

Abstract
The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) proteins are a family of ubiquitously expressed transcription factors that regulate the response to cellular stress. They mediate innate and adaptive immunity through the initiation of an inflammatory response to pro-inflammatory signals. The role of persistent inflammation in aiding tumor development has led to the NF-κB family of transcription factors being strongly implicated in promoting cancer. However, recent studies have now revealed that NF-κB can also function as a tumor suppressor through the induction of cellular senescence. Cellular senescence is a stable cell cycle arrest that normal cells undergo in response to a variety of intrinsic and extrinsic stimuli including: progressive telomere shortening, changes in telomeric structure, or other forms of genotoxic stress. Senescence can compromise tissue repair and regeneration, contributing to tissue and organismal aging via the accumulation of senescent cells, depletion of stem/progenitor cells and secretion of an array of inflammatory cytokines, chemokines, and matrix metalloproteinases. Senescence can also lead to the removal of potentially cancerous cells, thereby acting as a potent tumor suppressor mechanism. Herein, we review the evidence indicating a role for NF-κB in tumor suppression via cellular senescence and suggest that depending upon the subunit expressed, the biological context, and the type and intensity of the signal, NF-κB can indeed promote senescence growth arrest.
Keywords: cell senescence, growth arrest, NF-κB, SASP
Introduction to NF-κB
The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) family of transcription factors have been extensively studied over the last 25 years since they were discovered by binding to the immunoglobulin κ light chain gene enhancer (iκE).1 They comprise five family members: RELA (p65), RELB, c-REL, p105/p50 (NF-κB1), and p100/p52 (NF-κB2), which all share the Rel homology domain, permitting their dimerization and translocation to the nucleus.2,3 RELA, RELB, and c-REL also have a transactivation domain, whilst p105/p50 and p100/p52 contain ankyrin repeat domains (Figure 1). The primary mediator of NF-κB transcriptional activity in response to activators such as inflammatory cytokines and Toll-like receptor (TLR) signaling, is the RELA:p50 heterodimer, whereas other subunits have a more primary role in other contexts. The RELA:p50 heterodimer is the most readily detectable form of this complex.2,3
Figure 1.
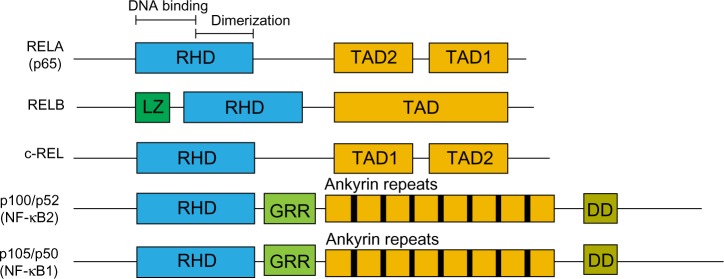
NF-κB protein family.
Notes: All family members share the RHD, composed of a DNA-binding region and a dimerization region. RELA and c-REL have two TAD whereas RELB contains only one. P100 and p105 precursor proteins are processed by the proteosome to the mature p52 and p50 respectively and lack a transactivation domain. Other indicated domains are: LZ; GRR; DD.
Abbreviations: DD, death domain; DNA, deoxyribonucleic acid; GRR, glycine-rich region; LZ, leucine zipper; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; RHD, Rel homology domain; TAD, transactivation domains.
The mechanism of action underlying the activation of NF-κB, falls into two broad categories: a canonical pathway and a non-canonical pathway (Figure 2). In the canonical pathway, NF-κB homo-and heterodimers are bound to inhibitors of NF-κB (IκB) in unstimulated cells, leading to them being retained in the cytosol.2 A variety of stimuli can induce the phosphorylation of IκB by the multi-subunit IκB kinase complex (IKKα, IKKβ, and IKKγ/NEMO), leading to ubiquitination and subsequent degradation of IκB by the 26S proteasome.2,4 This unmasks a nuclear localization signal, resulting in the nuclear translocation of NF-κB dimers and activation of a plethora of target genes. The canonical pathway is vital for the activation of innate immunity and inflammation.2
Figure 2.

Canonical and non-canonical pathways of NF-κB activation.
Notes: In the canonical pathway, a variety of inputs such as inflammatory cytokines induce the phosphorylation and activation of the IκB kinase (IKK) complex (IKKα, IKKβ, and IKKγ/NEMO). This phosphorylates IκB leading to its ubiquitination and degradation by the 26S proteasome leading to nuclear translocation of RELA:p50 dimers. The non-canonical pathway is activated by more specific inputs such as lymphotoxin-β and leads to NF-κB inducing kinase (NIK) phosphorylating and activating IKKα, a member of the multi-subunit IκB kinase complex. This leads to phosphorylation and subsequent ubiquitination of p100 resulting in its processing by the proteosome to produce transcriptionally active RELB:p52 dimers.
Abbreviations: NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; REL, reticuloendotheliosis virus; RIP, receptor-interacting serine/threonine protein kinase 1; TAB, transforming growth factor-beta activated kinase binding protein; TAK, transforming growth factor-beta activated kinase; Ub, ubiquitin.
In response to stimuli such as lymphotoxin β, an alternative non-canonical pathway is activated. This involves the NF-κB inducing kinase (NIK), which phosphorylates and activates IKKα, leading to the proteosomal processing of p100 with preferential nuclear translocation of RELB:p52 dimers.5,6 Although the canonical and non-canonical pathways have separate regulatory mechanisms, recent evidence suggests that crosstalk between these pathways is vital for NF-κB activation, as synthesis of RELB is controlled by RELA signaling.7,8
Physiological role of NF-κB
NF-κB mediates the cellular response to stimuli such as cytokines, free radicals, deoxyribonucleic acid (DNA) damage, oxidized low density lipoproteins, and bacterial or viral antigens, most of which are induced upon intrinsic or extrinsic cellular stress.9,10 It does this by binding to specific DNA sequences within target genes in the nucleus and can act as a transcriptional activator or repressor.11 This family of transcription factors (TFs) also has an important role in the development of the skeletal system and epithelium.12,13 They are required for proper organogenesis of several epithelial tissues such as the mammary gland; blockage of NF-κB activity in the mammary gland of mice leads to a severe lactation deficiency.14 Similarly, activation of NF-κB in axon-associated Schwann cells is essential for progression to a myelinating phenotype.15 However, as NF-κB proteins were first identified as factors that regulate B-lymphocyte-specific transcription, the role of NF-κB in the development, specialization, and maintenance of the immune system is more thoroughly documented.16–18 Hemopoiesis is characterized by apoptosis, differentiation, and proliferation, which are key processes controlled by the action of NF-κB. During hemopoiesis, cells of the lymphoid and myeloid lineages such as T cells, B cells, monocytes, macrophages, dendritic cells, natural killer cells, basophils, eosinophils, neutrophils, and mast cells are all formed. NF-κB contributes to hemopoiesis by promoting the survival and differentiation of precursors to a particular cell fate, throughout all stages of this process.19 These cells are important for both innate and adaptive immunity and can initiate an inflammatory response to pro-inflammatory signals such as cytokines and chemokines, many of which are transcriptionally activated by NF-κB.20
NF-κB also has an important role in normal B- cell maturation as purified B cells from p50/NF-kappa B knockout (p50−/−) mice have a reduced ability to mature to Ig secretion, germ-line CH gene activation, and undergo Ig class switching, as well as mitogenesis.18
As a result, deregulation or dysfunction of NF-κB is detrimental to organisms, and can induce pathologies such as chronic inflammation and autoimmune diseases, cardiovascular disease, neurodegenerative diseases, and type II diabetes.21 Persistent inflammation is also an important underlying condition that aids tumor development, thus suggesting a potential role for NF-κB in tumorigenesis.22,23 Accordingly, the NF-κB family of proteins is frequently implicated in cancer particularly since the Rel homology domain within these TFs is homologous to the viral oncogene, v-Rel, from the avian reticuloendotheliosis virus.22–24
The role of NF-κB in cancer
NF-κB proteins mediate proliferation, growth, and apoptosis. Mutations leading to aberrant activation of NF-κB have thus been implicated in various cancers.3,22–25 Indeed, many cancers, in particular melanomas, multiple myeloma, various types of leukemia, and B and T-cell lymphomas, are associated with the aberrant activation of NF-κB.25,26 This is in part due to the characteristic NF-κB pro-inflammatory response, but inhibition of apoptosis and promotion of metastasis and cell proliferation also play vital roles. NF-κB acts to inhibit apoptosis, a key hallmark of cancer, downstream of growth factor signaling such as epidermal growth factor (EGF).27 Here, activation of epidermal growth factor-receptor (EGFR) recruits phosphatidylinositide 3-kinase (PI3K) to the plasma membrane which in turn induces the de novo synthesis of phosphatidylinositol [3, 4, 5]-triphosphate; (PI[3,4,5]P3). This activates protein kinase B (AKT/PKB) with the help of phosphoinositide-dependent kinase-1 (PDK1) and mammalian target of rapamycin complex 2 (mTORC2).28 AKT has been suggested to activate NF-κB via IKK, resulting in the transcription of pro-survival genes, which prevent the death of cancer cells, particularly when EGFR or mTOR are constitutively activated due to mutation.26,29,30 Mutations leading to the activation of K-ras in the context of INK4a/ARF deficiency in mice lead to pancreatic cancer by persistent activation of the Notch and NF-κB signaling pathways.31 NF-κB can also be activated by hypoxia, which has been implicated in the promotion of angiogenesis for sustaining the growth of cancer cells.32 Moreover, NF-κB has been shown to contribute to initiation, promotion, and progression of tumorigenesis through its role in apoptosis, inflammation, and regulating matrix metalloproteinase-9 as well as inducing the epithelial to mesenchymal transition (EMT) via TWIST1 and SNAI1.33–37
Although it had been postulated that NF-κB can also function as a tumor suppressor for a number of years, through for example, its pro-apoptotic activities,38 recent studies have now revealed a new facet of this behavior through the induction of a stable cell growth arrest, otherwise known as cellular senescence.
Introduction to cellular senescence, aging, and cancer
Somatic cells typically undergo a finite number of divisions before they irreversibly exit the cell cycle; this replicative limit is known as the Hayflick limit.39 Cellular senescence occurs in response to a variety of intrinsic and extrinsic stimuli, some of which include: progressive telomere shortening/uncapping, oncogene activation, aberrant DNA damage, oxidative stress, and other forms of genotoxic and non-genotoxic stresses.40–42 Senescence results in cellular hypertrophy and a pro-inflammatory and hyper-secretory phenotype. Senescent cells show resistance to apoptosis, are metabolically active, and remain viable for long periods of time.43 They also exhibit dramatic changes in morphology whereby cells become enlarged and flattened, making the senescent phenotype unique, easy to distinguish, and thus easy to detect.44 Senescent cells may also be distinguished by the upregulation or increased activity of various biomarkers such as senescence-associated-β-galactosidase, plasminogen activator inhibitor (PAI)-1, fibronectin, p53, and the cyclin dependent kinase inhibitors p21CIP1/WAF1/SDI1 and p16INK4a.45
Although there are different categories of cellular senescence, such as replicative senescence or premature senescence, both trigger a DNA-damage response, resulting in activation of the p53 and the retinoblastoma protein (pRB) tumor suppressors.46 P53 initiates senescence by activating the expression of the cyclin dependent kinase inhibitor, p21CIP1/WAF1/SDI1, which in turn inhibits the cyclin E/cyclin dependent kinase 2 (CDK2) complex of the cell cycle. This prevents the phosphorylation and deactivation of the pRB family of pocket proteins permitting hypophosphorylated pRB to complex with the E2F family of heterodimeric TFs.47 In turn, pRB recruits histone deacetylases and remodeling factors to E2F responsive promoters, thereby inhibiting E2F-dependent S-phase gene expression.40,48 In response to non-genotoxic stress, the pRB pathway is activated independently of p53 via the upregulation of p16INK4A, which acts to inhibit cyclinD-CDK4/6 complexes from phosphorylating pRB.44 Equally, p53 may also induce senescence by alternative pathways, as it is a master TF that regulates a plethora of target genes affecting physiological pathways important for senescence such as E2F7, which promotes the repression of several E2F target genes.49 However, many of the pathways downstream of p53 still remain poorly defined (Figure 3).
Figure 3.

Schematic illustration of the pathways linking NF-κB to cellular senescence, cancer, and aging.
Notes: Inflammation, DNA damage, and oxidative/oncogenic stress all lead to the activation of IKKα/IKKβ resulting in the activation of NF-κB. NF-κB can inhibit tumorigenesis and promote aging by inducing a senescence growth arrest and SASP. Alternatively, depending upon the signal, NF-κB may promote tumorigenesis by activating cell cycle progression, blocking apoptosis, and inducing SASP for example.
Abbreviations: ATM, ataxia telangiectasia mutated kinase; CIP, cyclin dependent kinase interacting protein; CDK, cyclin dependent kinase; CycD, cyclin D; DNA, deoxyribonucleic acid; EGFR, epidermal growth factor receptor; FOXO, forkhead box; IGF1R, insulin like growth factor 1 receptor; HMGB1, high mobility group protein B1; IKK, IkB kinase; IL, interleukin; IR, insulin receptor; MAPK, mitogen activated protein kinase; miRNA, micro ribonucleic acid; NEMO, NF-kappa B essential modulator, also known as inhibitor of nuclear factor kappa B kinase subunit gamma; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; Pi3K, phosphatidylinositol 3-kinase; PIP, phosphatidylinositol phosphate; pRB, retinoblastoma protein; SASP, senescence associated secretory phenotype; TAB, transforming growth factor-beta activated kinase binding protein; TAK, transforming growth factor-beta activated kinase; TGF, transforming growth factor; TNF, tumor necrosis factor; TRAFS, TNF receptor associated factors.
Senescence can lead to organismal aging by compromising tissue renewal, repair, and regeneration via the accumulation of non-dividing cells and secretion of an array of inflammatory cytokines, chemokines, and matrix metalloproteinases.43,50 Accumulation of post-mitotic cells also leads to the depletion of stem and progenitor cell populations via the disruption of stem-cell homeostasis, thereby preventing the replenishment of old or damaged cells.51 Removal of senescent cells attenuates age-related tissue dysfunction and extends healthy lifespan. However, senescence can also act as a potent tumor suppressor mechanism through the removal of genetically unstable and pre-cancerous cells from the proliferative pool.43
Cancer is considered to be a disease of the cell cycle, as cancer cells lose the stringent controls normally present during cell cycle progression. This is often the result of mutations via DNA damage; such damage can also act as a signal for senescence and inhibition of the cell cycle, preventing tumor growth. Thus senescence may act as a quality control mechanism for eliminating less healthy cells from the population, thereby maintaining genome integrity and appropriate cellular function.52 Indeed, the induction of premature senescence in vivo has been shown to suppress tumor formation via the activation of key senescence pathways.53 Several studies suggest that senescence must first be overcome for the initiation of cancer or maintaining the growth of cancer cells, which can then proliferate indefinitely; a key hallmark of cancer.54–56 Interestingly, chemotherapy using antimitotic drugs has been shown to induce cellular senescence in cancer cells that have evaded apoptosis.57 The radiotherapy response of lung cancer cells has been shown to be mediated through both apoptosis and senescence.58 Hence, there has been a recent push in cancer therapeutics to target cellular senescence as a way of treating cancer, but the downstream effectors of cellular senescence are currently poorly defined. Moreover, vital questions remain to be answered about what makes this process of cell cycle arrest essentially irreversible and what are the players that underlie this stable arrest.
Role of NF-κB in promoting cellular senescence
The role of NF-κB in reinforcing cellular senescence is currently emerging and is rapidly being recognized to have a causal role in this growth arrest.59 Bernard and colleagues provided the first evidence indicating that NF-κB may inhibit growth by transiently overexpressing c-REL in HeLa cells; this led to a cell cycle arrest in all stages of the cell cycle, although the arrest was not demonstrated to be irreversible.60,61 Later studies by Hardy et al suggested a novel role for NF-κB in senescence growth arrest.62 They used cDNA microarrays to identify genes that were differentially expressed when conditionally immortal human mammary (HMF3A) fibroblasts underwent senescence, followed by in silico promoter analysis of the differential genes and electrophoretic mobility shift assays to show that NF-κB was activated upon senescence. Shortly after, Adler et al showed that NF-κB was strongly associated with and was required to enforce pathological aging in mice.63
Rovillain et al64 extended the Hardy et al study by using genome-wide expression profiling, in conjunction with inactivation of the p16-pRB and p53-p21 tumor suppressor pathways in the conditionally immortal HMF3A fibroblasts, and found that 91 NF-κB downstream targets were up regulated upon senescence arrest; many of these targets are associated with the senescence associated secretory phenotype (SASP). Moreover, suppression of NF-κB by short hairpin ribonucleic acid (shRNA) silencing, or by ectopic expression of the I-κB super-repressor of NF-κB, abrogated senescence arrest in these conditionally immortal fibroblasts.64
Interestingly, silencing of the bona-fide oncoprotein DEK leads to upregulation of NF-κB and its associated proteins resulting in a G2 cell cycle block and cellular senescence in CaSki cervical carcinoma cells.65 Senescence is thought to be reinforced by the activation of cell surface bound cytokine interleukin (IL)-1α, which in turn activates NF-κB, as the expression of IL-1α in culture is one of the earliest events to occur after growth arrest.66,67 Freund et al have proposed that p38 mitogen activated protein kinase (MAPK) induces senescence via NF-κB activation.68 Collectively, these, as well as other studies, have suggested a causal role for NF-κB in promoting cellular senescence.
NF-κB, DNA damage response, and senescence
Although it is not known how NF-κB activates senescence, links have been made between the DNA damage response, the best-known activator of cellular senescence, and NF-κB activation. Inhibition of NF-κB by depletion of one allele of p65, or pharmacological inhibition of IKK, has been reported to reduce oxidative damage and stress whilst delaying cellular senescence in progeroid mice.69 Further studies, in two different mouse models of accelerated aging, have shown that accumulation of prelamin A, at the nuclear lamina, triggers an ataxia telangiectasia mutated (ATM)- and NEMO- signaling pathway leading to activation of NF-κB and secretion of proinflammatory cytokines.70 Moreover, inhibition of NF-κB increases longevity of these mice by abrogating senescence.70 Lifespan extension in mice has also been recently achieved by preventing aging-related hypothalamic IKKβ and NF-κB activation, which normally inhibit gonadotropin-releasing hormone (GnRH).71 It is currently thought that genotoxic stress activates the NF-κB pathway, in mice, by stimulating Poly [ADP-ribose] polymerase 1 (PARP-1) and ATM, which induces the synthesis of poly adenosine diphosphate (ADP) ribose and the subsequent assembly of ATM and an IKKγ/PIASγ complex that activates NF-κB.72 Furthermore, studies in human dermal fibroblasts have revealed crosstalk between p53 and NF-κB to induce cellular senescence through the repression of cyclin D1 as a response to ultraviolet (UV) light-induced DNA damage.73
Since DNA-damage can also lead to other cell fates such as apoptosis or quiescence through activation of the p53 and pRB pathways, it remains to be elucidated how cellular senescence prevails over these cell fates in a particular context. As NF-κB has been shown in vitro and in vivo to block apoptosis in cancer cells, as well as normal cells, in response to DNA damage,74,75 it can be hypothesized that NF-κB has a role in committing the cell to cellular senescence by minimizing the choice of cell fates. Therefore, activation of ATM leads to a cell cycle arrest and NF-κB activation in response to DNA double strand breaks,75 whereas inhibition of NF-κB sensitizes transformed cells to apoptosis induced by ionizing radiation or chemotherapeutic drugs.76,77
Furthermore, inhibition of aurora kinases with MLN8054/MLN8237 in mice with metastatic melanoma tumors led to suppression of tumor growth by induction of polyploidy and the ATM/CHK2 DNA damage response, which mediated senescence and a NF-κB related SASP.78 However, blockade of IKKβ/NF-κB leads to reversal of MLN8237-induced senescence and SASP.78 This suggests that targeting cellular senescence through NF-κB could provide a potential anticancer therapy.
Senescence associated secretory phenotype and NF-κB
NF-κB activation has been coupled to the SASP.79 Senescent cells secrete a variety of inflammatory cytokines, proteases and growth factors such as: tumor necrosis factor (TNF) α and IL-6, monocyte chemoattractant protein-1, matrix metalloproteinases, and EGFs [44]. Campisi and colleagues have shown that a variety of senescent-inducing stimuli activate p38MAPK, which in turn induces SASP, by upregulating transcriptional activity of NF-κB.68 This is also sufficient for SASP induction in melanoma cells.79 Moreover, Chien et al recently observed that NF-κB signaling controlled the appearance of SASP in H-RasV12-induced senescence in human skin fibroblasts, and that RELA was significantly enriched in the chromatin of senescent fibroblasts.80
NF-κB is thought to promote SASP by activating the expression of IL-6 and IL-8, which also contribute to reinforcing senescence growth arrest. Some studies suggest that IL-1α is also required for this activation, as silencing of IL-1α significantly decreased the senescence associated secretion of IL-6/8 by reducing the DNA binding activity of NF-κB and CCAAT-enhancer-binding protein beta (c/EBPβ).81 Cells undergoing oncogene induced senescence also secrete multiple CXCR2-binding chemokines, in a NF-κB and c/EBPβ dependent manner.82 However, the occurrence of SASP in vivo, in response to NF-κB, remains to be elucidated and the role of SASP in senescence growth arrest is controversial, as SASP can also promote cancer.
SASP and cancer
Adding a further layer of complexity to the NF-κB friend/foe dilemma, senescence also has a role in tumor promotion through the secretion of inflammatory, growth promoting and remodeling factors that senescent cells secrete as SASP.67 Such factors promote the EMT, metastasis, and proliferation of neighboring cells. Metalloproteases, secreted as SASP, lead to the degradation of the basement membrane surrounding tissues, further promoting tumor metastasis. Hence, SASP creates a microenvironment that favors malignancy in neighboring cells when it is persistent, as is the case in aging cells.83 Interestingly, it has recently been shown that cultured medium, derived from senescent cells, is sufficient to induce the growth of cancer cells, but this was inhibited by the anti-diabetic drug, metformin, which inhibits SASP and downregulates NF-κB induced transcription.84 However, when SASP was inhibited by low levels of metformin, growth arrest still occurred, suggesting that other downstream targets are also important for the growth arrest. Interestingly, drug induced senescent cells have been shown to increase the proliferation of bystander cells in vitro but did not significantly alter tumor growth in vivo.85
SASP can also reinforce growth arrest and protect against cancer by activating the immune response, inducing the clearance of damaged and pre-cancerous cells.86 Accordingly, senescence may have evolved as an example of antagonistic pleiotrophy, since it provides beneficial traits during the reproductive age of an individual (tumor suppression via growth arrest), but causes deleterious effects later on in life (aging and cancer via SASP).43 We thus hypothesize that the dual role of NF-κB in tumor promotion and suppression may, in part, be due to its promotion of SASP (summarized in Figure 3).
Conclusion
The NF-κB family of transcription factors has gained considerable attention due to their roles in various stress-induced pathways. Central to this is the role of NF-κB in both promoting cancer and acting as a tumor suppressor via the induction of cellular senescence. We have summarized the evidence indicating a role of NF-κB in both of these pathologies and suggest that depending upon the biological context, type, and intensity of the signal, NF-κB can indeed promote senescence arrest in somatic cells. We propose that these opposing decisions are determined by a combination of differential NF-κB subunit expression and modification in combination with crosstalk from other signaling pathways such as p53. Future studies, aimed at providing a greater understanding of the preferential expression of different NF-κB subunits in a particular context, and their engagement with different signaling pathways, will shed light on the opposing roles of NF-κB in cancer.
Acknowledgments
We thank A Munro Neville and Ralph Andre for their critique. We are indebted to Richard Newton for graphics. SNM is a Natural Sciences MSci student, University College, London, UK.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–716. [PubMed] [Google Scholar]; J Immunol. 2006;177(11):7485–7496. [PubMed] [Google Scholar]
- 2.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perkins ND. The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer. 2012;12(2):121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 4.Monaco C, Andreakos E, Kiriakidis S, et al. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci U S A. 2004;101(15):5634–5639. doi: 10.1073/pnas.0401060101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basak S, Kim H, Kearns JD, et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128(2):369–381. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Chen ZJ. Regulation of NF-κB by ubiquitination. Curr Opin Immunol. 2013;25(1):4–12. doi: 10.1016/j.coi.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basak S, Shih VF, Hoffmann A. Generation and activation of multiple dimeric transcription factors within the NF-kappaB signaling system. Mol Cell Biol. 2008;28(10):3139–3150. doi: 10.1128/MCB.01469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu M, Qi X, Moreno JL, Farber DL, Keegan AD. NF-κB signaling participates in both RANKL- and IL-4-induced macrophage fusion: receptor cross-talk leads to alterations in NF-κB pathways. J Immunol. 2011;187(4):1797–1806. doi: 10.4049/jimmunol.1002628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian B, Nowak DE, Jamaluddin M, Wang S, Brasier AR. Identification of direct genomic targets downstream of the nuclear factor-kappaB transcription factor mediating tumor necrosis factor signaling. J Biol Chem. 2005;280(17):17435–17448. doi: 10.1074/jbc.M500437200. [DOI] [PubMed] [Google Scholar]
- 10.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25(51):6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 11.Basak S, Shih VF, Hoffmann A. Generation and activation of multiple dimeric transcription factors within the NF-kappaB signaling system. Mol Cell Biol. 2008;28(10):3139–3150. doi: 10.1128/MCB.01469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novack DV. Role of NF-κB in the skeleton. Cell Res. 2011;21(1):169–182. doi: 10.1038/cr.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schauer IG, Zhang J, Xing Z, et al. Interleukin-1β promotes ovarian tumorigenesis through a p53/NF-κB-mediated inflammatory response in stromal fibroblasts. Neoplasia. 2013;15(4):409–420. doi: 10.1593/neo.121228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao Y, Karin M. NF-kappaB in mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia. 2003;8(2):215–223. doi: 10.1023/a:1025905008934. [DOI] [PubMed] [Google Scholar]
- 15.Nickols JC, Valentine W, Kanwal S, Carter BD. Activation of the transcription factor NF-kappaB in Schwann cells is required for peripheral myelin formation. Nat Neurosci. 2003;6(2):161–167. doi: 10.1038/nn995. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Ohms SJ, Sun C, Fan J. NF-κB controls Il2 and Csf2 expression during T cell development and activation process. Mol Biol Rep. 2013;40(2):1685–1692. doi: 10.1007/s11033-012-2219-2. [DOI] [PubMed] [Google Scholar]
- 17.Kaileh M, Sen R. NF-κB function in B lymphocytes. Immunol Rev. 2012;246(1):254–271. doi: 10.1111/j.1600-065X.2012.01106.x. [DOI] [PubMed] [Google Scholar]
- 18.Snapper CM, Zelazowski P, Rosas FR, et al. B cells from p50/NF-kappa B knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J Immunol. 1996;156(1):183–191. [PubMed] [Google Scholar]
- 19.Gerondakis S, Banerjee A, Grigoriadis G, et al. NF-κB subunit specificity in hemopoiesis. Immunol Rev. 2012;246(1):272–285. doi: 10.1111/j.1600-065X.2011.01090.x. [DOI] [PubMed] [Google Scholar]
- 20.Thompson C, Cloutier A, Bossé Y, et al. CysLT1 receptor engagement induces activator protein-1- and NF-kappaB-dependent IL-8 expression. Am J Respir Cell Mol Biol. 2006;35(6):697–704. doi: 10.1165/rcmb.2005-0407OC. [DOI] [PubMed] [Google Scholar]
- 21.Xanthoulea S, Curfs DM, Hofker MH, de Winther MP. Nuclear factor kappa B signaling in macrophage function and atherogenesis. Curr Opin Lipidol. 2005;16(5):536–542. doi: 10.1097/01.mol.0000180167.15820.ae. [DOI] [PubMed] [Google Scholar]
- 22.Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1(5):a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. 2012;246(1):379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 24.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2(6):a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carbone C, Melisi D. NF-κB as a target for pancreatic cancer therapy. Expert Opin Ther Targets. 2012;16(Suppl 2):S1–S10. doi: 10.1517/14728222.2011.645806. [DOI] [PubMed] [Google Scholar]
- 26.Odqvist L, Sánchez-Beato M, Montes-Moreno S, et al. NIK controls classical and alternative NF-κB activation and is necessary for the survival of human T-cell lymphoma cells. Clin Cancer Res. 2013;19(9):2319–2330. doi: 10.1158/1078-0432.CCR-12-3151. [DOI] [PubMed] [Google Scholar]
- 27.Alberti C, Pinciroli P, Valeri B, et al. Ligand-dependent EGFR activation induces the co-expression of IL-6 and PAI-1 via the NFkB pathway in advanced-stage epithelial ovarian cancer. Oncogene. 2012;31(37):4139–4149. doi: 10.1038/onc.2011.572. [DOI] [PubMed] [Google Scholar]
- 28.Korkolopoulou P, Levidou G, Trigka EA, et al. A comprehensive immunohistochemical and molecular approach to the PI3K/AKT/mTOR (phosphoinositide 3-kinase/v-akt murine thymoma viral oncogene/mammalian target of rapamycin) pathway in bladder urothelial carcinoma. BJU Int. 2012;110(11 Pt C):E1237–E1248. doi: 10.1111/j.1464-410X.2012.11569.x. [DOI] [PubMed] [Google Scholar]
- 29.Parajuli B, Lee HG, Kwon SH, et al. Salinomycin inhibits Akt/NF-κB and induces apoptosis in cisplatin resistant ovarian cancer cells. Cancer Epidemiol. 2013;37(4):512–517. doi: 10.1016/j.canep.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 30.Howe EN, Cochrane DR, Cittelly DM, Richer JK. miR-200c targets a NF-κB up-regulated TrkB/NTF3 autocrine signaling loop to enhance anoikis sensitivity in triple negative breast cancer. PLoS ONE. 2012;7(11):e49987. doi: 10.1371/journal.pone.0049987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z, Banerjee S, Ahmad A, et al. Activated K-ras and INK4a/Arf deficiency cooperate during the development of pancreatic cancer by activation of Notch and NF-κB signaling pathways. PLoS ONE. 2011;6(6):e20537. doi: 10.1371/journal.pone.0020537. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Yu H, Mohan S, Natarajan M. Radiation-Triggered NF-κB Activation is Responsible for the Angiogenic Signaling Pathway and Neovascularization for Breast Cancer Cell Proliferation and Growth. Breast Cancer (Auckl) 2012;6:125–135. doi: 10.4137/BCBCR.S9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 34.Wang F, He W, Fanghui P, Wang L, Fan Q. NF-κBP65 promotes invasion and metastasis of oesophageal squamous cell cancer by regulating matrix metalloproteinase-9 and epithelial-to-mesenchymal transition. Cell Biol Int. 2013;37(8):780–788. doi: 10.1002/cbin.10089. [DOI] [PubMed] [Google Scholar]
- 35.Kirillova I, Chaisson M, Fausto N. Tumor necrosis factor induces DNA replication in hepatic cells through nuclear factor kappaB activation. Cell Growth Differ. 1999;10(12):819–828. [PubMed] [Google Scholar]
- 36.Meira LB, Bugni JM, Green SL, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118(7):2516–2525. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193(6):727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ. 2006;13(5):759–772. doi: 10.1038/sj.cdd.4401838. [DOI] [PubMed] [Google Scholar]
- 39.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 40.Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37(5):961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 41.Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21(1):43–48. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell. 2009;36(1):2–14. doi: 10.1016/j.molcel.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 43.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Ben-Porath I, Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J Clin Invest. 2004;113(1):8–13. doi: 10.1172/JCI200420663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24(22):2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13(3):332–337. doi: 10.1016/s0955-0674(00)00216-7. [DOI] [PubMed] [Google Scholar]
- 47.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13(1):77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 48.Di Micco R, Cicalese A, Fumagalli M, et al. DNA damage response activation in mouse embryonic fibroblasts undergoing replicative senescence and following spontaneous immortalization. Cell Cycle. 2008;7(22):3601–3606. doi: 10.4161/cc.7.22.7152. [DOI] [PubMed] [Google Scholar]
- 49.Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013 doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- 50.Doles J, Storer M, Cozzuto L, Roma G, Keyes WM. Age-associated inflammation inhibits epidermal stem cell function. Genes Dev. 2012;26(19):2144–2153. doi: 10.1101/gad.192294.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choudhery MS, Khan M, Mahmood R, Mehmood A, Khan SN, Riazuddin S. Bone marrow derived mesenchymal stem cells from aged mice have reduced wound healing, angiogenesis, proliferation and anti-apoptosis capabilities. Cell Biol Int. 2012;36(8):747–753. doi: 10.1042/CBI20110183. [DOI] [PubMed] [Google Scholar]
- 52.Vivarelli S, Wagstaff L, Piddini E. Cell wars: regulation of cell survival and proliferation by cell competition. Essays Biochem. 2012;53:69–82. doi: 10.1042/bse0530069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kriegl L, Neumann J, Vieth M, et al. Up and downregulation of p16(Ink4a) expression in BRAF-mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Mod Pathol. 2011;24(7):1015–1022. doi: 10.1038/modpathol.2011.43. [DOI] [PubMed] [Google Scholar]
- 55.Favaro E, Bensaad K, Chong MG, et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012;16(6):751–764. doi: 10.1016/j.cmet.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 56.Yang D, Li L, Liu H, et al. Induction of autophagy and senescence by knockdown of ROC1 E3 ubiquitin ligase to suppress the growth of liver cancer cells. Cell Death Differ. 2013;20(2):235–247. doi: 10.1038/cdd.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biggers JW, Nguyen T, Di X, et al. Autophagy, cell death and sustained senescence arrest in B16/F10 melanoma cells and HCT-116 colon carcinoma cells in response to the novel microtubule poison, JG-03-14. Cancer Chemother Pharmacol. 2013;71(2):441–455. doi: 10.1007/s00280-012-2024-6. [DOI] [PubMed] [Google Scholar]
- 58.Salim H, Akbar NS, Zong D, et al. miRNA-214 modulates radiotherapy response of non-small cell lung cancer cells through regulation of p38MAPK, apoptosis and senescence. Br J Cancer. 2012;107(8):1361–1373. doi: 10.1038/bjc.2012.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vaughan S, Jat PS. Deciphering the role of nuclear factor-κB in cellular senescence. Aging (Albany NY) 2011;3(10):913–919. doi: 10.18632/aging.100390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bernard D, Quatannens B, Begue A, Vandenbunder B, Abbadie C. Antiproliferative and antiapoptotic effects of crel may occur within the same cells via the up-regulation of manganese superoxide dismutase. Cancer Res. 2001;61(6):2656–2664. [PubMed] [Google Scholar]
- 61.Bernard D, Slomianny C, Vandenbunder B, Abbadie C. cRel induces mitochondrial alterations in correlation with proliferation arrest. Free Radic Biol Med. 2001;31(8):943–953. doi: 10.1016/s0891-5849(01)00668-2. [DOI] [PubMed] [Google Scholar]
- 62.Hardy K, Mansfield L, Mackay A, et al. Transcriptional networks and cellular senescence in human mammary fibroblasts. Mol Biol Cell. 2005;16(2):943–953. doi: 10.1091/mbc.E04-05-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007;21(24):3244–3257. doi: 10.1101/gad.1588507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rovillain E, Mansfield L, Caetano C, et al. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene. 2011;30(20):2356–2366. doi: 10.1038/onc.2010.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu K, Feng T, Liu J, Zhong M, Zhang S. Silencing of the DEK gene induces apoptosis and senescence in CaSki cervical carcinoma cells via the up-regulation of NF-κB p65. Biosci Rep. 2012;32(3):323–332. doi: 10.1042/BSR20100141. [DOI] [PubMed] [Google Scholar]
- 66.Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011;21(6):354–359. doi: 10.1016/j.semcancer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192(4):547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30(8):1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tilstra JS, Robinson AR, Wang J, et al. NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest. 2012;122(7):2601–2612. doi: 10.1172/JCI45785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Osorio FG, Bárcena C, Soria-Valles C, et al. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012;26(20):2311–2324. doi: 10.1101/gad.197954.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang G, Li J, Purkayastha S, et al. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature. 2013;497(7448):211–216. doi: 10.1038/nature12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hinz M, Stilmann M, Arslan SÇ, Khanna KK, Dittmar G, Scheidereit C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol Cell. 2010;40(1):63–74. doi: 10.1016/j.molcel.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 73.Lee YK, Cha HJ, Hong M, Yoon Y, Lee H, An S. Role of NF-κB-p53 crosstalk in ultraviolet A-induced cell death and G1 arrest in human dermal fibroblasts. Arch Dermatol Res. 2012;304(1):73–79. doi: 10.1007/s00403-011-1176-2. [DOI] [PubMed] [Google Scholar]
- 74.Poltz R, Naumann M. Dynamics of p53 and NF-κB regulation in response to DNA damage and identification of target proteins suitable for therapeutic intervention. BMC Syst Biol. 2012;6:125. doi: 10.1186/1752-0509-6-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bolderson E, Richard DJ, Zhou BB, Khanna KK. Recent advances in cancer therapy targeting proteins involved in DNA double-strand break repair. Clin Cancer Res. 2009;15(20):6314–6320. doi: 10.1158/1078-0432.CCR-09-0096. [DOI] [PubMed] [Google Scholar]
- 76.Deorukhkar A, Krishnan S. Targeting inflammatory pathways for tumor radiosensitization. Biochem Pharmacol. 2010;80(12):1904–1914. doi: 10.1016/j.bcp.2010.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cusack JC, Liu R, Baldwin AS. Inducible chemoresistance to 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothe cin (CPT-11) in colorectal cancer cells and a xenograft model is overcome by inhibition of nuclear factor-kappaB activation. Cancer Res. 2000;60(9):2323–2330. [PubMed] [Google Scholar]
- 78.Liu Y, Hawkins OE, Su Y, et al. Targeting aurora kinases limits tumour growth through DNA damage-mediated senescence and blockade of NF-κB impairs this drug-induced senescence. EMBO Mol Med. 2013;5(1):149–166. doi: 10.1002/emmm.201201378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ohanna M, Giuliano S, Bonet C, et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS) Genes Dev. 2011;25(12):1245–1261. doi: 10.1101/gad.625811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chien Y, Scuoppo C, Wang X, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25(20):2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106(40):17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Acosta JC, O’Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 83.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moiseeva O, Deschênes-Simard X, St-Germain E, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell. 2013;12(3):489–498. doi: 10.1111/acel.12075. [DOI] [PubMed] [Google Scholar]
- 85.Ewald J, Desotelle J, Almassi N, Jarrard D. Drug-induced senescence bystander proliferation in prostate cancer cells in vitro and in vivo. Br J Cancer. 2008;98(7):1244–1249. doi: 10.1038/sj.bjc.6604288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]