

Abstract
Notch signaling is an evolutionarily conserved pathway involved in cell fate control during development, stem cell self-renewal, and postnatal tissue differentiation. Roles for Notch in carcinogenesis, the biology of cancer stem cells, tumor angiogenesis, and epithelial-to-mesenchymal transition (EMT) have been reported. This review describes the role of Notch in the “stemness” program in cancer cells and in metastases, together with a brief update on the Notch inhibitors currently under investigation in oncology. These agents may be useful in targeting cancer stem cells and to reverse the EMT process.
Keywords: Notch signaling, EMT, cancer stem cells, mesenchymal stem cells, metastases, Notch inhibitors
Introduction
The Notch pathway is one of the most intensively studied candidate therapeutic targets in cancer stem-like cells (CSCs), and several investigational Notch inhibitors are being developed. Notch signaling has been reported to promote the self-renewal of CSC in several malignancies and to participate in tumor–stroma and tumor–endothelium interactions in CSC niches in primary and metastatic tumors.1,2 However, successful targeting of Notch signaling in CSCs will require a clear understanding of Notch regulation and the context-dependent interactions between Notch and other therapeutically relevant pathways. Understanding these interactions will increase our ability to design rational combination regimens that are more likely to prove safe and effective for primary and metastatic tumors. Additionally, to determine which patients are most likely to benefit from treatment with Notch-targeting therapeutics, reliable biomarkers to measure Notch pathway activity in CSCs from specific tumors will have to be identified and validated.
Notch receptors and ligands
Mammals express four transmembrane Notch receptors (Notch-1, Notch-2, Notch-3, and Notch-4)3 and five canonical transmembrane ligands (Delta-like [DLL] 1, DLL 3, DLL 4, Jagged-1, and Jagged-2) (Figure 1).4–7 Delta family ligands differ from Jagged family ligands because their smaller extracellular domains can mediate Notch activation in trans (from cell to cell) and Notch inhibition in cis (on the same cell). The relative affinity of Notch receptors for Delta and Jagged family ligands is controlled by receptor glycosylation, and specifically by the addition of Fuc-GlcNac (fucose-N-acetylglucosamine) moieties by a fucosyltransferase and Fringe family N-acetyl-glucosaminidyl-transferases. Cell-to-cell contact is generally necessary for the activation of Notch signaling. This usually results in coordinated modulation of genes involved in cell fate determination, such as proliferation, survival, or differentiation.7 Notch receptors undergo three proteolytic cleavages. First, Notch precursor proteins are processed in the trans-Golgi apparatus. A single polypeptide precursor is cleaved (S1) by a furin-like convertase to produce the mature Notch receptor, which is a heterodimer consisting of Notch extracellular (NEC) and Notch transmembrane (N™) subunits. Mature receptors are trafficked to the plasma membrane, where they await engagement with membrane-associated ligands. Upon ligand–receptor engagement, NEC is dissociated from N™ to be endocytosed with the ligand into the ligand-expressing cell. Subunit separation allows a second cleavage (S2) by a disintegrin and metalloproteinase domain-containing protein 10 or 17 (ADAM10 or ADAM17).8 ADAM10 is thought to cleave Notch in ligand-dependent activation, while ADAM17 may participate in the less clearly understood process of ligand-independent activation. The S2 cleavage releases a short extracellular peptide and generates a short-lived intermediate that is cleaved again (S3) by the γ-secretase complex. The S3 cleavage releases the intracellular portion of Notch (NIC).9 NIC translocates to the nucleus and binds to the CBF-1-Suppressor of Hairless/Lag1 ([CSL] also known as RBP-jκ), a constitutive transcriptional repressor, displacing corepressors and recruiting coactivators such as Mastermind-like (MAML) proteins, homologous to Drosophila Mastermind. The Notch–CSL–MAML complex in turn recruits multiple transcriptional regulators forming the “Notch transcriptional complex” (NTC).10,11 Notch activates many genes associated with differentiation and/or survival, including the Hairy/Enhancer of Split (HES) family and Hairy/enhancer-of-split related with YRPW motif-like protein (Hey) family of basic helix-loop-helix transcription factors,12 cyclin D1,13 and c-Myc (Figure 2).14 The genomic sites at which Notch activates transcription vary from cell to cell, and quite likely among different Notch paralogs. Other transcriptional regulators influence transcriptional regulation by Notch-1.15–17 The close-range cell–cell interaction necessary for Notch activation may be one of the signals whereby intercellular signals trigger epithelial-to-mesenchymal transition (EMT) in the tumor microenvironment.
Figure 1.
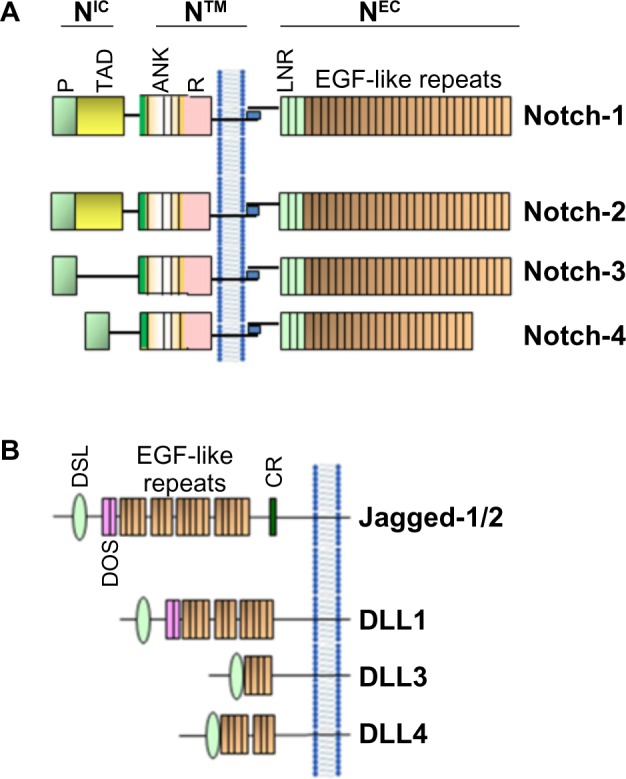
Schematic representation of Notch receptors (A) and Notch ligands (B) in mammals.
Notes: Mammals contain four Notch receptors (Notch-1–4) and five ligands (Jagged-1/2, homologous to Serrata in Drosophila) and DLL 1, 3, and 4 (homologous to Delta in Drosophila).
Abbreviations: ANK, ankyrin repeats; CR, cysteine-rich domain; DLL, Delta-like; DOS, Delta and OSM-11-like protein domain; DSL, Delta, Serrata, and LAG-2 domain; EGF, epidermal growth factor; LNR, cysteine-rich Lin 12-Notch repeats; P, PEST domain; R, RAM domain; TAD, transactivation domain; NIC, Notch intracellular domain; NEC, Notch extracellular domain; N™, Notch transmembrane domain.
Figure 2.
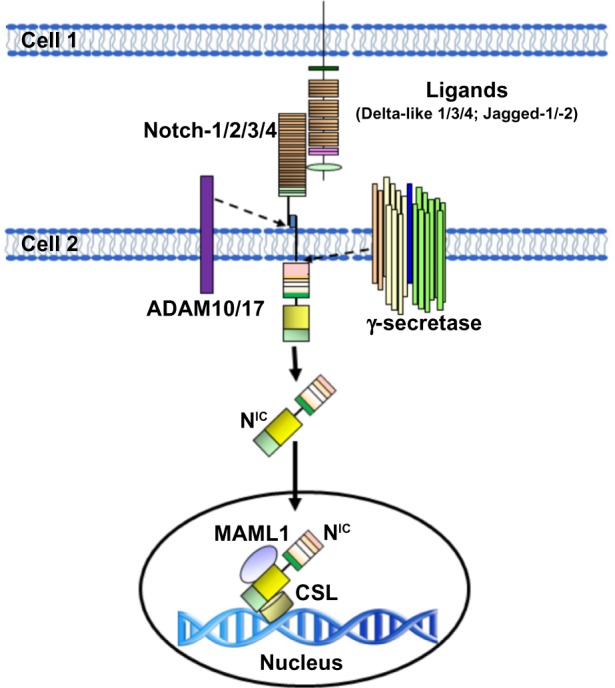
Schematic representation of the activation of Notch in mammal cells.
Notes: The Notch receptor is activated by binding to a ligand presented by a neighboring cell. Endocytosis and membrane trafficking regulate ligand and receptor availability at the cell surface. Ligand endocytosis is also thought to generate mechanical force to promote a conformational change in the bound Notch receptor. This conformational change exposes the site in Notch for cleavage by ADAM metalloproteases. This Notch cleavage generates the membrane-anchored Notch extracellular truncation fragment, a substrate for the γ-secretase complex. γ-secretase then cleaves the Notch transmembrane domain to release the NIC. γ-secretase cleavage can occur at the cell surface or in endosomal compartments, but cleavage at the membrane favors the production of a more stable form of NIC. NIC then enters the nucleus where it associates with the DNA-binding protein CSL. In the absence of NIC, CSL may associate with ubiquitous corepressor proteins and histone to repress transcription of some target genes. Upon NIC binding, allosteric changes may occur in CSL that facilitate displacement of transcriptional repressors. The transcriptional coactivator Mastermind-like protein 1 (MAML1) then recognizes the NIC/CSL interface, and this triprotein complex recruits additional coactivators to activate transcription.
Abbreviations: ADAM, A disintegrin and metalloprotease; CSL, CBF-1-Suppressor of Hairless/Lag1 (also known as RBP-jκ); NIC, Notch intracellular domain.
Notch signaling, EMT, and cancer stem cells
Many human cancers are thought to contain populations of cells that display stem cell-like properties. These properties include self-renewal, which drives tumorigenesis; resistance to cell death, which drives tumor progression; and differentiation, which contributes to cancer cell heterogeneity. There is increasing evidence that these CSCs mediate tumor metastasis and, by virtue of their relative resistance to chemotherapy and radiation therapy, may contribute to treatment failures and relapses following therapy.18
Self-renewal and cell fate determination of normal stem cells are regulated by both cell-autonomous (intrinsic) and non-cell-autonomous (extrinsic) pathways. The dysregulation of these pathways resulting in stem cell expansion may be a key event initiating carcinogenesis. Developmental pathways such as Notch play an important role in normal stem cell functions and are frequently deranged in cancers.19–22 Deregulated expression of Notch proteins, ligands, and targets, including overexpression and activation of Notch, has been described in a multitude of solid tumors, including cervical,23 head and neck,24 endometrial,25 renal,26 lung,27 pancreatic,28 ovarian,29 prostate,30 esophageal,31 oral,32 hepatocellular,33 and gastric34 carcinomas; osteosarcoma mesothelioma;35 melanoma;36 gliomas;37 and medulloblastomas.38 Dysregulation of Notch signaling has been reported in some hematological malignancies other than T-ALL. These include Hodgkin lymphomas, anaplastic large-cell non-Hodgkin lymphomas,39 some acute myeloid leukemias (AMLs),40 B-cell chronic lymphoid leukemias (B-CLLs),41 and multiple myeloma (MM)42,43 (for a recent review, see Pancewicz and Nicot44). In most cases, inappropriate activation of Notch signaling is oncogenic. In some cases, however, loss of function of Notch-1 has oncogenic effects. This has been demonstrated in the epidermis45,46 and, more recently, in a subset of head and neck squamous carcinomas. Notch signaling is essential to the orderly differentiation of squamous epithelia, and loss of Notch-1 causes loss of barrier in such epithelia.47 This in turn triggers an inflammatory response and cytokine cascade that may favor transformation. However, in the case of CSC, the literature supports a role of several Notch paralogs in the maintenance and survival of these cells.2 Extrinsic signals that regulate stem cell behavior originate in the stem cell microenvironment. Although there is still relatively little detailed information on the composition and function of cancer stem cell microenvironments in different malignancies, it is clear that tumor growth and metastasis are highly dependent on the tumor microenvironment. This microenvironment is comprised of tumor-associated fibroblasts, endothelial cells, adipocytes, and several types of immune cells, all of which have been demonstrated to play roles in tumor growth and metastasis.48 Several studies have demonstrated that loss of epithelial phenotype through EMT can promote the acquisition of a stem-like phenotype and drug resistance.49 Notch signaling regulates both the formation of CSCs and the acquisition of the EMT phenotype, which are associated with drug resistance.50,51 An epithelial gene signature has been associated with sensitivity to the epidermal growth factor receptor inhibitor erlotinib in lung cancer cells.52 Similar results have been reported in head and neck squamous cell carcinoma and hepatocellular carcinoma with gefitinib and cetuximab.53,54 Conversely, EMT has also been shown to promote resistance to conventional therapeutics, including paclitaxel, vincristine, and oxaliplatin.55 Recent studies have shown links between EMT and gemcitabine-resistant pancreatic cancer, oxaliplatin-resistant colorectal cancer, lapatinib-resistant breast cancer, and paclitaxel-resistant ovarian carcinoma.56–59 Therefore, elucidating mechanisms that govern the acquisition of EMT in cancer cells would likely be useful for devising targeted therapeutic approaches to overcome or prevent resistance to conventional cancer therapeutics.
Notch activation triggers mesenchymal transformation not only in epithelial but also in endothelial cells. These changes include downregulation of endothelial markers (vascular endothelial-cadherin, tyrosine kinase with immunoglobulin-like and epidermal growth factor-like domain [Tie]1, Tie2, platelet-endothelial cell adhesion molecule-1, and endothelial nitric oxide synthase) and upregulation of mesenchymal markers (α-SMA, fibronectin, and platelet-derived growth factor receptors).60 Jagged-1-mediated stimulation of endothelial cells induces phenotypic and functional changes consistent with EMT.60 Notch also cross-talks with several transcription and growth factors relevant to EMT, including Snail, Slug, and transforming growth factor (TGF)-β. Notch promotes EMT through the regulation of Snail. Overexpression of Notch-1 in immortalized endothelial cells in vitro induces Snail,61 which is thought to bind to E-boxes in the human E-cadherin promoter and repress E-cadherin gene expression.62 In addition, Notch could induce EMT by stabilizing Snail-1 protein under hypoxic condition.63 It has been reported that Slug is a direct target of Notch and that the Notch directly stimulates the Slug promoter, resulting in the upregulation of Slug and initiation of EMT.64 Slug was found to be essential for Notch-mediated repression of E-cadherin, resulting in β-catenin activation and EMT.65 It has been reported that TGF-β can induce the expression of Notch ligands66 and that TGF-β-induced EMT could be blocked by Hey-1 or Jagged-1 knockdown or by pharmacological inactivation of Notch.67 Notch-2 and Jagged-1 are highly upregulated in gemcitabine-resistant pancreatic cancer cells, which show acquisition of an EMT phenotype.68 Recently, EMT has been mechanistically linked with stem-like signatures in prostate cancer cells,69 with stem-like cells characterized by increased expression of Notch-1, Sox2, Nanog, Oct4, and Lin28B.69 An independent report has recently confirmed the importance of Notch and Hedgehog signaling in prostate CSCs.70
Epithelial cells from a primary prostate tumor can undergo EMT with activation of embryonic programs of epithelial plasticity, including Notch, and switch from a sessile, epithelial phenotype to a motile, mesenchymal phenotype.71 Growth factors and molecular alterations that contribute to EMT induction in primary tumors have been identified as important stimulators of skeletal metastasis formation.72 Aberrant expression of EMT markers N-cadherin, vimentin, platelet-derived growth factor-D, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), Notch-1, and zinc finger E-box-binding homeobox (ZEB)1 was observed in primary prostate cancers and bone metastatic lesions. Notch-1 was highly expressed in bone metastases compared to primary prostate cancers, suggesting that Notch-1 could play a role in prostate cancer bone metastasis through the induction of an EMT phenotype (Figure 3).73 Recent data from Zhu et al support this model, showing that Jagged-1 expression increases dramatically in high-grade and metastatic prostate cancers compared to primary lesions.74 Furthermore, Notch signaling is often and aberrantly activated by hypoxia, which induces EMT during tumor progression. Bone is one of the most frequently targeted organs for breast cancer metastasis, and regions of the bone are known to be hypoxic. This hypoxic niche in bone microenvironment is believed to promote self-renewal of hematopoietic stem cells. Xing et al have also shown that Jagged-2 was upregulated in bone marrow stroma under hypoxia, which significantly promoted EMT and self-renewal of breast CSCs,75 suggesting a critical role of Notch-induced EMT in tumor progression and metastasis.
Figure 3.

Role of Notch in tumor metastasis as an inducer of EMT.
Notes: Epithelial cells can undergo EMT with activation of embryonic programs of epithelial plasticity, including Notch. Aberrant expression of EMT markers N-cadherin, vimentin, platelet-derived growth factor-D, NF-κB, Notch-1, and ZEB 1 has been observed in metastatic lesions, together with high Notch receptor and ligand expression. Notch signaling is often and aberrantly activated by hypoxia that induces EMT during tumor progression. Thus, the hypoxic niche promotes EMT and self-renewal of breast CSCs, suggesting a critical role of Notch-induced EMT in tumor progression and metastasis.
Abbreviations: CSC, cancer stem-like cell; EMT, epithelial-to-mesenchymal transition; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; ZEB, zinc finger E-box-binding homeobox; NIC, Notch intracellular domain; MAML1, Mastermind-like protein 1; CSL, CBF-1-Suppressor of Hairless/Lag1 (also known as RBP-jκ).
Notch signaling and mesenchymal stem cells
Mesenchymal stem cells (MSCs) are multipotent cells with non-hematopoietic origin that constitute a minor population (0.01%) of nucleated cells in bone marrow.76 MSCs are a potential source of stem cells for cellular and genetic therapy, and can differentiate into multiple lineages such as chondrocytes, osteocytes, adipocytes, myocytes, and astrocytes. Recently, MSCs were found to play an important role in the tumor-supporting stroma.77,78 MSCs are known for their active mobilization from bone marrow and migration to sites of injury.79–81 Reports have suggested that bone marrow-derived MSCs are preferentially recruited to tumor stroma80 when compared to normal stroma,82 mainly by inflammatory factors in the tumor microenvironment. These reports increased interest in understanding the potential role of MSCs in tumor progression. MSCs are recruited to the tumor microenvironment in response to various cytokines, which are secreted by tumor cells and their associated stroma83–87 and act as precursors for pericytes and carcinoma cancer-associated fibroblasts.77,88,89 MSCs promote tumor cell proliferation indirectly through their immunosuppressive properties and directly through cancer cell supportive properties.90,91 An earlier study from Sanchez et al suggest that, under nutrient-deprived conditions, the MSCs associated with tumor stroma undergo autophagy, secreting antiapoptotic factors that protect breast cancer cells embedded in the stroma.78 These studies suggest that targeting tumor associated stromal cells along with tumor cells may provide more effective treatment strategies for breast cancer.92,93 Recent evidence also suggests that MSCs participate in tumor growth and metastasis and are the most prominent cell type within the stroma of many cancers. Subcutaneously implanted human mammary carcinomas coinjected with MSCs acquire an increased metastatic potential.94 An important factor in the function of tumor microenvironment is the cell–cell communication between stromal cells and cancer cells. The role of gap junctions in the transport of cellular communicators95 and juxtacrine regulation based on direct communication is well documented.96,97
The role of MSCs in inducing EMT in tumor cells has been the focus of a number of recent studies. Several possible mechanisms through which MSCs play a role in tumor microenvironment have been proposed. One such mechanism is exosomes secreted by MSCs, which promote EMT in gastric cancer cells.98 EMT processes endow epithelial tumor cells with properties that may facilitate CSC generation and survival, ie, increased invasive ability, increased resistance to apoptotic signals, and increased ability to potentiate angiogenesis. In vivo models of EMT-derived cells in primary tumors have enhanced metastatic potential.99 Inflammatory cells and cytokines, hypoxia-induced increase of reactive oxygen species in mitochondria, and MSC can all effectively drive the EMT of tumor cells.100 Passage by neoplastic epithelial cells through an EMT event allows these cells to approach a stem cell-like state. EMT programs are known to be induced by heterotypic signals that epithelial cells receive from their microenvironment. In response to stimulation by carcinoma cells, MSCs express greatly elevated levels of prostaglandin E2 (PGE2). The resulting PGE2, together with cytokines also induced in the MSCs, contributes to the entrance of nearby carcinoma cells into a stem cell-like state.101 An in vitro study using coculture models of MSCs and breast cancer cells showed that EMT is stimulated by increased expression of oncogenes and other genes associated with invasion, angiogenesis, and antiapoptosis.88,94 However, the nature of these heterotypic signals and the identities of the stromal cells that release them remain poorly understood.
Notch signaling is important for MSC differentiation and related to its role in EMT. MSC modified with miR-126 release proangiogenic factors and induce expression of proangiogenic Notch ligand DLL 4, enhancing angiogenesis.102 Moreover, Notch signaling regulates the expression of CXCR4 in MSCs, modulating their migration.103 Notch-1 has also been reported to mediate the induction of Tregs by MSCs.104 Tregs are thought to promote tumorigenesis by dampening antitumor immune responses. As with BMP and Wnt signaling in osteogenesis, Runx2 function is also influenced by Notch signaling. Notch-1IC can interact directly with Runx2 protein to repress terminal osteoblastic differentiation in vitro (Figure 4).105
Figure 4.
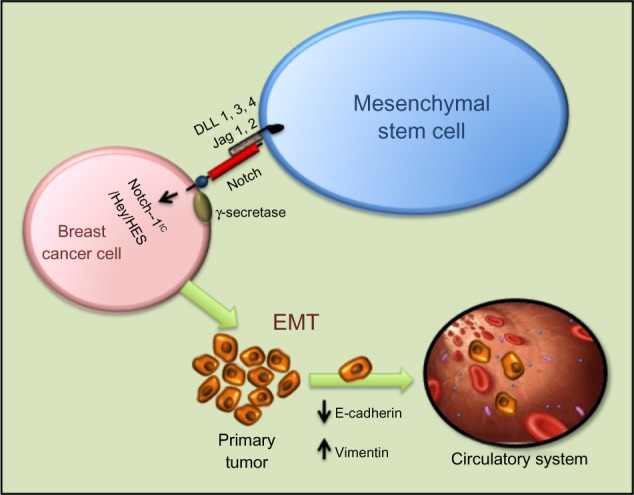
Intercellular interaction between mesenchymal stem cells and breast cancer cells through Notch can activate EMT through NIC, Hey, HES, and other activators. Mesenchymal stem cells modified with miR-126 release proangiogenic factors and induce expression of proangiogenic Notch ligand DLL 1, 3, 4 and Jagged-1/2 enhancing angiogenesis. Moreover, Notch signaling regulates the expression of CXCR4 in mesenchymal stem cells, modulating their migration.
Abbreviations: DLL, Delta-like; EMT, epithelial-to-mesenchymal transition; Notch-1IC, Notch-1 intracellular domain; Jag, Jagged; HES, Hairy/Enhancer of Split; Hey, Hairy/enhancer-of-split related with YRPW motif-like protein.
Notch signaling and tumor metastasis
Recent insights have linked Notch signaling to cancer metastasis.106
It is now well recognized that cancer progression not only requires deregulated signaling pathways and accumulated genetic alterations in cancer cells, but also relies on the support from tumor microenvironment.107,108 For example, in the case of breast cancer, tumor cells frequently metastasize to the bone and brain, where tissue microenvironment enhances the metastatic growth of cancer cells by providing growth factors and ligands that activate multiple metastasis-related pathways including Notch, Wnt, and Hedgehog.109–111
Elevated expression of Jagged-1 has been associated with an increased incidence of triple-negative breast cancer (TNBC) bone metastasis and tumor cells that overexpress Jagged-1-generated severe osteolytic lesions in mouse tibiae, suggesting a potential role of tumor-derived Jagged-1 in promoting bone metastasis.106 Interestingly, the same paper also showed that breast cancer cells with a high level of Jagged-1 promoted bone metastasis by activating Notch signaling in the osteoblasts, which in turn directly enhanced osteoclast differentiation by secreting interleukin (IL)-6. These results indicate a new paradigm for Notch signaling in breast cancer metastasis in which bone stromal cells respond to tumor-derived ligands and promote osteolysis in a paracrine manner. Furthermore, it has been shown that Notch signaling is activated in endothelial cells, promoting angiogenesis after interaction with cancer cells in head and neck squamous cell carcinomas.112 These studies indicate that reciprocal interactions of tumor and untransformed stromal cells in the microenvironment play critical roles in the activation of Notch signaling. Tumor microenvironment includes not only various types of stromal and immune cells but also several homeostatic factors, such as pH and oxygen concentration.113,114 Hypoxic breast cancer cells have been shown to enhance the capillary-like tube formation of endothelial cells in a Jagged-2-dependent manner. Knockdown of Jagged-2 by siRNA in cancer cells blocks angiogenesis of endothelial cells in coculture experiments.115
The brain is a frequent metastatic site for several types of tumors, including melanoma and lung and breast cancers, and metastatic tumor cells need to adapt to this totally different microenvironment. It has been demonstrated that brain metastatic TNBC cells excessively expressed IL-1β, which stimulates the surrounding astrocytes to express Jagged-1.116 Direct interaction of the reactivated astrocytes with CSCs resulted in significantly upregulated Notch signaling in CSCs. This in turn further enhanced the self-renewal of CSC, suggesting that there is a vicious circle paracrine loop of IL-1β and Notch signaling inducing one another through direct interaction between CSCs and astrocytes. This vicious circle promotes the growth of metastatic CSCs in the brain. The blood–brain barrier-permeable Notch inhibitor γ-secretase inhibitor (GSI) Compound E can significantly suppress brain metastasis in vivo.113 These results represent a novel paradigm for the understanding of how metastatic breast CSCs re-establish a niche for their self-renewal in a glial microenvironment entirely different from their tissue of origin, opening a new avenue by which to identify a novel and specific target for the brain metastatic disease (Figure 5).
Figure 5.
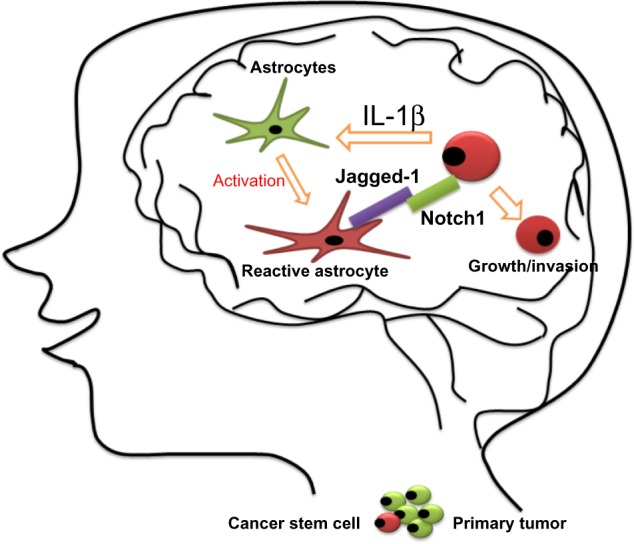
Proposed model for the growth of breast cancer stem cells in the brain.
Notes: Interleukin (IL)-1β secreted from metastatic cancer stem cells upregulates Jagged-1 on the reactivated astrocytes, which in turn promote self-renewal of cancer stem cells through the Jagged-1–Notch axis. Metastatic breast tumor cells in the brain highly express IL-1β, which then activates surrounding astrocytes. This activation significantly augments the expression of Jagged-1 in the astrocytes, and the direct interaction of the reactivated astrocytes and cancer stem cells significantly stimulated Notch signaling in cancer stem cells.
Targeting Notch signaling to reverse EMT and stemness in CSCs
Several classes of investigational Notch inhibitors have been developed. These include monoclonal antibodies against Notch receptors or ligands;117–123 decoys (soluble forms of the extracellular domain of Notch receptor or Notch ligands);124–127 blocking peptides;128 GSIs;129–135 or natural compounds.136–140 To date, GSIs are the most extensively explored. GSIs are less specific than biologics, but have the potential advantages of favorable biodistribution and pan-Notch inhibition. While γ-secretase has numerous substrates besides Notch receptors, the pharmacologic activity and toxicity of GSIs in vivo appears to be due largely to Notch inhibition.141,142 GSIs have been administered to patients in Phase I clinical trials, either as single agents or in combination with standard of care.143 As is the case for most stem cell pathway inhibitors, the development of Notch inhibitors will need to be guided by biology. Biomarkers indicative of Notch activity (and of its inhibition by investigational drugs) will have to be identified and validated in each indication. Additionally, mechanism-based combinations will have to be developed. This implies that standard clinical trial designs with single-agent investigational drug and tumor volume as the primary end point may not be the most appropriate strategy for clinical trials of Notch-targeting agents, or, for that matter, for other CSC-targeted drugs. For example, in Her2/Neu positive BT474 xenografts, the combination of two chemically different GSIs with trastuzumab dramatically inhibited tumor recurrence, producing complete cures in most animals treated with one drug and all animals treated with another.132 GSIs given as single agents had virtually no effect on tumor volume in this experimental model, nor did they enhance tumor volume regression induced by trastuzumab. Thus, these agents prevented tumor regression with no significant effect on tumor volume. This effect is most likely due to CSC blockade, and suggests that survival-based end points may be needed in the clinic, at least for some indications. Recurrence-free survival and/or good surrogate end points predictive of survival (eg, circulating tumor cells, tumor-sphere-forming cells) are likely to be more informative. These challenges do not diminish the tremendous therapeutic opportunity offered by a pathway that is essential for EMT and CSC maintenance, angiogenesis, and, in many cases, proliferation and survival of cancer cells.
The Notch pathway has tremendous potential as a new target in cancer therapy. Importantly, Notch may be a particularly powerful target for CSCs, which are resistant to standard treatments such as chemotherapy and radiation but seem especially sensitive to inhibition of stem cell pathways such as Notch. Although several Notch inhibitors are currently at the clinic,143 Notch inhibitors used as single agents do not always yield major responses based on tumor volume in all models. Several issues remain to be addressed:
GSIs can affect bulk tumor cells, CSCs, stroma, and angiogenesis. The effects observed in vivo depend on the relative importance of these cellular targets in each tumor and tumor model.
GSIs are not pharmacologically equivalent: they have different pharmacokinetics, potencies, and off-target effects, and should not be considered equivalent.
At least one Notch paralog, Notch-4, has been shown to be resistant to some GSIs.144 Whether this is true of all GSIs is unclear, but Notch-4-driven tumors may be resistant to some GSIs.
GSIs generally have gastrointestinal toxicity, which is Notch-mediated and results from goblet cell metaplasia of intestinal stem cells. This precludes long-term, sustained administration of these drugs. Intermittent administration schedules have been used in the clinic and in preclinical models. These regimens do dramatically decrease toxicity. However, it is unknown whether intermittent inhibition of Notch signaling is sufficient to achieve an anti-CSC effect. Tumor-selective delivery systems may be necessary to achieve sustained Notch inhibition within the tumor microenvironment.
Combination treatments, ideally based on mechanistic information, are likely to prove more successful than single-agent regiments. For instance, glucocorticoids decrease the intestinal side effects of GSIs in T-ALL models.145 In estrogen receptor alpha (ERα)-positive breast cancer, combinations of Notch inhibitors with endocrine therapy have shown promise in preclinical models146 and in two early-phase clinical trials.147,148 In a presurgical window study, the addition of GSI MK0752 to tamoxifen or letrozole decreased Ki67 in 17/20 patients compared to endocrine therapy alone.147 In the metastatic setting, a combination of exemestane and GSI RO4929097 yielded clinical responses in seven out of 14 patients.148 In both cases, no diarrhea was observed. This may be due to the fact that endocrine therapy ameliorates the gastrointestinal toxicity of GSIs.149 Similarly, in TNBC, combinations of GSIs and taxanes have shown synergistic efficacy.150
Non-GSI strategies to inhibit Notch signaling, including stapled peptides, decoys, monoclonal antibodies to Notch ligands or receptors, or inhibitors of downstream mediators may prove useful in some indications. Several classes of non-GSI Notch inhibitors are currently being developed. As our understanding of this group of targets and agents increases, it becomes clear that these issues are surmountable and there is growing optimism that Notch inhibition will become an exciting new approach to cancer.
Conclusion
Deregulation of Notch signaling has been associated with mobilization and spread of primary tumor cells to distant locations. EMT and mesenchymal–epithelial transition play important roles during tumor invasion, metastasis, and therapeutic resistance. EMT is also linked with the acquisition of stem cell-like characteristics. The concept of EMT inducing a CSC phenotype provides a possible mechanistic basis for metastasis, chemoresistance, tumor dormancy, and delayed recurrence. Notch signaling is one of a handful of embryonic pathways that control the generation and self-renewal of CSCs, at least in part through EMT. Significant efforts are underway to develop pharmacologic inhibitors of Notch signaling that can inhibit EMT and/or eradicate CSCs in common human malignancies.
Acknowledgments
This work was financially support by grants from the National Institute of Health (NIH): R01CA151851 (RP), R01CA124650 (KW), R01CA129000 (KW), and P01 AG2553101 (LM).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Gu JW, Rizzo P, Pannuti A, Golde T, Osborne B, Miele L. Notch signals in the endothelium and cancer “stem-like” cells: opportunities for cancer therapy. Vasc Cell. 2012;4:7. doi: 10.1186/2045-824X-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pannuti A, Foreman K, Rizzo P, et al. Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010;16(12):3141–3152. doi: 10.1158/1078-0432.CCR-09-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaumueller CM, Qi H, Zagouras P, Artavanis-Tsakonas S. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell. 1997;90(2):281–291. doi: 10.1016/s0092-8674(00)80336-0. [DOI] [PubMed] [Google Scholar]
- 4.Dunwoodie SL, Henrique D, Harrison SM, Beddington RS. Mouse Dll3: a novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development. 1997;124(6):3065–3076. doi: 10.1242/dev.124.16.3065. [DOI] [PubMed] [Google Scholar]
- 5.Lindsell CE, Shawber CJ, Boulter J, Weinmaster G. Jagged: a mammalian ligand that activates Notch1. Cell. 1995;80(6):909–917. doi: 10.1016/0092-8674(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 6.Shawber C, Boulter J, Lindsell CE, Weinmaster G. Jagged2: a serrate-like gene expressed during rat embryogenesis. Dev Biol. 1996;180(1):370–376. doi: 10.1006/dbio.1996.0310. [DOI] [PubMed] [Google Scholar]
- 7.Callahan R, Raafat A. Notch signaling in mammary gland tumorigenesis. J Mammary Gland Biol Neoplasia. 2001;6(1):23–36. doi: 10.1023/a:1009512414430. [DOI] [PubMed] [Google Scholar]
- 8.Brou C, Logeat F, Gupta N, et al. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5(2):207–216. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- 9.Saxena MT, Schroeter EH, Mumm JS, Kopan R. Murine notch homologs (N1–4) undergo presenilin-dependent proteolysis. J Biol Chem. 2001;276(43):40268–40273. doi: 10.1074/jbc.M107234200. [DOI] [PubMed] [Google Scholar]
- 10.Hsieh JJ, Zhou S, Chen L, Young DB, Hayward SD. CIR, a corepressor linking the DNA binding factor CBF1 to the histone deacetylase complex. Proc Natl Acad Sci U S A. 1999;96(1):23–28. doi: 10.1073/pnas.96.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu L, Aster JC, Blacklow SC, Lake R, Artavanis-Tsakonas S, Griffin JD. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat Genet. 2000;26(4):484–489. doi: 10.1038/82644. [DOI] [PubMed] [Google Scholar]
- 12.Maier MM, Gessler M. Comparative analysis of the human and mouse Hey1 promoter: Hey genes are new Notch target genes. Biochem Biophys Res Commun. 2000;275(2):652–660. doi: 10.1006/bbrc.2000.3354. [DOI] [PubMed] [Google Scholar]
- 13.Ronchini C, Capobianco AJ. Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic) Mol Cell Biol. 2001;21(17):5925–5934. doi: 10.1128/MCB.21.17.5925-5934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weng AP, Millholland JM, Yashiro-Ohtani Y, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20(15):2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Germar K, Dose M, Konstantinou T, et al. T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc Natl Acad Sci U S A. 2011;108(50):20060–20065. doi: 10.1073/pnas.1110230108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calderwood MA, Lee S, Holthaus AM, Blacklow SC, Kieff E, Johannsen E. Epstein-Barr virus nuclear protein 3C binds to the N-terminal (NTD) and beta trefoil domains (BTD) of RBP/CSL; only the NTD interaction is essential for lymphoblastoid cell growth. Virology. 2011;414(1):19–25. doi: 10.1016/j.virol.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miele L. Transcription factor RBPJ/CSL: a genome-wide look at transcriptional regulation. Proc Natl Acad Sci U S A. 2011;108(36):14715–14716. doi: 10.1073/pnas.1110570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kakarala M, Wicha MS. Implications of the cancer stem-cell hypothesis for breast cancer prevention and therapy. J Clin Oncol. 2008;26(17):2813–2820. doi: 10.1200/JCO.2008.16.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004;6(6):R605–R615. doi: 10.1186/bcr920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7(3):86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 23.Zagouras P, Stifani S, Blaumueller CM, Carcangiu ML, Artavanis-Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc Natl Acad Sci U S A. 1995;92(14):6414–6418. doi: 10.1073/pnas.92.14.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leethanakul C, Patel V, Gillespie J, et al. Distinct pattern of expression of differentiation and growth-related genes in squamous cell carcinomas of the head and neck revealed by the use of laser capture microdissection and cDNA arrays. Oncogene. 2000;19(28):3220–3224. doi: 10.1038/sj.onc.1203703. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki T, Aoki D, Susumu N, Udagawa Y, Nozawa S. Imbalanced expression of TAN-1 and human Notch4 in endometrial cancers. Int J Oncol. 2000;17(6):1131–1139. doi: 10.3892/ijo.17.6.1131. [DOI] [PubMed] [Google Scholar]
- 26.Rae FK, Stephenson SA, Nicol DL, Clements JA. Novel association of a diverse range of genes with renal cell carcinoma as identified by differential display. Int J Cancer. 2000;88(5):726–732. doi: 10.1002/1097-0215(20001201)88:5<726::aid-ijc7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 27.Dang TP, Gazdar AF, Virmani AK, et al. Chromosome 19 translocation, overexpression of Notch3, and human lung cancer. J Natl Cancer Inst. 2000;92(16):1355–1357. doi: 10.1093/jnci/92.16.1355. [DOI] [PubMed] [Google Scholar]
- 28.Miyamoto Y, Maitra A, Ghosh B, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3(6):565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 29.Hopfer O, Zwahlen D, Fey MF, Aebi S. The Notch pathway in ovarian carcinomas and adenomas. Br J Cancer. 2005;93(6):709–718. doi: 10.1038/sj.bjc.6602719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santagata S, Demichelis F, Riva A, et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res. 2004;64(19):6854–6857. doi: 10.1158/0008-5472.CAN-04-2500. [DOI] [PubMed] [Google Scholar]
- 31.Subramaniam D, Ponnurangam S, Ramamoorthy P, et al. Curcumin induces cell death in esophageal cancer cells through modulating Notch signaling. PloS One. 2012;7(2):e30590. doi: 10.1371/journal.pone.0030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao S, Xia J, Chen Z, Wang Z, et al. Inhibitory effect of curcumin on oral carcinoma CAL-27 cells via suppression of Notch-1 and NF-kappaB signaling pathways. J Cell Biochem. 2011;112(4):1055–1065. doi: 10.1002/jcb.23019. [DOI] [PubMed] [Google Scholar]
- 33.Wang C, Qi R, Li N, et al. Notch1 signaling sensitizes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human hepatocellular carcinoma cells by inhibiting Akt/Hdm2-mediated p53 degradation and up-regulating p53-dependent DR5 expression. J Biol Chem. 2009;284(24):16183–16190. doi: 10.1074/jbc.M109.002105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Yeh IT, Reddick RL, Kumar AP. Malignancy arising in seminal vesicles in the transgenic adenocarcinoma of mouse prostate (TRAMP) model. Prostate. 2009;69(7):755–760. doi: 10.1002/pros.20924. [DOI] [PubMed] [Google Scholar]
- 35.Bocchetta M, Miele L, Pass HI, Carbone M. Notch-1 induction, a novel activity of SV40 required for growth of SV40-transformed human mesothelial cells. Oncogene. 2003;22(1):81–89. doi: 10.1038/sj.onc.1206097. [DOI] [PubMed] [Google Scholar]
- 36.Balint K, Xiao M, Pinnix CC, et al. Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J Clin Invest. 2005;115(11):3166–3176. doi: 10.1172/JCI25001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purow BW, Haque RM, Noel MW, et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005;65(6):2353–2363. doi: 10.1158/0008-5472.CAN-04-1890. [DOI] [PubMed] [Google Scholar]
- 38.Fan X, Mikolaenko I, Elhassan I, et al. Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004;64(21):7787–7793. doi: 10.1158/0008-5472.CAN-04-1446. [DOI] [PubMed] [Google Scholar]
- 39.Jundt F, Anagnostopoulos I, Forster R, Mathas S, Stein H, Dorken B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood. 2002;99(9):3398–3403. doi: 10.1182/blood.v99.9.3398. [DOI] [PubMed] [Google Scholar]
- 40.Tohda S, Nara N. Expression of Notch1 and Jagged1 proteins in acute myeloid leukemia cells. Leuk Lymphoma. 2001;42(3):467–472. doi: 10.3109/10428190109064603. [DOI] [PubMed] [Google Scholar]
- 41.Hubmann R, Schwarzmeier JD, Shehata M, et al. Notch2 is involved in the overexpression of CD23 in B-cell chronic lymphocytic leukemia. Blood. 2002;99(10):3742–3747. doi: 10.1182/blood.v99.10.3742. [DOI] [PubMed] [Google Scholar]
- 42.Houde C, Li Y, Song L, et al. Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood. 2004;104(12):3697–3704. doi: 10.1182/blood-2003-12-4114. [DOI] [PubMed] [Google Scholar]
- 43.Jundt F, Probsting KS, Anagnostopoulos I, et al. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood. 2004;103(9):3511–3515. doi: 10.1182/blood-2003-07-2254. [DOI] [PubMed] [Google Scholar]
- 44.Pancewicz J, Nicot C. Current views on the role of Notch signaling and the pathogenesis of human leukemia. BMC Cancer. 2011;11:502. doi: 10.1186/1471-2407-11-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Restivo G, Nguyen BC, Dziunycz P, et al. IRF6 is a mediator of Notch pro-differentiation and tumour suppressive function in keratinocytes. EMBO J. 2011;30(22):4571–4585. doi: 10.1038/emboj.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicolas M, Wolfer A, Raj K, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33(3):416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 47.Dumortier A, Durham AD, Di Piazza M, et al. Atopic dermatitis-like disease and associated lethal myeloproliferative disorder arise from loss of Notch signaling in the murine skin. PloS One. 2010;5(2):e9258. doi: 10.1371/journal.pone.0009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;(2):139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 49.Kong D, Li Y, Wang Z, Sarkar FH. Cancer stem cells and epithelial-mesenchymal transition (EMT)-phenotypic cells: are they cousins or twins? Cancers (Basel) 2011;3(1):716–729. doi: 10.3390/cancers30100716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Z, Li Y, Banerjee S, Sarkar FH. Emerging role of Notch in stem cells and cancer. Cancer Lett. 2009;279(1):8–12. doi: 10.1016/j.canlet.2008.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z, Li Y, Kong D, Ahmad A, Banerjee S, Sarkar FH. Cross-talk between miRNA and Notch signaling pathways in tumor development and progression. Cancer Lett. 2010;292(2):141–148. doi: 10.1016/j.canlet.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yauch RL, Januario T, Eberhard DA, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11(24 Pt 1):8686–8698. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 53.Voulgari A, Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta. 2009;1796(2):75–90. doi: 10.1016/j.bbcan.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Frederick BA, Helfrich BA, Coldren CD, et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. 2007;6(6):1683–1691. doi: 10.1158/1535-7163.MCT-07-0138. [DOI] [PubMed] [Google Scholar]
- 55.Sabbah M, Emami S, Redeuilh G, et al. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist Updat. 2008;11(4–5):123–151. doi: 10.1016/j.drup.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 56.Kajiyama H, Shibata K, Terauchi M, et al. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. 2007;31(2):277–283. [PubMed] [Google Scholar]
- 57.Konecny GE, Venkatesan N, Yang G, et al. Activity of lapatinib a novel HER2 and EGFR dual kinase inhibitor in human endometrial cancer cells. Br J Cancer. 2008;98(6):1076–1084. doi: 10.1038/sj.bjc.6604278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol. 2007;14(12):3629–3637. doi: 10.1245/s10434-007-9583-5. [DOI] [PubMed] [Google Scholar]
- 59.Yang AD, Fan F, Camp ER, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006;12(14 Pt 1):4147–4153. doi: 10.1158/1078-0432.CCR-06-0038. [DOI] [PubMed] [Google Scholar]
- 60.Noseda M, McLean G, Niessen K, et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res. 2004;94(7):910–917. doi: 10.1161/01.RES.0000124300.76171.C9. [DOI] [PubMed] [Google Scholar]
- 61.Timmerman LA, Grego-Bessa J, Raya A, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18(1):99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Becker KF, Rosivatz E, Blechschmidt K, Kremmer E, Sarbia M, Hofler H. Analysis of the E-cadherin repressor Snail in primary human cancers. Cells Tissues Organs. 2007;185(1–3):204–212. doi: 10.1159/000101321. [DOI] [PubMed] [Google Scholar]
- 63.Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A. 2008;105(17):6392–6397. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol. 2008;182(2):315–325. doi: 10.1083/jcb.200710067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leong KG, Niessen K, Kulic I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007;204(12):2935–2948. doi: 10.1084/jem.20071082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Niimi H, Pardali K, Vanlandewijck M, Heldin CH, Moustakas A. Notch signaling is necessary for epithelial growth arrest by TGF-beta. J Cell Biol. 2007;176(5):695–707. doi: 10.1083/jcb.200612129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23(5):1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Z, Li Y, Kong D, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69(6):2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kong D, Banerjee S, Ahmad A, et al. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PloS One. 2010;5(8):e12445. doi: 10.1371/journal.pone.0012445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22(3):373–388. doi: 10.1016/j.ccr.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van der Pluijm G. Epithelial plasticity, cancer stem cells and bone metastasis formation. Bone. 2011;48:37–43. doi: 10.1016/j.bone.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 72.Papachristou DJ, Basdra EK, Papavassiliou AG. Bone metastases: molecular mechanisms and novel therapeutic interventions. Med Res Rev. 2012;32(3):611–636. doi: 10.1002/med.20224. [DOI] [PubMed] [Google Scholar]
- 73.Sethi S, Macoska J, Chen W, Sarkar FH. Molecular signature of epithelial-mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am J Transl Res. 2010;3(1):90–99. [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu H, Zhou X, Redfeld S, Lewin J, Miele L. Elevated Jagged-1 and Notch-1 expression in high grade and metastatic prostate cancers. Am J Transl Res. 2013;5(3):368–378. [PMC free article] [PubMed] [Google Scholar]
- 75.Xing F, Okuda H, Watabe M, et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene. 2011;30(39):4075–4086. doi: 10.1038/onc.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 77.Spaeth EL, Dembinski JL, Sasser AK, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PloS One. 2009;17(4):e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sanchez CG, Penfornis P, Oskowitz AZ, et al. Activation of autophagy in mesenchymal stem cells provides tumor stromal support. Carcinogenesis. 2011;32(7):964–972. doi: 10.1093/carcin/bgr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prockop DJ. Repair of tissues by adult stem/progenitor cells (MSCs): controversies, myths, and changing paradigms. Mol Ther. 2009;17(6):939–946. doi: 10.1038/mt.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hall B, Andreeff M, Marini F. The participation of mesenchymal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles. Handb Exp Pharmacol. 2007;(180):263–283. doi: 10.1007/978-3-540-68976-8_12. [DOI] [PubMed] [Google Scholar]
- 81.Vallabhaneni KC, Tkachuk S, Kiyan Y, et al. Urokinase receptor mediates mobilization, migration, and differentiation of mesenchymal stem cells. Cardiovasc Res. 2011;90(1):113–121. doi: 10.1093/cvr/cvq362. [DOI] [PubMed] [Google Scholar]
- 82.Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62(13):3603–3608. [PubMed] [Google Scholar]
- 83.Schichor C, Birnbaum T, Etminan N, et al. Vascular endothelial growth factor A contributes to glioma-induced migration of human marrow stromal cells (hMSC) Exp Neurol. 2006;199(2):301–310. doi: 10.1016/j.expneurol.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 84.Birnbaum T, Roider J, Schankin CJ, et al. Malignant gliomas actively recruit bone marrow stromal cells by secreting angiogenic cytokines. J Neurooncol. 2007;83(3):241–247. doi: 10.1007/s11060-007-9332-4. [DOI] [PubMed] [Google Scholar]
- 85.Feng B, Chen L. Review of mesenchymal stem cells and tumors: executioner or coconspirator? Cancer Biother Radiopharm. 2009;24(6):717–721. doi: 10.1089/cbr.2009.0652. [DOI] [PubMed] [Google Scholar]
- 86.Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15(10):730–738. doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 87.Dwyer RM, Potter-Beirne SM, Harrington KA, et al. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res. 2007;13(17):5020–5027. doi: 10.1158/1078-0432.CCR-07-0731. [DOI] [PubMed] [Google Scholar]
- 88.Kidd S, Spaeth E, Klopp A, Andreeff M, Hall B, Marini FC. The (in) auspicious role of mesenchymal stromal cells in cancer: be it friend or foe. Cytotherapy. 2008;10(7):657–667. doi: 10.1080/14653240802486517. [DOI] [PubMed] [Google Scholar]
- 89.Hogan NM, Dwyer RM, Joyce MR, Kerin MJ. Mesenchymal stem cells in the colorectal tumor microenvironment: recent progress and implications. Int J Cancer. 2012;131(1):1–7. doi: 10.1002/ijc.27458. [DOI] [PubMed] [Google Scholar]
- 90.Zhu W, Xu W, Jiang R, et al. Mesenchymal stem cells derived from bone marrow favor tumor cell growth in vivo. Exp Mol Pathol. 2006;80(3):267–274. doi: 10.1016/j.yexmp.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 91.Djouad F, Plence P, Bony C, et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003;102(10):3837–3844. doi: 10.1182/blood-2003-04-1193. [DOI] [PubMed] [Google Scholar]
- 92.Mao Y, Keller ET, Garfeld DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013;32(1–2):303–315. doi: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG); Davies C, Godwin J, et al. Relevance of breast cancer hormone receptors and other factors to the effiacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378(9793):771–784. doi: 10.1016/S0140-6736(11)60993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Martin FT, Dwyer RM, Kelly J, et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT) Breast Cancer Res Treat. 2010;124(2):317–326. doi: 10.1007/s10549-010-0734-1. [DOI] [PubMed] [Google Scholar]
- 95.Kandouz M, Batist G. Gap junctions and connexins as therapeutic targets in cancer. Expert Opin Ther Targets. 2010;14(7):681–692. doi: 10.1517/14728222.2010.487866. [DOI] [PubMed] [Google Scholar]
- 96.Mroue RM, El-Sabban ME, Talhouk RS. Connexins and the gap in context. Integr Biol (Camb) 2011;3(4):255–266. doi: 10.1039/c0ib00158a. [DOI] [PubMed] [Google Scholar]
- 97.Gilleron J, Carette D, Chevallier D, Segretain D, Pointis G. Molecular connexin partner remodeling orchestrates connexin traffic: from physiology to pathophysiology. Crit Rev Biochem Mol Biol. 2012;47(5):407–423. doi: 10.3109/10409238.2012.683482. [DOI] [PubMed] [Google Scholar]
- 98.Gu J, Qian H, Shen L, et al. Gastric cancer exosomes trigger differentiation of umbilical cord derived mesenchymal stem cells to carcinoma-associated fibroblasts through TGF-beta/Smad pathway. PloS One. 2012;7(12):e52465. doi: 10.1371/journal.pone.0052465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bonnomet A, Brysse A, Tachsidis A, et al. Epithelial-to-mesenchymal transitions and circulating tumor cells. J Mammary Gland Biol Neoplasia. 2010;15(2):261–273. doi: 10.1007/s10911-010-9174-0. [DOI] [PubMed] [Google Scholar]
- 100.Jing Y, Han Z, Zhang S, Liu Y, Wei L. Epithelial-mesenchymal transition in tumor microenvironment. Cell Biosci. 2011;1:29. doi: 10.1186/2045-3701-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li HJ, Reinhardt F, Herschman HR, Weinberg RA. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012;2(9):840–855. doi: 10.1158/2159-8290.CD-12-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang F, Zhu X, Hu XQ, et al. Mesenchymal stem cells modified with miR-126 release angiogenic factors and activate Notch ligand Delta-like-4, enhancing ischemic angiogenesis and cell survival. Int J Mol Med. 2013;31(2):484–492. doi: 10.3892/ijmm.2012.1200. [DOI] [PubMed] [Google Scholar]
- 103.Xie J, Wang W, Si JW, et al. Notch signaling regulates CXCR4 expression and the migration of mesenchymal stem cells. Cell Immunol. 2013;281(1):68–75. doi: 10.1016/j.cellimm.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 104.Del Papa B, Sportoletti P, Cecchini D, et al. Notch1 modulates mesenchymal stem cells mediated regulatory T-cell induction. Eur J Immunol. 2013;43(1):182–187. doi: 10.1002/eji.201242643. [DOI] [PubMed] [Google Scholar]
- 105.Engin F, Yao Z, Yang T, et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat Med. 2008;14(3):299–305. doi: 10.1038/nm1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011;19(2):192–205. doi: 10.1016/j.ccr.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27(45):5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McGowan PM, Simedrea C, Ribot EJ, et al. Notch1 inhibition alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer. Mol Cancer Res. 2011;9(7):834–844. doi: 10.1158/1541-7786.MCR-10-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rohner A, Spilker ME, Lam JL, et al. Effective targeting of Hedgehog signaling in a medulloblastoma model with PF-5274857, a potent and selective Smoothened antagonist that penetrates the blood-brain barrier. Mol Cancer Ther. 2012;11(1):57–65. doi: 10.1158/1535-7163.MCT-11-0691. [DOI] [PubMed] [Google Scholar]
- 111.Bleckmann A, Siam L, Klemm F, et al. Nuclear LEF1/TCF4 correlate with poor prognosis but not with nuclear β-catenin in cerebral metastasis of lung adenocarcinomas. Clin Exp Metastasis. 2012;30(4):471–482. doi: 10.1007/s10585-012-9552-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zeng Q, Li S, Chepeha DB, et al. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell. 2005;8(1):13–23. doi: 10.1016/j.ccr.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 113.Policastro LL, Ibañez IL, Notcovich C, Duran HA, Podhajcer OL. The tumor microenvironment: characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxid Redox Signal. 2012 Oct 2; doi: 10.1089/ars.2011.4367. Epub. [DOI] [PubMed] [Google Scholar]
- 114.Fiaschi T, Chiarugi P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. Int J Cell Biol. 2012;2012:762825. doi: 10.1155/2012/762825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pietras A, von Stedingk K, Lindgren D, Pahlman S, Axelson H. JAG2 induction in hypoxic tumor cells alters Notch signaling and enhances endothelial cell tube formation. Mol Cancer Res. 2011;9(5):626–636. doi: 10.1158/1541-7786.MCR-10-0508. [DOI] [PubMed] [Google Scholar]
- 116.Xing F, Kobayashi A, Okuda H, et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med. 2013;5(3):384–396. doi: 10.1002/emmm.201201623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ridgway J, Zhang G, Wu Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444(7122):1083–1087. doi: 10.1038/nature05313. [DOI] [PubMed] [Google Scholar]
- 118.Noguera-Troise I, Daly C, Papadopoulos NJ, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444(7122):1032–1037. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- 119.Scehnet JS, Jiang W, Kumar SR, et al. Inhibition of Dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood. 2007;109(11):4753–4760. doi: 10.1182/blood-2006-12-063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Thurston G, Noguera-Troise I, Yancopoulos GD. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer. 2007;7(5):327–331. doi: 10.1038/nrc2130. [DOI] [PubMed] [Google Scholar]
- 121.Yan M, Plowman GD. Delta-like 4/Notch signaling and its therapeutic implications. Clin Cancer Res. 2007;13(24):7243–7246. doi: 10.1158/1078-0432.CCR-07-1393. [DOI] [PubMed] [Google Scholar]
- 122.Reynolds ND, Lukacs NW, Long N, Karpus WJ. Delta-like ligand 4 regulates central nervous system T cell accumulation during experimental autoimmune encephalomyelitis. J Immunol. 2011;187(5):2803–2813. doi: 10.4049/jimmunol.1100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hayashi I, Takatori S, Urano Y, et al. Neutralization of the gamma-secretase activity by monoclonal antibody against extracellular domain of nicastrin. Oncogene. 2012;31(6):787–798. doi: 10.1038/onc.2011.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Funahashi Y, Hernandez SL, Das I, et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008;68(12):4727–4735. doi: 10.1158/0008-5472.CAN-07-6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Varnum-Finney B, Wu L, Yu M, et al. Immobilization of Notch ligand, Delta-1, is required for induction of notch signaling. J Cell Sci. 2000;113(Pt 23):4313–4318. doi: 10.1242/jcs.113.23.4313. [DOI] [PubMed] [Google Scholar]
- 126.Small D, Kovalenko D, Kacer D, et al. Soluble Jagged 1 represses the function of its transmembrane form to induce the formation of the Src-dependent chord-like phenotype. J Biol Chem. 2001;276(34):32022–32030. doi: 10.1074/jbc.M100933200. [DOI] [PubMed] [Google Scholar]
- 127.Smas CM, Chen L, Sul HS. Cleavage of membrane-associated pref-1 generates a soluble inhibitor of adipocyte differentiation. Mol Cell Biol. 1997;17(2):977–988. doi: 10.1128/mcb.17.2.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moellering RE, Cornejo M, Davis TN, et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462(7270):182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tammam J, Ware C, Efferson C, et al. Down-regulation of the Notch pathway mediated by a gamma-secretase inhibitor induces anti-tumour effects in mouse models of T-cell leukaemia. Br J Pharmacol. 2009;158(5):1183–1195. doi: 10.1111/j.1476-5381.2009.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wei P, Walls M, Qiu M, et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. 2010;9(6):1618–1628. doi: 10.1158/1535-7163.MCT-10-0034. [DOI] [PubMed] [Google Scholar]
- 131.Fouladi M, Stewart CF, Olson J, et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol. 2011;29(26):3529–3534. doi: 10.1200/JCO.2011.35.7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pandya K, Meeke K, Clementz AG, et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br J Cancer. 2011;105(6):796–806. doi: 10.1038/bjc.2011.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Macy ME, Sawczyn KK, Garrington TP, Graham DK, Gore L. Pediatric developmental therapies: interesting new drugs now in early-stage clinical trials. Curr Oncol Rep. 2008;10(6):477–490. doi: 10.1007/s11912-008-0073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zweidler-McKay PA. Notch signaling in pediatric malignancies. Curr Oncol Rep. 2008;10(6):459–468. doi: 10.1007/s11912-008-0071-2. [DOI] [PubMed] [Google Scholar]
- 135.Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8(10):806–823. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 136.Kallifatidis G, Labsch S, Rausch V, et al. Sulforaphane increases drug-mediated cytotoxicity toward cancer stem-like cells of pancreas and prostate. Mol Ther. 2011;19(1):188–195. doi: 10.1038/mt.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kawahara T, Kawaguchi-Ihara N, Okuhashi Y, Itoh M, Nara N, Tohda S. Cyclopamine and quercetin suppress the growth of leukemia and lymphoma cells. Anticancer Res. 2009;29(11):4629–4632. [PubMed] [Google Scholar]
- 138.Okuhashi Y, Itoh M, Nara N, Tohda S. Effects of combination of notch inhibitor plus hedgehog inhibitor or Wnt inhibitor on growth of leukemia cells. Anticancer Res. 2011;31(3):893–896. [PubMed] [Google Scholar]
- 139.Zhou W, Kallifatidis G, Baumann B, et al. Dietary polyphenol quercetin targets pancreatic cancer stem cells. Int J Oncol. 2010;37(3):551–561. doi: 10.3892/ijo_00000704. [DOI] [PubMed] [Google Scholar]
- 140.Wang Z, Zhang Y, Banerjee S, Li Y, Sarkar FH. Notch-1 down-regulation by curcumin is associated with the inhibition of cell growth and the induction of apoptosis in pancreatic cancer cells. Cancer. 2006;106(11):2503–2513. doi: 10.1002/cncr.21904. [DOI] [PubMed] [Google Scholar]
- 141.Luistro L, He W, Smith M, et al. Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res. 2009;69(19):7672–7680. doi: 10.1158/0008-5472.CAN-09-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rao SS, O’Neil J, Liberator CD, et al. Inhibition of NOTCH signaling by gamma secretase inhibitor engages the RB pathway and elicits cell cycle exit in T-cell acute lymphoblastic leukemia cells. Cancer Res. 2009;69(7):3060–3068. doi: 10.1158/0008-5472.CAN-08-4295. [DOI] [PubMed] [Google Scholar]
- 143.Espinoza I, Miele L. Notch inhibitors for cancer treatment. Pharmacol Ther. 2013;139(2):95–110. doi: 10.1016/j.pharmthera.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Harrison H, Farnie G, Howell SJ, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70(2):709–718. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Samon JB, Castillo-Martin M, Hadler M, et al. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther. 2012;11(7):1565–1575. doi: 10.1158/1535-7163.MCT-11-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rizzo P, Miao H, D’Souza G, et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008;68(13):5226–5235. doi: 10.1158/0008-5472.CAN-07-5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Albain K, Czerlanis C, Zlobin A, et al. Modulation of cancer stem cell biomarkers by the Notch inhibitor MK-0752 added to endocrine therapy for early stage ER+ breast cancer. Cancer Res. 2011;71:97s. [Google Scholar]
- 148.Means-Powell JA, Minton SE, Mayer IA, et al. A Phase Ib dose escalation trial of RO4929097 (a γ-secretase inhibitor) in combination with exemestane in patients with ER + metastatic breast cancer. Cancer Res. 2012;72:280s. doi: 10.1016/j.clbc.2021.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jieun Yun, Pannuti AP, Espinoza I, et al. Crosstalk between PKCα and Notch-4 in endocrine-resistant breast cancer cells. Oncogenesis. :2013. doi: 10.1038/oncsis.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schott AF, Landis MD, Dontu G, et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res. 2013;19(6):1512–1524. doi: 10.1158/1078-0432.CCR-11-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]