

Abstract
Olaparib has shown promising anticancer activity as a single agent in the treatment and maintenance of recurrent ovarian cancer in early clinical trials, but it is far from standard therapy. This article outlines the problem of relapsed ovarian cancer and the mechanisms of poly(ADP-ribose) polymerase inhibitors and reviews the recent literature pertaining to olaparib in ovarian cancer.
Keywords: anticancer, polymerase inhibitor
Introduction
Ovarian cancer is the most common cause of death from gynecologic cancer and is fourth on the list of cancer-related deaths in women.1 A significant proportion of women present at an advanced stage of disease, and 60% present at stage 3 to 4 disease. Five-year survival for all stages of ovarian cancer is 43%, and 10-year survival is 28%, but the 5-year survival for patients presenting with stage 3 disease is only 21.9%, and for stage 4 disease it is a dismal 5.6%.2 The vast majority (70%) of these cancers will be epithelial ovarian cancer, predominantly of serous type, but endometrioid, clear cell, and mucinous variants also exist.3 The lifetime incidence of a woman spontaneously developing ovarian cancer is 1.4%,4 but this incidence is significantly increased in carriers of germline mutations, mainly in either breast cancer gene 1 (BRCA1) (~40% lifetime risk) or 2 (BRCA2) (~10% lifetime risk) genes,5 which are implicated in 10%–15% of all epithelial ovarian cancer cases, including those women who have no family history of breast or ovarian cancer.6 Other gene candidates have also been demonstrated, but with increased rarity.7
Standard therapeutic approaches include optimal surgical debulking followed by adjuvant platinum doublet chemotherapy, usually in combination with a taxane. If immediate surgical resection is not possible, then neoadjuvant use of combination chemotherapy can be employed, with interval debulking as appropriate.8,9 These methods have high initial response rates, but only moderate progression-free survival (PFS) times, and three-quarters of patients with stage 3 and 4 disease will relapse. Recent developments include the use of bevacizumab (a monoclonal antibody against vascular endothelial growth factor receptor) to prolong the PFS and overall survival (OS) when used alongside standard chemotherapy and as maintenance treatment in a first-line setting. The success of this drug was particularly striking in those patients at high risk for relapse, in whom PFS increased from 14.5 months with standard therapy to 18.1 months with the addition of bevacizumab within the Gynecologic Cancer Intergroup International Collaboration on Ovarian Neoplasms 7 (ICON7) trial.10,11 OS was also affected (going from 28.8 to 36.6 months, respectively).12
In recurrent disease, the selection of subsequent therapy is influenced by the progression-free interval, number of lines and drugs used previously, performance status of the patient, and extent of relapse. In general, a PFS interval of greater than 6 months since platinum therapy would be considered to be platinum-sensitive disease and have a higher likelihood of a second response to a platinum-containing regimen. The International Collaborative Ovarian Neoplasm/Arbeitsgemeinschaft Gynäkologische Onkologie Studiengruppe Ovarialkarzinom (ICON 4/AGO-OVAR) 2.2 and other studies showed that a second challenge with a platinum/paclitaxel combination was effective, improving both PFS and OS.13 In those patients with significant adverse effects from their taxane exposure, gemcitabine can be used alongside platinum therapy.14 Recently published evidence also supports the use of bevacizumab in relapsed patients as part of the Study of Carboplatin and Gemcitabine Plus Bevacizumab in Patients With Ovary, Peritoneal, or Fallopian Tube Carcinoma (OCEANS) trial, and extended PFS (12.4 versus 8.4 months) and improved response rates (78.5% versus 57.4%) were seen when bevacizumab was added to carboplatin/gemcitabine chemotherapy.15 OS was unchanged, which was attributed to multiple further lines of therapy after participation in the OCEANS trial, including widespread use of bevacizumab in patients from the placebo group.
Patients considered to be platinum-resistant (<6 months since last platinum treatment) or refractory (disease progressed despite continuing platinum treatment) have a low rate of response to subsequent therapy, but liposomal doxorubicin, topotecan, or single-agent weekly paclitaxel are potential choices.16 Clinical trials of targeted therapy should also be considered, when available, in platinum-sensitive and platinum-resistant disease. In the past, therapies exploring the weaknesses of targets such as epidermal growth factor receptor, insulin-like growth factor 1, BRAF, mammalian target of rapamycin, and human epidermal growth factor receptor 2, as well as poly(ADP-ribose) polymerase (PARP), have been investigated.17–21
BRCA1, BRCA2, and “BRCAness”
BRCA1 and BRCA2 code for large proteins that participate in the repair of double-strand breaks in cellular DNA, via the homologous repair (HR) pathway.22,23 Cells from patients with the defect carry heterozygous mutations in either gene that are either germline or acquired. A further mutation in the remaining functional allele results in complete loss of either BRCA1 or BRCA2 function, resulting in aberrant double-strand deoxyribonucleic acid (DNA) repair and in chromosomal breaks, rearrangements, and other abnormalities, as well as associated genetic instability, causing cell death (Figure 1). Subsequent to the discovery of these two genes came an understanding that some sporadic epithelial ovarian cancers behaved in a very similar way to those known to bear either a BRCA1 or BRCA2 mutation, perhaps through expression of such genes as TP53 and FANCD224–27 (Figure 1). Recent work has also suggested a role for BRCA1 and BRCA2 and defective HR in sporadic ovarian cancer, with somatic mutation and epigenetic mechanisms eg (promoter methylation) implicated in as many as from 19% to 24% of unselected patients dependent on series, and potentially more (up to 50%) in high-grade serous cancer of the ovary.28,29 Overall, these patients tend to have a more favorable prognosis (Figure 2).
Figure 1.

Role of BRCA1 and BRCA2 and ATR in cancer susceptibility.
Notes: Cartoon showing factors required for double-stranded DNA repair; malfunction of any of the involved factors can potentially cause a BRCA-like effect (“BRCAness”). For example, germline mutations, such as CHEK2, PLAB, Rad50/51, FANC A/B/C (etc), BRIPI, and BRCA1/2, and somatic mutations, such as somatic BRCA mutations, non-BRCA double-stranded DNA repair gene mutation, transcription factor binding site, polymorphisms/mutations, and epigenetic factors (eg, BRCA promoter methylation).
Abbreviations: DNA, deoxyribonucleic acid; BRCA, breast cancer genes 1 and 2.
Figure 2.

Kaplan–Meier curves showing that BRCA1/2 mutations were associated with significantly improved progression-free survival time after surgery when compared with a BRCA wild-type ovarian cancer population.
Notes: 235 patients in total, 19% with mutant BRCA1/2. Median progression-free survival for BRCA mutants and nonmutants was 20.1 (CI, 15.6–43.8) and 13.8 (CI, 11.9–16.3) months, respectively. Reprinted with permission. © 2010 American Society of Clinical Oncology. All rights reserved. Hennessy B, et al. J Clin Oncol. 2010;28(22):3570–3576.29
Abbreviations: BRCA, breast cancer genes 1 and 2; CI, confidence interval.
Olaparib: rationale and preclinical development
DNA repair is a vital function for all cells to be able to proceed through the cell cycle and replicate without errors.30 DNA damage generally causes double-strand breaks, and HR is the main mechanism by which the double-strand breaks are repaired. However, HR is not the only method of double-strand DNA repair available to the cellular machinery: nonhomologous end joining and single-strand annealing can also be used, although these mechanisms are error-prone, resulting in loss of DNA and rearrangements, and, when repeatedly used, eventually result in overwhelming DNA damage, activation of cellular checkpoint mechanisms, cell-cycle exit, and cell death.31
Different mechanisms exist for repairing single-strand breaks. These include base excision repair, nucleotide excision repair, and mismatch repair. These processes are modulated by PARP,32 which binds to the break sites and recruits other elements of the DNA repair complex (Figure 3). If cells are unable to repair single-strand breaks before attempting to replicate, then double-strand breaks form.
Figure 3.

Simplified overview of the events involving PARP1, PARP2, or PARP3 in DNA repair pathways. (A) Base excision repair/single strand break repair, (B) homologous recombination, (C) double strand breaks.
Note: Reprinted from Biochem Pharmacol, 84(2), De Vos et al, The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art, pages 137–146. Copyright © 2012, with permission from Elsevier.32
Abbreviations: ADP, adenosine diphosphate; PARP, poly (ADP-ribose) polymerase; BER, base excision repair; SSBR, single strand break repair; HR, homologous repair; BRCA, breast cancer genes 1 and 2; DNA, deoxyribonucleic acid.
A specific inhibitor of PARP1 and PARP2 was developed by testing a series of substituted 4-benzyl-2H-phthalazin-1-ones. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one 47 (KU-0059436, AZD2281), now known as olaparib, was taken forward for further development as a nanomolar inhibitor of PARP with single-agent activity against BRCA-1-deficient cells.33 Inhibition of PARP-1 by olaparib prevents repair of single-strand breaks, which is of no consequence to normal cells that efficiently repair double-strand DNA via HR. In cells with deficiencies of HR where nonhomologous end joining and single-strand annealing are the only mechanisms of DNA repair, PARP inhibition produces stalled replication forks, increasing the number of double-strand breaks, which cannot be repaired in these cells homologous for BRCA mutations, leading to genetic chaos and cell death via apoptosis or senescence (Figure 4). Taking advantage of an abnormality within the cancer cells (homozygous loss of BRCA function) that is not present in the normal somatic cells of the body (they are heterozygous for the mutation and therefore produce sufficient functional BRCA protein) is a concept described as synthetic lethality.34,35
Figure 4.

Cartoon detailing DNA repair mechanisms in the absence of functional BRCA proteins.
Abbreviations: ds, double stranded; DNA, deoxyribonucleic acid; DSB, double-strand breaks; HR, homologous repair; NHEJ, nonhomologous end joining; SSA, single-strand annealing; BER, base excision repair; NER, nucleotide excision repair; MMR, mismatch repair; BRCA, breast cancer genes 1 and 2.
Synthetic lethality means there is much higher sensitivity to treatment with PARP inhibitors among cancer cells carrying homozygous mutations in either BRCA1 or BRCA2.36 In contrast, cells that were either wild-type or heterozygous for BRCA1 and BRCA2 showed no increase in cell death when treated. These findings indicate there should be minimal toxicity in test participants and, indeed, humans, offering scope for dose increments to produce the desired pharmacodynamic effect.37,38
Xenograft studies in NOD/SCID (nonobese diabetic/severe combined immunodeficiency) mice with patient-derived BRCA2-deficient ovarian tumor tissue showed significant reduction in the growth of tumors of those treated with either olaparib alone (19.7 ± 25.0 mm3 versus 97.3 ± 72.6 mm3) or carboplatin alone (4.4 ± 7.4 mm3 versus 97.3 ± 72.6 mm3), but the most striking response was in those mice treated with a combination of olaparib and carboplatin (1.2 ± 1.4 mm3 versus 97.3 ± 72.6 mm3) versus vehicle. Reimplantation of residual tumors after treatment did not result in any regeneration after 18 months for combination treatment, in comparison with just 6 weeks to develop palpable tumors after vehicle treatment. None of the treatment regimens caused the mice to lose significant weight or reduce oral intake, indicating the treatment was well tolerated.39
Much work was also carried out in BRCA1/2 carrier breast cancer cell lines but is beyond the scope of this review.40,41
Clinical trials
The first published trial in humans using olaparib was a Phase I study with an unselected dose-escalation (from 10 mg to 600 mg twice daily [bd]) and an expansion cohort in which only patients carrying BRCA1 or BRCA2 mutations were permitted and treated at 200 mg bd (Table 1).44 The maximum tolerated dose was set as 400 mg bd. PARP inhibition of greater than 90% was seen at doses of 60 mg bd and over.
Table 1.
Trials using olaparib, reported and currently underway
| Author | Trial Phase | Single-agent (bd) versus combination | Maintenance plt sens/res | PFS | OS/CBR | BRCA popn only? |
|---|---|---|---|---|---|---|
| Fong et al44 | I | Single agent Dose esc from 10 to 600 mg |
Plt res and sens | MTD: 400 mg PFS 7 months | CBR at 4 months: 69% plt sens, 46% plt res, and 23% plt ref | Yes (1 and 2) |
| Khan et al53 | I | Dacarbazine | Any solid tumor 20/36 pts melanoma | MTD: 100 mg bd with 600 mg/m2 dacarb 20 mg bd with 800 mg/m2 dacarb | PR 1/36 pts; CBR 7/36 | All comers |
| Samol et al54 | I | Topotecan | Any solid tumor | MTD: 100 mg bd × 3 d plus topo 1 mg/m2 × 3 d | Not progressed as subtherapeutic MTD (hem tox) | All comers |
| Dean et al55 | I | Bevacizumab | Any solid tumor (2 gyne) | MTD: 400 mg bd with 10 mg/kg/14 d bevacizumab | No response documented | All comers |
| Rajan et al56 | I | Cisplatin/gemcitabine | Any solid tumor | MTD: 100 mg bd d 1 only | PR in 2/21 | All comers |
| Audeh et al46 | II | 400 mg versus 100 mg | No maintenance Plt res and sens |
33% CR/PR for 9.6 months | 69% | Yes (1 and 2) |
| Kaye et al48 | II | 200 mg versus 400 mg versus PLD | No maintenance | 6.5 versus 8.8 versus 7.1 months | N/A | Yes (1 and 2) |
| Ledermann et al51 | II | Carbo/taxol then olaparib versus placebo maintenance | Maintenance Plat sens only |
Olaparib: 8.4 months versus placebo: 4.8 months | 29.7 versus 29.9 months | All comers |
| National Cancer Institute57 | I | Carboplatin | No maintenance Plt res and sens |
Recruiting | Recruiting | Yes (1 and 2) |
| ClinicalTrials.gov58 | I | Carboplatin | No maintenance Any gyne malig |
Recruiting | Recruiting | All comers |
| ClinicalTrials.gov59 | I | Carboplatin/paclitaxel | No maintenance Any solid tumor |
Closed in FU | Closed in FU | All comers |
| ClinicalTrials.gov60 | II | Carboplatin/paclitaxel | Maintenance Plat sens |
Closed PFS 12.2 versus 9.6 months (control) | Close OS 12 versus 11 months (control) | All comers |
| ClinicalTrials.gov61 | Ib | Weekly carboplatin/weekly paclitaxel | Any gyne malig | Recruiting | Recruiting | All comers |
| ClinicalTrials.gov62 | I | BKM120 (PI3 kinase inhibitor) | Plt res and sens | In setup | In setup | All comers |
| ClinicalTrials.gov63 | I | Cediranib | Plt sens | Recruiting | Recruiting | All comers |
Abbreviations:BRCA1/BRCA2, breast cancer genes 1 and 2; bd, twice a day; plt, platinum; sens, sensitive; res, resistant; ref, refractory; CBR, clinical benefit rate; popn, population; esc, escalation; MTD, maximum tolerated dose; dacarb, dacarbazine; PR, partial response; topo, topotecan; hem tox, hematological toxicity; CR, complete response; carbo, carboplatin; taxol, paclitaxel; gyne, gynecological; malig, malignancy; FU, follow up; PI3, PI3 kinase; N/A, not applicable.
Objective antitumor responses were seen only in those carrying BRCA1 or BRCA2 mutations and at doses of olaparib of 100 mg or greater bd. Eight (of 16) patients with ovarian cancer had a partial response by either Response Evaluation Criteria In Solid Tumors (RECIST)42 or Gynecologic Cancer InterGroup (GCIG) criteria,43 and a further patient had stabilization of disease for more than 4 months. The main toxicities were nausea, vomiting, fatigue, taste alteration, and anorexia. Only a 3%–5% incidence of myelosuppression was seen. The toxicity in the BRCA mutated population was not significantly different than in the noncarrier population.44 This trial provided proof-of-concept of synthetic lethality in a clinical setting, as well as proof that PARP inhibition could provide tumor responses as a single agent.
The trial data were further analyzed with regard to BRCA1/2 carriers and their platinum sensitivity. Fifty patients carrying germline BRCA1 and BRCA2 mutations were enrolled to the study (BRCA1, 41; BRCA2, 8; family history only, 1); 13 patients had platinum-sensitive disease, 24 patients had platinum-resistant disease, and 13 patients had platinum-refractory disease. Most patients (39/50) were treated in the expansion cohort, whereas the remainder (11/50) was treated within the dose-escalation cohort. No significant differences were seen between responses in either the BRCA1 or BRCA2 carrier groups. However, when the responses were examined with regard to platinum status, a statistically significant correlation with the platinum-free interval was demonstrated (Spearman rank [Rs], 0.33; 95% confidence interval [CI], 0.04–0.57). The overall clinical benefit rate (defined as those patients with an objective response by either RECIST or GCIG criteria or with stable disease for 4 or more months) was 69.2% in the platinum-sensitive, 45.8% in platinum-resistant, and 23.1% in the platinum-refractory groups. This did not translate to a difference in the duration of response between the groups, which was reported as approximately 7 months. The results of the study inferred that some mechanisms of sensitivity to platinum and PARP inhibition may be shared, but to confound this some patients with platinum-refractory disease had responses. Platinum sensitivity is therefore not the only factor involved in guiding the use of PARP inhibition with olaparib in a clinical setting.45
A Phase II study enrolled two cohorts of BRCA1-and BRCA2-positive women with recurrent advanced ovarian cancer who had previously received one or more lines of chemotherapy.46 This study was designed to look at whether equivalent responses would be seen at the maximum tolerated dose of 400 mg bd or at a lower, but still pharmacodynamically active, dose of 100 mg bd. An objective response was seen in 33% of patients in the 400 mg bd and 13% of the 100 mg bd group, with a median PFS of 5.8 months (95% CI 2.8–10.6) and 1.9 months (1.8–3.6), respectively. The toxicity profile was similar to that seen in the Phase I study but with more reported hematological toxicity, particularly anemia (15%–17%), although the majority of cases were grade 1 or 2 by Common Terminology Criteria for Adverse Events. The dose of 400 mg bd appeared to be more effective than 100 mg bd, but the prognostic characteristics of the latter group were described as less desirable. The study data supported the use of olaparib for recurrent pretreated ovarian cancer in both BRCA1 and BRCA2 germline mutation carriers. Responses were seen in both platinum-sensitive and resistant subgroups, although the study was not powered to look at this directly.46
Because pegylated liposomal doxorubicin (PLD) is a currently approved second-line therapeutic option for relapsed ovarian cancer, a Phase II trial to compare response to PLD versus olaparib was carried out.48 The data before this comparison study showed an overall response rate (ORR) to PLD therapy of 20% (with a PFS of 16 weeks) from a Phase III trial versus topotecan,47 in which the ORR to olaparib at 400 mg bd was 33% (response duration, 9.5 months) from the Phase I study of olaparib.44 The hypothesis therefore was that olaparib would be superior to PLD in BRCA1/2 mutated patients. The trial design consisted of three groups: PLD infusion at 50 mg/m2 every 28 days or olaparib 400 mg bd or olaparib 200 mg bd (as per the expansion cohort within the Phase I study) in a 1:1:1 ratio. Patients were stratified according to BRCA1 or 2 status and platinum sensitivity. Crossover to olaparib from PLD was permitted if a central assessment of response defined progressive disease. Median PFS times were 6.5 months (95% CI, 5.5–10.1 months), 8.8 months (95% CI, 5.4–9.2 months), and 7.1 months (95% CI, 3.7–10.7 months) for the olaparib 200 mg, olaparib 400 mg, and PLD groups, respectively. In the same group order, the ORRs were 25%, 31%, and 18%. Neither of these outcomes demonstrated statistically significant differences between olaparib at either dose versus PLD. Both treatments were generally well tolerated, although there was a tendency for more grade 3–4 adverse events with PLD use. The authors suggest that the better-than-expected PFS for PLD of 7.1 months confounded the ability of the study to detect a statistical benefit in favor of olaparib in BRCA1/2-mutated patients but is in agreement with recent work suggesting that this patient population derives greater benefit from anthracycline-based chemotherapy than unselected patients. 48–50
One of the most significant occurrences in ovarian cancer is the potential for multiple relapses over time. To investigate the role of olaparib in the maintenance and potential extension of PFS, a randomized, double-blind, placebo-controlled Phase II study was carried out in patients with platinum-sensitive recurrent serous ovarian cancer.51 Patients were required to have received two or more courses of platinum chemotherapy, with the most recent course inducing a RECIST- or GCIG-defined response. At entry to trial, the CA125 level had to be below the upper limit of normal. BRCA1/2 status was not required for inclusion. Patients were stratified by interval from last platinum regimen to progression, response to last regimen, and ancestry before randomization in a blinded 1:1 ratio to either olaparib 400 mg bd continuously or placebo within 8 weeks of completion of last chemotherapy regimen, until defined progression of disease. No crossover was permitted. Two hundred and sixty five patients were treated within the trial, with approximately half receiving olaparib. The median PFS was 8.4 months in the olaparib group versus 4.8 months in the placebo group (hazard ratio for progression or death, 0.35; 95% CI, 0.25–0.49; P < 0.001). Subgroup analysis demonstrated olaparib to be superior to placebo in all categories with regard to PFS. Complete response to the last chemotherapy regimen before trial entry significantly improved PFS (hazard ratio, 0.46; P < 0.001). OS was not affected (29.7 months in the olaparib group and 29.9 months in the placebo group). The olaparib group had slightly more nausea, vomiting, fatigue, and anemia, but generally did not require stoppage of trial therapy. Subanalysis of BRCA1/2 or, “BRCAness,” could not be done in the limited study data, which might have been valuable in selecting a group of patients for whom this maintenance therapy may result in an improved OS as well as PFS.51 Figure 5 shows the disappointing OS data in comparison to the promising PFS results obtained earlier. More recently, the formulation of olaparib has been changed from capsules to tablets, and this has required further initial studies of dose and efficacy. These trials have now completed recruitment and are currently being evaluated. Both the lack of OS benefit and the uncertainty around the optimal dosing of olaparib have led to a proposed Phase III maintenance trial being stopped from further development at this stage.52
Figure 5.
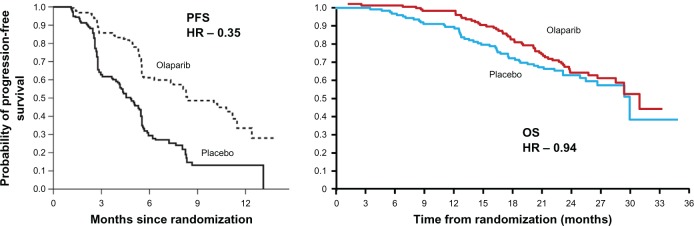
Progression-free survival (PFS) and overall survival (OS) following maintenance treatment with the PARP inhibitor, olaparib, in patients with relapsed ovarian cancer.
Abbreviations: HR, hazard ratio; PARP, poly(ADP-ribose) polymerase.
Multiple other Phase I combination studies with a variety of other agents have recently been reported, including dacarbazine,53 topotecan,54 bevacizumab,55 and cisplatin with gemcitabine.56 The topotecan and cisplatin combinations were particularly myelosuppressive and would require further dosing schedule modification before further work was undertaken.
Currently underway either Phase I, or Phase II in ovarian cancer, are studies of olaparib with carboplatin (Clinical Trials.gov identifiers NCT01445418, NCT01237067),57,58 carboplatin and paclitaxel (NCT00516724, NCT01081951, and NCT01650376),59–61 BKM120 (a PI3-kinase inhibitor, NCT01623349),62 and cediranib (a small molecule inhibitor of VEGF (vascular endothelial growth factor), NCT01116648).63 In addition to olaparib, many other PARP inhibitors are also under scrutiny (eg, rucaparib, veliparib) and are in development. It is clear that much interest surrounds the use of PARP inhibition in ovarian cancer, and the results of these trials will guide future direction with olaparib and other related compounds.
Resistance
Not all patients with BRCA1 or 2 somatic or germline mutations respond to PARP inhibition, and indeed, even patients carrying what appears to be the same mutation can demonstrate differences in responses. This is likely to be multifactorial in origin, but recent work has shown that secondary mutations within BRCA2 can restore function of the protein in patients who are clinically resistant to PARP inhibition;64 partial restoration of HR resulting from a loss of 53BP1,65,66 and functional Rad51 can provide escape mechanisms from growth inhibition by PARP inhibitors.67 As more work is carried out on these cellular pathways, a greater understanding of secondary resistance, and indeed potential biomarkers of initial response to PARP inhibition, will be delineated. There is already ongoing research in the role of predictive markers in PARP inhibition sensitivity: in vitro work has shown an increase in double-strand breaks with the addition of PARP inhibitors to radiation, using γ-H2AX foci, a well established marker of DNA double-strand breaks.68 Reduction of poly(ADP-ribose) levels has also been used as a surrogate to indicate PARP inhibitor activity.69
Conclusion
Olaparib has shown promising anticancer activity as a single agent in the treatment and maintenance of recurrent ovarian cancer in early clinical trials, but it is far from standard therapy, and much more work will be required to secure its optimal use. This patient group is particularly enriched in the BRCA1/2 germline mutation carriers but somatic mutations of BRCA and other defects of DNA repair mechanisms are also found in sporadic epithelial ovarian cancer, with an emphasis on high-grade serous type. Further work in combination studies with various chemotherapeutic agents and other targeted molecules is required to elucidate the best treatment strategies for this complex and deadly disease. As more is known regarding the molecular subgroups of ovarian carcinoma, the implications for platinum-sensitivity mechanisms, as well as acquired and inherent resistance to PARP inhibition, treatment can be increasingly tailored to the individual patient to maximize potential for response and increases in PFS and, ultimately, OS.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Cancer Research UK Cancer mortality for common cancers [webpage on the Internet] London, UK: Cancer Research UK; 2011Available from: http://www.cancerresearchuk.org/cancer-info/cancerstats/mortality/cancerdeathsAccessed October 10, 2012 [Google Scholar]
- 2.Cancer Research UK Ovarian cancer survival statistics [webpage on the Internet] London, UK: Cancer Research UK; 2012Available from: http://www.cancerresearchuk.org/cancer-info/cancerstats/types/ovary/survivalAccessed October 10, 2012 [Google Scholar]
- 3.Prat J. Ovarian carcinomas: five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. 2012;460(3):237–249. doi: 10.1007/s00428-012-1203-5. [DOI] [PubMed] [Google Scholar]
- 4.National Cancer Institute SEER stat fact sheets: ovary [webpage on the Internet] Bethesda, MD: 2010National Cancer Institute; Available from: http://seer.cancer.gov/statfacts/html/ovary.html#prevalenceAccessed October 10, 2012 [Google Scholar]
- 5.Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alsop K, Fereday S, Meldrum C, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30(21):2654–2663. doi: 10.1200/JCO.2011.39.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pennington KP, Swisher EM. Hereditary ovarian cancer: beyond the usual suspects. Gynecol Oncol. 2012;124(2):347–353. doi: 10.1016/j.ygyno.2011.12.415. [DOI] [PubMed] [Google Scholar]
- 8.Vergote I, Amant F, Kristensen G, Ehlen T, Reed NS, Casado A. Primary surgery or neoadjuvant chemotherapy followed by interval debulking surgery in advanced ovarian cancer. Eur J Cancer. 2011;47(Suppl 3):S88–S92. doi: 10.1016/S0959-8049(11)70152-6. [DOI] [PubMed] [Google Scholar]
- 9.Colombo N, Peiretti M, Parma G, et al. ESMO Guidelines Working Group Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v23–v30. doi: 10.1093/annonc/mdq244. [DOI] [PubMed] [Google Scholar]
- 10.Perren TJ, Swart AM, Pfisterer J, et al. ICON7 Investigators A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496. doi: 10.1056/NEJMoa1103799. [DOI] [PubMed] [Google Scholar]
- 11.Burger RA, Brady MF, Bookman MA, et al. Gynecologic Oncology Group Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483. doi: 10.1056/NEJMoa1104390. [DOI] [PubMed] [Google Scholar]
- 12.Kristensen G, Perren T, Qian W, et al. Result of interim analysis of overall survival in the GCIG ICON7 phase III randomised trial of bevacizumab in women with newly diagnosed ovarian cancer; Presented at: American Society of Clinical Oncology; June, 4–8, 2011; Chicago, IL, USA. Abstract LBA 5006. [Google Scholar]
- 13.Parmar MK, Ledermann JA, Colombo N, et al. ICON AGO Collaborators Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet. 2003;361(9375):2099–2106. doi: 10.1016/s0140-6736(03)13718-x. [DOI] [PubMed] [Google Scholar]
- 14.Pfisterer J, Plante M, Vergote I, et al. AGO-OVAR. NCIC CTG. EORTC GCG Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol. 2006;24(29):4699–4707. doi: 10.1200/JCO.2006.06.0913. [DOI] [PubMed] [Google Scholar]
- 15.Aghajanian C, Blank SV, Goff BA, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30(17):2039–2045. doi: 10.1200/JCO.2012.42.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall M, Rustin G. Recurrent ovarian cancer: when and how to treat. Curr Oncol Rep. 2011;13(6):459–471. doi: 10.1007/s11912-011-0199-3. [DOI] [PubMed] [Google Scholar]
- 17.Weroha SJ, Oberg AL, Ziegler KL, et al. Phase II trial of lapatinib and topotecan (LapTop) in patients with platinum-refractory/resistant ovarian and primary peritoneal carcinoma. Gynecol Oncol. 2011;122(1):116–120. doi: 10.1016/j.ygyno.2011.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia AA, Sill MW, Lankes HA, et al. A phase II evaluation of lapatinib in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a gynecologic oncology group study. Gynecol Oncol. 2012;124(3):569–574. doi: 10.1016/j.ygyno.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karp DD, Pollak MN, Cohen RB, et al. Safety, pharmacokinetics, and pharmacodynamics of the insulin-like growth factor type 1 receptor inhibitor figitumumab (CP-751,871) in combination with paclitaxel and carboplatin. J Thorac Oncol. 2009;4(11):1397–1403. doi: 10.1097/JTO.0b013e3181ba2f1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–861. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 22.Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 23.Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 24.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 25.Wysham WZ, Mhawech-Fauceglia P, Li H, et al. BRCAness profile of sporadic ovarian cancer predicts disease recurrence. PLoS One. 2012;7(1):e30042. doi: 10.1371/journal.pone.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rigakos G, Razis E. BRCAness: finding the Achilles heel in ovarian cancer. Oncologist. 2012;17(7):956–962. doi: 10.1634/theoncologist.2012-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26(13):1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Press JZ, De Luca A, Boyd N, et al. Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC Cancer. 2008;8:17. doi: 10.1186/1471-2407-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hennessy BT, Timms KM, Carey MS, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570–3576. doi: 10.1200/JCO.2009.27.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587–598. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 31.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4):497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 32.De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol. 2012;84(2):137–146. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 33.Menear KA, Adcock C, Boulter R, et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51(20):6581–6591. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 34.Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5(4):387–393. doi: 10.1016/j.molonc.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iglehart JD, Silver DP. Synthetic lethality – a new direction in cancer-drug development. N Engl J Med. 2009;361(2):189–191. doi: 10.1056/NEJMe0903044. [DOI] [PubMed] [Google Scholar]
- 36.Gottipati P, Vischioni B, Schultz N, et al. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010;70(13):5389–5398. doi: 10.1158/0008-5472.CAN-09-4716. [DOI] [PubMed] [Google Scholar]
- 37.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 38.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 39.Kortmann U, McAlpine JN, Xue H, et al. Tumor growth inhibition by olaparib in BRCA2 germline-mutated patient-derived ovarian cancer tissue xenografts. Clin Cancer Res. 2011;17(4):783–791. doi: 10.1158/1078-0432.CCR-10-1382. [DOI] [PubMed] [Google Scholar]
- 40.Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evers B, Drost R, Schut E, et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res. 2008;14(12):3916–3925. doi: 10.1158/1078-0432.CCR-07-4953. [DOI] [PubMed] [Google Scholar]
- 42.National Cancer Institute Cancer Therapy Evaluation Program Common terminology criteria for adverse events [webpage on the Internet] Bethesda, MD: National Cancer Institute; 2012Available from: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htmAccessed October 19, 2012 [Google Scholar]
- 43.Rustin GJ, Quinn M, Thigpen T, et al. Re: New guidelines to evaluate the response to treatment in solid tumors (ovarian cancer) J Natl Cancer Inst. 2004;96(6):487–488. doi: 10.1093/jnci/djh081. [DOI] [PubMed] [Google Scholar]
- 44.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 45.Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 46.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 47.Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol. 2001;19(14):3312–3322. doi: 10.1200/JCO.2001.19.14.3312. [DOI] [PubMed] [Google Scholar]
- 48.Kaye SB, Lubinski J, Matulonis U, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2012;30(4):372–379. doi: 10.1200/JCO.2011.36.9215. [DOI] [PubMed] [Google Scholar]
- 49.Graeser M, McCarthy A, Lord CJ, et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res. 2010;16(24):6159–6168. doi: 10.1158/1078-0432.CCR-10-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konstantinopoulos PA, Spentzos D, Karlan BY, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–3561. doi: 10.1200/JCO.2009.27.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392. doi: 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- 52.Carlson RH. Ovarian cancer: future of PARP inhibitor olaparib still uncertain in platinum-sensitive serous disease. Oncology Times. 2012;34(19):37–38. [Google Scholar]
- 53.Khan OA, Gore M, Lorigan P, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer. 2011;104(5):750–755. doi: 10.1038/bjc.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samol J, Ranson M, Scott E, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs. 2012;30(4):1493–1500. doi: 10.1007/s10637-011-9682-9. [DOI] [PubMed] [Google Scholar]
- 55.Dean E, Middleton MR, Pwint T, et al. Phase I study to assess the safety and tolerability of olaparib in combination with bevacizumab in patients with advanced solid tumours. Br J Cancer. 2012;106(3):468–474. doi: 10.1038/bjc.2011.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rajan A, Carter CA, Kelly RJ, et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin Cancer Res. 2012;18(8):2344–2351. doi: 10.1158/1078-0432.CCR-11-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.National Cancer Institute Olaparib and carboplatin to treat breast and ovarian cancer ClinicalTrialsgov [website on the Internet] Bethesda, MD: US National Library of Medicine; 2011Available from: http://www.clinicaltrials.gov/ct2/show/NCT01445418?term=olaparib&rank=37Accessed October 24, 2012 [Google Scholar]
- 58.ClinicalTrials.gov Olaparib in combination with carboplatin for refractory or recurrent women’s cancers [webpage on the Internet] Bethesda, MD: US National Institutes of Health; 2011Available from: http://www.clinicaltrials.gov/ct2/show/NCT01237067Accessed October 24, 2012 [Google Scholar]
- 59.ClinicalTrials.gov Study to assess the safety and tolerability of a PARP inhibitor in combination with carboplatin and/or paclitaxel [webpage on the Internet] Bethesda, MD: US National Institutes of Health; 2011Available from: http://www.clinicaltrials.gov/ct2/show/NCT00516724?term=olaparib&rank=19Accessed October 24, 2012 [Google Scholar]
- 60.ClinicalTrials.gov Study to compare the efficacy and safety of olaparib when given in combination with carboplatin and paclitaxel, compared with carboplatin and paclitaxel in patients with advanced ovarian cancer [webpage on the Internet] Bethesda, MD: US National Institutes of Health; 2011Available from: http://www.clinicaltrials.gov/ct2/show/NCT01081951?term=olaparib&rank=15Accessed October 24, 2012 [Google Scholar]
- 61.ClinicalTrials.gov Phase Ib study of olaparib plus weekly carboplatin and paclitaxel in relapsed ovarian cancer [webpage on the Internet] Bethesda, MD: US National Institutes of Health; 2011Available from: http://www.clinicaltrials.gov/ct2/show/NCT01650376?term=olaparib&rank=11Accessed October 24, 2012 [Google Scholar]
- 62.ClinicalTrials.gov Phase I of BKM120/olaparib for triple negative breast cancer or high grade serous ovarian cancer [webpage on the Internet] Bethesda, MD: US National Institutes of Health; 2011Available from: http://www.clinicaltrials.gov/ct2/show/NCT01623349?term=olaparib&rank=2Accessed October 24, 2012 [Google Scholar]
- 63.ClinicalTrials.gov Cediranib and olaparib in combination for recurrent ovarian or triple-negative breast cancer [webpage on the Internet] Bethesda, MD: US National Institutes of Health; 2011Available from: http://www.clinicaltrials.gov/ct2/show/NCT01116648?term=olaparib&rank=1Accessed October 24, 2012 [Google Scholar]
- 64.Barber LJ, Sandhu S, Chen L, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–429. doi: 10.1002/path.4140. [DOI] [PubMed] [Google Scholar]
- 65.Jaspers JE, Kersbergen A, Boon U, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81. doi: 10.1158/2159-8290.CD-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oplustilova L, Wolanin K, Mistrik M, et al. Evaluation of candidate bio-markers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle. 2012;11(20):3837–3850. doi: 10.4161/cc.22026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mukhopadhyay A, Plummer ER, Elattar A, et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: sensitivity to PARP inhibitors, platinum, and survival. Cancer Res. 2012;72(22):5675–5682. doi: 10.1158/0008-5472.CAN-12-0324. [DOI] [PubMed] [Google Scholar]
- 68.Nowsheen S, Bonner JA, Yang ES. The poly(ADP-Ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother Oncol. 2011;99(3):331–338. doi: 10.1016/j.radonc.2011.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Redon CE, Nakamura AJ, Zhang YW, et al. Histone gammaH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers. Clin Cancer Res. 2010;16(18):4532–4542. doi: 10.1158/1078-0432.CCR-10-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]