

Abstract
TaqMan Gene Expression assays were used to profile the mRNA expression of estrogen receptor (ERα and ERβ) and estrogen metabolism enzymes including cytosolic sulfotransferases (SULT1E1, SULT1A1, SULT2A1, and SULT2B1), steroid sulfatase (STS), aromatase (CYP19), 17β-hydroxysteroid dehydrogenases (17βHSD1 and 2), CYP1B1, and catechol-O-methyltransferase (COMT) in an MCF10A-derived lineage cell culture model for basal-like human breast cancer progression and in ERα-positive luminal MCF7 breast cancer cells. Low levels of ERα and ERβ mRNA were present in MCF10A-derived cell lines. SULT1E1 mRNA was more abundant in confluent relative to subconfluent MCF10A cells, a non-tumorigenic proliferative breast disease cell line. SULT1E1 was also expressed in preneoplastic MCF10AT1 and MCF10AT1K.cl2 cells, but was markedly repressed in neoplastic MCF10A-derived cell lines as well as in MCF7 cells. Steroid-metabolizing enzymes SULT1A1 and SULT2B1 were only expressed in MCF7 cells. STS and COMT were widely detected across cell lines. Pro-estrogenic 17βHSD1 mRNA was most abundant in neoplastic MCF10CA1a and MCF10DCIS.com cells, while 17βHSD2 mRNA was more prominent in parental MCF10A cells. CYP1B1 mRNA was most abundant in MCF7 cells. Treatment with the histone deacetylase inhibitor trichostatin A (TSA) induced SULT1E1 and CYP19 mRNA but suppressed CYP1B1, STS, COMT, 17βHSD1, and 17βHSD2 mRNA in MCF10A lineage cell lines. In MCF7 cells, TSA treatment suppressed ERα, CYP1B1, STS, COMT, SULT1A1, and SULT2B1 but induced ERβ, CYP19 and SULT2A1 mRNA expression. The results indicate that relative to the MCF7 breast cancer cell line, key determinants of breast estrogen metabolism are differentially regulated in the MCF10A-derived lineage model for breast cancer progression.
Keywords: Estrogen metabolism, Gene expression, MCF10A lineage, Breast cancer progression, Histone deacetylase inhibition
Introduction
The estrogen sensitive nature of breast cancer was observed by Beatson over 100 years ago [1]. Prolonged cumulative exposure to estrogen during a woman’s lifetime is a significant risk factor for the development of breast cancer [2]. Estrogen stimulates cell growth through estrogen receptor (ER)-mediated events, which can enhance opportunities for DNA mutation. In addition, reactive estrogen metabolites generated by cytochrome P450-mediated catalysis, such as estrogen catechols and quinones, can directly induce mutations [3]. Particularly in post-menopausal women who lack ovarian sources of estrogen, the factors that govern intra-tissue metabolism of biologically active estrogens (i.e., estrogen “intracrinology”) are important therapeutic targets and biomarkers for breast cancer progression [4]. Determinants affecting the amount and activity of estrogen in breast tissue include expression of the major forms of estrogen receptor (ERα and ERβ), cytosolic sulfotransferases (SULTs), steroid sulfatase (STS), aromatase (CYP19), 17β-hydroxysteroid dehydrogenase types 1 and 2 (17βHSD1 and 17βHSD2), CYP1B1, and catechol-O-methyltransferase (COMT; Fig. 1, adapted from [5–7]).
Fig. 1.
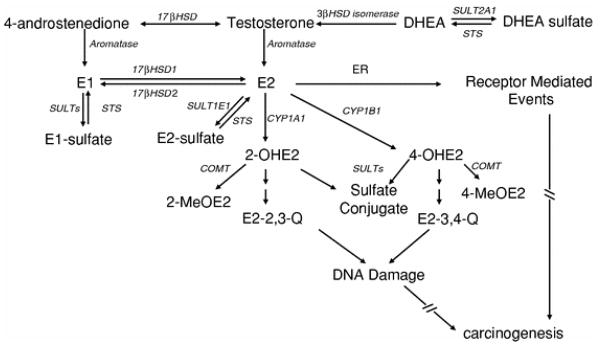
Pathways of estrogen metabolism, bioactivation, and action. CYP19 (aromatase) catalyzes the aromatization of androstenedione and testosterone to form estrone (E1) and 17β-estradiol (E2), which can bind to and activate estrogen receptor (ER). Of the cytosolic SULTs, SULT1E1 preferentially catalyzes the sulfation of E1 and E2 with high efficiency. Sulfated estrogens are ER-inactive. Steroid sulfatase (STS) catalyzes the deconjugation of sulfated steroids and favors the formation of biologically active estrogen. STS and 3-β-hydroxysteroid dehydrogenase (3βHSD isomerase) function in the formation of sex steroids from precursor hormones, such as dehydroepiandrosterone sulfate (DHEA sulfate), dehydroepiandrosterone (DHEA). 17βHSD1 reduces E1 to the more potent E2, while 17βHSD2 oxidizes E2 to E1. The 4-hydroxylation of E2 is catalyzed by CYP1B1; E2 2-hydroxylation is catalyzed by CYP1A1. Catecholestrogens auto-oxidize to form mutagenic orthoquinone electrophiles that can be detoxified via catechol-O-methyltransferase (COMT)-mediated conjugation
This study was designed to elucidate the expression of determinants of estrogen activity in a unique lineage model that captures the heterogeneity of breast cancer progression in humans. The parental MCF10A cell line was derived from spontaneously immortalized breast epithelial cells that were obtained from a donor with benign proliferative breast disease [8]. In vitro transformation of MCF10A cells with mutant c-Ha-ras conferred a measurable xenograft growth potential to these cells (termed MCF10AneoT), and serial passage of xenograft lesions led to the development of the preneoplastic MCF10AT1 cell line [9, 10]. When xenografted into immunodeficient mice, MCF10AT1 cells demonstrate an ~25% incidence of invasive cancers in xenograft lesions that develop over an extended period of time [10]. Serial passaging of MCF10AneoT xenograft lesions also resulted in establishment of the premalignant MCF10AT1K.cl2 cell line. The MCF10AT1K.cl2 variant retained an MCF10AT1-type pattern of xenograft growth but also displayed chromosomal aberrations more characteristic of genetic instability [11]. Sequential passage of a carcinoma arising from a MCF10AT1K.cl2 xenograft lesion produced an expanded series of aggressively neoplastic MCF10CA1 variant cell lines that rapidly produce tumors when xenografted, with an incidence of 100% [11]. The MCF10DCIS.com cell line was developed by cloning after serial passaging of an MCF10AT xenograft lesion [12]. Injection of MCF10DCIS.com cells into immunodeficient mice produces rapidly growing lesions that are consistent with the poor prognosis comedo-type ductal carcinoma in situ [12]. Overall, the MCF10A model captures the heterogeneity of human breast cancer and provides an opportunity to study both indolent and more aggressive breast cancer phenotypes.
The molecular phenotypic characteristics of the MCF10A lineage indicate that this is a model of basal breast cancer, in that the resulting MCF10CA1 cancer cell lines are triple negative (i.e., do not express ER, progesterone receptor or HER2/neu) [8, 13]. Nevertheless, xenograft studies have demonstrated that progression of preneoplastic MCF10AT1 cells is sensitive to estrogen manipulation [10, 14–16]. Estrogen has been shown to alter signal transduction in triple negative MDA-MB-231 (do not express ERα; do express ERβ) and SKBR3 cells (express neither ERα nor ERβ), causing activation of the extracellular signal regulated kinases, ERK1 and ERK2 [17]. Both estrogen and epidermal growth factor (EGF) induced phosphorylation of c-raf and ERK1/2 while stimulating the proliferation of SKBR3 cells [18], and the two agents were more effective in combination. Importantly, growth of ERα-negative breast cells was induced by the same 17β-estradiol (E2) concentration (10−9 M) that stimulates ERα-positive MCF7 cells, whereas a higher concentration (10−6 M) inhibited the growth of MDA-MB-231, and SKBR3 cells, and this inhibition was additive with heregulin [19].
Although MCF7 requires supplementation with E2 to mimic pre-menopausal human serum levels (typically 400–1,500 pg/ml) to achieve xenograft growth in nude mice, MCF10AT1 forms lesions that progress to atypical hyperplasia, ductal carcinoma in situ and invasive ductal carcinoma without E2 supplementation. Serum E2 levels in nude mice are typically less than 50 pg/ml [20], which mimics post-menopausal human serum levels [21]. However, MCF10AT1 cells form lesions consisting only of simple ducts in ovariectomized nude mice [22], which have serum E2 levels of ~5 pg/ml [23]. Therefore, MCF10AT1 xenografts respond to very low doses of E2 in vivo, suggesting that the levels of estrogen-processing enzymes in breast cells may play a critical role in determining growth response. Therefore, an analysis of the determinants of estrogenicity in MCF10A lineage cell lines was undertaken to provides insight into the dynamic changes in estrogen responsiveness that occur during basal breast cancer development.
Histone deacetylases (HDACs) are being increasingly recognized for their important roles as transcriptional modulators of pro-survival pathways during breast cancer progression [24]. Though HDAC inhibitors are currently being tested as clinical adjuncts in breast cancer therapy [25], their effects on the expression of estrogen metabolism machinery during breast cancer progression are unknown. Therefore, the effects of treatment with trichostatin A (TSA), a potent HDAC inhibitor, on ER and estrogen metabolism gene expression in the MCF10A lineage cell lines were also investigated.
Materials and methods
Materials
TaqMan Gene Expression reagents were purchased from Applied Biosystems (Foster City, CA). qPCR Human Reference Total RNA was purchased from Clontech Laboratories (Mountain View, CA). Culture media, sera, L-glutamine, sodium pyruvate, penicillin-streptomycin, anti-SULT1E1 antibody, recombinant human SULT1E1, Superscript II, and Lipofectamine 2000 were purchased from Invitrogen Corporation (Carlsbad, CA). EGF was purchased from BD Biosciences (San Jose, CA). Recombinant human insulin (Novolin R) was purchased from Novo Nordisk Pharmaceuticals, Inc. (Princeton, NJ). Cholera toxin, hydrocortisone and TSA were purchased from Sigma–Aldrich (St. Louis, MO). Horseradish peroxidase-conjugated goat anti-rabbit IgG was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ECL Plus Western Blotting Detection Reagents and Hybond-P membranes were purchased from GE Healthcare (Piscataway, NJ). Other materials were obtained from the sources indicated below.
Cell culture
MCF7 and MCF10A lineage (MCF10A, MCF10AT1, MCF10AT1K.cl2, MCF10CA1a, MCF10CA1d and MCF10DCIS.com) cell lines were obtained from the Cell Resources Facility of the Barbara Ann Karmanos Cancer Institute, Wayne State University. MCF10A lineage cell lines were cultured in phenol red-free Dulbecco’s Modified Eagle Medium/Ham’s F12 nutrient mixture (1:1) supplemented with 10 μg/ml insulin, 20 ng/ml EGF, 100 ng/ml cholera toxin, 0.5 μg/ml hydrocortisone, 5% horse serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. MCF7 cells were cultured in phenol red-free Minimum Essential Medium supplemented with 10 μg/ml insulin, 1 mM sodium pyruvate, 10% calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell lines were routinely maintained in T75 flasks in a 37°C humidified environment of 5% CO2/95% air. For experiments, 250,000 cells were plated into 60 mm dishes. For subconfluent cultures (~70% confluency), cells were harvested 2 days after plating. Confluency was reached approximately 5 days after plating. On day 7, confluent cultures were harvested for preparation of total RNA. Experiments were conducted at a cell density of 70% confluency unless otherwise indicated. For TSA studies, 24 h after plating, cells were treated with 0.1% dimethyl sulfoxide (DMSO control) or TSA.
TaqMan Gene Expression assays
Total RNA was prepared from individual dishes of cells using the RNeasy Mini Kit (Qiagen, Valencia, CA). RNA samples were reverse transcribed using Superscript II. Transcript levels were measured using the following Taq-Man Gene Expression Assays: Hs01046818_m1 (ERα), Hs00230957_m1 (ERβ), Hs00193690_m1 (SULT1E1), Hs00419411_m1 (SULT1A1), Hs01105284_m1 (SULT2B1), Hs00234219_m1 (SULT2A1), Hs00165853_m1 (STS), Hs00240671_m1 (CYP19), Hs00166219_g1 (17βHSD1), Hs00157993_m1 (17βHSD2), Hs00164383_m1 (CYP1B1), and Hs00241349_m1 (COMT). Each PCR reaction included 2 μl of cDNA template, a primer/probe (5-carboxyfluorescein fluor, minor groove binder quencher) set, a primer-limited primer/probe (VIC-minor groove binder) set for 18S rRNA and Universal PCR master mix, and amplifications were performed using an ABI Prism 7500 Sequence Detection System. Thermocycling parameters were 94°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. Cycle threshold (Ct) values were obtained using the SDS software package. For each sample, ΔCt was obtained by subtracting the Ct of 18S rRNA from the Ct of target mRNA. Then, ΔΔCt values were calculated by subtracting the ΔCt of the calibrator to which the other samples were compared from the ΔCt of each sample. Mean relative quantitative expression values were then calculated as 2−ΔΔCt.
Western blot and enzyme activity analyses
Cells in T75 flasks were washed with and scraped into ice-cold phosphate-buffered saline. Cells were pelleted and homogenized by sonication in buffer (200 μl per flask) consisting of 50 mM Tris–HCl, 25 mM sucrose, 1 mM EDTA and 1× Halt protease inhibitor (Thermo Fisher Scientific, Rockford, IL), pH 7.4. Homogenates were centrifuged at 20,000×g at 4°C for 20 min, and supernatants were used for western blot analysis. Protein concentrations were measured using the BCA Protein Assay (Thermo Fisher). Western blot analysis of SULT1E1 content was accomplished as described previously [26], using 30 μg of sample protein and polyclonal anti-SULT1E1 antibody. Uniform protein loading and transfer were verified using Ponceau S staining. SULT1E1 catalytic activity was measured in whole cell lysates prepared from MCF10CA1a cells as described [27].
Transient transfection analysis
The p2ERE-luc reporter was constructed by ligating a double-stranded oligonucleotide containing two vitellogenin estrogen response elements upstream of a minimal herpes simplex virus thymidine kinase promoter, which had been preligated into pGL3-Basic (Promega Corporation, Madison, WI). The sequences of the oligonucleotides are: 5′-CGCGTGTCCAAAGTCAGGTCACAGTGACCTG ATTCAGGTCACAGTGACCTGATCAAAGTTA-3′ and 5′-GATCTAACTTTGATCAGGTCACTGTGACCTGAAT CAGGTCACTGTGACCTGACTTTGGACA-3′.
MCF7 cells (400,000) were seeded into 12-well plates and cultured in 2 ml phenol red-free Minimum Essential Medium supplemented with 10 μg/ml insulin, 1 mM sodium pyruvate and 10% charcoal-stripped fetal bovine serum. The following day, Opti-MEM containing a pre-mixed complex of 4 μl of Lipofectamine 2000, 1.6 μg p2ERE-Luc and 1.25 ng pRL-SV40 (Promega) was added to each well. The following day, cultures (5 wells per group) were incubated with phenol red-free Minimum Essential Medium, supplemented as described above, containing 0.1% DMSO, 10 nM E2, 300 ng/ml TSA, or E2 and TSA in combination. After 48 h, the cells were harvested for measurement of firefly and Renilla luciferase activities using the Dual Luciferase Reporter Assay System (Promega) and a Dynex model MLX Luminometer.
Statistical analysis
Data were analyzed using the paired t-test or one-way analysis of variance followed by the Newman–Keuls test using Prism (GraphPad Software, San Diego, CA).
Results
The high catalytic efficiency of SULT1E1 toward E2 sulfation supports a major role for SULT1E1 in the in situ inactivation of E2 within the breast. SULT1E1 mRNA was detected in parental MCF10A cells, although the level of expression was highly dependent upon the confluency of the cultures. Relative to subconfluent MCF10A cells, the amount of SULT1E1 mRNA in confluent cells was significantly increased by ~16-fold (Fig. 2a). SULT1E1 mRNA expression was robust in MCF10AT1 cells, and, unlike MCF10A cells, was not significantly affected by confluency (Fig. 2a). SULT1E1 mRNA expression was also substantial in MCF10AT1K.cl2 cells, but was markedly diminished in MCF10CA1a, MCF10CA1d, MCF10DCIS.com, and MCF7 cells (Fig. 2a). SULT1E1 immunoreactive protein levels in the MCF10A lineage cell lines were in accord with the corresponding mRNA levels (Fig. 2b).
Fig. 2.

Expression of SULT1E1 mRNA and protein in the MCF10A-derived lineage cell culture model for breast cancer progression and in MCF7 cells. a A TaqMan Gene Expression assay was used to determine the relative levels of SULT1E1 mRNA in subconfluent (70%, white bars) and confluent (100%, black bars) MCF10A series cells (from left to right, MCF10A, MCF10AT1, MCF10AT1K.cl2, MCF10CA1a, MCF10CA1d and MCF10DCIS.com) and in MCF7 cells. SULT1E1 mRNA content is expressed relative to the level measured in subconfluent MCF10A cells, and all values represent the mean ± SEM of three independent cell culture experiments. Groups labeled with different letters are significantly different from each other (P < 0.05). b Representative Western blots showing the relative amounts of SULT1E1 immunoreactive protein in subconfluent MCF10A-derived cell lines
Since STS and other cytosolic SULTs have been implicated in the modulation of breast intracrinology [27–32], the expression patterns of STS and SULTs 1A1, 2A1, and 2B1 were characterized. STS mRNA was detected across the MCF10A series cell lines, as well as in MCF7 cells (Fig. 3a). SULT1A1 and SULT2B1 transcripts were detected only in MCF7 cells, while the mRNA for SULT2A1, an enzyme that is highly expressed in human liver and adrenal gland [28], was not detected in any of the breast cell lines (Fig. 3a).
Fig. 3.

Expression of estrogen metabolism enzymes in MCF10A series and MCF7 cell lines. TaqMan Gene Expression assays were used to determine the relative levels of STS, SULT1A1, SULT2B1, SULT2A1 (a), 17βHSD1, 17βHSD2 (b), CYP19, CYP1B1, and COMT (c) mRNA in MCF10A series cell lines and in MCF7 cells. The mRNA levels are expressed relative to their respective levels in MCF10A cells, MCF7 cells or a human RNA reference pool (HRP). All values represent the mean ± SEM of three independent cell culture experiments. For all transcripts except 17βHSD2 and CYP1B1: Groups labeled with different letters are significantly different from each other (P <0.05). For 17βHSD2 and CYP1B1: *** Significantly different from all other groups, P < 0.001. When the group labeled with *** was omitted from the analyses, groups labeled with different letters are significantly different from each other (P < 0.05). For STS and COMT, no significant differences among groups were detected. For SULT2A1 and CYP19, mRNA levels were undetected in all cell lines
17βHSD1 catalyzes the reduction of estrone (E1) to the more potent E2, while 17βHSD2 catalyzes the oxidation of E2 to E1 [33]. 17βHSD1 mRNA was detected in all cell lines but was highest in the MCF10CA1a and MCF10DCIS.com cells (Fig. 3b). By contrast, 17βHSD2 mRNA was expressed most abundantly in the parental MCF10A cell line (Fig. 3b).
The transcript for CYP19, which catalyzes the aromatization of androstenedione and testosterone to E1 and E2, respectively [34], was not detected in any cell line (Fig. 3c). CYP1B1 is the most active E2 hydroxylase [35], and high levels of E2 hydroxylation in estrogen-responsive tissues may play an important role in estrogen-related tumorigenesis [36]. By contrast, O-methylation catalyzed by COMT is an inactivation pathway for E2 catechols and quinones [36]. CYP1B1 mRNA levels were highest in MCF7 cells (Fig. 3c). Though CYP1B1 mRNA also appeared to be higher in MCF10AT1 and MCF10AT1K.cl2 cells than in the other MCF10A series cells (Fig. 3c), this difference was not maintained when the cultures were treated with DMSO (experiment shown in Fig. 6). COMT demonstrated relatively consistent mRNA expression across cell lines (Fig. 3c).
Fig. 6.

TSA treatment effects on estrogen metabolism enzyme expression in MCF10A series and MCF7cell lines. a Concentration-dependent effects of TSA treatment on SULT1E1 mRNA expression in MCF10CA1a cells. MCF10CA1a cells were treated for 24 h with 0.1% DMSO or with 75, 150 or 300 ng/ml TSA and harvested for measurement of SULT1E1 mRNA levels with a TaqMan Gene Expression assay. All values represent the mean ± SEM of three independent cell culture experiments relative to the amount measured in the DMSO-treated cells. *** Significantly different from the DMSO-treated group, P < 0.001. Inset Correlation of TSA-mediated changes in SULT1E1 enzymatic activity with changes in SULT1E1 mRNA levels. b MCF10A series and MCF7 cell lines were treated for 24 h with 0.1% DMSO or 300 ng/ml TSA and SULT1E1 mRNA levels were measured. SULT1E1 mRNA contents are expressed relative to the level measured in DMSO-treated MCF10A cells. All values represent the mean ± SEM of three independent experiments. * Significantly different from the corresponding DMSO-treated group (P < 0.05). c Cells were treated with as indicated in b and estrogen metabolism enzyme mRNA levels were measured. STS, COMT, CYP1B1, 17βHSD1, and 17βHSD2 mRNA contents are expressed relative to the respective levels measured in DMSO-treated MCF10A cells. SULT1A1 and SULT2B1 mRNA levels are expressed relative to the levels measured in DMSO-treated MCF7 cells. SULT2A1 mRNA levels are expressed relative to the level measured in TSA-treated MCF7 cells. CYP19 mRNA levels are expressed relative to the level measured in TSA-treated MCF10A cells. All values represent the mean ± SEM of three independent experiments. *, ** Significantly different from the corresponding DMSO-treated group, P < 0.05 and P < 0.01, respectively
ER expression is a major determinant of estrogenic activity. As expected, the ER-positive MCF7 cell line expressed ERα mRNA at a level that was>100-fold greater than that detected in any of the MCF10A series cell lines (Fig. 4). ERα immunoreactive protein is reportedly undetectable in MCF10A cells [13], and in the present study ERα mRNA was detected at a low level in the MCF10A lineage cells (Fig. 4). By comparison, ERβ mRNA levels were not abundant either in MCF7 cells or in the MCF10A-derived series of cell lines (Fig. 4).
Fig. 4.

ERα and ERβ mRNA expression in MCF10A series and MCF7 cell lines. TaqMan Gene Expression assays were used to determine the relative levels of ERα (top) and ERβ (bottom) mRNA. The mRNA contents of ERα and ERβ are expressed relative to their respective levels in MCF10A cells, and all values represent the mean ± SEM of three independent cell culture experiments. For ERα, groups labeled with different letters are significantly different from each other (P < 0.05). For ERβ, no significant differences among groups were detected
Since HDACs are important modulators of transcription and therapeutic targets in breast cancer [24, 25], the effects of TSA treatment were characterized on ER and estrogen metabolism enzyme expression. As previously reported [37], TSA treatment of MCF7 cells produced a dramatic decrease (>99%) in ERα mRNA expression (Fig. 5a). The loss of E2 agonistic activity following TSA treatment was confirmed using an ER-responsive reporter (Fig. 5b). By contrast, TSA treatment increased ERβ mRNA levels in MCF7 cells by ~11-fold (Fig. 5a). TSA produced less pronounced effects on ERα and ERβ mRNA expression in the MCF10A lineage cell lines (Fig. 5a).
Fig. 5.

Effects of TSA treatment on ERα and ERβ mRNA expression in MCF10A series and MCF7 cell lines and on E2-mediated activation of an estrogen-responsive reporter gene. a Cell lines were treated for 24 h with 0.1% DMSO (white bars) or 300 ng/ml TSA (black bars), and ERα and ERβ mRNA levels were measured with TaqMan Gene Expression assays. ERα and ERβ mRNA levels are expressed relative to the amounts measured in DMSO-treated MCF10A cells. All values represent the mean ± SEM of three independent cell culture experiments. *, ** Significantly different from the corresponding DMSO-treated group, P < 0.05 and P < 0.01, respectively. b MCF7 cells were transiently transfected with an estrogen-responsive luciferase reporter plasmid. After transfection, cells were treated with 0.1% DMSO, 10 nM E2, 300 ng/ml TSA, or E2 and TSA in combination for 48 h. After treatment, cells were harvested for the measurement of luciferase activities. Each bar represents the mean ± SEM of normalized (firefly/Renilla) luciferase measurements (5 wells per treatment group) relative to the activity measured in DMSO-treated cells. ** Significantly different from the E2-treated group P < 0.01
TSA also produced marked alterations in the mRNA expression of estrogen metabolism enzymes in the cell lines. In the neoplastic MCF10CA1a cell line, where SULT1E1 mRNA expression is substantially suppressed, TSA treatment produced a concentration-dependent increase in SULT1E1 mRNA expression that correlated with the induction of E2 sulfation activity (Fig. 6a). TSA treatment increased SULT1E1 mRNA content by at least two-fold (2.3- to 26.1-fold) in all MCF10A series cell lines except MCF10AT1, although the increase was statistically significant only for MCF10DCIS.com (Fig. 6b). TSA treatment also activated CYP19 expression in all of the cell lines (Fig. 6c). By contrast, TSA treatment uniformly suppressed STS and COMT expression (Fig. 6c). TSA treatment produced marked suppression of SULT1A1, SULT2B1 and CYP1B1 mRNA levels in MCF7 cells, while SULT2A1 expression was induced in TSA-treated MCF7 cells (Fig. 6c). TSA treatment also suppressed 17βHSD1 and 17βHSD2 expression in the MCF10A series cell lines that demonstrated constitutive expression (Fig. 6c).
Discussion
Selective ER modulators, selective ER down-regulators and inhibitors of estrogen-producing enzymes, such as CYP19, STS, and 17βHSD1, represent important classes of drugs for achieving estrogen blockade in the treatment of hormone-responsive breast cancer [38–42]. In addition, HDAC inhibitors, which have been shown to down-regulate ERα mRNA expression in MCF7 cells [37], are being tested in phase II clinical trials that include patients with relapsed/refractory breast cancer [25]. Despite an expanded range of targeted therapies, the critical alterations in breast intracrinology that predispose patients to breast cancer development remain to be elucidated.
In mammary tissue, stringent control of the powerful mitogen E2 is achieved through the tight balance of ERα and ERβ expression coupled with titration of E2 levels. Compared to ERβ, ERα is a high affinity receptor for estrogens [43]. Both ERα and ERβ bind to the same response elements but produce differential effects on target gene expression [44]. Though ERα is a recognized marker for E2-stimulated proliferation in breast cancer, the predictive role of ERβ is less clear, possibly due to the multiplicity of ERβ isoforms [45]. In the present study, ERα expression in the MCF10A lineage cell lines was low in comparison to its level in MCF7 cells, while ERβ mRNA expression was comparably low among the cell lines. As previously reported [37], TSA suppressed ERα and induced ERβ expression in MCF7 cells. However, TSA produced only modest effects on ERα and ERβ expression in the MCF10A series of cell lines.
The pro-estrogenic machinery of the breast includes STS, CYP19, and 17βHSD1 [34, 46]. Increased expression of STS occurs in 74% of breast cancer biopsies and its presence correlates with an adverse prognosis [32]. In the present analysis, STS mRNA levels were comparably detected across the MCF10A series of cell lines and in MCF7 cells, and TSA treatment consistently tended to repress STS expression.
CYP19 expression in breast tumor tissue facilitates a highly concentrated estrogen micro-environment. Like STS, elevated CYP19 expression in breast tumor tissue impairs prognosis [7]. Though CYP19 is more robustly expressed in breast stroma, the application of fine resolution techniques has revealed the presence of CYP19 in normal breast ductal epithelial cells and breast cancer cells, as well as in intra-tumor stromal cells and peri-tumoral adipose tissue [47]. In the absence of TSA treatment, CYP19 mRNA was not detected in MCF10A series cell lines or MCF7 cells. In contrast to a previous report demonstrating a repressive role for TSA on CYP19 expression in MCF7 cells [48], this investigation revealed a TSA-inducible effect on CYP19 expression in all of the cell lines examined.
There are at least fifteen 17βHSD enzymes that vary in catalytic range and efficiency [49]. 17βHSD1 and 17βHSD2 are involved in the interconversion of E2 with the less potent E1 [33]. Increased 17βHSD1 expression in breast cancer, either alone or in combination with CYP19, enhances estrogen concentrations and negatively impacts prognosis [50, 51], as does loss of 17βHSD2 expression [52]. In the present study, the mRNA expression of pro-estrogenic 17βHSD1 was detected in MCF10A cells but was more abundant in the neoplastic MCF10CA1a and MCF10DCIS.com variants. By contrast, 17βHSD2 mRNA was most prominent in parental MCF10A cells. TSA treatment produced only suppressive effects on 17βHSD1 and 17βHSD2 expression.
SULT1E1 is considered to be the predominant E2-inactivating enzyme in breast. A previous survey of cytosolic SULT expression concluded that human mammary epithelial cells expressed mainly SULT1E1, while breast cancer cell lines preferentially expressed SULT1A1 and only trace amounts of SULT1E1 and SULT2A1 [53]. In an independent study, SULT1E1 expression was reported in approximately 44% of human breast cancer biopsy specimens [32]. The present investigation revealed SULT1E1 mRNA expression in confluent MCF10A cells and in the preneoplastic MCF10AT1 and MCF10AT1K.cl2 cell lines. By contrast, SULT1E1 expression in the more neoplastically transformed MCF10A-derived cell lines and in MCF7 cells was markedly repressed. As previously described for CYP1A2 expression in MCF10A cells [54], SULT1E1 mRNA levels were more abundant in confluent than in proliferating MCF10A cultures. The constitutive expression of other steroid-metabolizing SULTs (i.e., SULT1A1, SULT2A1, and SULT2B1) was not a prominent feature across the MCF10A-derived cell lines.
4-Hydroxylated estrogen metabolites produced by CYP1B1 metabolism are particularly reactive and considered to be promutagenic [55]. CYP1B1 mRNA and protein expression has been reported in up to 73% of human breast cancer biopsies [56]. Increased CYP1B1 expression, coupled with the reduced expression of the detoxicating enzyme COMT, is associated with amplified breast cancer risk [55]. In the current analysis, CYP1B1 mRNA was present at relatively low levels in the MCF10A-derived cell lines, but was more abundant in MCF7 cells, where its constitutive expression has been previously described [57]. In contrast to a previous report suggesting a stimulatory effect of TSA treatment on CYP1B1 expression in MCF7 cells [58], the present study revealed TSA-mediated suppression for both CYP1B1 and COMT in MCF7 cells. In contrast to the relatively restricted expression of CYP1B1, COMT mRNA was widely detected across cell lines.
In aggregate, it appears that normal or early preneoplastic breast epithelial cells act to minimize the mitogenic effects of E2 by retaining the expression of SULT1E1, a major E2-inactivating enzyme, and by preserving the expression of the anti-estrogenic dehydrogenase, 17βHSD2. Particularly in the MCF10A lineage model for breast cancer progression where ERα levels are held to a minimum, the expression of key E2-metabolizing enzymes is crucial for the establishment of the breast intracrine environment. Within the progression model, the expression of pro-estrogenic STS and 17βHSD1 is maintained, while the capacity for in situ estrogen production through aromatization is restrained. Based on the current study, several positive aspects of HDAC inhibitor treatment in humans might be anticipated. These include the up-regulation of SULT1E1 in neoplastic breast epithelial cells and also down-regulation of pro-estrogenic metabolic enzymes such as STS and 17βHSD1. With HDAC inhibition, the prominent expression of CYP1B1 that is observed in MCF7 cells becomes down-regulated. However, as a counter-weight, the expression of the protective enzyme COMT also becomes down-regulated.
The results of this study demonstrate that established breast cancer cell lines, such as MCF7, do not necessarily reflect the changes in estrogen metabolism that occur in breast epithelial cells as they progress from benign proliferative breast disease toward neoplasia. In order to harness the promise and potential power of combined or sequential metabolically-targeted therapies in breast cancer intervention, it will be become essential to understand and control the real-time dynamics that determine the delicate balance of intracrine metabolism within the breast.
Acknowledgments
This work was supported by National Institutes of Health Grants ES05823 (to M.R.-M.), GM38953 (to C.N.F.) and Environmental Health Sciences Center Grant ES06639.
Contributor Information
Jiaqi Fu, Institute of Environmental Health Sciences, Wayne State University, Detroit, MI 48201, USA.
Amy M. Weise, Barbara Ann Karmanos Cancer Institute, Wayne State University, Detroit, MI 48202, USA
Josie L. Falany, Department of Pharmacology and Toxicology, University of Alabama at Birmingham, Birmingham, AL 35294, USA
Charles N. Falany, Department of Pharmacology and Toxicology, University of Alabama at Birmingham, Birmingham, AL 35294, USA
Bryan J. Thibodeau, Institute of Environmental Health Sciences, Wayne State University, Detroit, MI 48201, USA
Fred R. Miller, Barbara Ann Karmanos Cancer Institute, Wayne State University, Detroit, MI 48202, USA. Department of Pathology, Wayne State University, Detroit, MI 48201, USA
Thomas A. Kocarek, Institute of Environmental Health Sciences, Wayne State University, Detroit, MI 48201, USA
Melissa Runge-Morris, Email: m.runge-morris@wayne.edu, Institute of Environmental Health Sciences, Wayne State University, Detroit, MI 48201, USA.
References
- 1.Baumann CK, Castiglione-Gertsch M. Estrogen receptor modulators and down regulators: optimal use in postmenopausal women with breast cancer. Drugs. 2007;67:2335–2353. doi: 10.2165/00003495-200767160-00004. [DOI] [PubMed] [Google Scholar]
- 2.Hulka BS, Moorman PG. Breast cancer: hormones and other risk factors. Maturitas. 2001;38:103–113. doi: 10.1016/S0378-5122(00)00196-1. (discussion 113–116) [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Gaikwad NW, Olson K, Zahid M, Cavalieri EL, Rogan EG. Cytochrome P450 isoforms catalyze formation of catechol estrogen quinones that react with DNA. Metabolism. 2007;56:887–894. doi: 10.1016/j.metabol.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Sasano H, Suzuki T, Miki Y, Moriya T. Intracrinology of estrogens and androgens in breast carcinoma. J Steroid Biochem Mol Biol. 2008;108:181–185. doi: 10.1016/j.jsbmb.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 5.Adjei AA, Weinshilboum RM. Catecholestrogen sulfation: possible role in carcinogenesis. Biochem Biophys Res Commun. 2002;292:402–408. doi: 10.1006/bbrc.2002.6658. [DOI] [PubMed] [Google Scholar]
- 6.Dawling S, Roodi N, Parl FF. Methoxyestrogens exert feedback inhibition on cytochrome P450 1A1 and 1B1. Cancer Res. 2003;63:3127–3132. [PubMed] [Google Scholar]
- 7.Subramanian A, Salhab M, Mokbel K. Oestrogen producing enzymes and mammary carcinogenesis: a review. Breast Cancer Res Treat. 2008;111:191–202. doi: 10.1007/s10549-007-9788-0. [DOI] [PubMed] [Google Scholar]
- 8.Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- 9.Basolo F, Elliott J, Tait L, Chen XQ, Maloney T, Russo IH, Pauley R, Momiki S, Caamano J, Klein-Szanto AJ, et al. Transformation of human breast epithelial cells by c-Ha-ras oncogene. Mol Carcinog. 1991;4:25–35. doi: 10.1002/mc.2940040106. [DOI] [PubMed] [Google Scholar]
- 10.Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am J Pathol. 1996;148:313–319. [PMC free article] [PubMed] [Google Scholar]
- 11.Santner SJ, Dawson PJ, Tait L, Soule HD, Eliason J, Mohamed AN, Wolman SR, Heppner GH, Miller FR. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat. 2001;65:101–110. doi: 10.1023/A:1006461422273. [DOI] [PubMed] [Google Scholar]
- 12.Miller FR, Santner SJ, Tait L, Dawson PJ. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. J Natl Cancer Inst. 2000;92:1185–1186. doi: 10.1093/jnci/92.14.1185A. [DOI] [PubMed] [Google Scholar]
- 13.Shekhar PV, Chen ML, Werdell J, Heppner GH, Miller FR, Christman JK. Transcriptional activation of functional endogenous estrogen receptor gene expression in MCF10AT cells: a model for early breast cancer. Int J Oncol. 1998;13:907–915. doi: 10.3892/ijo.13.5.907. [DOI] [PubMed] [Google Scholar]
- 14.Miller FR, Soule HD, Tait L, Pauley RJ, Wolman SR, Dawson PJ, Heppner GH. Xenograft model of progressive human proliferative breast disease. J Natl Cancer Inst. 1993;85:1725–1732. doi: 10.1093/jnci/85.21.1725. [DOI] [PubMed] [Google Scholar]
- 15.Shekhar MP, Nangia-Makker P, Wolman SR, Tait L, Heppner GH, Visscher DW. Direct action of estrogen on sequence of progression of human preneoplastic breast disease. Am J Pathol. 1998;152:1129–1132. [PMC free article] [PubMed] [Google Scholar]
- 16.Strickland LB, Dawson PJ, Santner SJ, Miller FR. Progression of premalignant MCF10AT generates heterogeneous malignant variants with characteristic histologic types and immunohistochemical markers. Breast Cancer Res Treat. 2000;64:235–240. doi: 10.1023/A:1026562720218. [DOI] [PubMed] [Google Scholar]
- 17.Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/me.14.10.1649. [DOI] [PubMed] [Google Scholar]
- 18.Lim KT, Cosgrave N, Hill AD, Young LS. Nongenomic oestrogen signalling in oestrogen receptor negative breast cancer cells: a role for the angiotensin II receptor AT1. Breast Cancer Res. 2006;8:R33. doi: 10.1186/bcr1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoo JY, Lessor T, Hamburger AW. Inhibition of cell proliferation by 17beta-estradiol and heregulin beta1 in estrogen receptor negative human breast carcinoma cell lines. Breast Cancer Res Treat. 1998;51:71–81. doi: 10.1023/A:1006035603635. [DOI] [PubMed] [Google Scholar]
- 20.Laidlaw IJ, Clarke RB, Howell A, Owen AW, Potten CS, Anderson E. The proliferation of normal human breast tissue implanted into athymic nude mice is stimulated by estrogen but not progesterone. Endocrinology. 1995;136:164–171. doi: 10.1210/en.136.1.164. [DOI] [PubMed] [Google Scholar]
- 21.Schafer JM, Lee ES, O’Regan RM, Yao K, Jordan VC. Rapid development of tamoxifen-stimulated mutant p53 breast tumors (T47D) in athymic mice. Clin Cancer Res. 2000;6:4373–4380. [PubMed] [Google Scholar]
- 22.Visscher DW, Nanjia-Makker P, Heppner G, Shekhar PV. Tamoxifen suppresses histologic progression to atypia and DCIS in MCFIOAT xenografts, a model of early human breast cancer. Breast Cancer Res Treat. 2001;65:41–47. doi: 10.1023/A:1006490000659. [DOI] [PubMed] [Google Scholar]
- 23.Nkhata KJ, Ray A, Dogan S, Grande JP, Cleary MP. Mammary tumor development from T47-D human breast cancer cells in obese ovariectomized mice with and without estradiol supplements. Breast Cancer Res Treat. 2008;114:71–83. doi: 10.1007/s10549-008-9991-7. [DOI] [PubMed] [Google Scholar]
- 24.Kawai H, Li H, Avraham S, Jiang S, Avraham HK. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int J Cancer. 2003;107:353–358. doi: 10.1002/ijc.11403. [DOI] [PubMed] [Google Scholar]
- 25.Vansteenkiste J, Van Cutsem E, Dumez H, Chen C, Ricker JL, Randolph SS, Schoffski P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest New Drugs. 2008;26:483–488. doi: 10.1007/s10637-008-9131-6. [DOI] [PubMed] [Google Scholar]
- 26.Duanmu Z, Weckle A, Koukouritaki SB, Hines RN, Falany JL, Falany CN, Kocarek TA, Runge-Morris M. Developmental expression of aryl, estrogen, and hydroxysteroid sulfotransferases in pre- and post-natal human liver. J Pharmacol Exp Ther. 2006;316:1310–1317. doi: 10.1124/jpet.105.093633. [DOI] [PubMed] [Google Scholar]
- 27.Falany CN, Krasnykh V, Falany JL. Bacterial expression and characterization of a cDNA for human liver estrogen sulfotransferase. J Steroid Biochem Mol Biol. 1995;52:529–539. doi: 10.1016/0960-0760(95)00015-R. [DOI] [PubMed] [Google Scholar]
- 28.Comer KA, Falany CN. Immunological characterization of dehydroepiandrosterone sulfotransferase from human liver and adrenal. Mol Pharmacol. 1992;41:645–651. [PubMed] [Google Scholar]
- 29.Falany CN, He D, Dumas N, Frost AR, Falany JL. Human cytosolic sulfotransferase 2B1: isoform expression, tissue specificity and subcellular localization. J Steroid Biochem Mol Biol. 2006;102:214–221. doi: 10.1016/j.jsbmb.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Falany CN, Wheeler J, Oh TS, Falany JL. Steroid sulfation by expressed human cytosolic sulfotransferases. J Steroid Biochem Mol Biol. 1994;48:369–375. doi: 10.1016/0960-0760(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 31.Sasano H, Suzuki T, Nakata T, Moriya T. New development in intracrinology of breast carcinoma. Breast Cancer. 2006;13:129–136. doi: 10.2325/jbcs.13.129. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki T, Nakata T, Miki Y, Kaneko C, Moriya T, Ishida T, Akinaga S, Hirakawa H, Kimura M, Sasano H. Estrogen sulfotransferase and steroid sulfatase in human breast carcinoma. Cancer Res. 2003;63:2762–2770. [PubMed] [Google Scholar]
- 33.Miettinen MM, Mustonen MV, Poutanen MH, Isomaa VV, Vihko RK. Human 17beta-hydroxysteroid dehydrogenase type 1 and type 2 isoenzymes have opposite activities in cultured cells and characteristic cell- and tissue-specific expression. Biochem J. 1996;314(Pt 3):839–845. doi: 10.1042/bj3140839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki T, Miki Y, Akahira J, Moriya T, Ohuchi N, Sasano H. Aromatase in human breast carcinoma as a key regulator of intratumoral sex steroid concentrations. Endocr J. 2008;55:455–463. doi: 10.1507/endocrj.K07E-053. [DOI] [PubMed] [Google Scholar]
- 35.Chun YJ, Kim S. Discovery of cytochrome P450 1B1 inhibitors as new promising anti-cancer agents. Med Res Rev. 2003;23:657–668. doi: 10.1002/med.10050. [DOI] [PubMed] [Google Scholar]
- 36.Yager JD, Liehr JG. Molecular mechanisms of estrogen carcinogenesis. Annu Rev Pharmacol Toxicol. 1996;36:203–232. doi: 10.1146/annurev.pa.36.040196.001223. [DOI] [PubMed] [Google Scholar]
- 37.De los Santos M, Martinez-Iglesias O, Aranda A. Anti-estrogenic actions of histone deacetylase inhibitors in MCF-7 breast cancer cells. Endocr Relat Cancer. 2007;14:1021–1028. doi: 10.1677/ERC-07-0144. [DOI] [PubMed] [Google Scholar]
- 38.Brueggemeier RW. Update on the use of aromatase inhibitors in breast cancer. Expert Opin Pharmacother. 2006;7:1919–1930. doi: 10.1517/14656566.7.14.1919. [DOI] [PubMed] [Google Scholar]
- 39.Day JM, Foster PA, Tutill HJ, Parsons MF, Newman SP, Chander SK, Allan GM, Lawrence HR, Vicker N, Potter BV, Reed MJ, Purohit A. 17beta-hydroxysteroid dehydrogenase type 1, and not type 12, is a target for endocrine therapy of hormone-dependent breast cancer. Int J Cancer. 2008;122:1931–1940. doi: 10.1002/ijc.23350. [DOI] [PubMed] [Google Scholar]
- 40.Saji S, Kuroi K. Application of selective estrogen receptor modulators for breast cancer treatment according to their intrinsic nature. Breast Cancer. 2008;15:262–269. doi: 10.1007/s12282-008-0063-y. [DOI] [PubMed] [Google Scholar]
- 41.Stanway SJ, Purohit A, Woo LW, Sufi S, Vigushin D, Ward R, Wilson RH, Stanczyk FZ, Dobbs N, Kulinskaya E, Elliott M, Potter BV, Reed MJ, Coombes RC. Phase I study of STX 64 (667 Coumate) in breast cancer patients: the first study of a steroid sulfatase inhibitor. Clin Cancer Res. 2006;12:1585–1592. doi: 10.1158/1078-0432.CCR-05-1996. [DOI] [PubMed] [Google Scholar]
- 42.Wittmann BM, Sherk A, McDonnell DP. Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res. 2007;67:9549–9560. doi: 10.1158/0008-5472.CAN-07-1590. [DOI] [PubMed] [Google Scholar]
- 43.Barkhem T, Carlsson B, Nilsson Y, Enmark E, Gustafsson J, Nilsson S. Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists. Mol Pharmacol. 1998;54:105–112. doi: 10.1124/mol.54.1.105. [DOI] [PubMed] [Google Scholar]
- 44.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaaban AM, Green AR, Karthik S, Alizadeh Y, Hughes TA, Harkins L, Ellis IO, Robertson JF, Paish EC, Saunders PT, Groome NP, Speirs V. Nuclear and cytoplasmic expression of ERbeta1, ERbeta2, and ERbeta5 identifies distinct prognostic outcome for breast cancer patients. Clin Cancer Res. 2008;14:5228–5235. doi: 10.1158/1078-0432.CCR-07-4528. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki T, Miki Y, Moriya T, Akahira J, Hirakawa H, Ohuchi N, Sasano H. In situ production of sex steroids in human breast carcinoma. Med Mol Morphol. 2007;40:121–127. doi: 10.1007/s00795-007-0365-8. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki T, Miki Y, Ohuchi N, Sasano H. Intratumoral estrogen production in breast carcinoma: significance of aromatase. Breast Cancer. 2008;15:270–277. doi: 10.1007/s12282-008-0062-z. [DOI] [PubMed] [Google Scholar]
- 48.Luczak MW, Jagodziński PP. Trichostatin A down-regulates CYP19 transcript and protein levels in MCF-7 breast cancer cells. Biomed Pharmacother. 2008 doi: 10.1016/j.biopha.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 49.Day JM, Tutill HJ, Purohit A, Reed MJ. Design and validation of specific inhibitors of 17beta-hydroxysteroid dehydrogenases for therapeutic application in breast and prostate cancer, and in endometriosis. Endocr Relat Cancer. 2008;15:665–692. doi: 10.1677/ERC-08-0042. [DOI] [PubMed] [Google Scholar]
- 50.Miyoshi Y, Ando A, Shiba E, Taguchi T, Tamaki Y, Noguchi S. Involvement of up-regulation of 17beta-hydroxysteroid dehydrogenase type 1 in maintenance of intratumoral high estradiol levels in postmenopausal breast cancers. Int J Cancer. 2001;94:685–689. doi: 10.1002/ijc.1525. [DOI] [PubMed] [Google Scholar]
- 51.Salhab M, Reed MJ, Al Sarakbi W, Jiang WG, Mokbel K. The role of aromatase and 17-beta-hydroxysteroid dehydrogenase type 1 mRNA expression in predicting the clinical outcome of human breast cancer. Breast Cancer Res Treat. 2006;99:155–162. doi: 10.1007/s10549-006-9198-8. [DOI] [PubMed] [Google Scholar]
- 52.Gunnarsson C, Olsson BM, Stal O. Abnormal expression of 17beta-hydroxysteroid dehydrogenases in breast cancer predicts late recurrence. Cancer Res. 2001;61:8448–8451. [PubMed] [Google Scholar]
- 53.Falany JL, Falany CN. Expression of cytosolic sulfotransferases in normal mammary epithelial cells and breast cancer cell lines. Cancer Res. 1996;56:1551–1555. [PubMed] [Google Scholar]
- 54.Thomas RD, Green MR, Wilson C, Weckle AL, Duanmu Z, Kocarek TA, Runge-Morris M. Cytochrome P450 expression and metabolic activation of cooked food mutagen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in MCF10A breast epithelial cells. Chem Biol Interact. 2006;160:204–216. doi: 10.1016/j.cbi.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 55.Wen W, Ren Z, Shu XO, Cai Q, Ye C, Gao YT, Zheng W. Expression of cytochrome P450 1B1 and catechol-O-methyltransferase in breast tissue and their associations with breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2007;16:917–920. doi: 10.1158/1055-9965.EPI-06-1032. [DOI] [PubMed] [Google Scholar]
- 56.McKay JA, Melvin WT, Ah-See AK, Ewen SW, Greenlee WF, Marcus CB, Burke MD, Murray GI. Expression of cytochrome P450 CYP1B1 in breast cancer. FEBS Lett. 1995;374:270–272. doi: 10.1016/0014-5793(95)01126-Y. [DOI] [PubMed] [Google Scholar]
- 57.Nakajima M, Iwanari M, Yokoi T. Effects of histone deacetylation and DNA methylation on the constitutive and TCDD-inducible expressions of the human CYP1 family in MCF-7 and HeLa cells. Toxicol Lett. 2003;144:247–256. doi: 10.1016/S0378-4274(03)00216-9. [DOI] [PubMed] [Google Scholar]
- 58.Hooven LA, Mahadevan B, Keshava C, Johns C, Pereira C, Desai D, Amin S, Weston A, Baird WM. Effects of suberoylanilide hydroxamic acid and trichostatin A on induction of cytochrome P450 enzymes and benzo[a]pyrene DNA adduct formation in human cells. Bioorg Med Chem Lett. 2005;15:1283–1287. doi: 10.1016/j.bmcl.2005.01.032. [DOI] [PubMed] [Google Scholar]