

Abstract
Epigenetic signals are responsible for the establishment, maintenance and reversal of metastable transcriptional states that are fundamental for the cell's ability to “remember” past events, such as changes in the external environment or developmental cues. Complex epigenetic states are orchestrated by several converging and reinforcing signals, including transcription factors, non-coding RNAs, DNA methylation, and histone modifications. Although all these pathways modulate transcription from chromatin in vivo, the mechanisms by which epigenetic information is transmitted through cell division remain unclear. Because epigenetic states are metastable and change in response to the appropriate signals, a deeper understanding of their molecular framework will allow us to tackle the dysregulation of epigenetics in disease.
Adaptation to environmental changes and cell specialization in multi-cellular organisms require a complex orchestration of the transcriptional output of the genome. From the simplest prokaryote to the most sophisticated human neuron, cells have evolved forms of molecular memory of past stimuli that can often be transmitted through cell division. The maintenance of cell identity in multi-cellular organisms constitutes a classic example of such inheritable cellular memory: starting from the same zygotic genomes, subsets of progeny cells become engaged in distinct programs of gene expression that dictate their developmental trajectory and specific functions. Typically, cell identities are maintained for a lifetime, even when the differentiation signal was experienced only once, during embryonic development (1). This is no trivial achievement, as a complex pattern of gene expression must be faithfully transmitted to each progeny cell upon division.
We use the term epigenetics to classify those processes that ensure the inheritance of variation (-genetic) above and beyond (epi-) changes in the DNA sequence (Box 1). Unlike genetic alleles, epi-alleles do not differ in their DNA sequence; the epigenetic information resides in self-propagating molecular signatures that provide a memory of previously experienced stimuli, without irreversible changes in the genetic information. The nature of these molecular signatures and the manner by which they initiate, maintain, and reverse epigenetic states is the subject of this review.
Box 1. Epigenetics, what's in a name?
The term “epigenetics” was coined by C.H. Waddington in the 1940s, fusing the word “genetics” with “epigenesis”, the latter indicating the theory by which the adult form develops from the embryo through gradual steps, as opposed to being fully pre-formed in the zygote. Waddington intended to found a discipline to study the genetic control of developmental processes, merging the fields of embryology and genetics (78). More than a decade later D.L. Nanney defined “epigenetic systems” as “auxiliary mechanisms […] involved in determining which specificities [genes] are to be expressed in any particular cell”. Nanney also warned that “Cellular memory is not an absolute attribute” and that utilizing inheritance to define epigenetics might undermine the full understanding of the molecular pathways involved, which may also stabilize transcription patterns in non-dividing cells (79). With the discovery of inheritable patterns of DNA methylation, the idea that epigenetic traits were inherited as regulatory signals in addition to genetic information quickly took hold, and the definition of epigenetics became: “The study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence” (80).
The discovery of the regulatory role of histone post-translational modifications and their correlation with transcriptional states promoted a looser use of epigenetics, to mean any molecular signature found on chromosomes (especially histone marks) and broadly defined as: “the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states” (81). At the same time, a more narrow definition has been proposed as “a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence” (82).
Here, we intend epigenetics to signify “the inheritance of variation (-genetics) above and beyond (epi-) changes in the DNA sequence”. Rather than modifying this basic definition, we prefer to append to it appropriate modifiers. The chromosome-dependent definition given in (82) corresponds to what we call cis epigenetics, as opposed to self-propagating patterns that operate in trans. We convey the distinction between trans-generational inheritance and cellular memory by referring to meiotic vs. mitotic epigenetics, both cases of vertical epigenetics, as opposed to the RNA-mediated horizontal transmission of epigenetic states observed in plants (51).
Epigenetic signals in cis and trans
As long as a transcriptional response is self-sustaining in the absence of the originating stimulus, it can be categorized as epigenetic. This can be achieved by self-propagating trans-acting mechanisms, or by cis-acting molecular signatures physically associated with the DNA sequence they regulate.
Self-propagating transcriptional states that are maintained through feedback loops and networks of transcription factors (TFs) (2) are the most common type of trans epigenetic states (Figure 1A). These are often the system of choice for cellular memory in simple organisms, such as prokaryotes and single cell eukaryotes. If a TF activates its own transcription (or represses antagonistic networks), it yields an epigenetic state that is self-sustaining after the originating stimulus is removed. After each cell division, inherited TFs resume their trans function on regulatory DNA sequences. Some short RNAs (sRNAs) can also act as trans epigenetic signals (3, 4) (see also the Review by Bourc'his et al. in this Issue).
Figure 1. Cis and trans epigenetic signals.
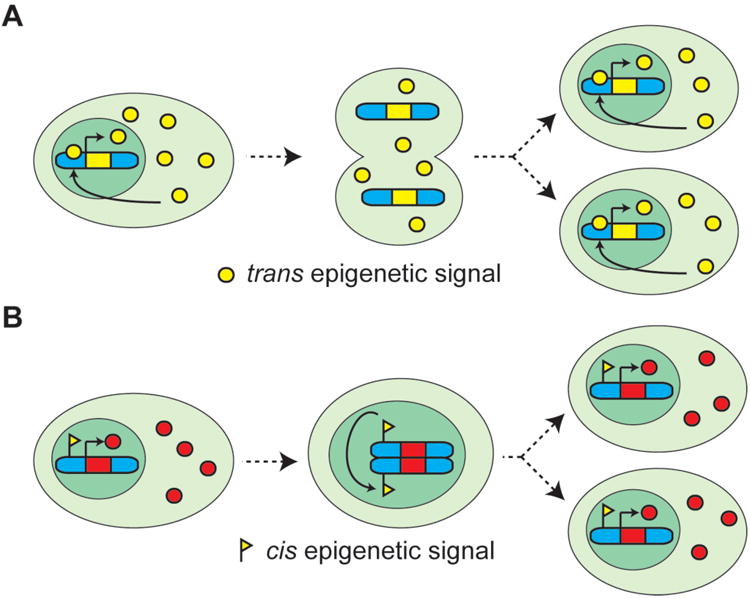
(A) A trans epigenetic signal (yellow circle) is transmitted by partitioning of the cytosol during cell division and maintained by feedback loops. As an example here we depict a simple regulatory loop in which the epigenetic signal induces its own expression. (B) Cis signals (yellow flag) are molecular signature physically associated with the DNA and inherited via chromosome segregation during cell division.
In contrast, cis epigenetic signals are physically associated and inherited along with the chromosome on which they act (Figure 1B); for example, as a covalent modification of the DNA itself, such as DNA methylation, or via alterations in the protein backbone of chromatin, histones. Histones can carry information in their primary sequence (histone variants), in post-translational modifications often present on their N- and C-terminal tails, or in their position (remodeling) relative to the DNA sequence (5-7). Cis epigenetic information might also be encoded in chromatin through stable association of non-histone proteins, higher-order chromatin structure, and nuclear localization.
It is often difficult to distinguish experimentally between trans and cis epigenetic signals. For example, initial observations implicated SWI/SNF chromatin remodelers in transcriptional memory at the S. cerevisiae GAL1 locus (8), but cell fusion experiments rigorously demonstrated that the site of memory was the cytosol (9), a case of trans epigenetics. However, if two identical DNA sequences are differentially regulated in the same nucleus, cis epigenetic mechanisms must be responsible. This is observed for mono-allelic gene expression in diploid cells, imprinting, and X inactivation in mammals, wherein large portions of the chromosome are inheritably silenced, while its homologue continues to transcribe in the same nucleus (10). In fact, X chromosome inactivation involves many putative epigenetic signals and provides an excellent experimental and didactical model to study epigenetics.
If trans-acting transcriptional memory systems were readily available during evolution, why did the appearance of multi-cellularity expand the repertoire of cis epigenetic signals? One possibility is that trans mechanisms were simply inadequate for tackling the increased complexity and number of transcriptional networks in a large multi-cellular organism. Epigenetic states that are encoded in cis need to be set only once, and many transcriptional patterns can be maintained by a relatively small number of common molecular pathways, without having to deploy trans-acting feedback loops for each gene network.
Establishment
Most epigenetic states are established by transiently expressed or transiently activated factors that respond to environmental stimuli, developmental cues, or internal events (e.g. the reactivation of a transposon). These establishment signals converge on chromatin to shape the transcriptional landscape, and are then converted into cis epigenetic signatures.
TFs orchestrate lineage specification programs and are leading candidates as establishment signals (11). In addition to recruiting factors that modulate transcription transiently, TFs also influence cis epigenetic states. In Drosophila, TFs encoded by the segmentation genes establish cellular fate during early embryo development and, although they disappear after only a few hours, cell identities are maintained into adulthood by the Polycomb group (PcG) of proteins that are associated with transcriptional repression, and the trithorax group (trxG) of proteins that are associated with transcriptional activation. Many members of both groups possess chromatin-modifying activities (12), and given that Drosophila contains little DNA methylation, the PcG/trxG system is likely to be directly involved in the transmission (maintenance) of epigenetic states.
It is not clear how the transition from TF-driven regulation to a cis epigenetic state takes place. One possibility is that TFs directly recruit chromatin-modifying enzymes to their genomic targets. For example, sequence-specific DNA-binding proteins are required for PcG-mediated memory in Drosophila and recruit PcG complexes to regulatory regions called Polycomb Responsive Elements (PREs) (13), but recruitment does not always translate into repression of the downstream locus (14), suggesting that PRE localization of PcG and trxG complexes is necessary but not sufficient for the maintenance of transcriptional memory. Similarly, several TFs physically interact with DNA methyltransferases and recruit them to target genes (15).
In addition, TFs might establish epigenetic states through the process of transcription itself. Transcription of Nesp is required for the establishment of DNA methylation at the imprinted Gnas locus downstream, suggesting a cis mechanism (16); and transcription of the non-coding PREs induces an inheritable active state (17). Transcription also guides the deposition of histone post-translational modifications, including H3K4me3, H3K36me3, H3K79me, and H2BK123ub (18), but these modifications have yet to pass the maintenance test for bona fide epigenetic signals (see below).
The transcription process affects chromatin structure, but it is oftentimes difficult to ascribe this effect to the physical passage of RNA polymerase II (RNAPII) or to the synthesis of non-coding RNA (ncRNAs). Non-coding regions of the genome are heavily transcribed, giving rise to a constellation of ncRNAs that often have regulatory functions (19). While early investigations focused on post-transcriptional gene silencing by microRNAs and other sRNAs, pioneering work in S. pombe and A. thaliana established that sRNAs also impact epigenetic states (3). Plants in particular use sRNAs to repress transposable elements and regulate gene expression through the process of RNA-dependent DNA methylation (20). Other sRNAs with proposed effects on cis epigenetic states are PIWI-interacting RNAs (piRNAs) (21), and short RNAs that bind to (and perhaps mediate the recruitment of) PcG proteins (22). Even more classes of sRNAs may be involved in chromatin regulation, although a functional link has not yet been demonstrated; these include promoter- and termini-associated RNAs, tiny RNAs, and endo-siRNAs (19).
Short ncRNAs are well suited for a role in bridging chromatin modifiers with the genome (see also the Review by Bourc'his et al. in this issue), but to fulfill this function they must interact in sequence-specific fashion with chromatin. We envision three modes of sequence recognition: 1) RNA:RNA interactions with nascent transcripts (3) (Figure 2A); 2) RNA:ssDNA heteroduplex (Figure 2B); and 3) RNA:dsDNA triplex (23) (Figure 2C). These recognition modes are not mutually exclusive: piRNAs guide PIWI to Drosophila chromatin both by RNA:RNA and RNA:DNA interactions (24). Recruitment of chromatin modifiers might occur directly or via adaptor proteins, as in the case of Stc1, an S. pombe LIM domain-containing protein, that bridges Ago1 and its associated sRNAs to the H3K9 methyltransferase complex CLRC (25).
Figure 2. RNA-directed deposition of epigenetic signals.
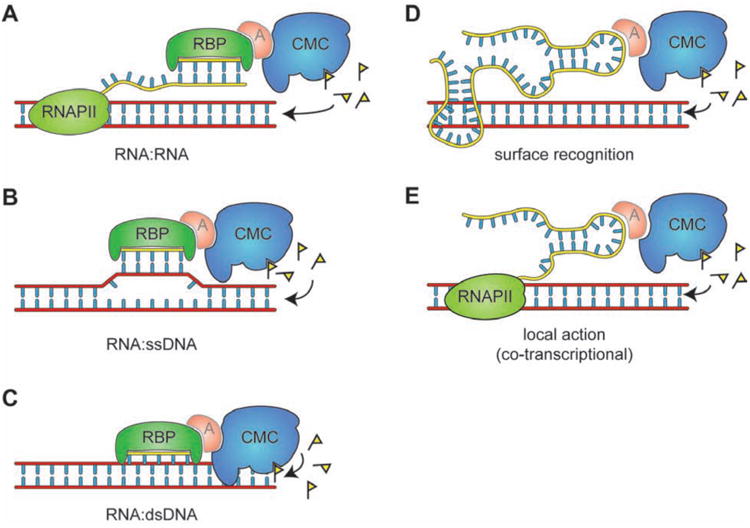
(A-C) Sequence-specific recognition models for both sRNAs (depicted) and lncRNAs. (A) Interaction of an sRNA with complementary sequences on a nascent transcriptl. (B) Interaction of an sRNA with ssDNA to form a heteroduplex. (C) Recognition of sequence motifs by sRNA in a closed dsDNA duplex. (D-E) additional recruitment models for lncRNAs. (D) A folded lncRNA recognizes a DNA sequence via complex surface-mediated interactions. (E) A lncRNA acting locally in a co-transcriptional fashion by being anchored to chromatin by RNA PII. RBP, RNA binding protein; A, hypothetical adaptor protein; CMC, chromatin-modifying complex.
Long ncRNA (lncRNAs) may also function as establishment signals for epigenetic states (26). Because of their size, these molecules can fold into complex structures with molecular surfaces dedicated to protein binding, while retaining the ability to recognize nucleotide sequences by base-pair interactions as described above for sRNAs (27), although their tertiary structure may also allow for recognition modes that do not involve direct base pair interactions (Figure 2D). Some lncRNAs, such as HOTAIR appear to act globally to recruit chromatin modifiers to target loci (28). Other lncRNAs act locally, and establish cis epigenetic states on the genomic region from which they are transcribed. This second category include lncRNAs involved in the classic epigenetic phenomena of parental imprinting and X chromosome inactivation, and their localized effect may be explained by two mechanisms: 1) their 5′ terminus may fold and interact with chromatin-modifying complexes while the 3′ terminus is still tethered to the encoding locus by a transcribing RNAPII (Figure 2E); and 2) antisense transcription of a lncRNA might interfere with transcription of a neighboring gene, or create double-stranded RNA species that are processed locally and establish cis epigenetic signal(s) to silence the locus.
Reinforcement and spreading
Cis epigenetic states, such as those presumably encoded by histone post-translational modifications and DNA methylation, can be reinforced locally or spread to adjacent areas to form larger chromatin domains. Feedback loops exist, in which enzymes responsible for the installment of a histone modification also interact with factors that bind to it. Examples of such histone modifier/binder pairs are SUV39H1 and HP1 for H3K9me, PR-SET7 and L3MBTL1 for H4K20me1, and EZH2 and EED for H3K27me (29-31). The local reinforcement may be necessary, because histone modifications are not permanent and may be removed by dedicated enzymes or histone turnover.
On the other hand, spreading in cis may be required to extend the reach of epigenetic regulation beyond the confined area in which establishment took place. Spreading of chromatin domains is the basis of classical epigenetic phenomena such as position-effect variegation in Drosophila and formation of silent domains in S. cerevisiae (10), and was also observed for artificially established H3K27me3 domains in human cells (32).
Epigenetic states can also be reinforced by cross-talk among histone modifications and DNA methylation (33). De novo DNA methyltransferases and associated factors bind to unmethylated H3K4 (34), whereas the H3K4 methyltransferase MLL preferentially binds to unmethylated DNA (35), providing a molecular explanation for the anti-correlation between H3K4me and DNA methylation levels (36). H3K9me is a pre-requisite for all DNA methylation in N. crassa, and CHG DNA methylation in A. thaliana (37). In mouse, the H3K9 methyltransferases Suv39h1/2 and G9a can direct de novo DNA methylation to certain loci, although the catalytic activity of G9a is not always necessary (38-40). This interplay suggests that, when present, DNA methylation may serve as a reinforcing signal for pre-existing—but less stable—epigenetic signatures such as histone modifications.
Transmission
Three independent criteria should determine whether a certain molecular signal is indeed epigenetic: 1) mechanism for propagation, i.e. pathways that explain how the molecular signature is faithfully reproduced after DNA replication/cell division; 2) evidence of transmission, i.e. the demonstration of self-sustaining transmission to the progeny; and 3) effect on gene expression, i.e. a bona fide epigenetic signal should be sufficient to cause a transcriptional outcome1 reminiscent of that caused by the establishing stimulus.
DNA methylation satisfies all three requirements: 1) Because of the semiconservative nature of DNA replication, a DNA sequence carrying symmetrical methylation marks on both strands gives rise to two hemi-methylated double strands, which can be restored to fully methylated status by maintenance methyltransferases (37) (Figure 3A); 2) in vitro methylated DNA remains methylated after several rounds of DNA replication in vivo (41); and 3) methylation regulates transcription.
Figure 3. Transmission of epigenetic states.
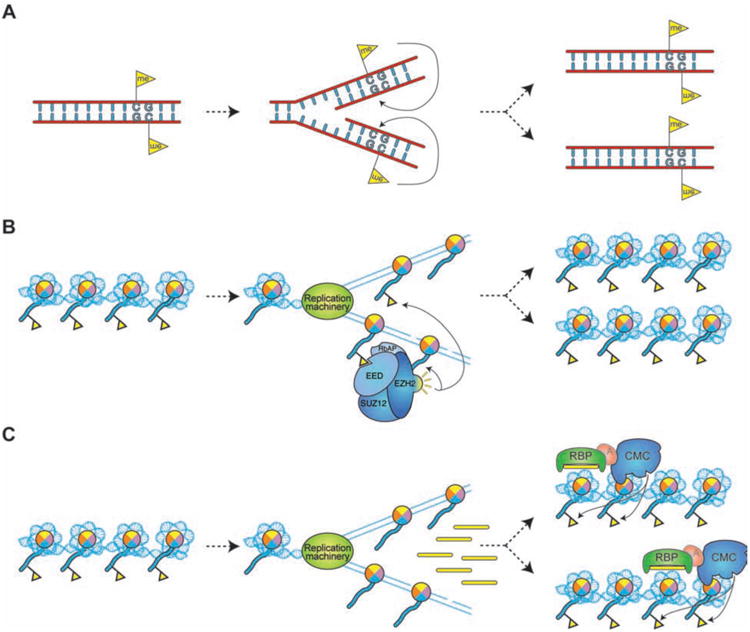
(A) Transmission of DNA methylation patterns after DNA replication. (B) Hypothetical model for the maintenance of a histone-associated epigenetic signal, using H3K27me3 (yellow flag) as an example. H3K27me3 is diluted during DNA replication by the deposition of unmodified octamers. Binding of EED to H3K27me3 stimulates the enzymatic activity of EZH2, which places more H3K27me3 marks on neighboring nucleosomes, thus restoring a full epigenetic signature on both chromatids (48). (C) Maintenance of a chromatin domain via a “secondary” signal. S phase transcription of heterochromatic repeats in S. pombe generates sRNA species that recruit chromatin-modifying complexes to reestablish heterochromatic signatures at the target loci.
The case for histone post-translational modifications is less clear, and each mark should be considered separately. Some modifications exhibit strong correlation with transcriptional states (6); however, correlation does not imply causation, and experimental evidence for the epigenetic inheritance of histone modifications remains scarce.
Propagation mechanisms (criterion #1) have been proposed for several histone modifications in the form of the same histone modifier/binder interactions involved in signal reinforcement and spreading (Figure 3B). This model assumes that the information to reestablish chromatin domains is transferred from the parental nucleosomes containing such modifications to those deposited on the two daughter strands. However, it remains unclear whether and how parental histones (and their associated modifications) are reassembled after DNA replication in vivo. Alternatively, domains of histone modifications could be propagated by way of an intermediary (secondary) epigenetic signal. This appears to be the case for H3K9me in S. pombe heterochromatin, where S phase-restricted transcription of repetitive sequences generates sRNAs that direct the re-establishment of H3K9me after replication (42) (Figure 3C).
Whether histone post-translational modifications are transmitted (criterion #2) remains largely unknown. This question can be addressed by artificially recruiting a histone modifier to chromatin using the GAL4/UAS system, and then measuring the persistence of the resultant histone modification through cell division after terminating the expression of the histone modifier. The GAL4/UAS system can also be used to demonstrate that histone modifications cause—not only correlate with—a transcriptional response (criterion #3) (30). To date only short-term (4 days) transmission of H3K27me3 in cultured human cells has been observed (32, 43), but doubts remain regarding incomplete repression of the GAL4-fused histone modifier. In addition, Polycomb Repressive Complex 1 remains bound to chromatin (independently of histone modifications) during DNA replication in vitro (44), and MLL (a trxG protein) appears to associate with mitotic chromosomes (45), suggesting that some chromatin modifiers may also function as cis epigenetic signals.
Some epigenetic information is transmitted through meiosis and gamete formation in multi-cellular organisms, giving rise to trans-generational inheritance. Many epigenetic signals appear capable of meiotic transmission, including maternally deposited TFs and piRNAs (46), RNAs involved in paramutation in mice (47), histone modifications in sperm chromatin (48), and DNA methylation in plants (49).
Finally, epigenetic signals are also transmitted cell-to-cell in a horizontal fashion (see Bourc'his et al. in this issue). This phenomenon is at the basis of trans-generational inheritance of RNAi in C. elegans and occurs in plants, both in the germline, and in the soma (50, 51), two cases in which sequence information is transmitted across cells in order to silence transposable elements. As no genomic DNA is exchanged between these cells, the epigenetic information must be transmitted in trans. In fact, it takes the form of sRNAs that direct DNA methylation of genomic sequences, converting a trans epigenetic signal into a cis epigenetic state (51).
Reversal
Epigenetic regulatory mechanisms are conservative in that no information is lost and, given the appropriate signal, an epigenetic state can transition to a different one, as exemplified by the generation of induced pluripotent stem (iPS) cells from mouse embryonic fibroblasts (MEFs) by transient overexpression of cocktails of transcription factors (52). Although embryonic stem (ES) cells and MEFs have very different expression profiles, H3K4me and H3K27me distribution, and DNA methylation patterns, MEF-derived iPS cells closely resemble ES cells both at the transcriptional and epigenetic levels (53). These observations not only demonstrate the plasticity of epigenetic signals, but also confirm the inter-dependence between cell identity and epigenetic states.
Reversible chromatin changes and antagonism between TFs provide the basis for cellular plasticity. Opposing transcription factor networks reinforced by feedback loops direct the specification of haematopoietic and embryonic lineages (11, 54). Histone modification profiles are also the result of a delicate balance between antagonistic pairs of histone-modifying enzymes: histone acetyl-transferases vs. deacetylases and histone methyl-transferases vs. demethylases (6).
Nonetheless, the forced transition between two metastable epigenetic states requires a considerable “activation energy” as evidenced by the poor efficiency of epigenetic reprogramming by nuclear transfer or overexpression of pluripotency factors. Cells that fail to fully overcome this barrier are trapped in an intermediate state, likely due to a failure in resetting epigenetic signatures (53). For example, improper silencing by DNA hypermethylation and histone hypoacetylation of the imprinted Dlk1-Dio3 cluster correlates with the failure of many iPS lines to generate chimaeras (55). Thus, small molecule inhibitors of histone modifiers and DNA methyl-transferases that stimulate reprogramming may do so by facilitating the creation of an “epigenetic tabula rasa” (56).
The most dramatic epigenetic plasticity is observed during early embryo development and germ line specification in mammals. At fertilization, the condensed paternal genome is hyper-methylated, until a wave of DNA demethylation restores it to an active state (57). Some imprinted genes are not affected, suggesting that they must be marked with a different epigenetic signal (58). During development, germ cells undergo a second wave of DNA methylation (59), possibly in response to environmental cues in the gonads, demonstrating that even the most stable epigenetic signal can be reversed by the appropriate stimuli.
These two cases in which genome-wide DNA methylation is rapidly lost, likely involve the most elusive mode of epigenetic plasticity: DNA demethylation (60). Two mechanisms for DNA demethylation appear possible: 1) active demethylation through oxidation; and 2) base excision repair (BER). Hydroxylation of 5-methylcytosine by TET1-3 (61, 62) argues in favor of the first mechanism, although the resulting 5-hydroxymethylcytosine must be further converted to unmethylated cytosine via presently unknown mechanisms. In plants, genetic evidence supports DNA demethylation via BER (63), but the importance of this pathway in animals is unclear. Activation-induced deaminase (AID) is involved in DNA demethylation in mammalian cells (64, 65); yet, its inability to act on dsDNA (66) and the lack of developmental defects in Aid-/- mice argue against a central role in DNA demethylation and development (67) (see also the Review by Feng et al. in this issue).
Epigenetic states then are stable enough to maintain cell identity, but also reversible enough to allow, when the proper signals are delivered, transitions among states. For example, the epigenetic plasticity of germ cells allows for cell specialization during gamete development, while retaining the potential for returning to the ground state of totipotency after fertilization.
Future directions
Epigenetic signals have been traditionally studied in the context of development and transposon control, but we believe that epigenetic mechanisms will be the crux of many other biological processes. For example, brain function and memory formation may involve epigenetic signals (68, 69). Although most neurons do not divide, they are very long-lived and must maintain a defined transcriptional state for an extremely long time. By our own definition, this phenomenon is not truly epigenetic, yet signals that evolved to maintain epigenetic states through cell divisions might have been co-opted by neurons to stabilize transcriptional profiles after the stimulus has disappeared (69).
Another area in which a better understanding of epigenetic signals may benefit research is that of stem cell function. Stem cells divide asymmetrically, giving rise to one self-renewing cell and one cell committed to differentiate (70). This is usually attributed to external cues and asymmetric partitioning of trans-acting factors, but evidence suggests that chromatids (and any cis epigenetic signal they may carry) can also be segregated asymmetrically (71). Thus, asymmetric partitioning of epigenetic signals may participate in cell fate decisions, as proposed in the “silent sister” hypothesis (72)
Epigenetics is also likely to impact aging, given that epi-mutations, especially in DNA methylation, may contribute to the aging phenotype (73). Epigenetic transmission is more error prone than DNA replication (41), and likely accumulates significant amounts of inheritable errors over a lifetime. Pathways that impinge on chromatin states are also implicated in aging (74), but we are far from understanding the molecular mechanisms involved.
Conserved molecular machineries manage genetic information in all organisms, yet different epigenetic signals predominate in different species. For this reason, epigenetic research has been conducted on an unusually broad spectrum of model organisms. If we had only studied Drosophila, we would know little about DNA methylation. Extending our studies to new model organisms will undoubtedly be beneficial. Two emerging models appear very promising: the Planarian Schmidtea mediterranea, which displays incredible epigenetic flexibility in regenerating a complete adult from small patches of dissected tissue (75); and social insects, in which a single genome gives rise to epigenetically distinct castes that display dramatic physiological, morphological and behavioral differences (76, 77).
Acknowledgments
We thank L. Vales for critical reading of this manuscript. Work in the Reinberg laboratory is supported by HHMI and NIH grants GM064844 and GM037120. R.B. is a fellow of the Helen Hay Whitney Foundation.
Footnotes
More generally, any phenotypic change may be transmitted epigenetically. For sake of simplicity in this review we only discuss epigenetic control of gene expression. For an example of epigenetic transmission of altered protein function see the Review by Lindquist and colleagues in this Issue.
References and notes
- 1.Ringrose L, Paro R. Annu Rev Genet. 2004 Dec 1;38:413. doi: 10.1146/annurev.genet.38.072902.091907. [DOI] [PubMed] [Google Scholar]
- 2.Alon U. An Introduction to Systems Biology: Design Principles of Biological Circuits. Chapman and Hall/CRC; 2006. [Google Scholar]
- 3.Moazed D. Nature. 2009 Jan 22;457:413. doi: 10.1038/nature07756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennecke J, et al. Cell. 2007 Mar;128:1089. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 5.Talbert PB, Henikoff S. Nat Rev Mol Cell Biol. 2010 Apr;11:264. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 6.Kouzarides T. Cell. 2007 Feb 27;128:693. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Cairns BR. Nature. 2009 Sep 10;461:193. doi: 10.1038/nature08450. [DOI] [PubMed] [Google Scholar]
- 8.Kundu S, Horn PJ, Peterson CL. Genes Dev. 2007 Apr 15;21:997. doi: 10.1101/gad.1506607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zacharioudakis I, Gligoris T, Tzamarias D. Curr Biol. 2007 Dec 4;17:2041. doi: 10.1016/j.cub.2007.10.044. [DOI] [PubMed] [Google Scholar]
- 10.Allis CD, Jenuwein T, Reinberg D. In: Epigenetics. 1st. Caparros M, editor. Cold Spring Harbor Laboratory Press; 2007. p. 502. [Google Scholar]
- 11.Graf T, Enver T. Nature. 2009 Dec 3;462:587. doi: 10.1038/nature08533. [DOI] [PubMed] [Google Scholar]
- 12.Simon JA, Kingston RE. Nat Rev Mol Cell Biol. 2009 Oct;10:697. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 13.Ringrose L, Paro R. Development. 2007 Jan;134:223. doi: 10.1242/dev.02723. [DOI] [PubMed] [Google Scholar]
- 14.Papp B, Müller J. Genes Dev. 2006 Aug 3;20:2041. doi: 10.1101/gad.388706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hervouet E, Vallette FM, Cartron PF. Epigenetics. 2009 Oct;4:487. doi: 10.4161/epi.4.7.9883. [DOI] [PubMed] [Google Scholar]
- 16.Chotalia M, et al. Genes Dev. 2009 Jan 1;23:105. doi: 10.1101/gad.495809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitt S, Prestel M, Paro R. Genes Dev. 2005 Mar 15;19:697. doi: 10.1101/gad.326205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carey Li, Workman JL. Cell. 2007 Feb 27;128:707. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 19.Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. J Pathol. 2010 Jan;220:126. doi: 10.1002/path.2638. [DOI] [PubMed] [Google Scholar]
- 20.Matzke M, Kanno T, Huettel B, Daxinger L, Matzke AJ. Curr Opin Plant Biol. 2007 Oct;10:512. doi: 10.1016/j.pbi.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 21.Aravin AA, et al. Mol Cell. 2008 Sep 26;31:785. doi: 10.1016/j.molcel.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanhere A, et al. Mol Cell. 2010 Jun 11;38:675. doi: 10.1016/j.molcel.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Escude C, et al. Nucleic Acids Res. 1993 Dec 11;21:5547. doi: 10.1093/nar/21.24.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brower-Toland B, et al. Genes Dev. 2007 Sep 15;21:2300. doi: 10.1101/gad.1564307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayne EH, et al. Cell. 2010 Mar 5;140:666. doi: 10.1016/j.cell.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JT. Genes Dev. 2009 Aug 15;23:1831. doi: 10.1101/gad.1811209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koziol MJ, Rinn JL. Curr Opin Genet Dev. 2010 Apr;20:142. doi: 10.1016/j.gde.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rinn JL, et al. Cell. 2007 Jun 29;129:1311. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stewart MD, Li J, Wong J. Mol Cell Biol. 2005 Apr;25:2525. doi: 10.1128/MCB.25.7.2525-2538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalakonda, et al. Oncogene. 2008 Jul 17;27:4293. doi: 10.1038/onc.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margueron R, et al. Nature. 2009 Oct 8;461:762. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen KH, et al. Nat Cell Biol. 2008 Nov 1;10:1291. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- 33.Cedar H, Bergman Y. Nat Rev Genet. 2009 May;10:295. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 34.Ooi SKT, et al. Nature. 2007 Aug 9;448:714. [Google Scholar]
- 35.Birke M, et al. Nucleic Acids Res. 2002 Feb 15;30:958. doi: 10.1093/nar/30.4.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meissner A, et al. Nature. 2008 Aug 7;454:766. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goll MG, Bestor MGGaTH, Bestor TH. Annu Rev Biochem. 2005 Jun 15;74:481. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 38.Lehnertz B, et al. Curr Biol. 2003 Jul 15;13:1192. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 39.Epsztejn-Litman S, et al. Nat Struct Mol Biol. 2008 Nov;15:1176. doi: 10.1038/nsmb.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong KB, et al. EMBO J. 2008 Oct 22;27:2691. doi: 10.1038/emboj.2008.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wigler M, Levy D, Perucho M. Cell. 1981 Apr;24:33. doi: 10.1016/0092-8674(81)90498-0. [DOI] [PubMed] [Google Scholar]
- 42.Chen ES, et al. Nature. 2008 Feb 7;451:734. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- 43.Sarma K, Margueron R, Ivanov A, Pirrotta V, Reinberg D. Mol Cell Biol. 2008 Apr;28:2718. doi: 10.1128/MCB.02017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Francis NJ, Follmer NE, Simon MD, Aghia G, Butler JD. Cell. 2009 Apr 3;137:110. doi: 10.1016/j.cell.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blobel GA, et al. Mol Cell. 2009 Dec 25;36:970. doi: 10.1016/j.molcel.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brennecke J, et al. Science. 2008 Nov 28;322:1387. doi: 10.1126/science.1165171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rassoulzadegan M, et al. Nature. 2006 May 26;441:469. doi: 10.1038/nature04674. [DOI] [PubMed] [Google Scholar]
- 48.Hammoud SS, et al. Nature. 2009 Jul 23;460:473. doi: 10.1038/nature08162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saze H. Semin Cell Dev Biol. 2008 Dec;19:527. doi: 10.1016/j.semcdb.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 50.Slotkin RK, et al. Cell. 2009 Feb 6;136:461. doi: 10.1016/j.cell.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Molnar A, et al. Science. 2010 May 14;328:872. doi: 10.1126/science.1187959. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi K, Yamanaka S. Cell. 2006 Aug 25;126:663. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 53.Mikkelsen TS, et al. Nature. 2008 Jul 3;454:49. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arnold SJ, Robertson EJ. Nat Rev Mol Cell Biol. 2009 Feb;10:91. doi: 10.1038/nrm2618. [DOI] [PubMed] [Google Scholar]
- 55.Stadtfeld M, et al. Nature. 2010 May 13;465:175. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huangfu D, et al. Nat Biotechnol. 2008 Jul;26:795. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Nature. 2000 Feb 3;403:501. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 58.Reik W, Dean W, Walter J. Science. 2001 Aug 10;293:1089. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 59.Hajkova P, et al. Mech Dev. 2002 Sep;117:15. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 60.Ooi SK, Bestor TH. Cell. 2008 Jun 27;133:1145. doi: 10.1016/j.cell.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 61.Tahiliani M, et al. Science. 2009 May 15;324:930. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ito S, et al. Nature. 2010 Aug 26;466:1129. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu JK. Annu Rev Genet. 2009;43:143. doi: 10.1146/annurev-genet-102108-134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhutani N, et al. Nature. 2010 Feb 25;463:1042. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Popp C, et al. Nature. 2010 Feb 25;463:1101. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bransteitter R, Pham P, Scharff MD, Goodman MF. Proc Natl Acad Sci U S A. 2003 Apr 1;100:4102. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muramatsu M, et al. Cell. 2000 Sep 1;102:553. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 68.Si K, Lindquist S, Kandel E. Cold Spring Harb Symp Quant Biol. 2004;69:497. doi: 10.1101/sqb.2004.69.497. [DOI] [PubMed] [Google Scholar]
- 69.Dulac C. Nature. 2010 Jun 10;465:728. doi: 10.1038/nature09231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morrison SJ, Kimble J. Nature. 2006 Jun 29;441:1068. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 71.Rando TA. Cell. 2007 Jun 29;129:1239. doi: 10.1016/j.cell.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 72.Lansdorp PM. Cell. 2007 Jun 29;129:1244. doi: 10.1016/j.cell.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 73.Gravina S, Vijg J. Pflugers Arch. 2010 Jan;459:247. doi: 10.1007/s00424-009-0730-7. [DOI] [PubMed] [Google Scholar]
- 74.Pegoraro G, Misteli T. Aging (Albany NY) 2009 Dec;1:1017. doi: 10.18632/aging.100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanchez Alvarado A. Cell. 2006 Jan 27;124:241. doi: 10.1016/j.cell.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 76.Consortium HGS. Nature. 2006 Oct 26;443:931. [Google Scholar]
- 77.Bonasio R, et al. Science. 2010 Aug 27;329:1068. doi: 10.1126/science.1192428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Waddington C. Endeavour. 1942;1:18. [Google Scholar]
- 79.Nanney DL. Proc Natl Acad Sci U S A. 1958 Jul 15;44:712. doi: 10.1073/pnas.44.7.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Riggs A, Martienssen R, Russo V. In: Epigenetic mechanisms of gene regulation. Russo V, Martienssen R, Riggs A, editors. Vol. 32 Cold Spring Harbor Laboratory Press; 1996. [Google Scholar]
- 81.Bird A. Nature. 2007 May 23;447:396. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 82.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. Genes Dev. 2009 Apr 1;23:781. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]