoradiography exposure, the conditions were as followed: 1.5 ug of chromatin, 100 ng of reconstituted PRC2 complex, peptide (5–40 μM), 3H-SAM (0.3 μM) was used. For scintillation counting, the assay was performed as followed: 1.5 ug of chromatin, 50 ng of reconstituted PRC2 complex, peptide (100 μM), SAM (24.8 μM, 3H-SAM/SAM 1/30 ratio) and 15 min, unless otherwise stated in the figure legend.
SF9 culture, infection and complex purification
As previously described38.
Antibodies
H3K27me1 (Millipore), H3K27me2 (Abcam ab24684 and ab6002), H3K27me2 and H3K27me3 (Gift from Thomas Jenuwein), total H3 (Abcam, ab1791), Flag (Sigma), Myc 9E10 (chemicon). Previously described antibodies were used for Eed38 and E(Z)31.
Fly strains and mutants
The Df(1)y1w67c23 strain (yw) was used as wild type control and for P-mediated germ-line transformation. For transgene insertion at specific genomic sites, we used fly strains in which the •C31 integrase gene is inserted on the X chromosome and attP landing sites are located in 68E or 86Fb, gifts from K. Basler39. Mutant strains for esc6 and esc6, escld01514 were used as described previously15 and detailed crossing schemes to test transgene function are given in Supplementary Figures S9 and S10. ChIP with larval tissues were done using flies homozygous for esc>Myc-ESC in homozygous esc6, escld01514 background or flies homozygous for esc>Myc-ESC Phe77Ala in homozygous esc6, escld01514 (selected using a CyO, GFP balancer).
Transposon construction
ESC mutant transgenes were produced using the esc>Myc-ESC construct15 as starting material. The esc>Myc-ESC Phe77Ala and esc>Myc-ESC Tyr338Ala constructs for conventional P-mediated germ-line transformation were assembled in the pCaS-escp construct15. To generate transgenic lines for esc>Myc-ESC or esc>Myc-ESC Phe345Ala at the same chromosomal locations, we utilized the •C31 recombinase-mediated cassette exchange technique (RMCE)40. For recombinase-mediated cassette exchange, pCaSpeRattB plasmid was first generated by excising with BamHI the UAS and hsp70 minimal promoter from pUASTattB (a gift of K. Basler) and replacing them with a PCR-amplified multicloning site with BglII cohesive ends. A fragment containing the esc promoter was excised from the esc>Myc-ESC construct by NotI and KpnI and inserted in the NotI-KpnI site of pCaSpeRattB to generate pCaB-escp. The esc>Myc-ESC and esc>Myc-ESC Phe345Ala constructs for RMCE were constructed by inserting the wild type esc cDNA or esc cDNA with the relevant mutation in the KpnI site of the the pCaB-escp.
Western blotting and immunoprecipitation
Drosophila larval extracts were prepared as previously described15. Briefly, approximately 30 3rd instar larvae, frozen in liquid nitrogen, were pulverized in protein lysis buffer I (50 mM Tris pH 6.8, 100 mM DTT, 2% SDS, 5 mM EDTA, 1 mM PMSF, 10% glycerol) using a micropestle. After heating at 95¼C for 5 min, and centrifuging 10 min, supernatants were used for western blot analysis. Ovary extracts were made by homogenizing 60 sets of ovaries in extraction buffer (30 mM HEPES-KOH pH 7.6, 150 mM KOAc, 2 mM MgOAc, 5 mM DTT, 0.1% NP40 and Protein Inhibitor cocktail (Roche)) using a micropestle and the extracts cleared by centrifugation. Immunoprecipitation was performed using anti-Myc and protein G sepharose beads (GE Healthcare). The beads were washed five times for 5 min in extraction buffer, boiled in sample buffer and analysed by western blotting. Rabbit anti-H3 and rabbit anti-H3K27me2 or -me3 were used with anti-rabbit IgG-APase as a secondary antibody. The rabbit anti-E(Z) was used with goat anti-rabbit IgG-HRP and mouse anti-Myc was used with goat anti-mouse IgG-HRP Light chain-specific.
ChIP with Drosophila larvae
ChIP was done essentially as previously described with slight modifications30. Approximately 300 mg of larvae were taken for two independent experiments. The frozen larvae were first pulverized using a mortar and pestle in liquid N2, then homogenized with 10 strokes of a Dounce homogenizer in cross-linking solution (1.8% Formaldehyde, 50 mM HEPES 8.0, 1 mM EDTA, 0.5 mM EGTA, 100 mM NaCl). The homogenates were incubated by rotating at room temperature for 15 min. The fixation was stopped by washing for 5 min in PBS/0.01% Triton X-100/0.125M Glycine three times with mixing. After centrifugation at 1500xg for 3 min at 4°C, the pellets were washed for 10 min in 1 ml of wash buffer A (10 mM Hepes 7.6, 10 mM EDTA, 0.5 mM EGTA, 0.25% Triton X-100) and subsequently for 10 min in 1 ml wash buffer B (10 mM Hepes 7.6, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.01% Triton X-100) by mixing gently. The washed pellets were resuspended in sonication buffer (10 mM Hepes 7.6, 1 mM EDTA, 0.5 mM EGTA). Sonication was performed with a Bioruptor as described previously31. After sonication, samples were supplemented with N-lauroylsarcosine (0.5% final) and incubated for 10 min at 4°C with gentle mixing. Soluble chromatin was fractionated by centrifugation at top speed for 10 min and transferred to new eppendorf tubes. The chromatin was aliquoted, quick-frozen in liquid N2 and stored at −80°C. All steps for immunoprecipitation were performed at 4°C. An aliquot of sonicated chromatin was first precleared by mixing with Protein G Sepharose beads (GE helthcare) or Protein A Sepharose beads (Sigma) for 1 hr. After centrifugation, precleared chromatin was incubated with anti-Myc9E10 (Chemicon), anti-EZ or anti-H3K27me3 (Abcam, ab6002) overnight. Protein G or Protein A Sepharose beads were then added to allow binding to the antibody for 2 hr and then washed 5 times with RIPA buffer, once with LiCl buffer (10 mM Tris-Cl (pH 8.0), 250 mM LiCl, 0.5% NP-40, 0.5% Sodium deoxycholate and 1 mM EDTA) and twice with TE. The beads were resuspended in 100 μl of TE, and treated with 0.1 mg/ml RNase A at 37°C for 30 min. After supplementing with SDS (0.5% final), the beads were treated with 0.5 mg/ml Proteinase K at 37°C overnight and subsequently at 65°C for 6 hr. The immunoprecipitated DNA was recovered by phenol-chloroform extraction and ethanol precipitation, and then dissolved in H2O. Control mock immunoprecipitations were done in the same way except that no antibodies were added to the reaction mixture. Real-time PCR quantification of immunoprecipitated DNA was performed as previously described31. The input DNA extracted from the same sonicated chromatin aliquots as above was used to plot a standard curve. Primers were as follow: for bxd PRE (FM4 and FM6), FM4.1, 5′-AGCAATTTGTCACCGCAAGG-3′, FM4.2, 5′-GGATTTTGAGTGCGTTCTTCC-3′, FM6.1, 5′-CCAACGGAAAAGCGAGTGG-3′, and FM6.2, 5′-GCACTAAACCCCATAAAAGTC-3′; for PBX enhancer, PBX-enh-5′, GAAAACACACAAGTGCAG-3′ and PBX-enh-3′, 5′-GGAGATCCTAAAACATGC-3′; for UBX promoter, U-up1.1, 5′-ATTCGCGAGATACCAATGCC-3′ and U-up1.2, 5′-ATTCGCGAGATACCAATGCC-3′; for white locus, W2.1, 5′-ATGCCACGACATCTGACC-3′ and w2.3, 5′-AATGCCAGACGCTTCCTTTC-3′. The quantity obtained by real-time PCR was corrected to obtain the percentage of input.
Supplementary Material
Acknowledgments
We thank Ms. D. McCabe for technical assistance, Dr. K. Basler (University of Zurich) for fly stocks and pUASTattB plasmid, Dr T. Jenuwein for antibodies and Dr J Muller for dPRC2 baculovirus. We thank Jonathan Millar and Alex Gould for suggestions and discussions, David Allis for insightful discussions on histone specificity, Philip Walker for technical assistance and Joe Brock, NIMR Photographics, for assistance with figures. This work was supported by the following grants: Fellowship from the Deutsche Akademie der Naturforscher Leopoldina (#LPDS 2009-5) to PV, NIH grant GM064844 and GM37120 and HHMI to DR. Work in the SJG laboratory is funded by the MRC. Work in the VP laboratory was supported by the Division of Life Sciences of Rutgers University. Structural data have been deposited with the Protein Databank; Eed/NDSB – 3IJC, Eed/H3K27 – 3IIW, Eed/H1K26 – 3IIY, Eed/H3K9 – 3IJ0 & Eed/H4K20 – 3IJ1.
References
- 1.Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome Regulation by Polycomb and Trithorax Proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Czermin B, et al. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–96. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 3.Cao R, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 4.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller J, et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 6.Nekrasov M, et al. Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at Polycomb target genes. Embo J. 2007 doi: 10.1038/sj.emboj.7601837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarma K, Margueron R, Ivanov A, Pirrotta V, Reinberg D. Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol. 2008;28:2718–31. doi: 10.1128/MCB.02017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han Z, et al. Structural basis of EZH2 recognition by EED. Structure. 2007;15:1306–15. doi: 10.1016/j.str.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Schiefner A, et al. Cation-pi interactions as determinants for binding of the compatible solutes glycine betaine and proline betaine by the periplasmic ligand-binding protein ProX from Escherichia coli. J Biol Chem. 2004;279:5588–96. doi: 10.1074/jbc.M309771200. [DOI] [PubMed] [Google Scholar]
- 10.Kouzarides T. Chromatin Modifications and Their Function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Nekrasov M, Wild B, Muller J. Nucleosome binding and histone methyltransferase activity of Drosophila PRC2. EMBO Rep. 2005;6:348–53. doi: 10.1038/sj.embor.7400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simon MD, et al. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–12. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tie F, Stratton CA, Kurzhals RL, Harte PJ. The N terminus of Drosophila ESC binds directly to histone H3 and is required for E(Z)-dependent trimethylation of H3 lysine 27. Mol Cell Biol. 2007;27:2014–26. doi: 10.1128/MCB.01822-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Struhl G, Brower D. Early role of the esc+ gene product in the determination of segments in Drosophila. Cell. 1982;31:285–92. doi: 10.1016/0092-8674(82)90428-7. [DOI] [PubMed] [Google Scholar]
- 15.Ohno K, McCabe D, Czermin B, Imhof A, Pirrotta V. ESC, ESCL and their roles in Polycomb Group mechanisms. Mech Dev. 2008;125:527–41. doi: 10.1016/j.mod.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Kurzhals RL, Tie F, Stratton CA, Harte PJ. Drosophila ESC-like can substitute for ESC and becomes required for Polycomb silencing if ESC is absent. Dev Biol. 2008;313:293–306. doi: 10.1016/j.ydbio.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen KH, et al. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10:1291–300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- 18.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of Histone H3 Lysine-4 Methylation by the Double Tudor Domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 19.Li H, et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–5. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pena PV, et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–3. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol Cell. 2009;33:181–91. doi: 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 22.Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell. 1996;87:95–104. doi: 10.1016/s0092-8674(00)81326-4. [DOI] [PubMed] [Google Scholar]
- 23.Murzina NV, et al. Structural basis for the recognition of histone H4 by the histone-chaperone RbAp46. Structure. 2008;16:1077–85. doi: 10.1016/j.str.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz YB, Pirrotta V. Polycomb complexes and epigenetic states. Curr Opin Cell Biol. 2008;20:266–73. doi: 10.1016/j.ceb.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 25.Ringrose L, Paro R. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development. 2007;134:223–32. doi: 10.1242/dev.02723. [DOI] [PubMed] [Google Scholar]
- 26.Papp B, Muller J. Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes Dev. 2006;20:2041–54. doi: 10.1101/gad.388706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz YB, et al. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat Genet. 2006;38:700–5. doi: 10.1038/ng1817. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;267 doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–61. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–72. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perrakis A, Sixma TK, Wilson KS, Lamzin VS. wARP: improvement and extension of crystallographic phases by weighted averaging of multiple-refined dummy atomic models. Acta Crystallogr D Biol Crystallogr. 1997;53:448–55. doi: 10.1107/S0907444997005696. [DOI] [PubMed] [Google Scholar]
- 32.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallographica Section A. 1994;50:157–163. [Google Scholar]
- 33.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallographica Section D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 34.Brünger AT, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallographica Section D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47 (Pt 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, Richmond TJ. Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol. 1997;272:301–11. doi: 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- 38.Margueron R, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32:503–18. doi: 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A. 2007;104:3312–7. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bateman JR, Lee AM, Wu CT. Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics. 2006;173:769–77. doi: 10.1534/genetics.106.056945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.